
General Disorders of the Musculoskeletal System > 10 –
Musculoskeletal Neoplasms and Disorders That Resemble Neoplasms
present difficult diagnostic and treatment dilemmas. The anatomic
location, tissue of origin, clinical presentation, and behavior of
these lesions varies greatly. They may appear in any region of the
musculoskeletal system; consist of or involve almost any tissue
including bone, cartilage, fibrous tissue, bone marrow, lymphoid
tissue, nerve, and blood vessel; and arise in patients of any age. They
may destroy normal tissue and cause dramatic signs and symptoms
including intolerable pain, massive swelling, severe disability, and
pathologic fracture, or they may have little effect on normal tissues
and remain asymptomatic. They vary in natural history from lesions that
spontaneously regress to those that rapidly spread and lead to death
despite early diagnosis and aggressive treatment. Despite their
diversity, benign and malignant neoplasms and lesions that resemble
neoplasms can have similar clinical and radiographic presentations.
frequently, cause minimal discomfort or disability, and can be treated
without surgery. Musculoskeletal neoplasms present more complex
problems. Because of their rarity and variability in presentation and
behavior, few physicians have extensive experience with these problems.
A standardized approach cannot always be applied to each patient
with
symptoms, signs, or imaging studies that suggest the presence of a
musculoskeletal neoplasm. In many instances, optimal care requires a
multidisciplinary team of physicians, including orthopaedic surgeons,
radiologists, pathologists, medical oncologists, and radiation
oncologists. For these reasons, orthopaedic surgeons and other
specialists with additional training in the treatment of malignant and
aggressive benign neoplasms of the musculoskeletal system should
provide the definitive care for most patients with these rare, complex
problems.
easily distinguished from patients with disorders that resemble
neoplasms when they first seek medical attention. These lesions often
come to the patient’s or physician’s attention because of nondiagnostic
symptoms, signs, and abnormalities on imaging studies; as a result most
patients with musculoskeletal neoplasms first present to generalists
rather than orthopaedic surgeons with special experience in the
treatment of these problems. It is important for the initial treating
physician to determine whether a lesion is present and whether to refer
the patient to an orthopaedic surgeon. The orthopaedist then must
decide whether to recommend observation and symptomatic treatment,
further laboratory and imaging studies, biopsy, or definitive treatment.
identify musculoskeletal neoplasms and disorders resembling neoplasms
and make initial decisions concerning diagnosis and treatment, this
chapter first reviews the clinical presentation of these disorders.
Subsequent sections summarize the more common disorders that resemble
neoplasms, as well as benign, malignant, and metastatic neoplasms of
the musculoskeletal system (Table 10-1 and 10-2).*
|
TABLE 10-1. Common Musculoskeletal Disorders That Resemble Neoplasms and Benign Neoplasms
|
||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||||||||||||||||||||||||||||||||||||||||||
lesions that resemble neoplasms usually come to the attention of the
patient or physician because of pain, swelling, or loss of
musculoskeletal function. For benign, indolent lesions, a physical
examination or imaging study performed for other reasons may identify
the presence of a neoplasm.
ask specific questions to clarify the patient’s symptoms and
presentation. For bone lesions, pain is usually the primary symptom,
but the type and
pattern
of pain varies. Musculoskeletal lesions can cause symptoms that range
from excruciating, sharply localized pain to vague discomfort or a
sense of fullness, weakness, abnormal sensation, or stiffness. When
aggressive benign or malignant musculoskeletal neoplasms cause pain,
patients commonly describe the pain as a progressive, deep aching that
interferes with normal activities, awakens them at night, or disrupts
sleep. Activity-related pain alone is more consistent with inflammation
or injury. Often, rest does not relieve tumor-related pain, and
occasionally the pain may be referred to a more distal location. For
example, a tumor involving the hip may either cause radiating pain from
the thigh to the knee or pain limited to the ipsilateral knee. A tumor
of the cervical spine may cause radiating pain down the arm, and
neoplasms of the lumbar spine or pelvis may cause discomfort in the
buttock, thigh, or leg. The physician should not be fooled by referred
pain when performing a clinical or radiographic evaluation. For
example, it is not unusual for a patient with a proximal femoral lesion
to complain of knee pain, and subsequently have extensive studies of
the knee before radiographs of the proximal femur reveal the presence
of the lesion responsible for their symptoms.
|
TABLE 10-2. Common Malignant Musculoskeletal Neoplasms and Metastatic Neoplasms
|
||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||||||||||||||||||||||
generally painful, and this often leads to denial by the patient and a
delay in diagnosis. A thorough clinical history can often provide clues
to whether a lesion is benign or malignant, especially in the soft
tissues. It is important to ask how long the pain or a mass has been
noted. A long-standing mass is less likely to be malignant, but rare
malignant lesions, such as a synovial sarcoma, grow slowly over months
or years. An enlarging mass is more worrisome than a small, stable
mass, but some soft tissue sarcomas such as an epithelioid sarcoma or
clear cell sarcoma can present as small nodules along the tendon
sheaths of the hands and feet. A history of trauma might signify the
presence of myositis ossificans. It is important to ask about a
personal history of cancer, as carcinomas can metastasize to the bone
or soft tissues. A family history of masses might suggest an inherited
disorder such as neurofibromatosis.
swelling or a firm, well-defined mass. Others produce only a slight
increase in limb circumference. Infrequently, the reaction of normal
tissues to a tumor near a synovial joint causes an effusion, and
bleeding into a tumor produces a hematoma, but physicians should be
alert to the underlying problem. Tumors confined within bone cannot be
palpated, and deep soft tissue tumors may produce little or no increase
in limb circumference. This is a particular problem with deep soft
tissue tumors of the pelvis, thigh, hip, and shoulder. These lesions
can reach substantial size without producing an easily palpated mass,
particularly in obese or muscular patients. For these reasons, lack of
a palpable mass or measurable swelling does not eliminate the
possibility of a musculoskeletal neoplasm. Masses worrisome for
malignancy are large (>5 cm), firm, deep, fixed, and proximal in the
extremities. A rock-hard mass is typical of a benign,
but
aggressive, desmoid tumor. Small superficial nodules in the hand
include the possibility of an epithelioid sarcoma, and nodules along
the tendon sheath may be a clear cell sarcoma. It is important to
examine the entire extremity for additional masses, as malignant soft
tissue tumors can have satellite lesions or regional nodal metastasis.
present with a primary complaint of loss of musculoskeletal function.
The loss of function associated with tumors may result from pain,
neurologic deficit, pathologic fracture or restriction of joint motion.
Neurologic deficits may progress slowly as in a patient with a large
soft tissue sarcoma of the posterior thigh gradually compressing the
sciatic nerve. More commonly, the neurologic deficits may occur acutely
as in the sudden compression of the spinal cord or nerve roots
resulting from rapid enlargement of a primary or metastatic vertebral
tumor.
any patient that develops a fracture following minor trauma. If the
mechanism of injury does not correlate with the fracture, there should
be a high suspicion of an underlying pathologic process. Some patients
report pain at the site of the fracture before the injury. Plain
radiographs often show a bony irregularity or lytic lesion, but these
irregularities may be difficult to identify. The neoplasm may cause a
diffuse decrease in bone mass and density instead of a localized
abnormality. Thus, even when the radiographs do not show an obvious
lesion, patients who sustain a fracture following minimal trauma should
be carefully evaluated for the presence of a neoplasm. If a pathologic
fracture secondary to an underlying malignant bone tumor is not
recognized, the surgical treatment could compromise the limb or life of
the patient.
neoplasms cannot be identified with routine laboratory studies. There
are few serum markers available of diagnostic or prognostic
significance for patients with musculoskeletal tumors. Elevated white
blood cell counts and erythrocyte sedimentation rates (ESR) are usually
indicative of infection, but these values can be elevated in tumors
such as Ewing’s sarcoma. Often alkaline phosphatase is elevated in
metastatic bone disease or Paget’s disease. Patients with multiple
myeloma often have a marked anemia and an abnormal serum and urine
protein electrophoresis.
diagnose benign bone lesions. Many have a classically described
appearance, and more elaborate imaging is not helpful. Occasionally,
radiographs performed for a different reason reveal an unsuspected bone
lesion. The majority of these incidentally noted findings are benign.
For patients with a soft tissue mass, the plain radiographs may reveal
helpful soft tissue shadows or calcifications within the lesion, such
as are occasionally found in hemangiomas and synovial sarcomas. For
patients with bone lesions, it is important to evaluate radiographs in
two planes, and for those with metastatic disease, the entire bone
should be imaged to rule out the possibility of additional lesions.
skeletal lesions without performing multiple radiographs. In rapidly
growing lesions with minimal osteoblastic response such as multiple
myeloma and some metastatic bone tumors, the bone scan is not reliable
and a skeletal survey should be performed instead. For patients with
multiple osteochondromas or Langerhans cell histiocytosis, skeletal
surveys are also preferred over bone scans in order to better define
the characteristics of specific lesions. In recent years, FDG-PET scans
are gaining in importance for staging patients with metastatic disease
and following the effects of systemic treatment.
(CT) and magnetic resonance imaging (MRI) are frequently used to
evaluate musculoskeletal neoplasms. The CT scan is useful in defining
the bony anatomy, the integrity of the cortex surrounding a lesion, and
calcification within a lesion. The MRI scan is useful when evaluating
patients with malignant bone or soft tissue tumors. The multiple
magnetic sequences now available can accurately delineate the extent of
marrow involvement of bone tumors and the effect of soft tissue masses
on surrounding visceral or neurovascular structures.
patient with a suspected bone or soft tissue lesion is complete, the
physician should have a general idea of whether the lesion is benign or
malignant.
Unless
the radiographic appearance is classic for a benign lesion, a biopsy is
needed to make a definitive diagnosis. Only a radiologist or
orthopaedic surgeon with training and expertise in performing biopsies
should do this procedure. Options include a fine needle aspiration,
core biopsy, or an open incisional biopsy. The diagnosis of these rare
musculoskeletal lesions should be made at institutions with trained
musculoskeletal pathologists, especially in situations where needle
biopsies are utilized. All biopsies should be performed with oncologic
principles in mind to minimize contamination of surrounding tissues and
allow the best chance of limb-sparing surgery if the lesion is
malignant.
tumor simulators, occur more frequently than neoplasms. The symptoms,
signs, and imaging studies of metabolic bone diseases, musculoskeletal
injuries, infections, developmental disorders and diseases of unknown
etiology may closely resemble neoplasms. For example, a stress fracture
can mimic a bone-forming neoplasm, and myositis ossificans can resemble
a sarcoma. Osteomyelitis and Ewing’s sarcoma are often difficult to
distinguish based on the history, physical findings, and plain
radiographs.
osteomalacia, and various forms of hyperparathyroidism, decrease bone
density and strength and thereby increase the probability of fracture.
Some pathologic fractures associated with nonneoplastic causes of
osteopenia may be difficult to distinguish from pathologic fractures
due to diffuse lysis from multiple myeloma or metastatic carcinoma.
vertebral bodies in patients with osteopenia occur frequently and can
present difficult diagnostic problems. Common nonneoplastic conditions
that cause osteopenia and predispose older patients to vertebral
compression fractures include osteoporosis and osteomalacia; neoplasms,
especially multiple myeloma and metastatic carcinoma, may produce
similar clinical, physical, and radiographic findings. Many vertebral
compression fractures associated with nonneoplastic causes of
osteopenia require only symptomatic treatment, but untreated vertebral
fractures due to neoplastic disease can lead to progressive pain and
neurologic compromise.
may present as a pathologic fracture or after radiographs are taken for
evaluation of another problem and show decreased bone density. Clinical
evaluation and standard laboratory tests can help identify the probable
cause of diffuse osteopenia. Well-nourished elderly patients with
diffuse osteopenia but a normal blood count, erythrocyte sedimentation
rate (ESR), serum calcium, and serum phosphorus usually have
osteoporosis, but a bone biopsy may be necessary to rule out
osteomalacia and make a definitive diagnosis. Elevated serum calcium
and depressed serum phosphorus suggest the possibility of
hyperparathyroidism, and an elevated ESR with anemia suggests the
possibly of neoplastic disease such as multiple myeloma.
reaction of bone without apparent disruption of the cortex. It usually
causes pain and may cause radiographic changes that imitate
bone-forming tumors. They occur in children, adults, and the elderly.
In children, stress fractures may resemble osteosarcomas; in older
people stress fractures may resemble metastatic tumors. Biopsy of a
stress fracture reveals cellular tissue with extensive new bone
formation that may be difficult to distinguish from tissue found in a
malignant bone-forming neoplasm.
of bone or a localized reaction of bone to repetitive loading. Common
sites include the metatarsals, tibia, fibula, femur, pelvis, and lamina
of the lumbar vertebrae. Stress fractures often occur in young active
individuals with normal bone, but they also present in patients with
osteopenia from osteoporosis or osteomalacia and in those with
neoplasms. The pain associated with a stress fracture usually increases
with activity and decreases with rest. In the early phases of a stress
fracture or stress reaction of bone, plain radiographic studies may not
show obvious abnormalities, but a bone scan will demonstrate increased
uptake at the site of new bone formation. At later stages plain
radiographs may or may not reveal a fracture line, but they usually
show periosteal new bone formation that
extends
as a dense line into the medullary cavity of the bone. Stress fractures
usually heal with restriction of activity or immobilization.
expansile, destructive bone lesions called brown tumors. They mimic
neoplasms because of diffuse bone resorption similar to neoplastic and
non-neoplastic osteopenia. The focal, destructive lesions occur within
the diaphysis or metaphysis of long bones where they resorb the
medullary cancellous bone and expand the bone cortex. They may reach a
large size and lead to pathologic fractures. Because of their
radiographic appearance and histologic picture of multiple giant cells,
hemorrhage, and fibrous tissue, brown tumors can be mistaken for giant
cell tumors (GCTs) of bone. Unlike patients with GCTs, those with
hyperparathyroidism often have a history consistent with
hyperparathyroidism—an elevated serum calcium, depressed serum
phosphorous, elevated parathyroid hormone, and radiographic changes of
diffuse bone resorption. The bone disease associated with
hyperparathyroidism usually resolves following successful treatment of
the underlying disorder.
 |
|
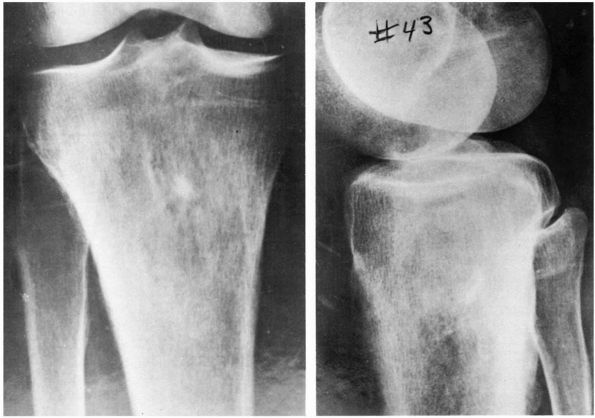
FIGURE 10-1. Radiographs showing osteomyelitis of the proximal tibia with irregular bone destruction and formation. (see color image)
|
bacteria and can cause pain, systemic symptoms, physical findings and
radiographic changes (bone destruction combined with new bone formation
and prominent periosteal reaction) that closely resemble tumors
including osteoid osteoma, eosinophilic granuloma, Ewing’s sarcoma,
lymphoma, and osteosarcoma (Figure 10-1, Figure 10-2). Histologic examination of infected bone reveals inflammatory cells, necrotic bone, and new bone formation (Figure 10-3).
Some patients with osteomyelitis have systemic symptoms including
fever, weight loss, and fatigue, while others are nearly asymptomatic.
metaphysis of long bones, but it can occur in any location. A
definitive diagnosis of osteomyelitis depends on a positive culture of
the infecting organism. Biopsies of presumably infected bone show
nonspecific inflammation with regions of new bone formation that could
result from infection by a variety of organisms, but these same
processes can be found near bone tumors. In addition to antibiotic
therapy, treatment of osteomyelitis usually requires surgical
debridement.
 |
|
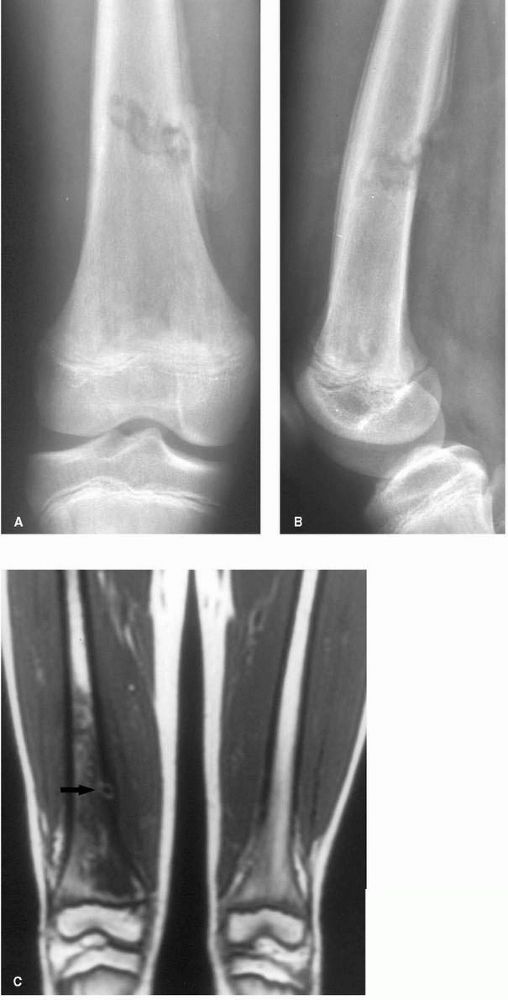
FIGURE 10-2. (A) and (B) Radiographs showing osteomyelitis of the distal femur. Periosteal new bone increases the diameter and density of the bone. (C) An MRI reveals the extensive marrow signal change (arrow). (see color image)
|
 |
|
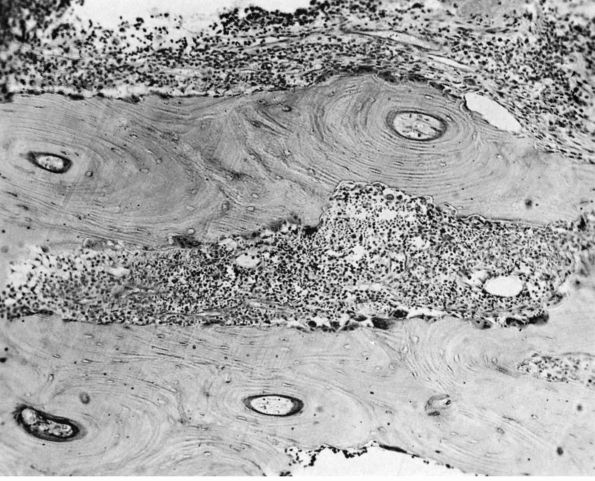
FIGURE 10-3.
Low power histologic section of infected bone. Inflammatory cells fill the marrow space, and osteoclasts resorb adjacent bone. The areas with no osteoclasts indicate necrotic bone. (see color image) |
draining sinus may develop from the underlying site. Some patients,
after years of sinus drainage, develop squamous cell carcinoma in the
sinus tract. These malignancies can be detected by a change in the
amount or odor of the drainage, a friable, vascular, enlarging mass,
and subsequent bone destruction. Treatment of these secondary
carcinomas requires wide resection or amputation.
disorder that causes rapid periosteal new bone formation, occurs most
frequently in children under 6 months of age. Common sites include the
diaphysis of long bones, ribs, mandible, and scapula. Fever,
leukocytosis, and an increased ESR often accompany the periosteal new
bone formation. Tenderness of the involved bones and palpable swelling
of the periosteum precede radiographic changes. Once new bone formation
begins, radiographs show multiple layers of periosteal new bone (Figure 10-4).
The clinical presentation and radiographic changes resemble
osteomyelitis, syphilis, Vitamin A toxicity, Vitamin C deficiency,
trauma, and Ewing’s sarcoma. The patient may have multiple remissions
and exacerbations, but the disorder eventually resolves and the bones
remodel to a normal configuration.
increased bone resorption and bone formation, produces characteristic
bone lesions that cause pain, deformity, and fracture. It either occurs
in one bone (monostotic Paget’s disease) or multiple bones (polyostotic
Paget’s disease). It varies in severity from an isolated, asymptomatic
bone lesion to crippling deformities of multiple bones. The clinical
and radiographic presentation of Paget’s disease may resemble neoplasms
such as metastatic carcinoma. Paget’s disease rarely occurs before age
20, and most patients are over age 50. It can affect any part of the
skeleton, although it frequently appears in the pelvis, femur, skull,
tibia, and spine. It is one of the few disorders that causes bone
enlargement.
phases: a purely lytic phase, a mixed lytic and blastic phase, and a
blastic phase (Figure 10-5). The early bone
lysis results from extensive osteoclastic bone resorption. The blastic
changes correspond to a decrease in bone resorption and increase in
formation of new dense bone with an irregular mosaic pattern. The
earliest phase of Paget’s disease appears radiographically as a wedge,
flame,
or
V-shaped region of bone lysis at the end of a long bone. Later the
cortical bone and medullary trabeculae become more dense, enlarged, and
irregular. Some regions of the bone become extremely dense,
obliterating the areas of lysis. Histologic examination of pagetoid
bone reveals an irregular or “mosaic” pattern of bone formation and
fibrovascular tissue filling the marrow spaces (Color Figure 10-1).
 |
|
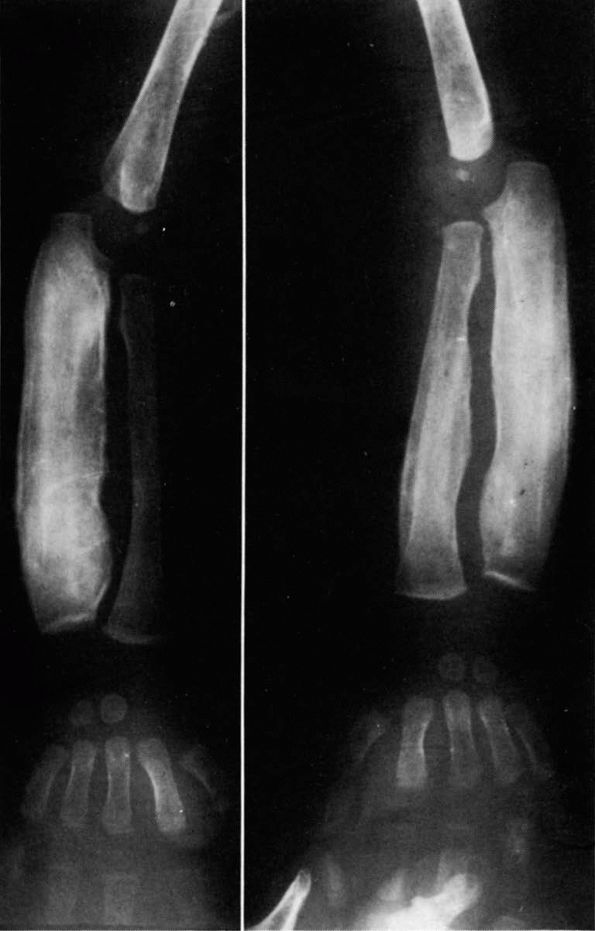
FIGURE 10-4.
Radiographs showing characteristic laminations of subperiosteal new bone about the radius and ulna in a patient with infantile cortical hyperostosis. (see color image) |
result it deforms and fractures more easily. Patients with Paget’s
disease may develop primary malignancies such as osteosarcoma in the
abnormal bone that spread rapidly and portend an extremely poor
prognosis. Increasing localized pain, a soft tissue mass, radiographic
evidence of bone destruction (Figure 10-6), or a pathologic fracture suggest the possibility of malignant degeneration.
medullary canal, and their actual incidence is unknown. They
occasionally cause pain. Avascular necrosis may occur following
traumatic or surgical interruption of the blood supply to a portion of
the bone or in association with corticosteroid use, radiation therapy
or sickle cell anemia. Idiopathic bone infarcts occur in the absence of
any underlying medical condition or known cause. Common sites for bone
infarcts include the femoral head, femoral condyle, talus, carpal
navicular, and metaphyses of the long bones, but the process can occur
in any region of the skeleton.
increased density within the medullary canal that resembles an
enchondroma or low grade chondrosarcoma. They have a classic,
serpiginous appearance on MRI scan. Necrosis of small regions of bone
marrow may be painful but does not significantly weaken the bone.
However, necrosis of load-bearing subchondral regions with subsequent
vascular invasion and resorption of necrotic bone can lead to
structural collapse, particularly in the femoral head, humeral head,
and talus. Degenerative joint disease develops following collapse of
the articular surface. Rarely, a malignant fibrous histiocytoma can
develop in or near bone infarcts, especially the idiopathic,
metaphyseal infarcts of long bones.
 |
|
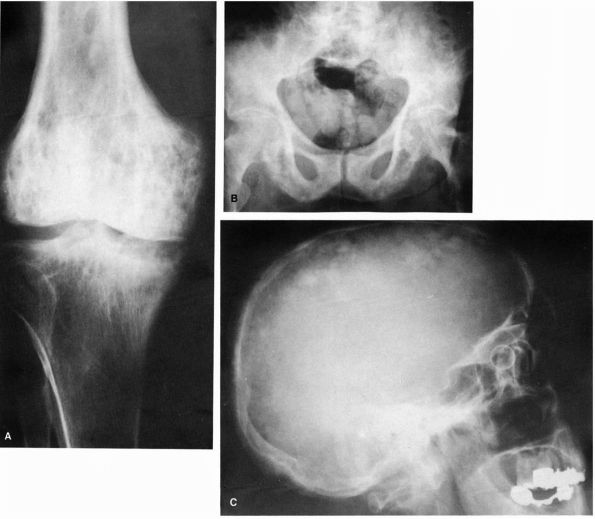
FIGURE 10-5. Radiographs showing the lytic and blastic changes of Paget’s disease in the (A) distal femur, (B) pelvis, and (C) skull. (see color image)
|
symptomatic treatment, but subchondral infarcts that cause articular
surface collapse often require surgical intervention. The indications
and long-term results of surgical procedures intended to revascularize
or decompress large areas of subchondral bone necrosis are
controversial.
lamellar bone within the medullary cavity. On radiographic studies they
are rounded lesions less than two centimeters in size located within
cancellous bone (Figure 10-7). Occasionally
they resemble osteoblastic metastasis. Because they are asymptomatic,
bone islands are usually identified as incidental findings on plain
radiographs or other imaging studies. They do not require treatment.
fluid-filled cavities within bone lined by a thin layer of fibrous
tissue. They occur most commonly in children less than 15 years of age.
Approximately 50% occur in the proximal humeral metaphysis. Other
common sites include the proximal femur
and
iliac wing. They may cause slight expansion of bone and thinning of the
cortex. As a result patients often present with a pathologic fracture
through the cyst. Some cysts may be discovered incidentally when
radiographs are taken for other reasons.
 |
|
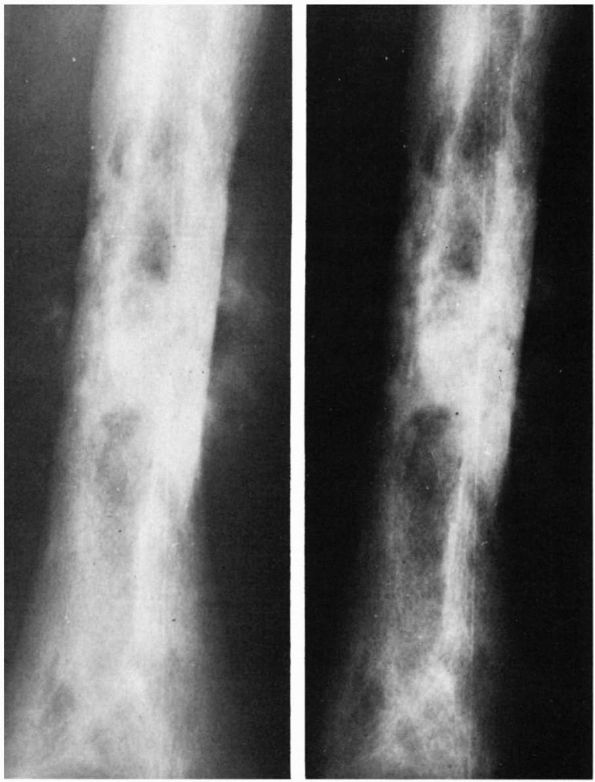
FIGURE 10-6.
Radiographs showing a femoral osteosarcoma arising in pagetoid bone. The tumor has formed new bone and extended through the cortex. (see color image) |
As new bone in skeletally immature patients grows away from the cyst,
the lesion may eventually reside in the diaphysis. Unicameral cysts
that present as central, lytic lesions of the proximal humerus or
proximal femur in young children have a classic appearance that does
not require further imaging or biopsy. However, cysts in other
locations or those that lack characteristic radiographic features may
mimic other musculoskeletal lesions including aneurysmal bone cysts,
fibrous dysplasia, and osteosarcoma. More aggressive lesions can be
eliminated based on the histologic appearance of a large cystic lesion
with innocuous cells in the lining tissue. Simple cysts resolve as
patients reach skeletal maturity, but they can cause multiple
pathologic fractures during growth, especially when they are noted at a
very young age. The fractures cause skeletal deformity, especially when
they occur in the proximal femur. Most fractures through simple cysts
heal rapidly with closed treatment (Figure 10-9).
The current recommended treatment of simple cysts includes observation
with restriction of activity and steroid injections. Intralesional
curettage and bone grafting is generally reserved for large cysts at
high risk for fracture in the proximal femur.
 |
|
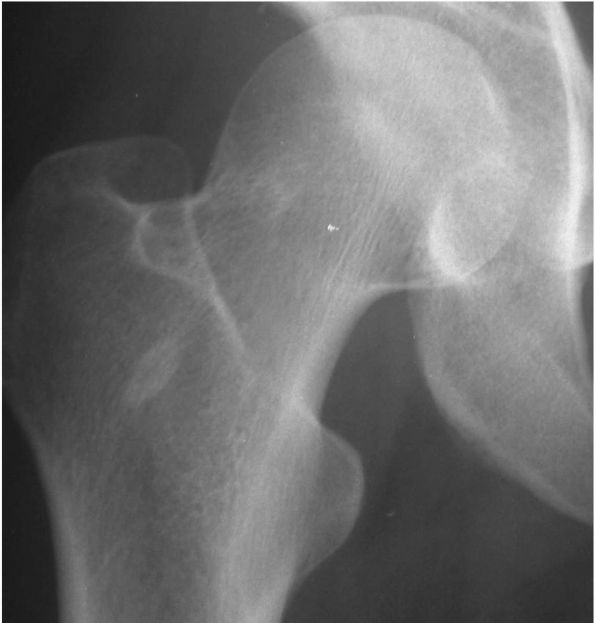
FIGURE 10-7. Radiograph showing a small bone island in the proximal femur. It is sclerotic and incites no surrounding reaction. (see color image)
|
cavities lined by fibrous septae that include giant cells and areas of
osteoid but no true endothelial cells (Color Figure 10-2).
Approximately 85% of patients with ABCs present before age 20. The most
common symptoms are pain, swelling, and tenderness on palpation of the
involved bone. ABCs commonly involve the metaphysis of long bones,
posterior elements of the vertebrae, pelvis, or scapula, but they can
occur throughout the skeleton (Figure 10-10).
ABCs can grow rapidly and frequently cause pathologic fractures. When
located in the spine, they can cause neurologic compromise.
expansion of the involved bone and occasional periosteal new bone
formation (Figure 10-11). An MRI scan often
reveals fluid-fluid levels. Aggressive benign neoplasms such as giant
cell tumors and primary malignant neoplasms such as telangiectatic
osteosarcoma can have a similar radiographic appearance.
 |
|
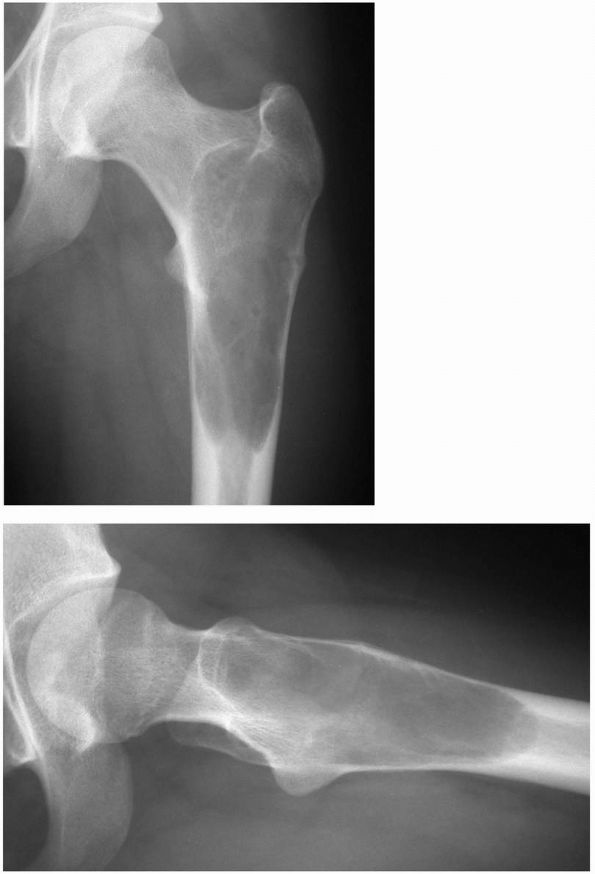
FIGURE 10-8.
Radiographs showing a unicameral (simple) bone cyst of the proximal femur at risk for pathologic fracture. Notice the symmetrical metadiaphyseal location of the cyst. (see color image) |
 |
|
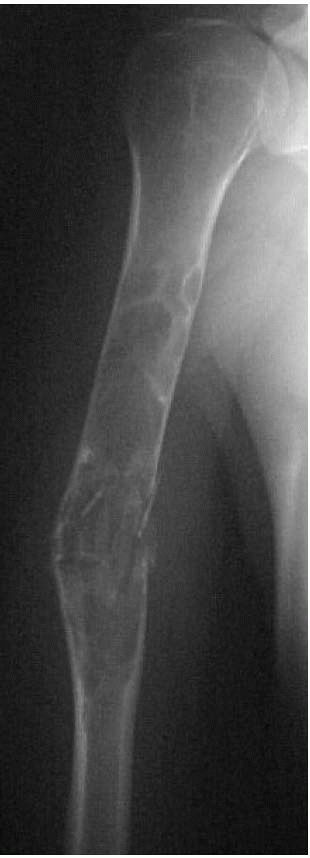
FIGURE 10-9. Radiograph showing an extensive simple bone cyst of the humerus with a pathologic fracture and mild displacement. (see color image)
|
lesions including fibrous dysplasia, giant cell tumor, simple bone
cyst, eosinophilic granuloma, nonossifying fibroma, chondroblastoma,
osteoblastoma, and occasionally osteosarcoma. They may stop expanding
and begin to ossify after reaching a certain size or they may regress
spontaneously. Standard treatment is a confirmatory biopsy followed by
intralesional curettage and bone grafting.
and nonossifying fibromas are lytic bone lesions that consist of
fibroblasts arranged in whirled bundles with scattered giant cells and
regions of histiocytes. They are commonly discovered incidentally on
plain radiographs. They rarely appear in children younger than 2 years
of age or in adults over 20 years of age. They develop in the
metaphysis and, as the bone grows, they gradually seem to move toward
the diaphysis. When patients near skeletal maturity, the lesions ossify
or completely disappear. Most authors refer to large fibrous cortical
defects that extend into the intramedullary region of the bone as
nonossifying fibromas.
Except for rare pathologic fractures, they do not cause pain. The
diagnosis can usually be made based on their distinctive radiographic
appearance. These lesions heal spontaneously, so they do not require
treatment unless a pathologic fracture has or is likely to occur. In
these situations, intralesional curettage and bone grafting allows
definitive healing of the lesion.
and is among the most common lesions of bone. It can weaken bone and
lead to a pathologic fracture, and it may have a radiographic
appearance that resembles a more aggressive tumor. In some patients it
involves a single bone (monostotic fibrous dysplasia), but in others it
involves multiple bones (polyostotic fibrous dysplasia). The severity
of the disorder ranges from isolated small lesions that remain
asymptomatic to lesions that cause repeated pathologic fractures and
deformity of multiple bones. Although most authors consider fibrous
dysplasia a developmental disorder rather than a neoplasm, it can
destroy or expand normal bone and frequently recurs following curettage
and bone grafting.
 |
|
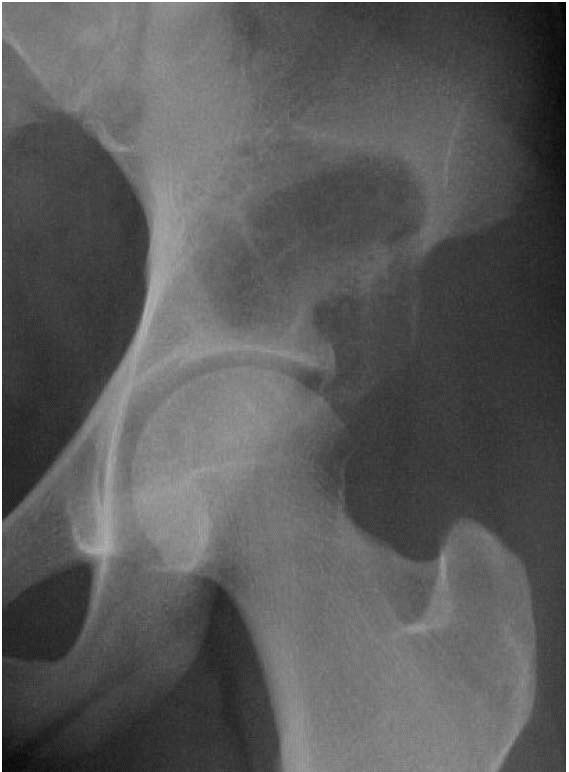
FIGURE 10-10. Radiograph of the hip showing an expansile, lytic lesion in the acetabulum consistent with an aneurysmal bone cyst. (see color image)
|
discovered because of pain, pathologic fracture, or skeletal deformity.
Plain radiographs show circumscribed areas of decreased bone density
having a ground glass or “shower door glass” appearance. Because
fibrous dysplasia can thin and expand the cortex concentrically or
eccentrically, the lesions may resemble unicameral or multilocular
cysts (Figure 10-14).
When fibrous dysplasia involves the proximal femur, the resulting
weakening of the bone may lead to progressive microfractures and is
referred to as a “Shepherd’s crook deformity” (Figure 10-16).
progressively enlarge. The lesions often become less active or inactive
at skeletal maturity, but they can grow in adults. In some patients,
polyostotic fibrous dysplasia occurs in association with precocious
puberty and darkly pigmented skin lesions in a disorder called
McCune-Albright syndrome. Several case reports have described the
appearance of malignant tumors in regions of fibrous dysplasia,
although malignant transformation is extremely rare. Most patients with
small lesions of fibrous dysplasia do not require treatment. Patients
with pathologic fractures or progressive skeletal deformity require
curettage, bone grafting, and skeletal stabilization. Recently,
bisphosphonates have been used with documented pain relief in a subset
of patients.
 |
|
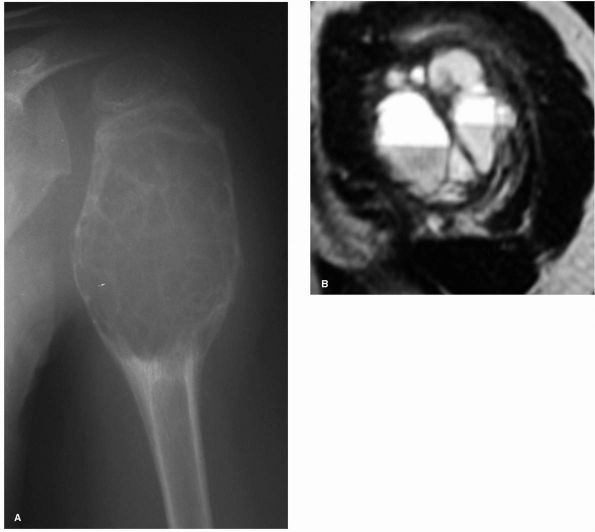
FIGURE 10-11. A large aneurysmal bone cyst in the proximal humerus is shown by (A) radiograph and (B) MRI scan. Note the expansile nature of the lesion as well as the fluid-fluid levels.
|
masses that resemble neoplasms. The hemorrhage that results from these
injuries causes swelling, and the muscle that retracts from the site of
a tear may form a discrete mass. Initially the muscle damage,
inflammation, and hemorrhage causes pain and weakness. As the tissue
heals the pain resolves, but a mass can remain for several months.
ruptures blood vessels and damages muscle cells. Common sites of muscle
contusion include the deltoid, brachialis, biceps, and quadriceps
muscle bellies. Muscle tears usually result from contraction of the
muscle against resistance. They often occur at the musculotendinous
junction, and common sites include the hamstrings, quadriceps, and
biceps muscles.
become organized and remain as firm, intramuscular masses. These
organized hematomas may mimic intramuscular neoplasms on physical
examination and MRI scans. Most patients identify a specific traumatic
episode followed by pain, swelling, weakness, and ecchymosis, but not
all patients have a definite history of trauma. In individuals with
muscle tears, the physical examination often reveals a muscle defect or
retraction. Muscle tears and intramuscular hematomas can be treated by
gentle stretching and restriction of heavy activity. In the absence of
trauma, deep intramuscular hematomas are unlikely and may be difficult
to distinguish from neoplasms without performing a biopsy. Further
workup is necessary if the MRI appearance is indeterminate or the mass
is enlarging.
 |
|
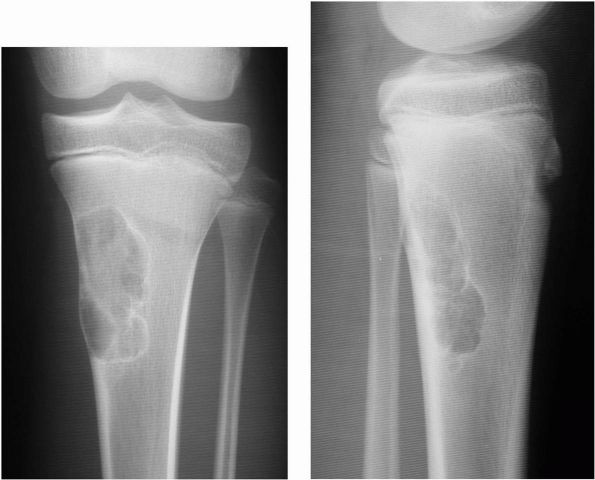
FIGURE 10-12. Radiographs showing a nonossifying fibroma in the proximal tibia. Note the sclerotic rim and eccentric location of the lesion.
|
 |
|
FIGURE 10-13.
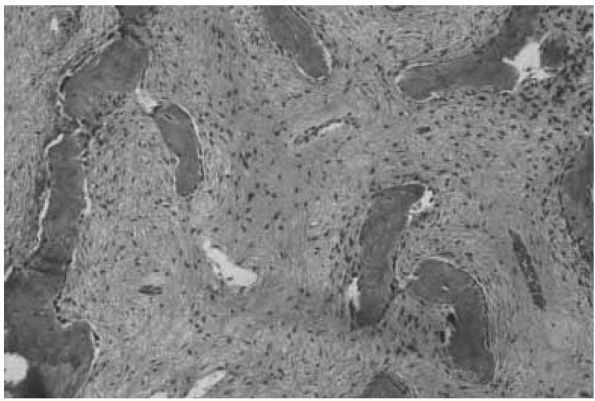
A histologic section of fibrous dysplasia reveals woven bone trabeculae surrounded by a fibrous tissue stroma. Unlike other bone-forming lesions, fibrous dysplasia forms metaplastic bone directly from the stroma. |
 |
|
FIGURE 10-14.
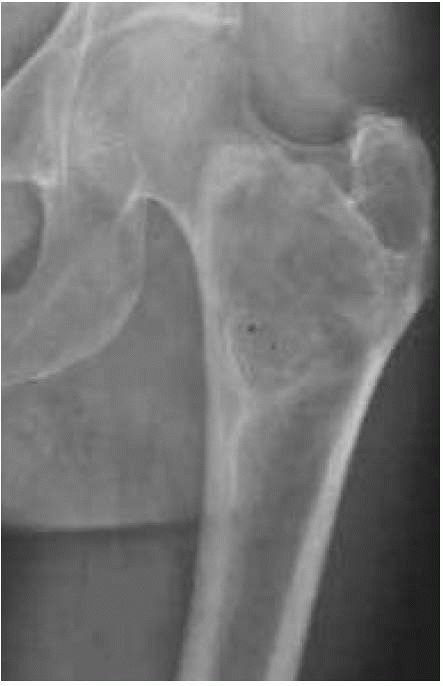
A radiograph reveals fibrous dysplasia of the proximal femur. Note the mottled density of the neoplastic bone. It has a well-circumscribed benign appearance. |
 |
|
FIGURE 10-15.
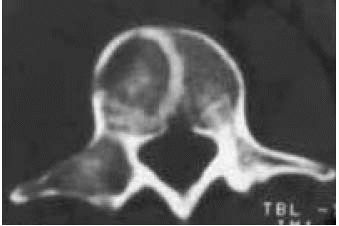
A CT scan of the spine reveals an area of fibrous dysplasia in the vertebral body. The lesion is well-circumscribed with no soft tissue mass. |
causes myositis ossificans, formation of benign bone, cartilage, and
fibrous tissue within contused muscle. This poorly understood condition
begins following muscle damage with hemorrhage and inflammation. During
the initial stages it causes pain or tenderness along with diffuse
swelling and may be confused with a malignant soft tissue tumor,
parosteal osteosarcoma, or benign lesions such as nodular fasciitis. As
the inflammation subsides, pain and tenderness decrease, but a firm
mass containing bone usually remains.
adolescents and young adults. Common sites include the quadriceps,
adductors, deltoid, and brachialis muscles. Mineralization begins
within the lesion approximately 4 to 6 weeks after injury and proceeds
from the periphery toward the center.
area of soft tissue swelling that becomes progressively mineralized to
contain bone (Figure 10-17). Microscopic examination reveals a distinct zonal pattern reflecting the gradations of cellular maturation (Figure 10-18).
The inner region of the lesion contains immature, rapidly proliferating
fibroblasts along with inflammatory cells and occasional giant cells. A
zone of poorly defined osteoid trabeculae with fibroblasts and
osteoblasts surrounds this region and, in the peripheral areas, the
osteoid mineralizes into mature lamellar bone. Current initial
treatment of myositis ossificans includes restriction of activity and
gentle stretching to prevent contractures. The mass is only removed in
symptomatic cases after the appearance is completely mature.
 |
|
FIGURE 10-16.
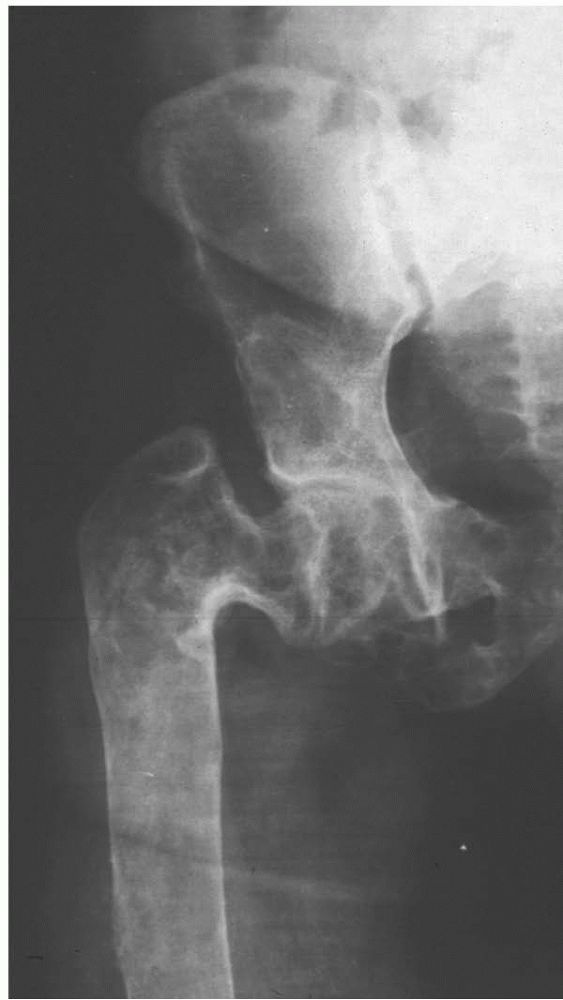
Extensive fibrous dysplasia of the proximal femur produces a characteristic Shepherd’s crook deformity from multiple microfractures over time. |
and inflammatory cells that produce a tender soft tissue mass most
often in the subcutaneous tissues and infrequently within muscle. It
develops most commonly in adolescents and young adults and occasionally
enlarges rapidly. The cause of this rare disorder remains unknown, but
it appears to be an inflammatory process instead of a neoplasm. Simple
excision of the mass is usually sufficient to provide a definitive
diagnosis and local control.
 |
|
FIGURE 10-17. A lateral radiograph reveals myositis ossificans in the posterior thigh musculature. It gradually ossifies as it matures.
|
death, hemorrhage, and inflammation. As the inflammation subsides it
can leave a firm plaquelike mass consisting of scar tissue and fat that
persists long after the injury. A history of injury and a subcutaneous
location of the mass usually suggests the diagnosis of traumatic fat
necrosis.
causes proliferation of nerve tissue that can form firm, usually
mobile, nodules. These traumatic neuromas may grow to moderate size and
cause intense discomfort. A history of trauma or previous surgery
combined with paresthesias or a Tinel’s sign in the region of the mass
helps establish the clinical diagnosis of posttraumatic neuroma.
Neuromas that develop in the surgical site after resection of a soft
tissue neoplasm may be difficult to distinguish from recurrent tumor
without a biopsy.
uncommon lesions develop from open wounds or by direct extension of
infection from
adjacent
structures including bones and joints. They can result from
hematogenous spread of organisms, especially in immunocompromised
patients or those with diabetes mellitus. Most bacterial soft tissue
abscesses cause exquisite tenderness, erythema, and fever, but most
tuberculous abscesses (cold abscesses) cause minimal tenderness and may
not produce systemic symptoms. Indolent abscesses may be difficult to
distinguish from a neoplasm without a biopsy. The treatment of a
musculoskeletal soft tissue abscess includes tissue cultures, surgical
drainage, and antibiotics.
 |
|
FIGURE 10-18. Light micrographs of myositis ossificans. (A)
The cellular inner zone illustrates numerous cells with occasional atypical mitotic figures and variations in size and shape of the cells. The histologic appearance is sarcomatous. (B) The middle zone shows osteoid formation with a fibrovascular background. The cellular pattern is uniform. (C) The outer zone illustrates mature, well-oriented peripheral bone. The fibrous stroma appears more mature than at the center of the lesion. (Courtesy of Dr. William Bacon) |
collections of translucent fluid or gelatinous myxoid tissue surrounded
by fibrous tissue. They can occur in patients of any age and are
located in a superficial location adjacent to synovial joints or tendon
sheaths. They are commonly found near the wrist, hand, and knee.
Occasionally those that develop near the knee grow to a large size and
dissect through the surrounding soft tissues. Enlargement of the limb
or swelling caused by these unusual ganglia may suggest the presence of
a neoplasm. Although aspiration can remove the fluid from a ganglion
cyst, surgical resection is currently the most predictable method of
eradicating symptomatic lesions.
consisting of an abundant, gelatinous, myxoid matrix containing few
cells and occasional cystic areas that resemble ganglia. Clinically,
they are painless, fluctuant, mobile intramuscular masses that occur in
patients between the ages of 40 and 70 years. When multiple myxomas are
associated with monostotic or polyostotic fibrous dysplasia, it is
referred to as Mazabraud’s syndrome.
years. Because of their deep, intramuscular location in the thigh,
shoulder, buttock, or arm, they cannot be easily distinguished from
soft tissue sarcomas by clinical evaluation. They are well defined and
have low signal relative to muscle on T1-weighted MR images. Treatment
of symptomatic or enlarging lesions is by simple excision.
commonly than malignant neoplasms. Benign lesions result from cell
proliferation and matrix synthesis that produces new tissue, but their
behavior varies considerably. Many enlarge to a certain size during
skeletal growth and then remain unchanged indefinitely; thus, they
might be considered developmental disorders rather than neoplasms.
Lesions that follow this pattern include osteochondromas, enchondromas,
lymphangiomas, and hemangiomas. Benign lesions including GCTs of tendon
sheath, elastofibromas, and pigmented villonodular synovitis may
represent inflammatory or reactive disorders, but lesions such as GCTs
of bone and osteoblastomas are more aggressive neoplasms.
proliferative lesions require varied treatments. Most osteochondromas
and enchondromas do not require surgical treatment, but GCTs and
osteoblastomas require intralesional curettage or resection. The more
aggressive benign tumors invade and destroy normal tissue and may be
difficult to distinguish from low-grade malignant neoplasms. Some of
these benign lesions such as GCT and chondroblastoma can metastasize to
the lungs despite their histologic appearance.
bone. They form on endosteal and periosteal bone surfaces and occur
most commonly in the mandible, flat bones of the skull, and tibia in
adolescents and young adults. On plain radiographs they appear as dense
nodules of bone. Patients may notice a firm bony mass or the lesion may
be detected as an incidental finding on plain radiographic studies.
Osteomas do not require treatment.
consisting of a central nidus less than 2 cm in diameter containing
capillaries, osteoclasts, and osteoblasts forming large volumes of
disorganized osteoid (Color Figure 10-3). A
larger region of reactive new bone formation matures to become dense,
lamellar bone around the central region. A thin rim of granulation
tissue may separate the central osteoid-forming region from the dense,
reactive bone.
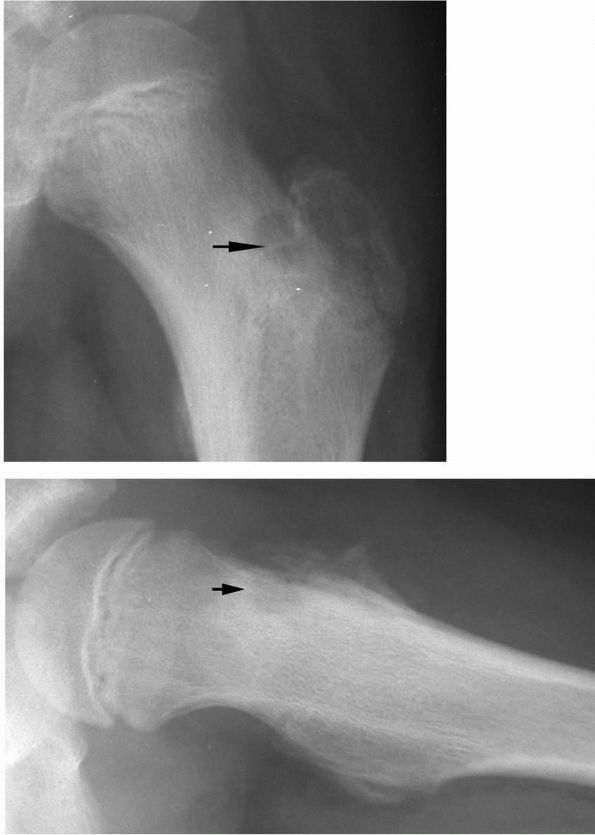
adults less than 30 years old. They cause considerable pain,
classically worse at night. Typically aspirin provides better pain
relief than other medications. Osteoid osteomas occur most frequently
in the diaphysis and metaphysis of long bones, but they can develop in
any bone. When they occur near synovial joints, effusions, muscle
spasms, and contractures may be apparent. The pain will eventually
resolve if aspirin or anti-inflammatory medications are taken for a
prolonged period of time, but most lesions are now treated with
radiofrequency ablation (RFA). In some vertebral lesions where RFA is
unsafe near the spinal cord, resection or surgical burring of the area
may be indicated.
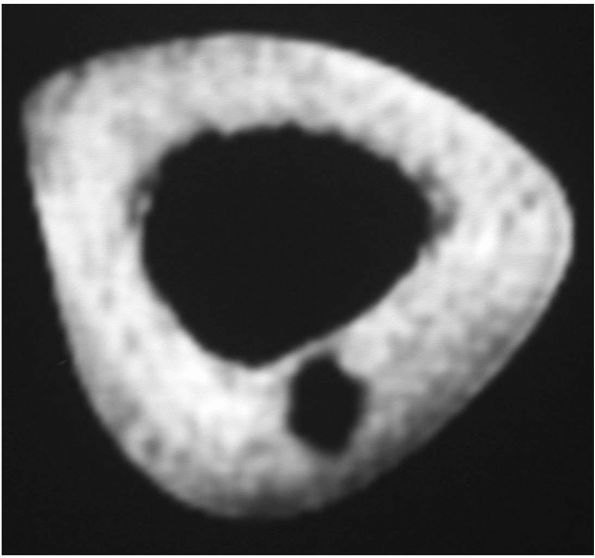
Occasionally, they appear only as small, lucent areas surrounded by
dense bone as the increased density of the reactive bone hides the
central lesion on plain radiographs. In these instances, CT scans are
the procedure of choice to identify the nidus (Figure 10-20).
They are benign, bone-forming neoplasms larger than osteoid osteomas
that can expand and destroy bone. Most osteoblastomas are painful and
occur in patients less than 30 years of age. The pain is usually less
severe than that associated with an osteoid osteoma. They occur in the
metaphysis or diaphysis of long bones or the posterior elements of the
vertebrae (Figure 10-22, Figure 10-23).
thin rim of reactive bone. The central lucent area often contains areas
of mineralization. The differential diagnosis of an osteoblastoma
includes osteosarcoma, giant cell tumor, and osteoid osteoma.
enlargement to rapid aggressive growth that resembles the behavior of
an osteosarcoma. Occasionally they cause pathologic fractures. Surgical
resection or meticulous intralesional curettage provides local control.
 |
|
FIGURE 10-19. Radiographs of the proximal femur reveal a small lucency in the lateral femoral neck (arrow) consistent with an osteoid osteoma. There is mild surrounding sclerosis.
|
 |
|
FIGURE 10-20. The nidus of an osteoid osteoma is best visualized on a thin-cut CT scan.
|
one of the most common benign bone tumors. It consists of a bony base
or stalk with a cartilage cap that projects from the normal bone away
from a nearby joint (Figure 10-24). A fibrous
tissue capsule or bursa typically covers the cartilage surface.
Osteochondromas may develop from proliferation of cartilage-forming
periosteal cells or from a defect in the fibrous tissue surrounding a
physis and therefore likely represent a developmental disorder instead
of a neoplasm. Common locations include the metaphysis of the proximal
tibia, distal femur, distal tibia, distal fibula, proximal femur, and
proximal humerus. They also can develop from flat bones of the pelvis
and scapula.
 |
|
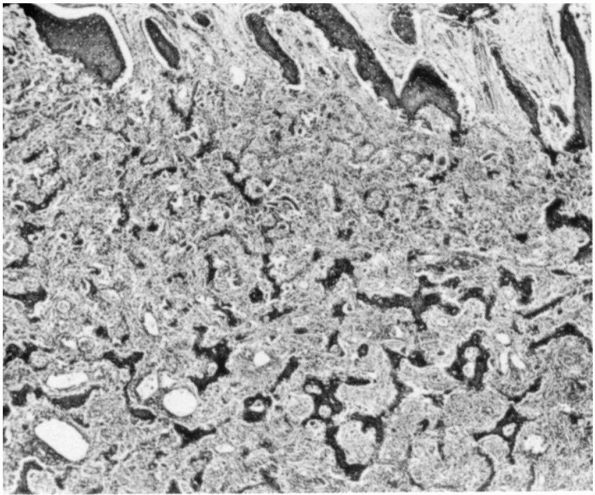
FIGURE 10-21. Low power histologic section of an osteoblastoma shows new bone trabeculae lined by neoplastic cells.
|
 |
|
FIGURE 10-22.
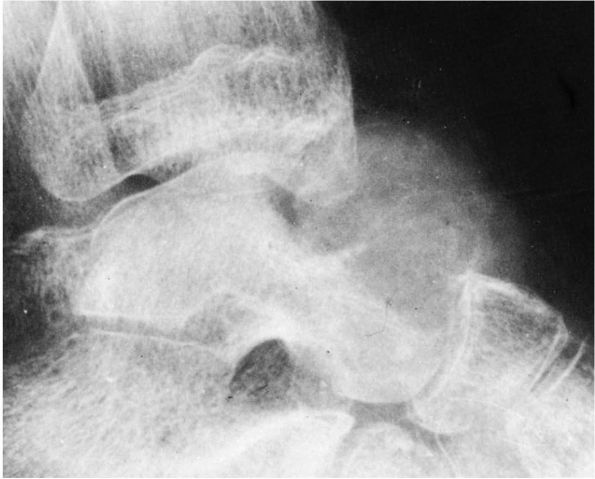
A radiograph of an osteoblastoma reveals an osteolytic, expanding tumor that has eroded the cortex of the talus and expanded into the surrounding soft tissues. It is surrounded by a thin shell of bone. Scattered, minute opacities throughout the tumor represent new bone. (Pochaczevsky R, Ven YM, Sherman RS. The roentgen appearance of benign osteoblastoma. Radiology 1960;75:429) |
firm, fixed, asymptomatic bony masses. Most affected patients have a
solitary osteochondroma, but some individuals have a hereditary
disorder that causes multiple osteochondromas. This disorder, multiple
hereditary exostoses (MHE), is transmitted as an autosomal dominant
trait with a high degree of penetrance and can cause marked skeletal
deformity and disability. Most patients recognize the presence of
multiple lesions before 20 years of age. Severely affected people
develop considerable skeletal deformity.
diagnosis of an osteochondroma. The bony base of the lesion extends
directly from the medullary canal of normal bone (Figure 10-24).
During skeletal growth the lesions enlarge with the surrounding bone,
and they stabilize with skeletal maturity. As the bone component of an
osteochondroma forms by enchondral ossification, growing
osteochondromas typically have a
large
cartilaginous component. As the lesions mature, the cartilage component
decreases until the osteochondroma consists primarily of bone.
 |
|
FIGURE 10-23.
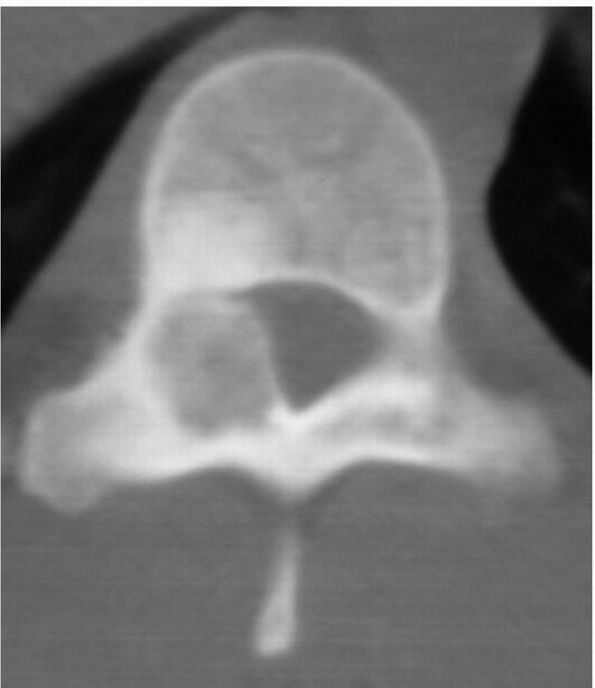
A CT scan of the spine reveals an osteoblastoma appearing in the posterior elements, a common location for this aggressive lesion. |
bony stalk or develops an overlying soft tissue bursa. In these
instances it may cause pain. Enlargement of an osteochondroma may cause
adjacent nerve compression or skeletal deformity. Rarely, a
chondrosarcoma develops from an osteochondroma. This is more common in
patients with multiple lesions. Malignant transformation occurs more
frequently in osteochondromas of flat bones, particularly the pelvis
and scapula. This phenomenon should be suspected when an osteochondroma
causes pain or enlarges in an adult. The main treatment for an
osteochondroma is observation until the patient reaches skeletal
maturity. Patients with MHE are followed yearly with skeletal surveys.
Surgical resection is only indicated when there is neurologic
compromise, abnormal growth, skeletal deformity, or decreased motion of
the adjacent joint.
located in the medullary cavity of otherwise normal bones. It
frequently occurs in the bones of the hands and feet but may appear in
any bone including the femur, tibia, and humerus (Figure 10-25).
It is generally considered an asymptomatic, indolent lesion. It is most
frequently noted when obtaining radiographs to evaluate impingement of
the shoulder or degenerative arthritis of the knee.
lucent region that may be mineralized. Enchondromas resemble bone
infarcts. Further imaging studies are not necessary, but enchondromas
normally show increased activity on a bone
scan.
During skeletal growth the lesions may slowly enlarge. Following
completion of normal growth, they cease to enlarge and the cartilage
component calcifies to give a stippled radiographic appearance (Figure 10-26).
In extremely rare cases, a chondrosarcoma can develop from an
enchondroma. Because enchondromas are usually asymptomatic and do not
enlarge after skeletal maturity, a lesion that causes pain or enlarges
in an adult strongly suggests the possibility of malignant
transformation. It is important to eliminate other causes for pain
before attributing symptoms to an enchondroma.
 |
|
FIGURE 10-24.
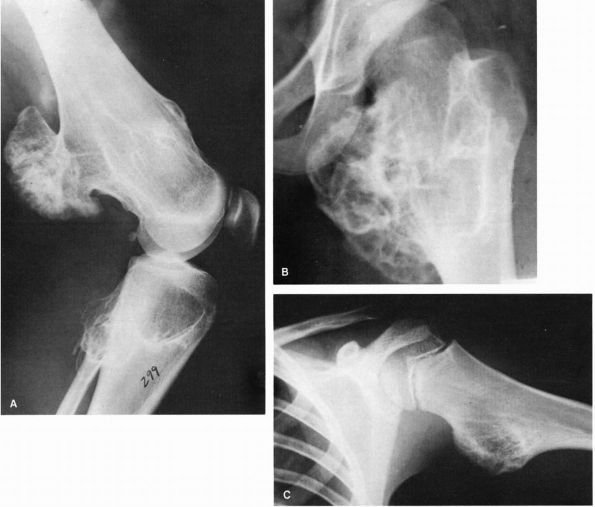
Plain radiographs showing typical osteochondromas of long bone metaphyses. Notice that the bony bases of the lesions extend directly from normal bone and that the medullary cavity of the normal bone extends into the lesion. Plain radiographs do not show the cartilage portion of an osteochondroma, so the lesions may be larger than visualized on the plain radiographic images. Osteochondromas are shown of the (A) distal femoral and proximal tibial metaphyses in a patient with multiple hereditary exostoses, (B) the proximal femoral metaphysis, and (C) the proximal humeral metaphysis. |
enchondromas, which causes severe deformity and stunting of growth.
These patients have an increased probability of malignant
transformation of an enchondroma. Maffucci’s syndrome is a condition
where multiple enchondromas occur in association with multiple
hemangiomas. These patients have an increased probability of developing
a chondrosarcoma or other visceral malignancies. Enchondromas generally
do not require surgical treatment.
consisting of hyaline cartilage. It forms between the cortical bone and
overlying periosteum,
often
creating an indentation in the bone surface and a smooth bulge of
periosteum-covered cartilage that projects into the soft tissues. Most
patients are young or middle-aged adults. Presumably periosteal
chondromas develop from proliferation of cartilage-forming periosteal
cells. They occur most frequently in the proximal humeral metaphysis,
phalanges, metacarpals, and metatarsals.
 |
|
FIGURE 10-25.
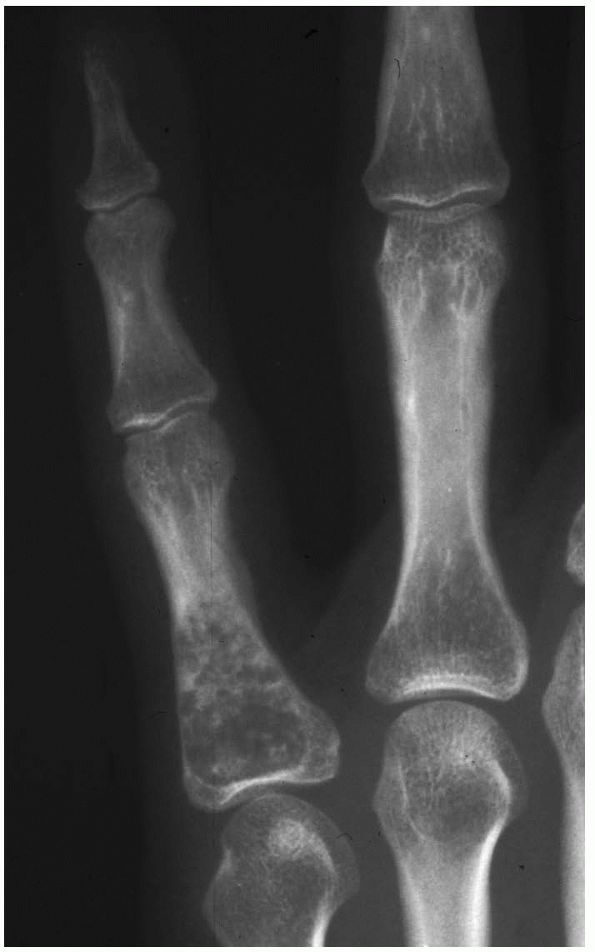
A radiograph reveals an enchondroma of the proximal phalanx with a stippled appearance. These lesions are the most common bone tumors found in the hand. They can be expansile and often fracture. |
an incidental radiographic finding. Radiographs show a scalloped
depression in the bone cortex and may show the faint image of a soft
tissue mass containing speckled regions of calcification. Periosteal
chondromas can slowly enlarge, but they have not been shown to be
aggressive. For symptomatic or enlarging lesions, surgical resection
provides definitive local control.
 |
|
FIGURE 10-26. A radiograph of the proximal humerus reveals a calcified enchondroma. There is no expansion of the bone or soft tissue mass.
|
of regions or “islands” of densely packed polyhedral cells called
chondroblasts admixed with fibrous tissue and chondrocytes forming a
cartilage matrix (Color Figure 10-4). In some
areas the cartilage matrix mineralizes creating a distinctive “chicken
wire” pattern, while other regions contain large numbers of giant
cells. Chondroblastomas involve the epiphysis of long bones in patients
with open physes. They occur most commonly in the proximal humerus,
distal femur, proximal tibia, and proximal femur. Most patients present
with pain and local tenderness. Occasionally they have swelling,
limitation of joint motion, and an effusion.
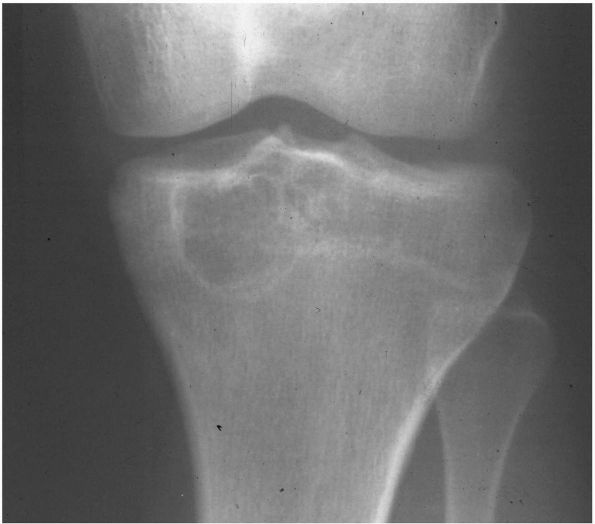
lucency with punctate calcifications. A sclerotic rim surrounds the
lucent area (Figure 10-27). The lesions rarely involve more than half of the epiphysis and only occasionally extend into the
metaphysis. Intralesional curettage and bone grafting is indicated for
a chondroblastoma, but occasionally these lesions recur in the bone or
surrounding soft tissues. Rarely chondroblastomas metastasize to the
lungs.
 |
|
FIGURE 10-27.
A radiograph of the proximal tibia reveals a lytic lesion in the epiphysis with a sclerotic rim consistent with a chondroblastoma. |
cartilage tumor consisting of fibrous, cartilaginous and myxoid tissues
in variable proportions (Figure 10-28). Most
present in older adolescents and young adults. It occurs most commonly
in the metaphysis of the tibia as well as the pelvis and scapula (Figure 10-29).
CMFs may be discovered as an incidental finding on a plain radiograph
or because of mild to moderate pain. In some locations they form a
palpable mass, but they rarely cause pathologic fractures. Plain
radiographs demonstrate an eccentrically located lytic lesion with
smooth sclerotic margins. Treatment by intralesional curettage or
resection provides local control.
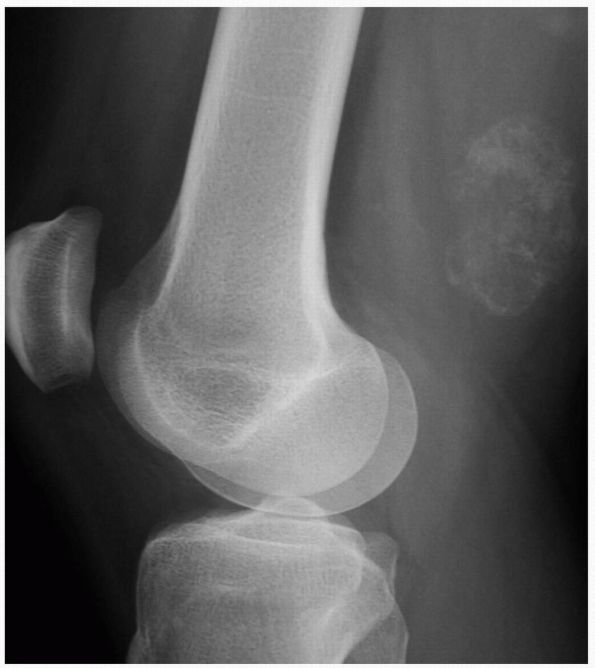
consisting of osteoclast-like giant cells, fibroblast-like stromal
cells, and blood vessels (Figure 10-30).
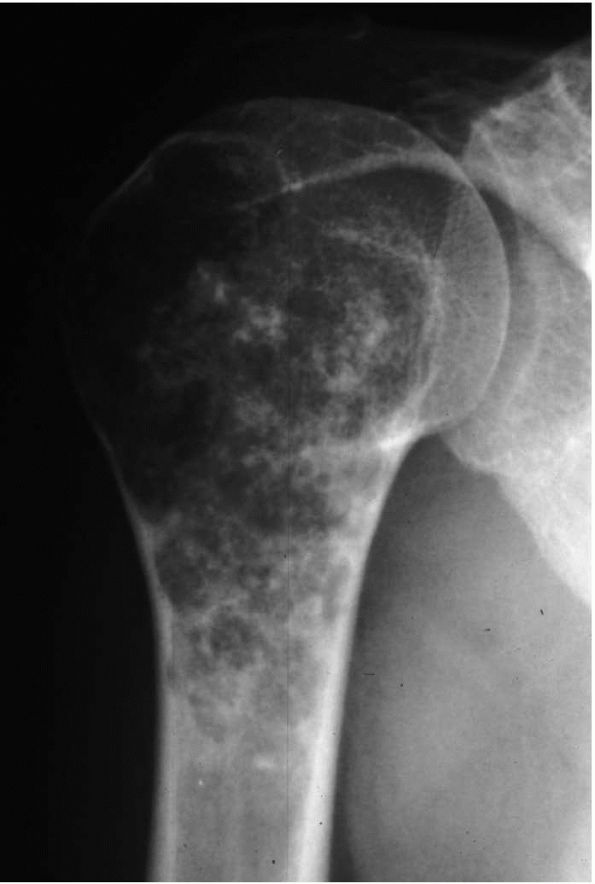
Patients with GCTs are usually between 30 and 45 years of age with a
female predominance. Unlike chondroblastomas, most GCTs occur after
physeal closure in the involved bone. They can cause pain, swelling,
and pathologic fracture, and aggressive lesions cause extensive
destruction of normal tissue. They occur in the epiphysis of long bones
and extend into the metaphysis. GCTs abut the subchondral surface of
the adjacent joint. Common sites include the tibial plateau, femoral
condyle, distal radius, and humeral head (Figure 10-31).
 |
|
FIGURE 10-28.
Light micrograph showing a chondromyxoid fibroma. The lesion consists of lobules of myxoid cartilage matrix surrounded by vascular fibrous tissue. The cell density of the lobules increases towards the periphery. |
 |
|
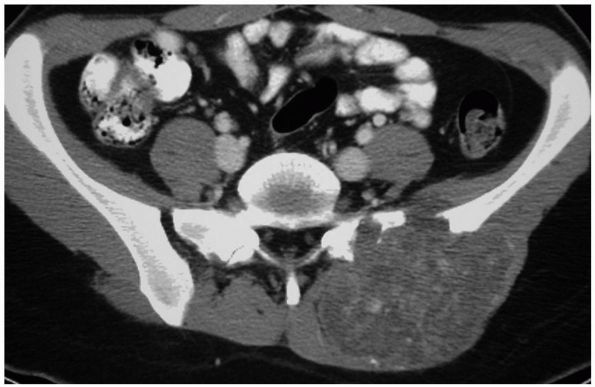
FIGURE 10-29.
A CT scan of the pelvis reveals an aggressive lesion in the posterior ilium and lateral sacrum. There are visible calcifications within the lesion. This is a common location for a chondromyxoid fibroma. |
 |
|
FIGURE 10-30. Histologic section of a giant cell tumor reveals multinucleated giant cells, fibroblast-like stromal cells, and blood vessels.
|
The differential diagnosis of GCT includes ABC, chondroblastoma, Brown
tumor of hyperparathyroidism, and nonossifying fibroma. The
radiographic appearance of aggressive GCTs can resemble a sarcoma.
Rarely, giant cell tumors appear in multiple sites or produce benign
lung metastases.
intralesional curettage using a high-speed burr followed by cementation
or bone grafting. Frequently, adjuvants such as phenol, cryotherapy, or
argon beam coagulation are used to extend the zone of treatment. There
is a 10 to 20% risk of local recurrence. In aggressive lesions with
extensive bone destruction, resection of the joint is necessary
followed by reconstruction using either an allograft or metal
prosthesis.
 |
|
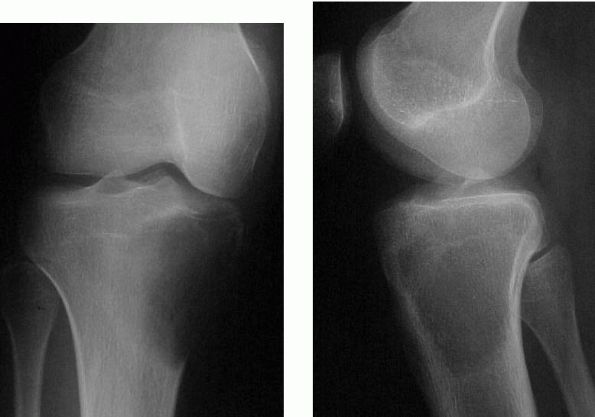
FIGURE 10-31.
Radiographs of the proximal tibia reveal an eccentric, lytic lesion with no sclerotic border located in the epiphysis or metaphysis. It is consistent with a giant cell tumor. |
dense benign fibrous tissue. These rare lesions behave as low-grade
malignant neoplasms and may be mistaken for a fibrosarcoma of bone.
They can aggressively destroy bone and invade surrounding soft tissues.
Desmoplastic fibromas usually appear in patients younger than 30 years
of age. They may cause aching pain, swelling, and rarely, pathologic
fractures.
occasionally produce bone expansion. Although they do not metastasize
and their clinical behavior varies, they frequently recur unless
excised with a margin of normal tissue.
and often occur in vertebral bodies. They may represent a developmental
disorder or a neoplasm. Generally, they remain within the medullary
cavity. Plain radiographs reveal regions of generalized decreased bone
density with abnormally prominent bone trabeculae. Most of these
lesions
are asymptomatic and detected as an incidental finding, thereby
requiring no surgical treatment. However, occasionally vertebral
hemangiomas are associated with pain and neurologic deficits.
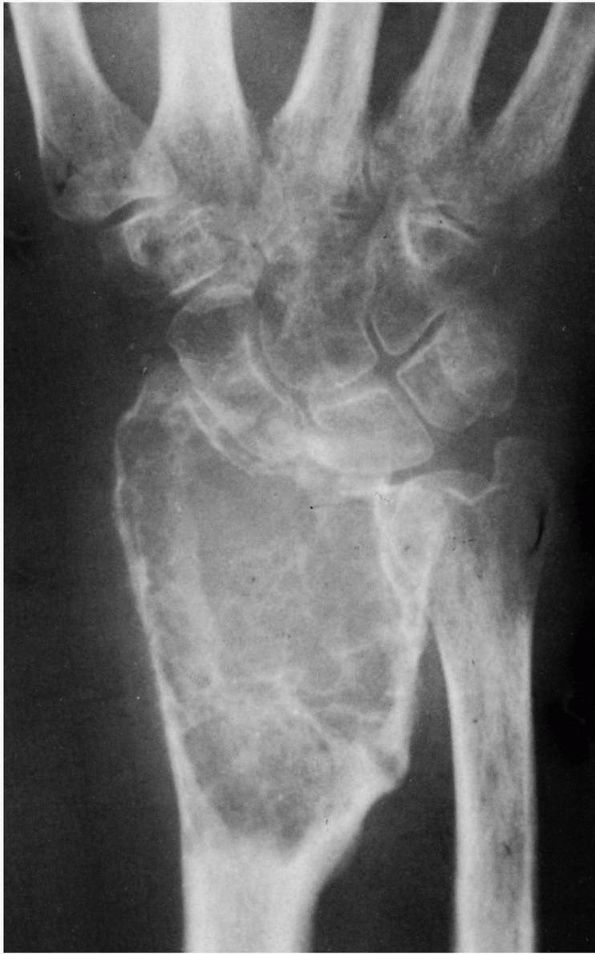 |
|
FIGURE 10-32. A giant cell tumor of the distal radius has expanded the cortex.
|
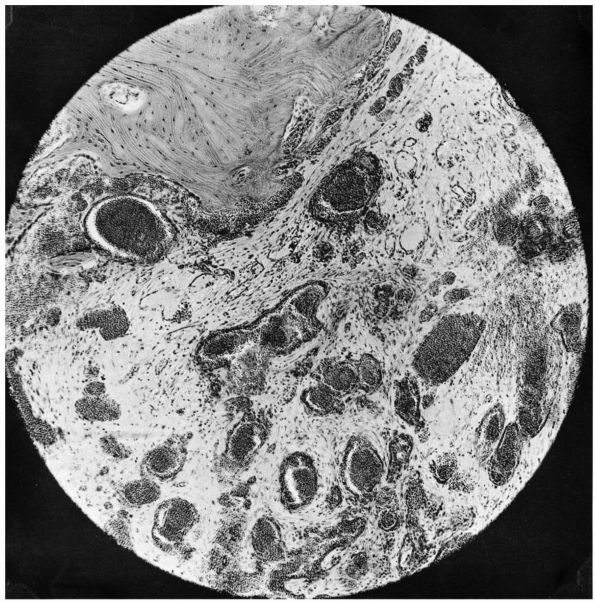 |
|
FIGURE 10-33. A histologic section of an intraosseous hemangioma reveals blood vessels admixed with fibrous tissue within the bone.
|
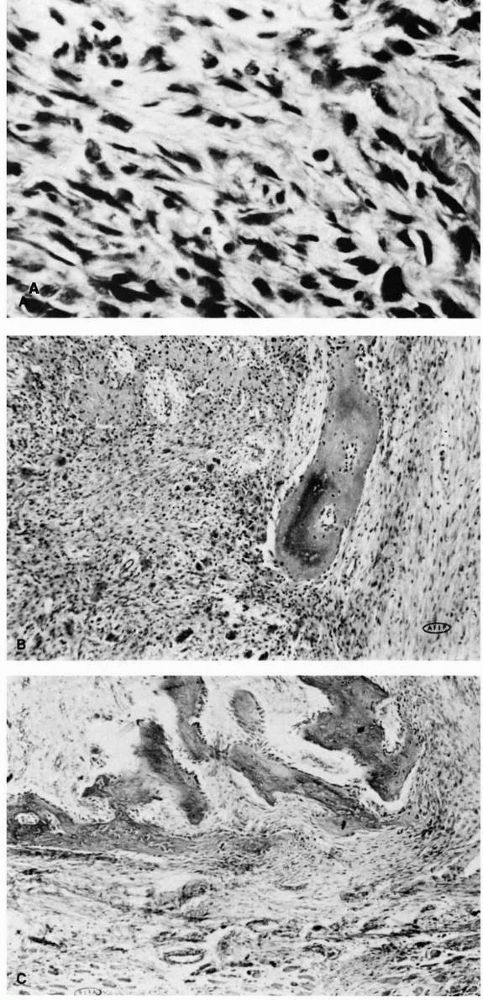
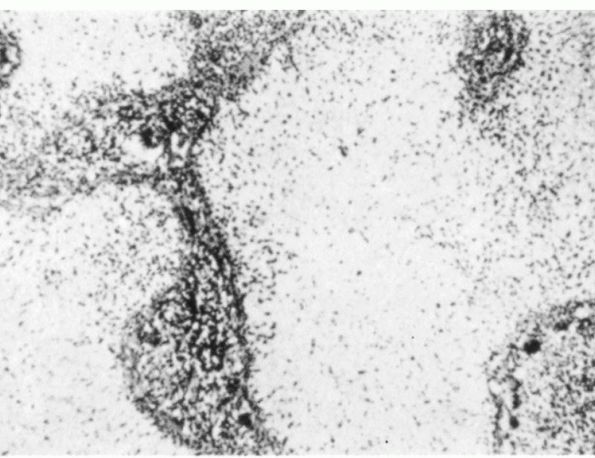
consist of histiocytes admixed with eosinophils, lymphocytes, and
neutrophils (Figure 10-34). They cause bone
destruction and a surrounding reaction that mimics benign and malignant
neoplasms as well as osteomyelitis. They can lead to pathologic
fractures and, in children, collapse of a vertebral body, called
vertebra plana (Figure 10-35). Plain
radiographs of the lesions vary from sharply circumscribed, round or
oval lucent regions to extensive, permeative bone destruction (Figure 10-36).
previously separated LCH into three overlapping disorders. Two
disorders, Letterer-Siwe and Hand-Schüller-Christian, involved multiple
tissues and affected young children. A solitary bony lesion
(eosinophilic granuloma) was more common in older children and young
adults. More recently, LCH has been referred to as a spectrum of
disease with varying degrees of severity and bone
or
soft tissue involvement. The more severe forms of LCH cause systemic
illness in young children, and these patients rarely present with
skeletal involvement alone. In contrast, patients with a solitary
eosinophilic granuloma seek medical attention because of bone pain, a
pathologic fracture, or because the lesion was discovered incidentally.
 |
|
FIGURE 10-34. Cytologic features of Langerhans cell histiocytosis. (A)
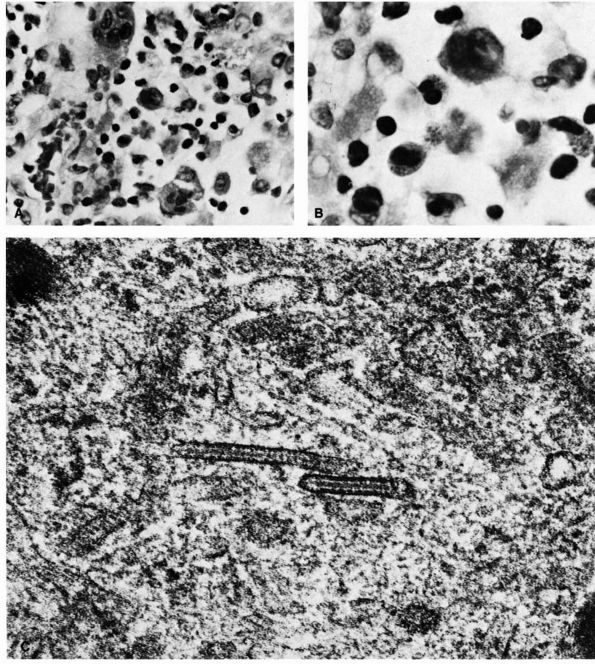
Smear of aspiration specimen shows an admixture of abundant histiocytes, containing either single or multiple nuclei. These nuclei often contain lipid-filled vacuoles and hemosiderin-like pigment. Also shown are eosinophils, lymphocytes, and neutrophils (×460). (B) Higher magnification. The histiocytes are irregularly shaped with ill-defined outlines, and they contain an eccentric, large, indented, finely creased nucleus with delicate chromatin, surrounded by abundant, delicate, pink-staining cytoplasm that contains granular material. The histiocytes sometimes have a loose syncytial appearance, often possess long cytoplasmic processess, and appear to fuse to form giant cells (×1150). (C) Ultramicroscopic features of Langerhans cell histiocytosis shows characteristic Langerhans granules. The electron micrograph shows the tubular inclusions within the cytoplasm of a typical histiocyte of eosinophilic granuloma. These are invariable features of this disease. They may be found in any condition in which the pathologic process is associated with a reactive histiocytosis (×140,000). (Katz R, Silva EG, de Santos LA et al. Diagnosis of eosinophilic granuloma of bone by cytology, histology, and electron microscopy of transcutaneous bone-aspiration biopsy. J Bone Joint Surg 1980;62A:1284) |
at less than 2 years of age with an acute onset of the disease that may
include hepatosplenomegaly, lymphadenopathy, rash, bleeding diathesis,
anemia, and occasionally exophthalmos and diabetes insipidus. Patients
with a more chronic from of disseminated histiocytosis usually present
before age 5 and may develop otitis media, diabetes insipidus,
exophthalmos, fever, hepatosplenomegaly, lymphadenopathy, anemia, and
disturbances of liver function. These disseminated forms of the disease
have
historically had a high mortality rate despite treatment. However, new
chemotherapy regimens have been successful in improving the overall
survival of these patients in recent years.
 |
|
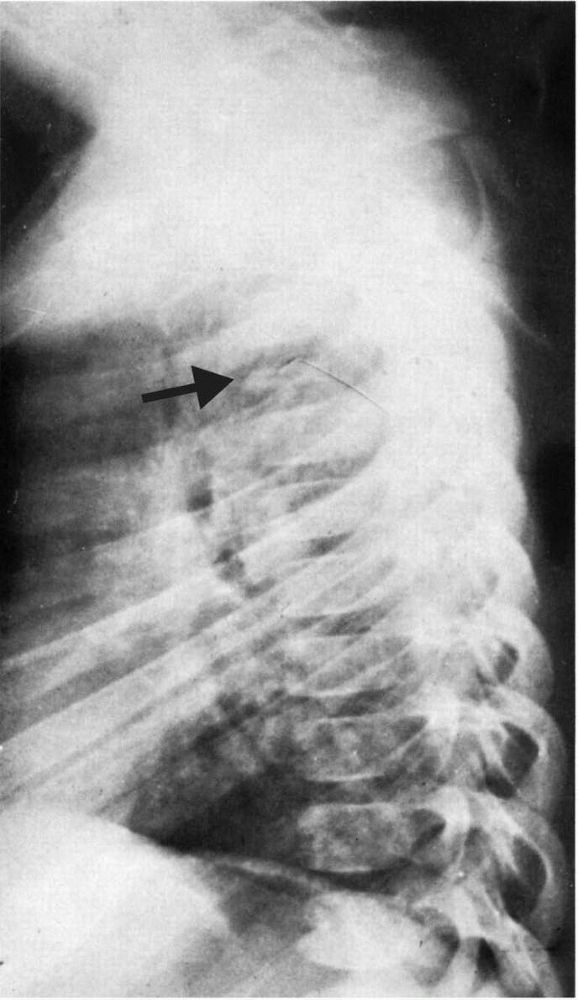
FIGURE 10-35.
Radiograph showing an eosinophilic granuloma that has caused collapse of the T5 vertebral body (vertebra plana). These lesions usually resolve spontaneously and, if a sufficient period of growth remains, the vertebral body regains normal size. |
adolescents, and young adults, although most patients present before
age 10. The primary symptom is localized pain. It occurs in the
diaphysis or metaphysis of long bones and can stimulate a periosteal
reaction that resembles osteomyelitis or Ewing’s sarcoma. Patients may
have a solitary lesion, multiple lesions simultaneously, or a
succession of lesions over years. Some patients that present with an
isolated bone lesion later develop multiple lesions or more diffuse
systemic involvement. Eosinophilic granulomas involving bone heal
spontaneously, but establishing the diagnosis, relieving pain, or
preventing pathologic fracture may require intervention before this
occurs. Steroid injection is the treatment of choice with intralesional
curettage reserved for persistent lesions.
 |
|
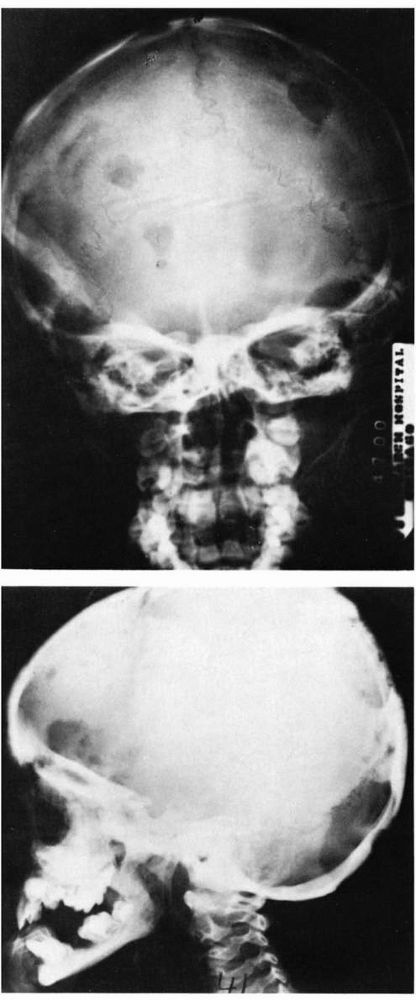
FIGURE 10-36.
Radiographs showing disseminated Langerhans cell histiocytosis involving the skull. The lesions are seen as well-circumscribed lytic bone defects. |
mature fat. It rarely develops in individuals less than 20 years of
age, and it becomes more common in the fifth and sixth decades. Lipomas
occur
in both the superficial subcutaneous and deep soft tissues. Superficial
lipomas occur frequently on the back, shoulder, neck, and proximal
regions of the arms and legs. Lipomas that develop deep in the muscle
fascia (usually within muscle) can occur in almost any muscle but
appear most frequently in the large muscles of the limbs, back, and
pelvis. Lipomas present as asymptomatic, slowly growing, mobile soft
tissue masses. Occasionally large lipomas compress peripheral nerves
and cause pain.
masses, but an MRI shows a classic appearance of a lesion bright on
T1-weighted images that also suppresses with fat suppression sequences (Figure 10-37).
Left untreated, some tumors become large enough that patients have
difficulty finding suitable clothing. Simple excision successfully
removes the lipoma with a low risk of recurrence.
 |
|
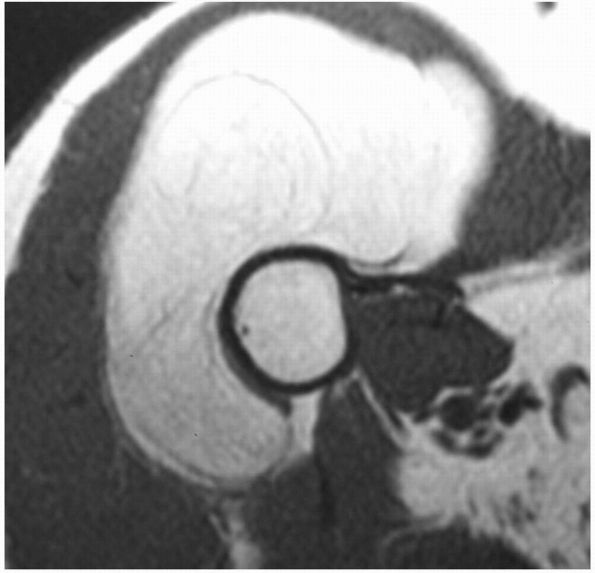
FIGURE 10-37.
An axial MRI of the proximal arm reveals a lipoma deep to the deltoid muscle. It has a signal similar to the superficial adipose tissue. |
vessels. It is not clear if they result from a disturbance in tissue
development or actual neoplasia. More than 80% of these lesions first
appear in people younger than 30. They present as slowly enlarging soft
tissue masses, and many patients have pain or vague discomfort in the
region of the hemangioma for years before diagnosis. Hemangiomas occur
most frequently in the skin and subcutaneous tissues where they form
soft masses with a blue appearance. These superficial lesions may be
present at birth or develop during childhood and adolescence. The
clinical appearance of these lesions is diagnostic.
malignant neoplasms. They consist of blood vessels, fibrous tissue, and
adipose tissue. They may appear in any muscle but most commonly involve
the lower extremity, particularly the thigh muscles. They do not cause
skin discoloration unless they extend into the subcutaneous tissues.
Small deep lesions may be difficult to identify without performing an
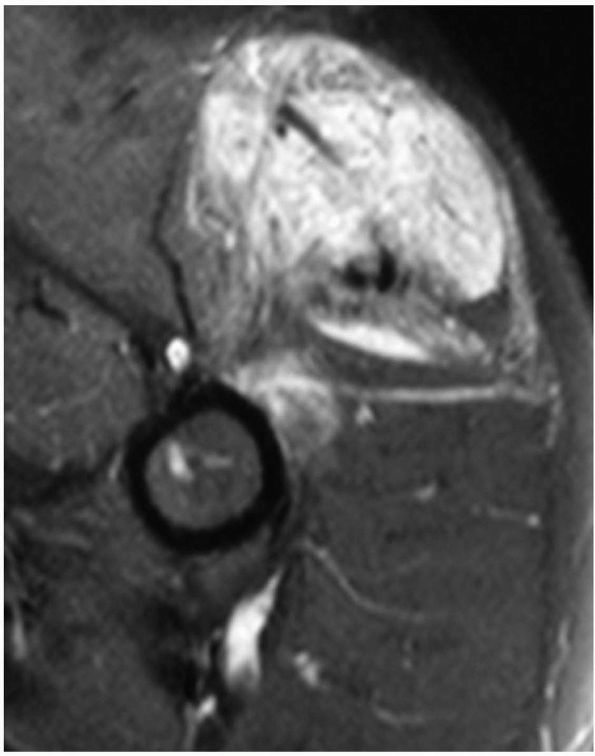
MRI of the involved muscle (Figure 10-38).
Although many hemangiomas remain confined to muscle, these lesions can
erode adjacent bone or surround nerves and blood vessels.
these lesions do not metastasize. They have not been shown to become
malignant even in patients with large lesions, and they generally do
not enlarge after skeletal maturity. Recurrence of the hemangioma often
follows surgical treatment. In some cases, successful surgical excision
of small, superficial hemangiomas can be performed, but a more recent
option is embolization or sclerotherapy of symptomatic lesions. This
treatment is usually performed by an interventional radiologist. Other
options for treatment of symptomatic, inoperable hemangiomas include
anti-inflammatory use and compression stockings.
 |
|
FIGURE 10-38.
An axial MRI of the proximal arm reveals a soft tissue hemangioma. It contains both fluid and adipose elements and is diffusely present within the muscle. |
throughout the soft tissues of an extremity. This disorder, congenital
hemangiomatosis, increases the circumference and length of the involved
extremity and may cause significant deformity. During childhood the
discrepancy in size between the normal and involved extremities may
increase and, in some patients, amputation may be appropriate.
surrounded by layers of polygonal cells called pericytes. Fine
unmyelinated nerve fibers run between the blood vessels. These tumors
occur in young adults and usually form soft, vascular masses no more
than a few millimeters in diameter. Patients with glomus tumors
characteristically report episodes of intense pain in the region of the
tumor. Many of these episodes follow minor trauma. The tumor can be
found in multiple locations, but it typically develops in the nail bed
of a finger or toe where it may be seen through the nail as a small,
blue nodule. Surgical excision eradicates most glomus tumors.
vessels and lymphoid aggregates that rarely affects the musculoskeletal
system. These lesions are discovered in newborns or infants. More than
50% present at birth, and over 90% present by the age of 2 years.
Parents or physicians can recognize the presence of a lymphangioma
because of a fluctuant soft tissue mass that may fluctuate in size. The
lesions rarely cause pain at rest but may be associated with discomfort
following trauma or exercise. Large lymphangiomas may cause
disfigurement or interfere with musculoskeletal function. Most
lymphangiomas involving the trunk or limbs occur in the superficial
tissues of the axilla or upper extremity. It is not certain if these
lesions form by neoplastic proliferation of lymph vessels or represent
hamartomas or lymphangiectasis. They are benign but may enlarge by
accumulation of fluid.
attempts to excise large lesions frequently lead to complications
including infection and wound healing problems.
nerve sheath cells. It can occur at all ages but reaches a peak
incidence in patients between the ages of 20 and 50 years. It most
commonly develops in the larger nerves of the head, neck, and flexor
surfaces of the upper and lower extremities. It grows slowly and rarely
causes pain or neurologic deficits unless it becomes quite large.
Palpation or compression of a neurolemmoma usually causes paresthesias.
Malignant transformation of a neurolemmoma is rare. Neurolemmomas tend
to expand within a nerve sheath without infiltrating between nerve
fibers, so they can often be safely removed without sacrificing nerve
function.
clinical and histologic features. Presumably, they both originate from
the Schwann cell but, theoretically, neurolemmomas consist of a more
homogeneous population of cells and have distinct capsules. They can
both develop from major nerves and have similar clinical presentations
but, unlike neurolemmomas, neurofibromas also develop from small,
unmyelinated nerves. In some instances it is difficult to identify a
clear relationship between a neurofibroma and a nerve. Neurofibromas
also tend to grow in and around individual nerve fibers rather than
compressing or displacing them as is the case with neurolemmomas. This
entwining growth often makes it difficult to resect a neurofibroma
without causing neurologic damage.
individuals between the ages of 20 and 30 and appear as superficial
lesions in the dermis or as deep soft tissue masses. They usually
remain asymptomatic. However, some may grow slowly and cause a
neurologic deficit. Occasionally, a solitary neurofibroma becomes
malignant. Surgical excision of a neurofibroma is curative.
acoustic neuromas, café-au-lait spots and skeletal deformities
including scoliosis, kyphosis, pseudarthrosis and asymmetrical
enlargement of limbs. Neurofibromatous tissue may form discrete masses
like those seen in patients with solitary neurofibromas or plexiform
neurofibromas, which are diffuse lesions that extend throughout normal
tissues without a definite margin (Figure 10-40).
inheritance pattern with a high degree of penetrance, but approximately
50% of patients present as new mutations. The severity of
neurofibromatosis varies among patients: some have café-au-lait spots
with or without neurofibromas, and others have extensive, disfiguring
neurofibromas and severe
skeletal deformity (Figure 10-41, Figure 10-42).
In severely affected patients or those with a family history of
neurofibromatosis, the diagnosis is often apparent at birth. In others,
multiple neurofibromas appear during childhood or adolescence and
progressively enlarge.
 |
|
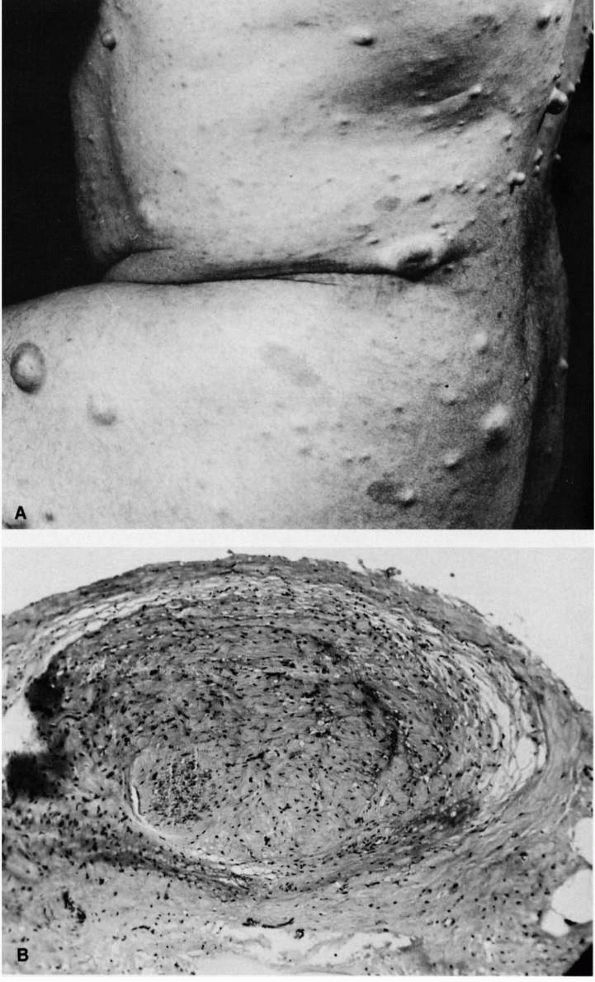
FIGURE 10-39. Neurofibromatosis. (A) Typical multiple subcutaneous tumor nodules. These nodules are composed of nerve tissue covered by areolar tissue and skin. (B)
Low power magnification of enlarged nerve fiber removed from a plexiform neurofibromatous mass. (McCarroll HR. Clinical manifestations of congenital neurofibromatosis. J Bone Joint Surg 1950;82A:601) |
 |
|
FIGURE 10-40.
Neurofibromatosis. Multiple nerve root involvement at cauda equine. The patient died at 42 years of age of sarcomatous degeneration of a neurofibroma of the brain. (Courtesy of Pathology Department, Mount Sinai Hospital, Chicago) |
deformities, patients with neurofibromatosis have a higher risk of
secondary malignant transformation of a lesion. Patients who have had
the disease for over 10 years are at the greatest risk. Rapid
enlargement of a neurofibroma or increasing pain in a neurofibroma
suggest the possibility of malignant change. Despite radical surgical
excision and aggressive adjunctive treatment, the prognosis for
patients with malignant nerve sheath tumors in the setting of
neurofibromatosis remains poor.
tumors in the soft tissues consist of dense, proliferating fibrous
tissue. They rarely affect infants or the elderly and occur most
commonly in people between 25 and 35 years of age. They present as
rock-hard, painless masses. With time they may cause discomfort, nerve
compression, and muscle weakness. Large lesions can restrict joint
motion. The tumors are usually located deep in the musculature of the
shoulder, chest wall, back, or thigh. They often lie within muscle, but
they can extend along fascial planes and appear in multiple sites
within a single limb. These lesions do not metastasize, but they can
aggressively invade normal tissue including bone, and they occasionally
develop in old scars.
behavior. Their aggressiveness has led some authors to call them
well-differentiated fibrosarcomas,
but
an occasional desmoid tumor ceases growing or even spontaneously
regresses. If possible these tumors should be treated by wide surgical
excision, which is often difficult. With less than a wide excision, the
lesions frequently recur. Radiation therapy has been used in cases with
an inadequate surgical margin or at the time of local recurrence. More
recently there are low-dose chemotherapy regimens available that may
provide benefit and allow surgery to be avoided in anatomically
difficult areas. Amputation is to be avoided if possible as this is a
benign tumor.
 |
|
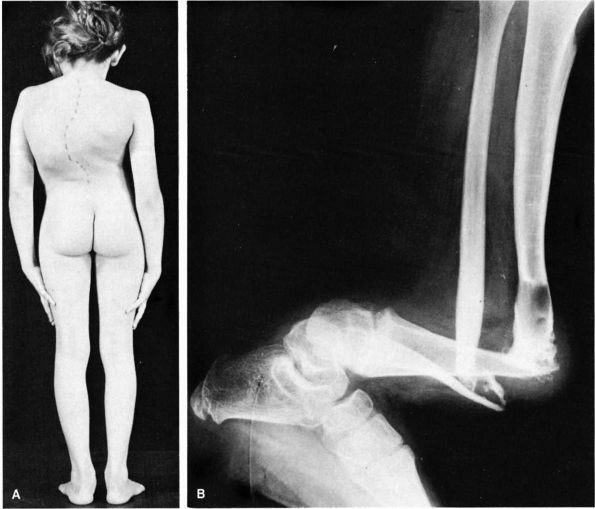
FIGURE 10-41. Neurofibromatosis. (A)
Congenital thoracic scoliosis in a child. The angulation is acute and usually rapidly progressive and disabling. Fusion should be done promptly. (B) Pseudoarthrosis of tibia at site of a cystic lesion. (McCarroll HR. Clinical manifestations of congenital neurofibromatosis. J Bone Joint Surg 1950;82A:601) |
tissue containing elastic fibers that occurs in the soft tissues
between the scapula and chest wall of elderly people. It may result
from repetitive mechanical trauma rather than a neoplastic process, but
this hypothesis has not been proven. This lesion causes mild
tenderness, pain, and occasional restriction of scapulothoracic motion.
It does not adhere to the skin and can often be palpated in the lower
subscapular area deep to the rhomboid and latissimus dorsi muscles
where it is firmly fixed to the chest wall. An elastofibroma grows
slowly and can be treated by surgical excision.
multinucleated giant cells, inflammatory cells, histiocytes, and
fibroblasts. It develops in or near synovial joints, bursae, and tendon
sheaths and may represent a reactive inflammatory process or a benign
neoplasm. It most frequently develops in people between 30 and 50 years
of age. It commonly appears in the hand and less commonly in the foot,
ankle, and knee. Physical examination reveals a firm, small, lobulated
mass fixed to the underlying tissues or tendon sheaths. Occasionally it
can erode bone. This tumor may grow slowly and recur following surgical
excision.
 |
|
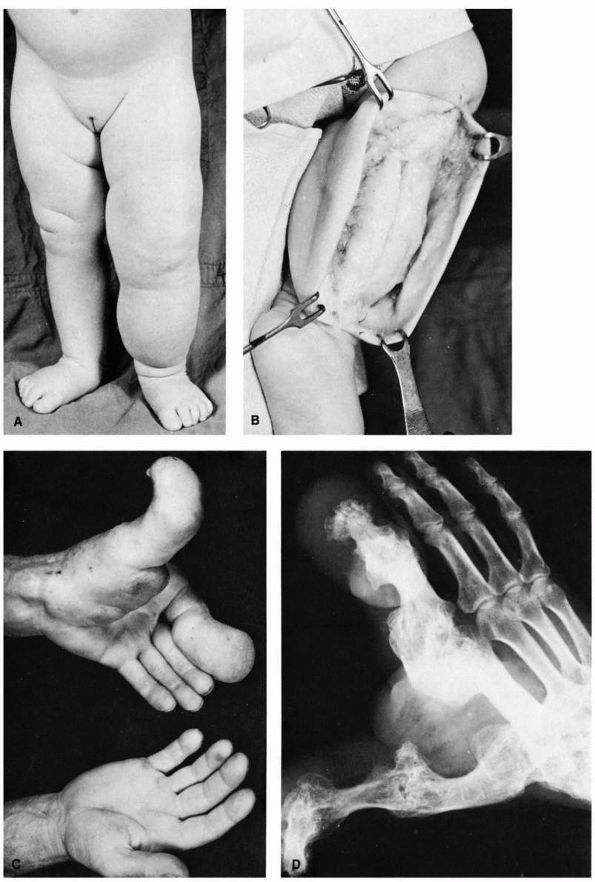
FIGURE 10-42. Neurofibromatosis. (A) Diffuse soft tissue hypertrophy and increased length of the lower extremity. (B)
A typical tumor mass of hypertrophied soft tissue is exposed at operation. It is not encapsulated and is superficial to the deep fascia. (C) and (D) Hypertrophy of the left thumb and index finger and corresponding portion of the hand. (McCarroll HR. Clinical manifestations of congenital neurofibromatosis. J Bone Joint Surg 1950;82A:601) |
proliferating synovial tissue containing histiocytes, fibroblasts,
multinucleated giant cells, and capillaries that can destroy dense
fibrous tissue, form soft tissue masses, and invade bone. Like GCT of
tendon sheath, PVNS may represent a reactive inflammatory process or a
benign neoplasm. It occurs most commonly in adolescents and young
adults in large synovial joints including the knee, hip, and ankle,
although it also occurs in smaller synovial joints, tendon sheaths, and
bursae. It can occur in a focal or diffuse form. Most patients present
with a swollen joint and give a history of recurrent effusions. Plain
radiographs initially show erosions on both sides of a joint (Figure 10-43).
There is periarticular bone destruction and degenerative joint disease
in the later stages. The MRI appearance of PVNS is a low signal on both
T1- and T2-weighted images. Occasionally it presents as solitary or
multiple soft tissue nodules near a joint and can resemble GCT of
tendon sheath.
the diffuse type. Local recurrence is common. Adjuvant chemotherapy and radiation have been tried with variable success.
 |
|
FIGURE 10-43.
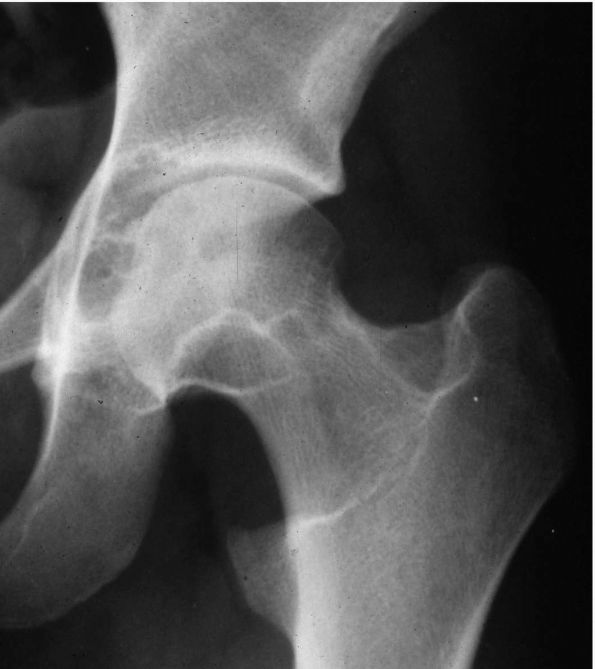
A radiograph of the hip reveals lytic lesions on both sides of the joint and mild degenerative changes consistent with an active synovial process such as pigmented villonodular synovitis (PVNS). |
cartilage nodules within the synovium of large joints. It occurs most
frequently in young adults. Patients present with pain, mechanical
symptoms, and an enlarging mass. Most patients develop loose fragments
of cartilage within the joint, but some patients form an enlarging mass
of proliferating chondrocytes in the periarticular tissues. Radiographs
reveal speckled calcifications within the joint or in the soft tissues
around a joint. The current treatment of synovial chondromatosis
includes complete synovectomy and removal of any cartilaginous loose
bodies.
less frequently than benign lesions. Unlike their benign counterparts,
malignant tumors have the capacity to cause disseminated disease and
death. Some malignant neoplasms develop in or near previously existing
benign lesions such as osteochondromas (multiple hereditary exostoses),
enchondromas (Ollier’s disease and Maffucci’s syndrome), chronic
draining osteomyelitis, bone infarcts, Paget’s disease, and
neurofibromas (neurofibromatosis). The development of a malignant
neoplasm presumably results from overexpression of preexisting
malignant potential (Ollier’s disease). In spontaneous cases of
malignancy, the development presumably results from an alteration in
the normal cells near the lesion (bone infarcts).
presentation. For example, multiple myeloma frequently causes
generalized weakness and bone pain without a mass in elderly,
debilitated people, whereas osteosarcoma commonly causes sharply
localized bone pain with a soft tissue mass in children and adolescents
without systemic symptoms. Current treatment of these neoplasms also
varies from systemic chemotherapy and radiation used for multiple
myeloma to preoperative chemotherapy and surgery used for osteosarcoma.
sarcomas vary less in clinical presentation and current treatment. Most
present as a painless mass. Wide surgical excision of primary bone and
soft tissue tumors is the recommended treatment with or without
adjuvant chemotherapy and radiation.
The disease rarely occurs before the fifth decade of life. In many
patients the malignant cells extend throughout the bone marrow (BM), so
a BM aspiration provides diagnostic tissue. Most patients have moderate
to severe anemia, an elevated ESR, and an abnormal serum protein
electrophoresis and immunoelectrophoresis. Occasionally, patients
present with a solitary focus of plasma cells referred to as a
plasmacytoma. These individuals usually develop diffuse disease after a
latent period of up to 5 to 10 years.
is diffuse or localized to regions of bone destruction. Because the
disease often involves the vertebral bodies, patients can develop back
pain and vertebral compression fractures. In addition, many affected
individuals have systemic symptoms including weakness, fatigability,
and weight loss. The radiographic features of multiple myeloma vary
from multiple “punched out” areas of bone destruction often in the
skull and pelvis (Figure 10-44), to diffuse
osteopenia difficult to distinguish from that secondary to metabolic
bone disease. In the early stages of multiple myeloma, radiographs may
not show any changes. An osteoblastic reaction to multiple myeloma is
rare, so bone scans may be falsely negative. Pathologic fractures due
to multiple myeloma commonly occur in the vertebral bodies but may
occur throughout the skeleton. Radiation alone can effectively treat a
plasmacytoma, and chemotherapy with radiation is the treatment of
choice for disseminated disease. Recently, stem cell transplant has
been used with success in many patients.
 |
|
FIGURE 10-44. Radiograph showing the multiple punctate lucent defects in the pelvis of a patient with multiple myeloma.
|
and occurs in middle-aged patients. The appearance of a lymphoma, also
called a reticulum cell sarcoma, frequently resembles a Ewing’s
sarcoma. However, a lymphoma contains a network or reticulum of fine
matrix fibers that appear as thin dark threads when stained with a
silver or reticulum stain (Figure 10-45).
fracture. Plain radiographs show extensive, permeative lesions that
often extend throughout the bone. The regions of bone destruction have
an irregular appearance that blends with normal bone. Occasionally
plain radiographs appear normal, but a bone scan is positive and an MRI
scan shows a large soft tissue mass. The majority of lymphomas involve
bone as part of a systemic presentation. These patients are treated
with chemotherapy as well as radiation to the bony site. Isolated
lymphoma of bone is rare but can be treated with radiation with or
without surgery.
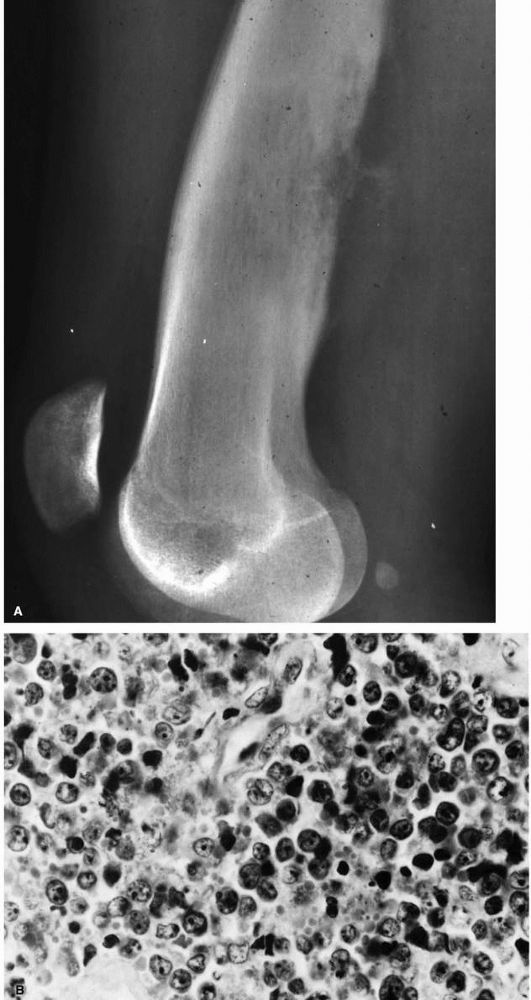 |
|
FIGURE 10-45. (A) A lateral radiograph of the distal femur in a patient with lymphoma reveals subtle permeative destruction. (B) A histologic section shows the population of round cells that form the neoplasm. (Histology courtesy of Dr. A.G. Huvos)
|
consists of densely packed, distinctive, small round cells of uncertain
origin. The light microscopic appearance is similar to other small
round cell tumors; however, recent molecular tests have a high
sensitivity for an accurate diagnosis. Ewing’s sarcoma occurs more
frequently in males and in patients younger than 20 years of age. The
tumor may appear in any bone or occasionally in the soft tissues, but
most present in the pelvis and lower extremities. The tumor usually
causes pain and may enlarge rapidly to produce significant soft tissue
swelling and occasional pathologic fractures. Some patients with
Ewing’s sarcoma have systemic symptoms such as fever, generalized
weakness, and laboratory abnormalities including anemia and an elevated
ESR. It is important to differentiate the clinical presentation from
osteomyelitis.
In long bones Ewing’s sarcoma usually involves the diaphysis, but it
may extend into the metaphysis. In flat bones such as the pelvis or
scapula, it can involve the entire bone. The radiographic appearance of
Ewing’s sarcoma may resemble osteomyelitis, lymphoma, osteosarcoma,
eosinophilic granuloma, and metastatic carcinoma. The tumors often
break through the cortex and periosteum with invasion of the soft
tissues at the time of diagnosis (Figure 10-46).
systemic chemotherapy. Local control is usually obtained with wide
surgical resection. Radiation is also effective and is indicated for
unresectable lesions.
The matrix of a typical osteosarcoma consists of osteoid that may or
may not mineralize. Other osteosarcomas synthesize a cartilaginous or
fibrous matrix, and a rare subtype forms large vascular channels. These
neoplasms occur most frequently in the second decade of life with an
additional peak in patients 70 to 80 years of age. Osteosarcomas cause
progressively increasing pain. At the time of diagnosis many extend
through the bone cortex and produce a firm soft tissue mass (Figure 10-47). They can
develop in association with Paget’s disease or following radiation
therapy and, in these situations, have an extremely poor prognosis.
Osteosarcomas develop in the metaphysis of long bones, especially the
distal femur, proximal tibia, and proximal humerus, although they can
be found in any bone (Figure 10-48).
Less commonly osteosarcomas can develop on the surface of the bone
(periosteal and parosteal osteosarcomas). A rare type of osteosarcoma
develops in the soft tissues (extraosseous osteosarcoma).
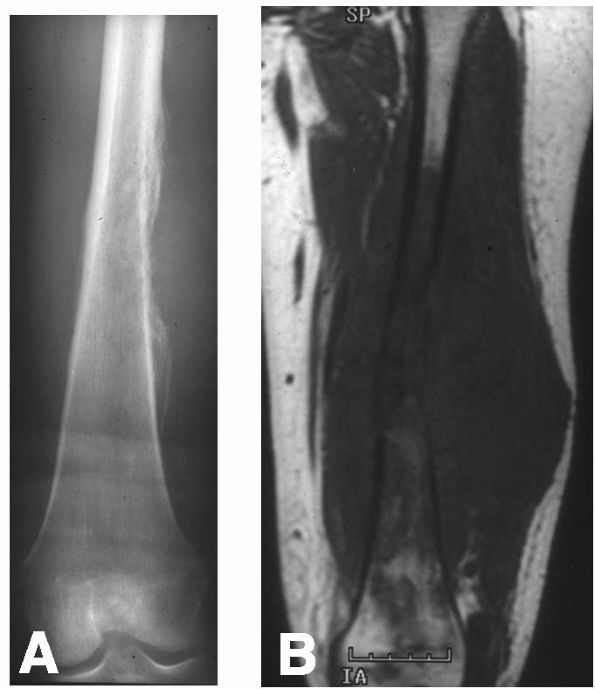 |
|
FIGURE 10-46. (A) A radiograph and (B)
an MRI scan of a patient with Ewing’s sarcoma of the femur. Note the subtle destruction and periosteal reaction on the radiograph as well as the large soft tissue mass on the MRI scan. |
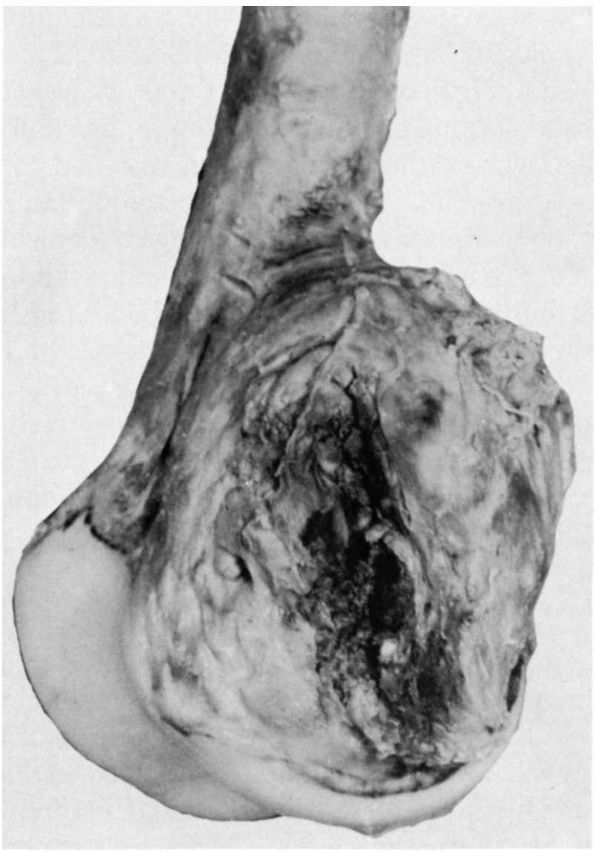 |
|
FIGURE 10-47.
Photograph of a classic intramedullary osteosarcoma of the distal femur. Notice how the neoplasm has broken through the bone and formed a large soft tissue mass. |
although, tumor osteoid does not always mineralize, so occasionally
there is no evidence of new bone formation. The radiographic appearance
of a telangiectatic osteosarcoma containing large vascular channels may
resemble the bone changes seen in an ABC. Parosteal osteosarcomas
mineralize so densely that they appear as sclerotic masses on the
surface of the bone.
aggressively. They rapidly destroy normal bone, invade soft tissue, and
frequently metastasize to the lungs. The current treatment for these
tumors is systemic multiagent chemotherapy followed by surgical
resection with a wide margin.
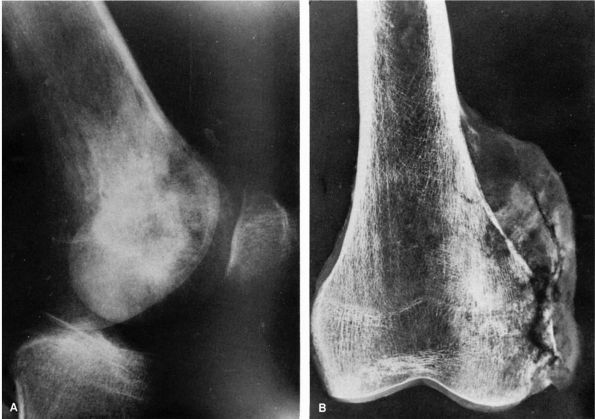 |
|
FIGURE 10-48. Radiographs of a distal femoral osteosarcoma. (A)
Lateral view of the distal femur showing the irregular bone destruction and bone formation that gives the bone a mottled appearance. (B) A radiograph of the resected specimen after removal of the soft tissues. This view reveals destruction of the cortex and development of a soft tissue mass. Notice the new bone formation in the soft tissue mass. |
of long bones most often in young adults. They erode the cortex
producing a shallow cavity and reactive periosteal new bone formation.
Some produce radiographic changes that resemble a periosteal chondroma,
but the osteosarcoma creates a more irregular bony margin that suggests
the presence of a malignant tumor. As a periosteal osteosarcoma grows,
it extends into the soft tissues, and ossification may appear within
the enlarging mass. Eventually the lesions grow into the medullary
cavity. Patients with a periosteal osteosarcoma often present after
they notice a firm, fixed, slowly enlarging mass. This type of
osteosarcoma is an intermediate grade malignancy. Surgical resection
with or without chemotherapy is the standard treatment.
classic intramedullary or periosteal osteosarcoma. Most parosteal
osteosarcomas involve the
femur, humerus, or tibia with the vast majority occurring on the posterior aspect of the distal femur (Figure 10-49).
These tumors grow slowly over years, and the patient may notice
swelling but little discomfort. Plain radiographs reveal dense mineral
deposits within the tumor. It lies directly on the cortical surface and
occasionally extends into the medullary canal. The radiographic
differential diagnosis includes myositis ossificans, periosteal
osteosarcoma, and osteochondroma. Wide surgical excision without
chemotherapy is necessary for local control.
contain regions of ossification. It occurs in young adults near the
shoulder and pelvis. These rare lesions are treated by surgical
resection with or without adjuvant chemotherapy and radiation similar
to the treatment of a soft tissue sarcoma.
It may develop from an enchondroma or osteochondroma but occurs more
commonly in bones with no known preceding cartilage lesion.
Chondrosarcomas occur in adults and the elderly most commonly in the
pelvis, scapula, humerus, femur, and tibia. Patients with multiple
hereditary exostoses, Ollier’s disease and Maffucci’s syndrome have a
higher risk for malignant transformation of a cartilaginous lesion.
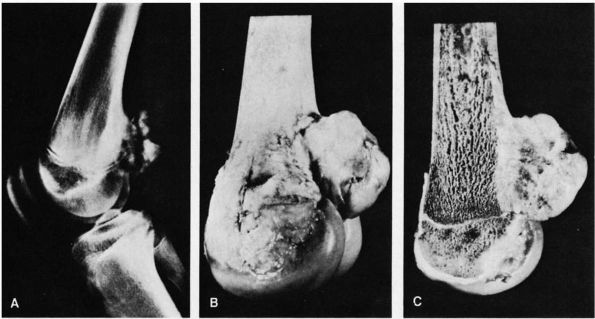 |
|
FIGURE 10-49. Grade I lesion of parosteal osteosarcoma. (A)
Radiographs show an irregular ossified mass with prominent lucency indicating a large amount of fibrous and cartilaginous tissue. The subjacent cortex is sclerotic. (B) Typical external appearance. This is the most common location of the tumor on the posterior aspect of the distal end of the femur at the popliteal area. (C) Sagittal section shows that this tumor merges imperceptibly with the cortex along its entire base. The underlying cortical bone is thickened. (Ahuja SC, Villacin AB, Smith J et al. Juxtacortical (parosteal) osteogenic sarcoma. J Bone Joint Surg 1977;59A:632) |
some enlarge rapidly, aggressively invade normal tissue, metastasize,
and cause death, but most are low grade tumors that enlarge slowly,
cause little damage to adjacent tissues, and metastasize only after
many years. They vary in their histologic appearance from lesions that
resemble normal hyaline cartilage to lesions consisting of poorly
differentiated cells that fail to produce a typical hyaline cartilage
matrix. These differences in the histologic appearance of
chondrosarcoma led to a three-tiered classification of chondrosarcoma
based on its resemblance to normal hyaline cartilage (Figure 10-50).
The grade of the tumor is a strong predictor of its behavior. Rare
subtypes include mesenchymal, dedifferentiated, and clear cell
chondrosarcoma.
chondrosarcoma) or from the surface of the bone (peripheral
chondrosarcoma). Plain radiographs reveal bone destruction as well as
mineralization within the tumor (Figure 10-51).
Peripheral chondrosarcomas may resemble osteochondromas on plain
radiographs, although they commonly show bone destruction and irregular
calcification of the cartilage cap. MRI and CT scans of these lesions
may show increased thickness of the cartilage cap suggesting the
presence of malignancy. Because plain radiographs do not accurately
show the cartilage component of these tumors, they may be considerably
larger than they appear. Central chondrosarcomas appear as regions of
bone destruction with areas of calcification. Frequently they extend
further in the medullary cavity than the plain radiographs suggest.
They can break through the cortex and form large soft tissue masses.
There is usually no reactive bone formation.
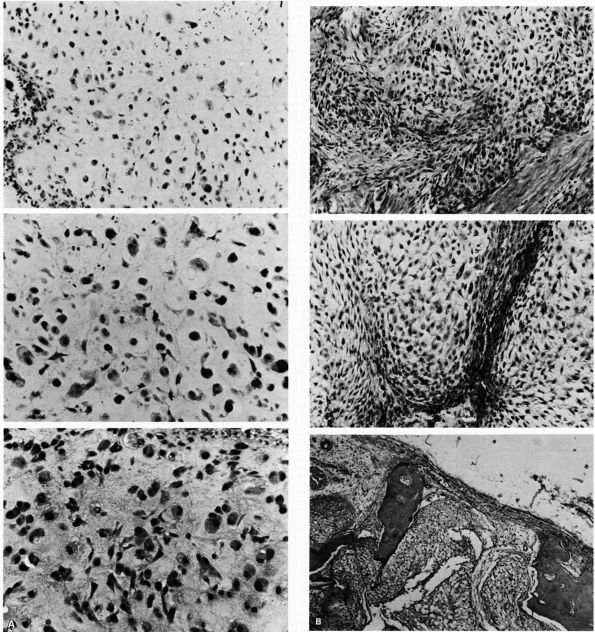 |
|
FIGURE 10-50. Light micrographs showing the variability in the histologic appearance of chondrosarcomas. (A) The histologic appearance of grade I and grade II chondrosarcomas. (Top and middle) Grade I changes include increased cell density and increased variability cell size, shape and staining. (Bottom) Grade II changes include more advanced cellular pleomorphism. (B) The histologic appearance of grade III chondrosarcomas. (Top and center) Dedifferentiation toward fibrosarcoma at the periphery of chondrosarcomatous lobules. (Bottom)
Lobules of malignant cartilage penetrate bone, demonstrating the ability of these lesions to destroy normal bone. (Dahlin DC, Salvador AH. Chondrosarcomas of bones of the hands and feet: a study of 30 cases. Cancer 1974:34:755) |
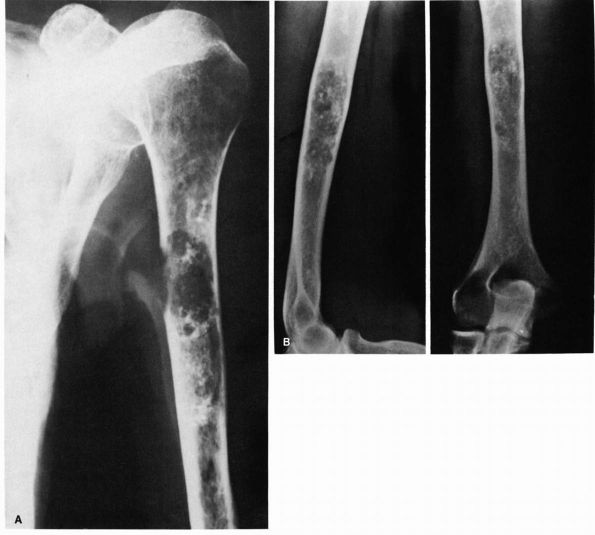 |
|
FIGURE 10-51. Plain radiographs showing central chondrosarcomas. These lesions cause bone destruction and produce regions of mineralization. (A)
A central chondrosarcoma of the humerus showing an irregular area of central bone destruction with mineralization and erosion of the medial cortex. (B) Radiographic features include osteolysis, scalloping endosteal erosion of inner cortex, central areas of calcification, and no reactive bone formation about the periphery. |
cause little or no discomfort despite reaching large size, especially
within the pelvis (Figure 10-52). Peripheral
and central chondrosarcomas that extend into the soft tissues form
firm, fixed masses. In some patients, a mass may have been present for
years. Wide surgical excision is necessary and offers the best
possibility of cure, as radiation and chemotherapy have not proven to
be effective methods of treatment for chondrosarcoma. Locally recurrent
disease is common and may be difficult to treat.
bone tumors. Like a fibrosarcoma of soft tissue, it consists of
fibroblasts and a collagenous matrix. These tumors occur over a wide
age range and may appear in any part of the skeleton. Most of them
cause pain and swelling. They may occur in association with Paget’s
disease, fibrous dysplasia, bone infarcts, or following radiation
therapy. Plain radiographs show irregular, mottled bone destruction
with occasional regions of reactive new bone formation around the
periphery. Wide surgical excision is the currently accepted treatment,
and neoadjuvant chemotherapy has been used for some patients.
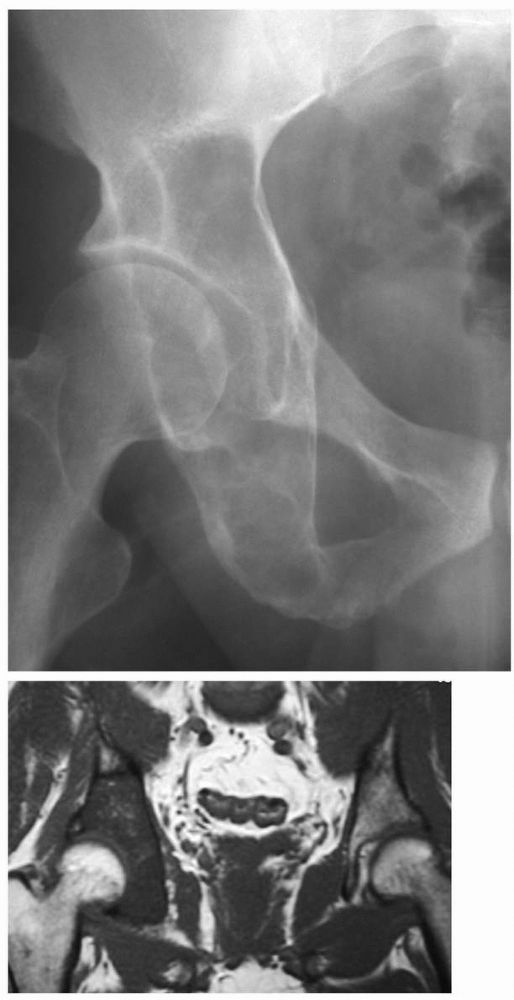 |
|
FIGURE 10-52. (A) A plain radiograph and (B)
MRI of the pelvis reveal a lesion in the right periacetabular region consistent with a chondrosarcoma. These tumors frequently occur in the pelvis as lytic lesions and can become destructive with large soft tissue masses that may go unrecognized given their deep location. |
malignant tumor consisting of the same cell types found in MFH of soft
tissues such as giant cells, fibroblasts and histiocytes. It is much
more commonly found in the soft tissues but can occur in bone. These
tumors occur in patients of any age and have also been reported in
association with Paget’s disease, bone infarcts, and radiation therapy.
Patients have pain, and plain radiographs show irregular bone
destruction. They are treated with the same chemotherapy regimen as is
used for osteosarcoma. Wide surgical resection is indicated following
neoadjuvant chemotherapy.
capillaries formed by malignant cells, but they vary in their
predominant cell type. In hemangioendotheliomas, capillary endothelial
cells are the predominant cell type, whereas in hemangiopericytomas,
pericytes are the predominant cell type. Hemangioendotheliomas occur
more frequently in bone, and hemangiopericytomas occur more frequently
in the soft tissues. An angiosarcoma refers to the most malignant form
of hemangioendothelioma or hemangiopericytoma.
the skeleton, and occasionally occur in multiple sites within the same
limb. They usually produce pain, and radiographs show evidence of bone
destruction. Surgical resection and chemotherapy are indicated for high
grade solitary lesions. Radiation therapy is frequently the sole
treatment for hemangioendothelioma involving multiple areas within the
same bone.
or strands of epithelial-like cells surrounded by fibrous tissue (Color
Figure 10-8). It is almost exclusively found in
the tibial diaphysis. It tends to follow a prolonged course and occurs
in individuals between late adolescence and middle age. Most
adamantinomas cause mild to moderate discomfort and a palpable mass
over the anterior tibial border. Radiographically, they cause
irregular, bubbly bone destruction of the anterior cortex (Figure 10-53). The lesion may be confined to the cortex of the bone.
treatment. The diaphysis is most commonly affected, so an intercalary
allograft is often the reconstruction of choice. Chemotherapy and
radiation have no role in treatment. Patients can develop late
pulmonary metastasis.
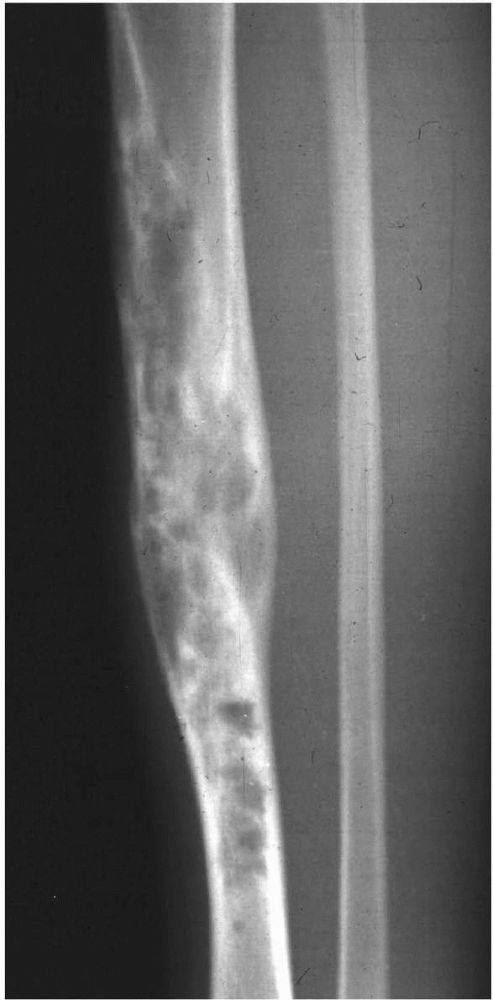 |
|
FIGURE 10-53. A radiograph of the tibia reveals a lytic, bubbly lesion involving the cortex consistent with an adamantinoma.
|
It may occur throughout the spine but is most frequently noted in the
sacrococcygeal region or at the base of the skull. These tumors rarely
occur in individuals less than 30 years of age. Most chordomas grow
slowly, and patients present with pain, bowel dysfunction, or
neurologic symptoms. Plain radiographs of chordomas typically show
irregular midline bone destruction. Small lesions are often difficult
to recognize on plain radiographs due to overlying shadows. A CT or MRI
scan is preferred to better define the anatomical extent of the tumor (Figure 10-54).
Chordomas often have a large presacral mass that can be identified on a
rectal examination. A wide surgical margin is necessary to achieve
local control, but this is difficult given the anatomic location. Nerve
roots or visceral structures involved by the chordoma should be
sacrificed to obtain an adequate resection. Radiation can be helpful
for areas where margins are positive or for locally recurrent disease.
Chemotherapy has not been effective in the treatment of this tumor.
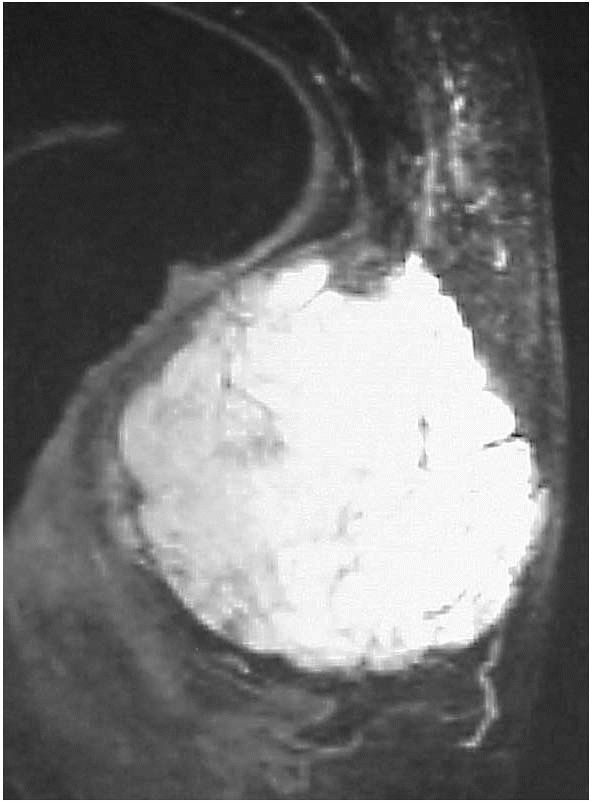 |
|
FIGURE 10-54. A sagittal MRI reveals a large lesion emanating from the lower sacrum with a large presacral mass consistent with a chordoma.
|
and neurofibrosarcoma, are named for their presumed cell of origin.
However, for many lesions such as synovial sarcoma and epithelioid
sarcoma, the cell of origin is unknown. Other soft tissue sarcomas
consist of undifferentiated cells that cannot be identified with a
specific tissue. Although soft tissue sarcomas differ in basic cell
type, they have similar clinical presentations, radiographic
appearances, and treatment. They usually present as firm, painless
masses. MRI scans are extremely accurate in elucidating the size and
anatomic location of a soft tissue tumor, but they cannot reliably
distinguish between histologic subtypes or between benign and malignant
neoplasms. Soft tissue sarcomas appear as heterogeneous masses that are
often large and deep to fascial planes (Figure 10-55).
The current treatment of soft tissue sarcomas consists of wide surgical
resection and preoperative or postoperative radiation. The use of
chemotherapy is the standard of care for patients with rhabdomyosarcoma
but is controversial for adults with soft tissue sarcomas. There is a
high risk of pulmonary metastasis for large, deep, high grade tumors.
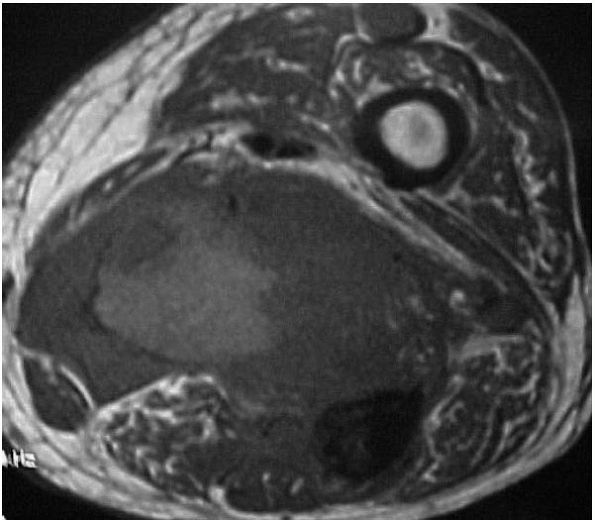 |
|
FIGURE 10-55.
An MRI of the thigh reveals a large, inhomogeneous soft tissue mass consistent with the appearance of multiple different histologic subtypes of soft tissue sarcoma. |
tissue sarcoma and, similar to its rare bony counterpart, consists of
malignant fibroblasts and histiocytes as well as giant cells and
occasionally inflammatory cells. The majority of soft tissue MFHs
present as painless, enlarging masses in people between 50 and 70 years
of age. They occur most frequently in the deep soft tissues of the
lower extremity.
variable histologic appearance. They are the second most common soft
tissue sarcoma. They frequently reach a large size and have a peak
incidence in people between 40 and 60 years of age. Most liposarcomas
of the extremities lie deep within the soft tissues, especially in the
quadriceps muscle and popliteal fossa. Other common locations include
the shoulder and calf. Patients usually detect liposarcomas because of
a mass or increase in size of the limb. The clinical behavior of these
tumors varies based on their grade.
fibroblasts. They occur in patients of any age but appear most
frequently in those between 30 and 55 years of age. They can occur
throughout the soft tissues, most frequently in the thigh. Usually they
present deep within the soft tissues and as an enlarging mass. Only
rarely do they cause significant discomfort before reaching a large
size. Occasionally fibrosarcomas arise in tissues previously treated
with radiation or in burn scars.
originate from the synovium. These tumors consist of two
morphologically distinct cell types: epithelial-like cells that assume
a cuboidal or columnar form and fibroblast-like spindle cells. The
combination of these two cell types creates a distinctive biphasic
pattern: the spindle cells surround islands and strands of epithelial
cells (Color Figure 10-10). Some synovial sarcomas only contain the spindle cells and are referred to as monophasic synovial sarcomas.
between 15 and 35 years of age and rarely appear in individuals over 50
years of age. They present as discrete masses or diffuse swelling, and
they often cause discomfort. These tumors grow quite slowly, and many
patients have symptoms for years before biopsy or excision demonstrates
the presence of the tumor. Synovial sarcomas appear most frequently in
the extremities, usually in periarticular regions or near tendon
sheaths, bursae and joint capsules. It is extremely uncommon to find
synovial sarcomas within a joint.
calcification within a synovial sarcoma. These tumors can metastasize
to regional lymph nodes. In some series, chemotherapy adds to the
effectiveness of surgery and radiation.
peripheral nerve sheath tumors) arise from peripheral nerve sheath
cells or from neurofibromas. Most occur in patients between 20 and 50
years old. They present as enlarging masses that may or may not cause
pain. Patients with neurofibromatosis (von Recklinghausen’s disease)
have a significantly increased risk of developing a neurofibrosarcoma,
and this underlying disease may predispose them to a worse prognosis
than those who develop this tumor spontaneously. Like other soft tissue
sarcomas, surgical excision is the best treatment for this disorder.
Unfortunately, some of the tumors assume a diffuse form that makes wide
resection almost impossible. In addition, when major neurologic
structures such as the brachial plexus are involved, amputation may be
the best surgical option.
consist of cells that form myofibers. There are different histologic
subtypes with different prognoses. They are the most common soft tissue
sarcomas in children and adolescents. Most of these tumors present as
enlarging, deep masses closely associated with muscle and can
metastasize to regional lymph nodes. They rarely cause pain or
tenderness. A combination of surgical excision, radiation therapy, and
chemotherapy are used to treat patients with this tumor.
their bony counterparts, malignant vascular tumors of soft tissue form
capillaries and vary in their predominant cell type.
Hemangiopericytomas occur more frequently in the soft tissues than in
bone. Some malignant vascular neoplasms arise in the superficial
tissues and skin, but others develop in the deep soft tissues and
closely resemble other soft tissue sarcomas. In some vascular sarcomas,
a wide surgical margin is difficult to achieve given their diffuse
nature.
mesenchymal origin, inflammatory cells, and capillaries. These
neoplasms occur most commonly in young adults and often develop in the
hand or foot. They are the most common soft tissue sarcomas of the
hand. Epithelioid sarcomas present as small, tender masses fixed to
tendon sheaths, periarticular tissues, and fascia. Because their
clinical presentation differs from other soft tissue tumors and because
there are inflammatory cells in the lesion, these tumors are frequently
mistaken for nonspecific inflammatory processes even after a biopsy.
The current treatment of these tumors is wide surgical excision.
Regional lymph node metastases are common, and patients are at high
risk for eventual lung metastasis.
with clear cytoplasm resembling swollen chondrocytes. It occurs in
older adolescents and young adults and presents as a slowly growing,
fixed mass. It is the most common malignant soft tissue tumor of the
foot. Like epithelioid sarcomas, clear cell sarcomas may involve
periarticular tissues and tendon sheaths and spread to regional lymph
nodes. The current treatment is wide excision, and there are no
effective options for chemotherapy. Patients with this disease have a
poor prognosis unless the tumor is discovered before it metastasizes.
frequently than primary bone tumors. Malignant cells grow within the
bone, replace normal bone marrow, and usually cause bone destruction.
Prostate and some breast cancer metastases stimulate neoplastic bone
formation. Nearly all malignant tumors have the ability to metastasize
to bone, but carcinomas of the breast, lung, prostate, kidney, and
thyroid are the most common. Approximately 50% of patients with breast,
lung, or prostate cancer eventually develop bone metastasis.
Examination of the skeletal metastases reveals cells similar to those
in the primary neoplasm (Figure 10-56, Figure 10-57, Figure 10-58). Soft tissue sarcomas and primary bone tumors can metastasize to bone, but this is extremely rare.
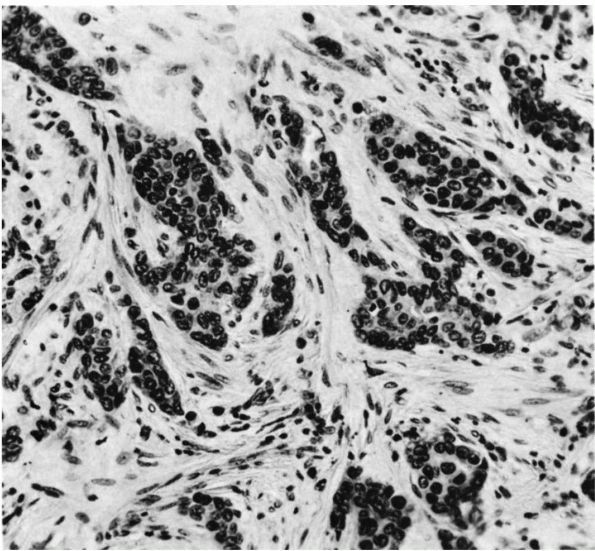 |
|
FIGURE 10-56.
A histologic section showing a metastatic carcinoma of the breast. The lesion consists of islands of neoplastic epithelial cells surrounded by reactive fibrous tissue. |
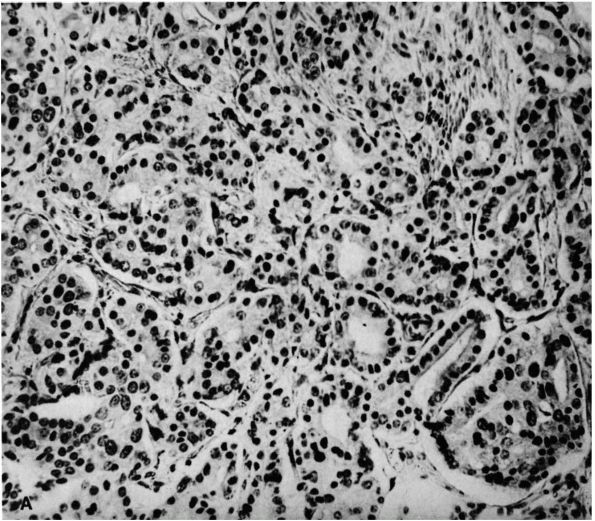 |
|
FIGURE 10-57. A histologic section showing metastatic prostate carcinoma. The lesion consists of gland-forming epithelial cells.
|
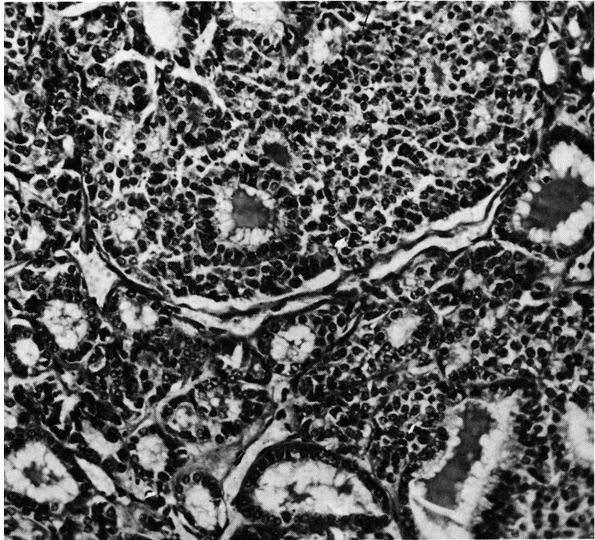 |
|
FIGURE 10-58.
A histologic section of metastatic carcinoma of the thyroid gland. Note the filled spaces with a similar appearance to native thyroid tissue. |
Although bone metastases occur primarily in adults over 45 years of
age, neuroblastomas can metastasize to bone in children. Most patients
with bone pain or a pathologic fracture from skeletal metastases have a
known primary malignant tumor, but occasionally the skeletal metastasis
is the first indication of a primary malignancy. For this reason, the
possibility of metastatic carcinoma should be investigated in
middle-aged and older patients with a destructive bone lesion.
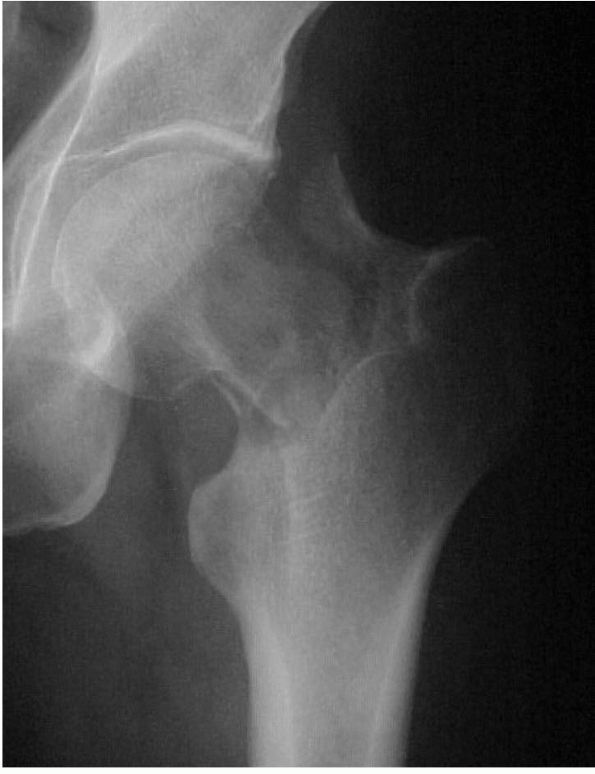 |
|
FIGURE 10-59. A radiograph of the hip showing a pathologic fracture from metastatic renal cell carcinoma.
|
 |
|
FIGURE 10-60.
Thoracic and lumbar vertebra with multiple metastasis from breast carcinoma. The roughly spherical metastatic lesions have replaced bone marrow in part of every vertebral body. |
destroy 30 to 50% of the bone before a plain radiograph reveals the
lesion. A technetium bone scan offers a more sensitive method of
detecting metastatic disease, although it may not be reliable for
rapidly destructive lesions such as multiple myeloma or renal cell
carcinoma. Most metastases destroy bone, but those from prostate and
breast cancers can stimulate bone formation and appear as regions of
increased bone density on plain radiographs. If plain radiographs or
bone scans fail to demonstrate suspected bone metastasis, an MRI
scan
may provide more detailed information. In a small subset of these
patients, biopsy of the bone lesion shows a poorly differentiated
metastatic carcinoma, and physical examination, imaging studies and
laboratory studies will not identify the primary lesion.
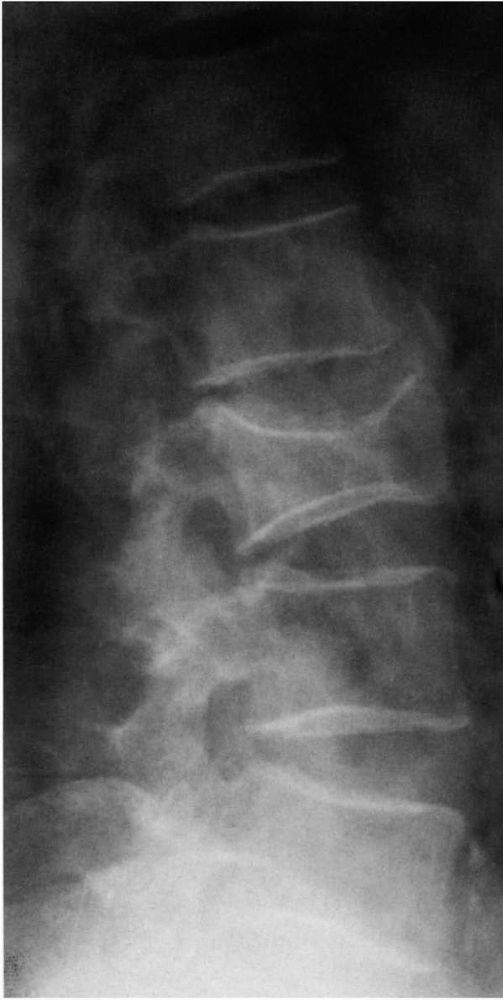 |
|
FIGURE 10-61. Radiograph showing a pathologic compression fracture of a vertebral body caused by metastatic breast carcinoma.
|
preserve musculoskeletal function and relieve pain. Options include
prophylactic internal fixation or prosthetic reconstruction of involved
bones to prevent pathologic fractures. New implants and techniques are
available to provide durable reconstructions. Methyl methacrylate is
often used as a local adjuvant to add strength to the metal construct.
Postoperatively, radiation is often effective in maintaining local
control. Bisphosphonates are now used routinely in patients with lytic
bone metastasis and inhibit osteoclast-mediated bone destruction.
can involve all areas of the musculoskeletal system. They vary in
clinical behavior from developmental disturbances that resolve
spontaneously to benign neoplasms that can grow and destroy bone to
highly malignant tumors that cause death, despite aggressive treatment.
suspect neoplastic disease in a patient with vague discomfort,
swelling, loss of musculoskeletal function, or incidental radiographic
abnormalities. In addition, common benign lesions of the
musculoskeletal system may initially resemble malignant lesions.
imaging studies that suggest the presence of a musculoskeletal tumor
should have a careful evaluation. Based on this evaluation the
physician must decide if the patient can be observed and treated
symptomatically or if the patient should have further testing,
referral, or treatment. Conditions requiring immediate treatment
include pathologic fractures, impending pathologic fractures,
neurologic deficits, and uncontrolled pain. In most instances, the
definitive evaluation and biopsy of a lesion that may be malignant
should be performed by an orthopaedic surgeon with experience in the
management of these problems.
author reviews the clinical presentation and surgical treatment of bone
and soft tissue neoplasms. The first section of the book discusses the
presentation, natural history, and staging of musculoskeletal
neoplasms. The second section explains how different anatomic locations
affect the presentation, progression. and treatment of these neoplasms,
and the last section covers specific types of neoplasms.
authors provide in-depth discussions of benign and malignant neoplasms
of the musculoskeletal soft tissues and lesions of the soft tissues
that resemble neoplasms.
FJ, McCarthy EF. Pathology of Bone and Joint Disorders with Clinical
and Radiographic Correlation. Philadelphia: W. B. Saunders, 1998. An
orthopaedic oncologist and musculoskeletal pathologist have combined
talents to provide an excellent teaching guide for diagnosis of
musculoskeletal conditions. This book is limited to disorders of bone
including neoplasms, infection, fractures, and metabolic bone disease.
It is well illustrated with radiographs and classic histology for all
conditions discussed.
detailed text presents the clinical, radiologic, and pathologic
characteristics of bone tumors. The author also summarizes the course
and treatment of each neoplasm.
authors of this book provide an excellent review of the diagnosis and
treatment of metastatic bone disease. They include discussions of
clinical findings, laboratory tests, imaging studies, and biopsy in the
sections on diagnosis. The chapters devoted to treatment include the
general principles of surgical and nonsurgical management and the
specific approaches to metastases in different parts of the skeleton
and to particular types of metastatic neoplasms.
editors compiled a group of experts in the field to provide the most
recent comprehensive overview of the diagnosis and treatment of benign
and malignant bone and soft tissue tumors. Surgical approaches are
outlined for each area of the musculoskeletal system. There is a
separate section on metastatic bone disease.
updated well-illustrated atlas summarizes the symptoms, signs,
radiographic features, pathologic features, differential diagnosis, and
treatment of orthopaedic tumors and lesions that resemble tumors.