
the most common neuromuscular disorders. These disorders present some
of the most challenging problems in orthopaedics.
picture of cerebral palsy (CP) is static, the orthopaedic
manifestations are often evolving, due to the effects of abnormal
muscle tone and movement disorders over the individual’s lifetime. For
instance, one of the first features of CP may be delay in the normal
motor milestones of development. For example, some eventually spastic
patients may first present with hypotonia and be considered a “floppy
baby.” Later the child will demonstrate retardation in normal motor
development. In general, most children are able to sit independently by
6 months and the normal range of walking onset is from 9 to 18 months.
Patients with CP will have delay in these milestones that is concordant
to their level of brain injury. Hemiplegic patients may
walk
in the upper range of normal gait onset or have delays of several
months. Diplegic patients may not walk until 2 to 3 years of age;
severely involved diplegic patients or quadriplegic patients may never
walk. Understandably, families are focused on the potential for
ambulation for their children. Bleck has described a rating system
based on retention of certain neonatal reflexes beyond one year of age;
this system can be used to predict eventual walking. One point is given
for the presence of asymmetric tonic neck reflex, neck-righting reflex,
Moro reflex, an absent parachute reaction, an absent foot-placement
reaction, and extensor thrust. If more than two are present the
prognosis for ambulation is poor. In general, if a child is unable to
sit unsupported by 4 years of age, the potential of independent
ambulation is low.
function and development of every aspect of the musculoskeletal system.
In general, the more severe fixed skeletal deformities result in those
individuals with spasticity. A child with significant components of
movement disorders such as athetosis tends to not develop fixed
contractures. These children will move their extremities about and tend
to provide excellent range of motion therapy on their own. Results of
orthopaedic treatment are less predictable in patients with movement
disorders then in those with pure spasticity. The bulk of treatment
options, which we will discuss, are directed toward those patients with
spasticity as a major feature of their CP.
individualized for each patient and may consist of a variety of
different modalities. Rehabilitative methods include physical and
occupational therapy, casting or orthotic use, seating programs, and
medical management to reduce spasticity (oral baclofen or valium) or
injection of phenol around the motor nerves or botulinum A toxin
injection. The later modality has revolutionized the care of spasticity
in younger patients with cerebral palsy. Botulinum A toxin is a potent
neurotoxin produced by clostridium botulinum that induces a prolonged,
but reversible, paralysis of skeletal muscle. The toxin diffuses
locally and prevents the release of acetylcholine at the neuromuscular
junction. The effects on the neuromuscular junction are not permanent,
but rather reversible, further suggesting the possibility for botulinum
A toxin as a temporary agent. The effect of the toxin is related to the
dose, the concentration of the dose injected, and the location within
the muscles. With time, the temporary blockade is reversed with new
sprouting of nerve fibers, which then tend to retract once the original
toxin effect at the neuromuscular blockade is reversed. In general,
botulinum A toxin lasts for 3 to 4 months with better results seen in
younger patients and patients with initial treatment. Efficacy tends to
wane with repeated doses and increased age of the patients. Although
commonly used to combat dynamic contractures (spasticity) in an attempt
to avoid the morbidity from surgical release of muscle contractures,
some clinicians have attempted to expand the indications of botulinum A
toxin to individuals with muscle contractures. Unfortunately it is
unknown whether the combination of chemical muscle relaxation
(botulinum A toxin) and progressive stretch (serial casting) will
actually make muscles longer.
selective dorsal root rhizotomy (SDR) and intrathecal baclofen (ITB)
pump placement. Selective dorsal root rhizotomy (SDR) is useful in the
4- to 5-year-old diplegic patients with pure spasticity and no fixed
joint contractures who are ambulatory. These children are usually a
result of premature delivery and with a higher association of low birth
weights. The indications for SDR have regional differences; some
centers have broad experience while others tend to utilize orthopaedic
surgery as the primary method to improve function. The primary benefit
of SDR is to reduce spasticity and improve the quality and smoothness
of gait. This translates into more efficient gait with less oxygen
need. These individuals may still require orthopaedic surgery for bony
abnormalities or residual contractures. Patients who undergo SDR should
be admitted for intensive therapy for approximately one month.
Potential complications include development of spinal deformity in 5 to
10% of patients, of which, very few require eventual spinal
stabilization. Surgical treatment of muscle contractures or residual
bony malalignment should be delayed for a year after SDR.
patients with spasticity and may be more effective in patients who have
movement disorders as a component of their involvement. Use of
intrathecal baclofen was first described in 1984 and was shown to
reduce spasticity in selected patients with spinal cord injury and
multiple sclerosis. Long-term ITB was introduced several years later
and is considered safe and effective in reducing spasticity in patients
with CP. The advantages of ITB pump placement are its relative
reversibility
(it
can be removed) and the ability to increase or decrease the effect of
baclofen. The disadvantages include risks of infection, trunk weakness,
spinal deformity, and the need to refill the pumps with baclofen.
Although designed to reduce lower extremity spasticity, both SDR and
ITB pump placement may concurrently decrease some upper extremity
spasticity.
his or her potential for ambulation, sitting, or other activities of
daily living. For instance, a child who is severely involved usually
has diminished function and the orthopaedist may contribute care that
is designed to balance sitting and to prevent contractures that
preclude comfortable sitting. Conversely, a mild hemiplegic patient may
benefit from sophisticated tendon transfers, which may essentially
normalize hand and foot function. In general, more severely involved
patients will present with abnormalities of the entire skeletal system
while more minimally involved individuals have fewer problems with the
spine and hips and present with problems more distal in the extremities.
severely involved patients with cerebral palsy. In a recent survey, we
documented an 87% incidence of scoliosis in 77 severely involved adult
patients with CP. The incidence in hemiplegic patients is much less and
approaches that seen in adolescent idiopathic scoliosis. Spinal
deformity may also develop as a sequelae of ITB pump placement or SDR (Figure 8-1).
The most common spinal deformity is thoracolumbar scoliosis with
attendant pelvic obliquity. In the sagittal plane, these patients may
also have fairly significant lordosis, which is due to concurrent hip
flexion contractures and spasticity of the paraspinous muscles. The
deformity differs from curves encountered in patients with idiopathic
scoliosis. Cerebral palsy curves tend to be long C-shaped curves
without compensatory curves. As such, patients tend have unbalanced
sitting posture.
and his or her level of function. In general, sitting programs,
bolsters, and orthoses have a minimal to no effect in slowing the
evolution or eventual progression of scoliosis. They may have a role in
preventing rapid progression and allow one to delay surgical treatment
in immature patients. The decision to perform surgery is often
difficult and two important indications: (1) severe curves greater than
50° and sitting imbalance that may reduce sitting endurance and
increase pain, and (2) the patient should be of sufficient health and
function in order to gain maximal improvement. Surgery is recommended
for those individuals with head control and some cognitive abilities
such as caregiver or environmental recognition. Surgical treatment may
not be needed in patients who are cognitively devastated and is
contraindicated in patients with severe hip extension contractures.
Such patients will not benefit from improved axial alignment as the
hips preclude sitting balance.
complex and involves a great deal of time discussing the benefits
versus the risks of surgery. Families should be given the opportunity
to decide if the risks outweigh the benefits, and each decision is
individualized. In general, the main benefits are to prevent curve
progression and to improve sitting balance. It could be argued that the
latter doesn’t directly improve the quality of life of severely
involved patients. On the other hand, patients with severe scoliosis
and sitting imbalance are often difficult to care for. If spine surgery
improves the ability of these patients to sit, it is less likely that
they will be bed bound as they age.
pelvic obliquity. Rarely one will encounter less involved ambulatory
patients with curves similar to those seen in idiopathic scoliosis; in
these patients surgical stabilization that corrects the curve without
necessary stabilization to the pelvis is recommended. Fixation is
performed with rods, sublaminar wires, and judicious use of pedicle
screws and hook fixation. Pelvic fixation may be performed with
Galveston fixation or iliac and sacral screws. Many have found good
results with the unit rod construct. Anterior release and interbody
fusion is indicated in patients with severe curves that do not reduce
on unbending or traction films (> 45°) or if pelvic obliquity is
greater than 15° on preoperative traction films (Figure 8-2).
In younger patients, anterior fusion may be indicated to prevent
crankshaft phenomenon due to anterior growth. Others have demonstrated
that posterior segmental fixation and fusion may prevent clinically
significant spinal deformity as a result of continued anterior growth.
Anterior release and interbody fusion is usually performed on the same
day that the posterior surgery is performed. Spinal cord monitoring
with somatosensory evoked potentials is utilized in nonambulatory
patients with voluntary bowel and
bladder
function. However, it is not used in severely involved nonambulatory
individuals with no motor control, as the signals are often difficult
to evaluate in those severely affected individuals. Practically
speaking, improved spinal alignment is more beneficial even if the
severely affected individual without any voluntary muscle control
suffers some neural compromise.
 |
|
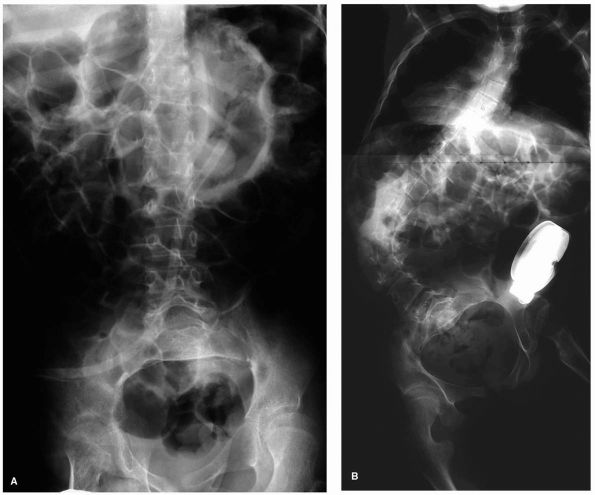
FIGURE 8-1. (A)
AP radiograph of a 15-year-old male with severe spastic quadriplegia. Prior to placement of baclofen pump demonstrating a left thoracic curve with a Cobb angle of 10°. (B) AP radiograph 17 months after baclofen pump placement demonstrating a left thoracic curve with a Cobb angle of 76°. |
spinal surgery is long and includes risk of infection, blood loss and
need for transfusion, and anesthetic complications such as aspiration
pneumonia. The complication rates, and especially rates of infection,
seem to be higher in patients who are more severely involved and with
poor nutritional status. Careful nutritional assessment is needed to
optimize the nutritional status prior to surgery. Some patients begin
central venous nutrition in the immediate postoperative period. Often
these patients are chronically malnourished, and it is difficult to get
adequate caloric and protein intake following large surgeries. Dying
within 1 year of surgery may result from complications from surgery at
a rate of about 1 to 3%; this is certainly more
common
in the more involved patients with CP spinal deformity. Other
complications include pressure sores from new pelvic position and
superior mesenteric artery syndrome. The later is a result of
significant surgical correction and poor nutrition. These patients will
present with vomiting, obstruction, and potentially fatal electrolyte
abnormalities with prolonged vomiting. Treatment of obstruction is with
hyperalimentation via a feeding tube placed past the obstruction.
 |
|
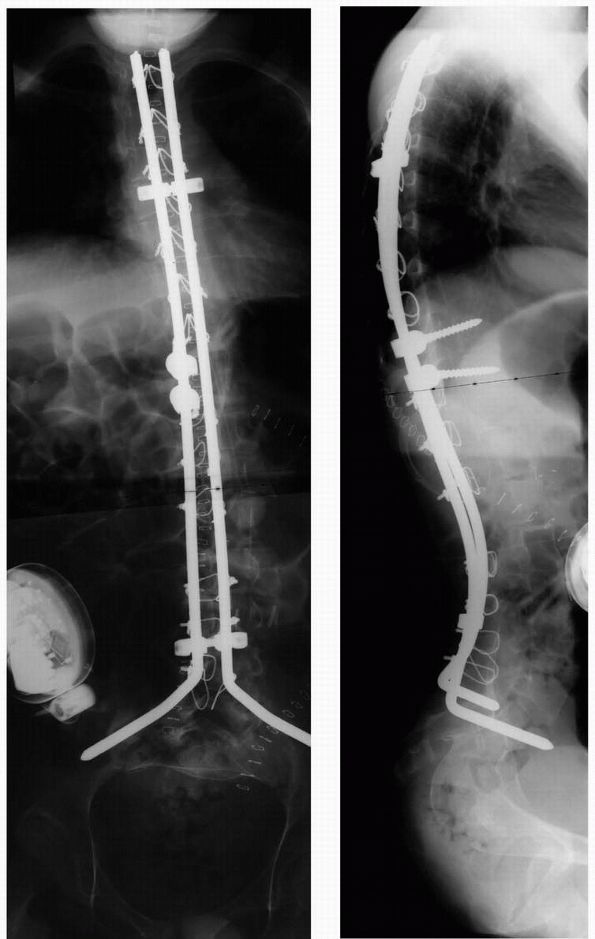
FIGURE 8-2. AP and lateral radiograph after anterior release and posterior instrumentation and fusion.
|
the most challenging to control. These patients will development muscle
spasms in the lengthened tendons that result in increased pain in cut
muscles leading to more spasm and pain. The use of epidural catheter
placement and infusion with local anesthetics, narcotics or clonidine
produces good results. Supplemental intravenous narcotics and
benzodiazapenes are helpful to keep these patients comfortable in the
challenging postoperative period.
yet a broad spectrum of hip abnormalities may develop as a result of
abnormal muscle forces. These abnormalities may range from hip
contractures to hip dislocation. The development of hip deformity in
spastic patients is thought to be secondary to asymmetric muscle forces
produced across the hip by the hip adductors, iliopsoas, and
hamstrings. Persistent femoral anteversion, coxa valga, acetabular
dysplasia and pelvic obliquity often result from retained neonatal
reflexes and abnormal muscle forces and have also been implicated in
the evolution of hip subluxation. The increased muscle forces lead to
hip migration in a posterior and superior direction. Hip migration
leads to femoral head deformity and acetabular dysplasia. One percent
of patients may have dislocation in an anterior direction as a result
of overactive hip extensors (gluteus maximus and hamstring muscles) and
hip abductors. It may be
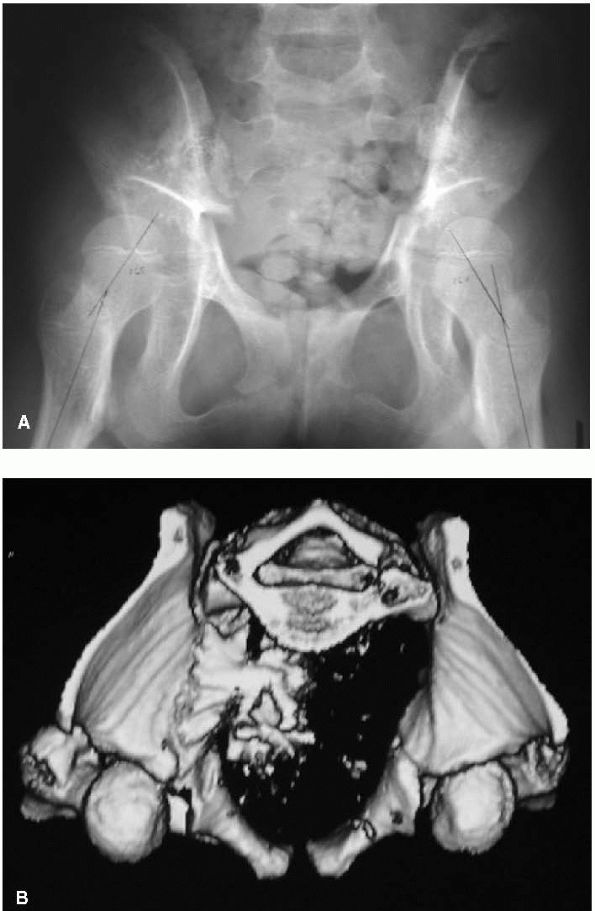
hard to recognize anterior instability on standard radiographs but is better documented on CT studies (Figure 8-3).
may result in a serious problem for affected patients. The incidence of
hip subluxation or dislocation in these patients varies from 2.6 to 75%
seen in more severely involved patients with spastic quadriplegic
patients. Uncorrected hip subluxation or dislocation may lead to later
problems with pain, perineal care, and sitting balance. Patients with
anterior dislocation may be more likely to have pain than posterior
dislocations. Studies estimate the rates of eventual pain as a result
of hip dislocation to be from 0 to 50%. In a recently completed study
of severely involved adults with CP, 15% of hips were dislocated and
radiographic arthritis was detected in 23% of hips.
 |
|
FIGURE 8-3. (A)
AP radiograph of a 9-year-old girl with cerebral palsy. Radiograph demonstrates capacious acetabulum with deficient anterior coverage. (B) CT scan confirms anterior displacement of both femoral heads. |
with CP and include contractures that hinder activities of daily
living, acetabular or femoral dysplasia, and hip subluxation and hip
dislocation with or without adaptive or degenerative changes in the
pelvis and femur. As a result, surgical procedures are recommended in
children with progressive hip displacement in an attempt to prevent
progression and to treat the present state of dysplasia and possible
hip dislocation. Surgery can be classified as reconstruction procedures where the hip is stabilized or reduced with a combination of soft tissue releases, femoral or pelvic osteotomy. Salvage procedures are used in patients
with long-standing and painful dislocation with or without arthrosis.
in hips of CP patients varies depending on the degree of subluxation,
patient age, range of motion, and presence of pain or arthrosis. Soft
tissue releases such as adductor release or transfer, psoas release,
and abductor advancement have been variably recommended for hips
considered likely to progress to subluxation or dislocation. Adductor
release is performed through a transverse groin incision; the adductor
longus is sectioned at its origin once the gracilis has been
identified. Lengthening of the gracilis follows and portions of the
brevis may also be released in order to obtain 45 to 50° of hip
abduction. If a hip flexion contracture greater then 20° exists, the
iliopsoas muscle insertion may also be released off of the lesser
trochanter through the interval of the pectineus and the adductor
brevis. In ambulatory patients it’s better to lengthen the psoas tendon
more proximally. “Over the brim” isolated psoas lengthening provides
better hip flexion power (retained iliacus function) as opposed to
direct removal off of the lesser trochanter (loss of both psoas and
iliacus). Finally, it’s important to remember that the medial
hamstrings are often hip adductors and may need to be concurrently
lengthened. Following soft tissue releases use nighttime bracing for up
to a year. Patients should be followed for several years as the
contractures may recur and progressive hip subluxation may become
evident.
success of stabilization with only soft tissue procedures rapidly
decreases. The decision to perform femoral and acetabular procedures is
dependent on the amount of femoral head coverage and age. Complete hip
dislocation at any age will require bony surgery for stabilization.
Stabilization of the hip is obtained via femoral osteotomy, acetabular
osteotomy, or augmentation, or a combination of these procedures may be
recommended. Femoral osteotomy is recommended for older patients (<
6 years of age) with greater than 50% uncovering of the femoral head.
Concurrent pelvic osteotomy is recommened in older patients with
greater than 50% uncovering. The goals of femoral osteotomy are to
reduce the neck shaft angle by placing the head in varus, shortening of
the femur and with external rotation of the distal shaft. The neck
shaft angle is reoriented 95 to 105° in nonambulatory patients and 110
to 115° in ambulatory patients. A great deal of soft tissue and muscle
tension can be obtained by removing a 2-cm wedge of bone. This may
obviate the need for hamstring release. Derotation of the femur and
reducing the persistent femoral anteversion may be of some benefit in
patients with significant femoral internal torsion. In patients with
asymmetric hip subluxation, one may perform bilateral femoral
osteotomy, to reduce the displaced hip and shorten and equalize the
less-dysplastic side. It may be wise to perform bilateral soft tissue
releases if contractures occur in the contralateral hip. Contralateral
hip subluxation can occur if one fails to recognize abnormal tone in
the contralateral hip. A windswept hip appearance can occur by
releasing muscles only in the more severed hip.
coverage of the femoral head by correcting the dysplasia mentioned
above. These patients tend to have capacious acetabuli with focal areas
of deficiency. Pelvic osteotomies such as the Dega, Pemberton, and the
San Diego osteotomy are effective at reducing the volume of the
acetabulum and improving coverage. The Pemberton works well to improve
anterior coverage occasionally seen in those patients with anterior
dislocations. Triple innominate osteotomies are effective for extensive
and congruent dysplasia.
procedures are chosen and include proximal femoral resection and
interposition of muscle, valgus femoral osteotomy with or without
femoral head resection, hip fusion, or hip replacement. Proximal
femoral resections with muscle interposition have good reliability in
reducing pain in nonambulatory patients but may not completely remove
all of the discomfort. Resection and interposition arthroplasty is
performed through an extended lateral position. Careful extraperiosteal
dissection is performed; the proximal femur is resected at a level that
is equal to a line drawn across the ischial tuberosities. More proximal
resection of only the femoral head and without interposition of muscle
will likely lead to proximal migration and continued preoperative pain
levels. The vastus lateralis and rectus femoris is pulled over the
proximal femoral shaft while the gluteus medius and psoas are sutured
into the hip capsule. The patients will benefit from indomethacin or
other anti-inflammatory medicines to reduce heterotopic bone.
Alternatively a single treatment of 700 rads of radiotherapy is used.
Patients that undergo proximal femoral resection may be stabilized in
traction, with external fixation for several weeks in order to
allow soft tissue healing. A pica cast application with two external fixation half pins in the distal femur is recommended.
patients and is most useful in painful hips with severe adduction
contractures. Hip replacement is most effective in marginal to better
ambulatory patients who require the ability to position the hip in
extension for ambulation and flexion for sitting. Hip fusions have less
utility in these patients.
with CP. Muscle spasticity due to the abnormal stretch reflex is the
primary problem. During physical examination, patients with only
spasticity will have increased tone; however, the joints can be
positioned adequately at near normal extremes of flexion and extension
by prolonged and slow stretch. However, with time, muscle contracture
results from the prolonged spasticity. For example, hamstring muscle
contracture is detected when the knee cannot be fully extended with the
hip flexed at 90°. Hamstring contracture without knee contracture is
present if the knee can be fully extended with the hip extended.
Finally prolonged muscle contracture may lead to fixed contracture of
the joint. In our previous example, a knee contracture can develop from
long-standing hamstring contracture. The evidence showed restricted
knee motion despite placing the hip in any position of flexion or
extension. In this instance, isolated hamstring lengthening would be
expected to have minimal effect on intrinsic knee stiffness. Other
procedures such as arthrotomy or extension osteotomy would be needed to
fully extend the knee.
spasticity and contractures according to each patient’s functional
levels. Ambulatory patients are classified as community
ambulators if they are able to ambulate with minimal assistance in
public. These individuals occasionally require the use of orthotics. Household ambulatory patients require wheelchairs for travel outside the home but can use walkers or crutches to be independent at home. Functional ambulators may stand to transfer and take a few short steps with maximal assist for balance and power. Nonambulatory
patients require full assistance for transfer from the bed to chair and
back. It is intuitive to consider that treatment is more beneficial in
community and household ambulatory patients. Yet on the other hand, it
is important to maintain as much standing as possible in functional
ambulators. This is of paramount importance for parents who see that
their ability to assist and care for the children would decrease with
aging and the increase in size of the children. In these children it is
of benefit for them to have standing abilities maintained by avoiding
severe hip and knee flexion contractures.
household ambulatory patients with CP is often challenging and
treatment has traditionally been selected based on physical exam and
observational gait analysis (OGA). In the early 1980s, methods of
three-dimensional gait analysis (GA) became popular and have evolved
into sophisticated measures of gait. Today, instrumented GA is
performed in motion analysis laboratories and usually consists of
physical exam, videotaping for OGA, and calculation of time-distance
parameters. Kinematic assessment of joint motion and kinetic evaluation
for powers and moments is obtained with the use of reflective markers,
multiple recording cameras, and refined computer software and force
plate data. Finally, surface or fine wire electrodes are often applied
for electromyographic measurement of muscle activity.
ability to document and quantify preoperative abnormalities in all
planes. Such precise assessment theoretically enables the surgeon to
detect all of the pathologic and compensatory components of gait and to
plan and perform all of the procedures required for their correction
during the same anesthetic session. When performed postoperatively,
gait analysis generates objective data that allows for assessment of
treatment and guides further treatment in similar patients. Detractors
of modern gait analysis believe that past methods are perfectly
adequate for assessment of gait abnormalities and possible treatment
interventions. These physicians also cite cost (over $2,000 for each
gait analysis) and the difficulty in reproducing similar data in the
same patient.
be more problematic than others. For instance, diarthrodial muscles
such as the psoas, hamstring, rectus femoris, and gastrocnemius muscles
cause greater difficulty with ambulation. These muscles cross two
joints and are more likely to invoke abnormal stretch reflexes due to
their increased muscle excursion during gait. Common gait abnormalities
as a result of contracture or spasticity
include
equinus gait or back-kneeing in stance (gastrocsoleus), crouch gait
(hamstring and/or psoas), scissor gait (adductors), and stiff-kneed
gait (rectus and hamstring). Selective lengthening or transfer (rectus
femoris) of the pathologic muscles at one operative setting is
reasonable once a child reaches 7 to 8 years of age. Earlier surgery
tends to result in recurrence prior to maturity, and younger children
tend to respond well to nonoperative modalities such as botulinum A
toxin injections and casting or bracing.
significant deforming muscles at the same time. A good example of this
principle exists in a patient with tight tendoachilles and hamstrings
and hip flexion contractures. If the tendoachilles was solely
lengthened, the patient would lose knee extension moment and would tend
to crouch more due to the persistent knee flexion deformity of the
hamstrings. Conversely, release of the hamstrings only would lead to
severe back-kneed gait. Further hamstring release in the face of severe
hip flexion contracture would lead to increase in pelvic tilt.
leading to deformity, such as in the patient with equinus in gait. This
deformity may be treated via lengthening of the Achilles tendon (in
effect, lengthening the soleus and the gastrocnemius) or just the
gastrocnemius fascia. The first surgery is more appropriate when an
equinus contracture exists whether the knee is in either full extension
or in 90° of flexion. Isolated gastrocnemius lengthening by the Strayer
or Vulpius method is better considered in that patient whose foot can
only be dorsiflexed past neutral with the knee flexed. (By flexing the
knee the gastrocnemius is relaxed and the test isolates the soleus
muscle.) In general, medial hamstring contractures are treated in
ambulatory patients by Z-lengthening of the semitendinosis and gracilis
and a fascial lengthening of the semimembranosis. Consideration for
transfer of the gracilis and semi tendinosis is reasonable in the face
of weak hip extensors or marginal hip flexion contracture. Lengthening
of the fascia of the biceps tendon is reasonable in patients with a
concurrent knee flexion contracture or who are nonambulatory. In these
later patients it is perfectly acceptable to simply cut the semi
tendinosis and gracilis without Z-lengthening them.
older patients with recurrent flexion deformities of the hip and knee
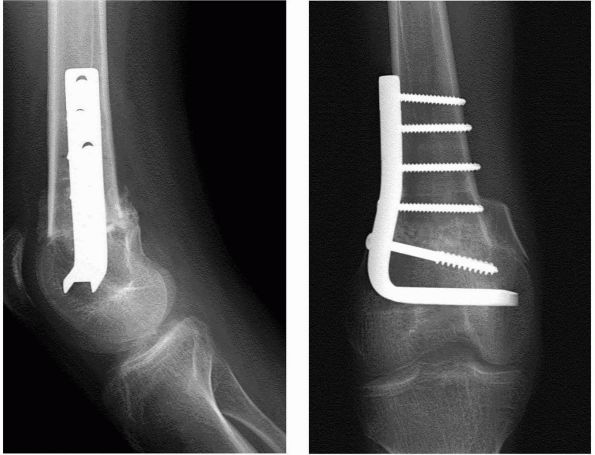
(extension osteotomy) (Figure 8-4) or in
patients with rotational malalignment (external rotational osteotomy of
the femur or internal osteotomy of the tibia). In these individuals
rotational abnormalities can be a hindrance to gait. For instance,
excessive femoral anteversion can lead to significant knee valgus in
stance phase as well as in toeing with the feet hitting in swing phase.
External rotation deformity at the tibia will lead to lever arm
dysfunction. In this scenario, the externally rotated foot presents a
shorter moment arm, thus diminishing the knee extension moment in
stance phase as the leg proceeds from the second to the third ankle
rocker.
involve an equinus component and may have a varus or valgus deformity
of the hind foot. Varus or supination deformity tends to predominate in
hemiplegic patients and some diplegic patients. This is due to various
combinations of overactivity of the posterior tibialis or supination of
the forefoot as a result of anterior tibialis spasticity. It can be
difficult to determine which muscle is the deforming force. If the
posterior tibialis is the deforming force, the foot seems to have more
hind foot varus without foot supination that is seen when the anterior
tibialis forces. Spastic posterior tibialis may be appreciated by
palpating the taunt tendon behind the malleolus. Fine wire EMG can help
determine whether the anterior tibialis or the posterior tibialis is
the deforming force. In normal gait, the posterior tibialis is most
active in stance phase and should be relatively quiescent in swing
phase. The tibialis anterior dorsiflexes the foot at the start of swing
phase due to concentric contraction. In addition, it also fires in the
early stance phase, allowing eccentric lengthening and lowering of the
foot to the floor. Deviations from these patterns on EMG will delineate
the most likely cause of the foot inversion.
selective lengthening or transfer of all or part of the offending
muscles. The most common treatment of equinovarus deformity includes
lengthening of the gastrocsoleus (if the foot is in equinus
irregardless of knee position) or the gastrocnemius (if no equinus
exists when the knee is flexed). The two most common methods of
correcting the foot inversion is via a split posterior tibialis
transfer to the peroneus brevis or with a combination of posterior
tibialis lengthening and split anterior tibialis transfer. In the
latter procedure, the lateral half of the anterior
tibialis
is placed into the cuboid bone. Each procedure has their proponents but
in general each case must be individualized as other methods including
isolated posterior tibialis lengthening or transposition may be more
appropriate. In general, patients should be treated with a
postoperative ankle-foot orthosis (AFO) for at least 1 year. Although
rare, severe equinovarus deformities may exist in the nonambulatory
patients. Surgical treatment is indicated for lateral pressure sores or
due to an inability to provide proper shoe wear. In these cases triple
arthrodesis is a good method to insure a plantargrade foot with low
recurrence rate.
 |
|
FIGURE 8-4. AP and lateral radiograph of distal femoral osteotomy for knee flexion contracture in an older patient with cerebral palsy.
|
quadriplegic patients; these deformities usually do not require
treatment beyond an orthosis. Surgical treatment may be indicated in
cases of extreme deformity with foot sores and pain. Flat foot
deformities may also be seen in ambulatory patients with spastic
diplegia. In these cases, the equinus contracture will lead to collapse
of the midfoot at Chopart’s joints and an increase in forefoot
abduction. The collapse causes a vertical positioning of the talus with
potential for pressure sores; the abducted foot will contribute to
lever arm dysfunction as the foot is externally rotated on the shank of
the tibia. This inefficiency may hinder knee extension in the later
stages of stance phase. Most patients tolerate the planovalgus foot
deformities with little problems, and an accommodative
ankle-foot-orthosis (AFO) is sufficient to prevent problems. Surgical
treatment may be necessary in rare patients with skin breakdown or
pain; treatment options usually require Achilles contracture release in
addition to
subtalar
arthrodesis, sliding calcaneal osteotomy, or lengthening osteotomy of
the calcaneus. In the later operation the calcaneus is cut between the
anterior and medial facets with insertion of a block of bone. The
lengthening of the lateral column of the foot induces reduction of the
talonavicular joint. Long-term results for this operation are pending
and early recurrences may be prevented when the lateral lengthening is
combined with medial reefing of the posterior tibialis and
talonavicular joint. The flat foot deformity is associated with
excessive pronation of the first ray with the development of hallux
valgus deformity. This deformity is rarely symptomatic; however, in
rare cases metatarsal phalangeal joint arthrodesis may be needed.
Standard hallux valgus operations are usually prone to failure and
recurrence.
diagnosing and treating lower extremity problems in CP patients, yet an
increased interest in hand and upper extremity function has
concurrently developed. Patients with upper extremity spasticity yield
a variety of deformities that may affect function as well as cosmesis
and hygiene. The latter issues are of relevant importance in the more
severely affected individuals. Severe muscle contractures lead to
thumb-in-palm deformities and finger and wrist flexion deformities that
may result in poor hygiene and skin maceration. Upper extremity
function may be diminished in the more functional hemiplegic or
diplegic patients at different levels. At the shoulder, patients can
have spontaneous abduction when excited or running. At the elbow,
patients usually have a dynamic flexion contracture with activities or
when excited; the forearm can be pronated relative to the upper arm.
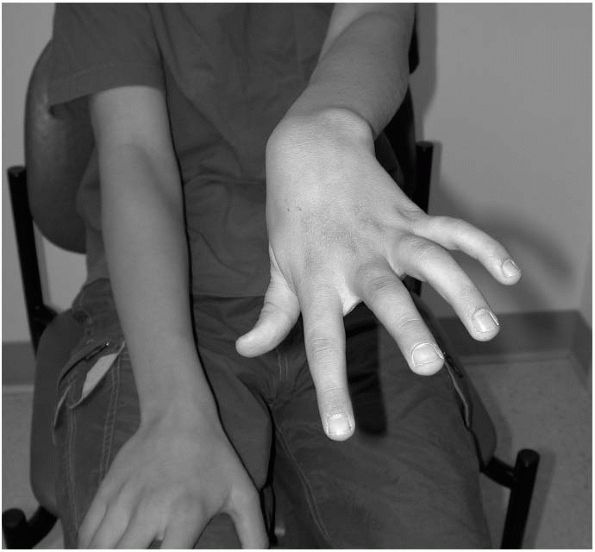
The wrist is often flexed and ulnarly deviated due to increased
activity in the flexor carpi ulnaris or other wrist flexors (Figure 8-5).
Treatment is needed to correct functional problems such as elbow
flexion deformity, diminished pronation, wrist flexion and ulnar
deviation, extrinsic finger contractures, and intrinsic muscle
imbalances. The latter may result in thumb adduction and finger swan
necking.
fixed deformities. Botulinum A toxin may also benefit these patients.
The latter is usually more successful in the upper than the lower
extremity and is a result of the ability to place sufficient quantities
of toxin in the smaller muscles. Surgical treatment of contractures
with lengthening or transfers of muscles and tendons improves hand
function one to two levels. Less functional patients with severe or
fixed deformities may be treated to improve hygiene and cosmesis with
surgical release, transfer, or arthrodesis. Concurrent flexion
deformities of the elbow, wrist, and fingers may be treated with a
flexor-pronator slide. In this procedure the common flexor origin may
retract a great deal when an extensive extraperiosteal release is
performed with dissection of the neurovascular structures. The flexor
pollicis longus may require an isolated lengthening in the face of
severe flexion deformity. Wrist arthrodesis is useful in older patients
or in patients with recurrent or severe flexion deformity. Through a
dorsal approach, a proximal row carpectomy is performed and will
functionally lengthen the wrist and finger flexors. Occasionally, a
volar approach and Z-lengthening or release of the wrist flexors,
profundus tendons, or the superficialis tendons may also be needed. A
dorsal plate is placed across the distal radius, the capitate, and the
third metacarpal and will provide stabilization while the morselized
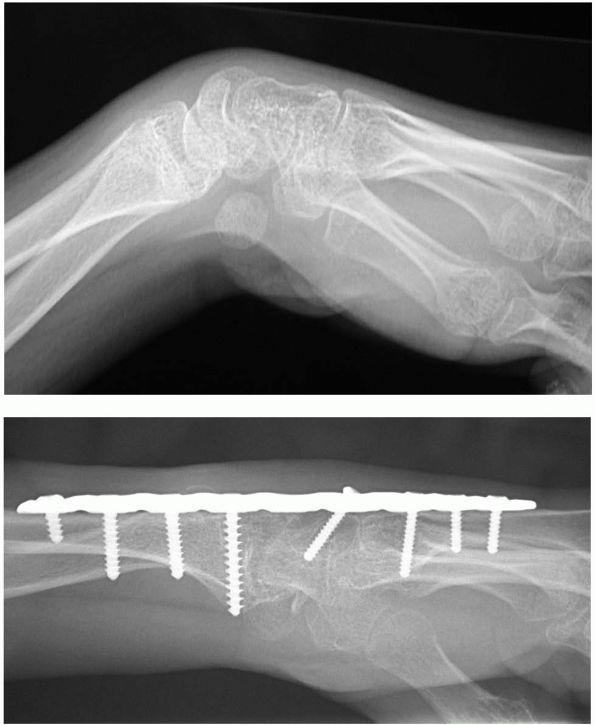
scaphoid, lunate, and triquitrium provides bone graft (Figure 8-6).
Release of thumb adductors and intrinsic flexors improves the position
of the thumb out from the palm. Augmentation of the thumb abductors
prevents recurrence of thumb in palm deformity.
 |
|
FIGURE 8-5. The flexed, pronated, and ulnarly deviated hand is commonly seen in patients with hemiplegia.
|
hemiplegic patients in order to improve function. Prior to surgical
intervention, careful serial physical examinations with an experienced
hand therapist helps identify the functional problems. Use of dual
videotaping and EMG monitoring
helps
delineate which muscles are firing during specific activities. Prior to
surgery, selective nerve blockade may simulate potential results of
muscle transfer or release. In general, surgery should be delayed until
the patient is able to cooperate with postoperative occupational
therapy. In addition, there may be higher chances of recurrence or
overcorrection in younger patients. Adolescent patients tend to be
excellent candidates as they have the maturity to decide if treatment
is in their best interest and, therefore, desire maximum improvement in
function through postoperative occupational therapy.
may be treated with fascial lengthening of the brachialis and
Z-lengthening of the biceps tendon. Concomitant release of the lacertus
fibrosis increases the ability of the biceps to improve supination.
Pronation contracture is treated with isolated release of the pronator
teres or pronator teres transfer. The latter is usually efficacious
when the patient has active pronation without fixed contracture.
Transfer through the interosseous membrane to the dorsum of the radius
converts the pronator into a supinator. Other tendon transfers, such as
dorsal transfer of the flexor carpi ulnaris may also increase
supination power.
 |
|
FIGURE 8-6.
Preoperative and postoperative radiographs in a skeletally mature individual with severe cerebral palsy that underwent proximal row carpectomy and wrist arthrodesis. |
of the wrist flexors (simple lengthening of the flexor carpi radialis
and ulnaris) or increasing the wrist extensor power via augmentation
(central transfer of the extensor carpi ulnaris or brachioradialis).
Dorsal transfer of flexor carpi ulnaris is indicated when a patient has
weak wrist or finger extension in addition to overactive wrist flexion.
Transfer to the finger extensors is indicated when the patient has weak
release and to the extensor carpi radialis brevis when they have weak
grasp. Care is needed to ensure that concurrent finger flexion
contractures are not present and, if so, are treated with fractional
lengthening.
adduction (type 1), thumb adduction with MP joint contracture (type 2),
thumb adduction with MP hyperextension (type 3), and thumb adduction
with flexion deformity of both the MP and IP joints (type 4). Treatment
of thumb deformity requires release of contracted tissues and
augmentation of the weakened thumb abductors and extensors. Adduction
is released with a 2- or 4-flap Z-plasty of the first web space and
subsequent release of the adductor pollicis muscle and tendon as well
as the first dorsal interosseus muscle off of the first metacarpal. If
the IP joint of the thumb is contracted, Z-lengthening of the flexor
pollicis longus is indicated. Hyperextension deformity of the MP is
treatable with MP capsulodesis or MP arthrodesis. Alternatively, this
deformity may stabilize after rebalancing of the thumb muscles. Tendon
transfers to increase thumb abduction include exterior pollicis longus
EPL re-routing through the first dorsal compartment, transfer of the
extensor pollicis brevis to the abductor pollicis longus (APL) tendon,
or transfer of the brachioradialis to the APL. Finger deformities such
as flexion contracture may be treated with release of the flexor
digitorum superficialis or fascial lengthening. Swan neck deformities
are treated with lateral band transfer or flexor digitorum
superficialis tenodesis.
MMC patients. In general, deformities in MMC result from muscle
imbalance due to paralysis of muscles. In CP, joint deformities are
usually a result of asymmetric spasticity in addition to weakness.
Furthermore, most patients with MMC have low to normal intelligence
despite severe motor impairment and, therefore, are able to communicate
effectively with the health care team. On the other hand, MMC patients
tend to have comorbidities such as frequent urinary tract infections
and therefore higher rate of opportunistic infections. These patients
may have unrecognized hormone imbalances and the potential for obesity
and increased energy consumption during ambulation. Patients usually
require self-catheterization for urinary retention; spine fusions may
make it impossible for female patients to perform this task. Fusions
may predispose recurrent skin and joint problems as the inherent
flexibility of joints is sacrificed. As such, adjacent and insensate
skin and joints must accommodate and are therefore predisposed to
breakdown (skin sores and bony Charcot changes). Spine fusions may also
predispose to decubitus ulcers due to stiffening of the spine and
pelvis.
concerned about the potential for independent ambulation. A functional
level of the third lumbar nerve root (medial hamstring function) is
needed to have the possibility of some independent ambulation.
Furthermore, the level of motor function is predictive for independent
ambulation and for the different orthopaedic manifestations. For
instance, patients with a motor level in the thoracic spine will never
walk independently and will have hip instability with the potential for
spinal deformity. Upper lumbar levels (L1 and L2) may have some
ambulation in the early years but as they age and grow this is usually
lost. Spinal deformity is less common but hip instability is more
common than in thoracic-level deformities. Lower lumbar levels (L3 and
L4) maintain ambulatory potential for longer periods of time and have a
slightly lower rate of hip instability issues than in the upper lumbar
level patients. Finally, L5 and sacral level patients have good
ambulatory potential throughout life but are hampered by inefficient
gait from weak hip extensors and abductors (L5 level) and foot
deformities (sacral levels).
not static; there is a tendency to undergo progressive degeneration.
This is partially due to society’s acceptance of people with special
needs and the ease at which affected individuals are able to live. As
patients age and their weight increases, it becomes increasingly more
difficult to ambulate with crutches and braces and more acceptable to
use wheelchairs. In addition, degeneration of the central nervous
system may be present due to several possible causes. These causes
include Arnold-Chiari malformations, tethered cord, shunt malfunctions
and hydrocephalus, and syrinx development. The neurological
deterioration can be sudden or very slow and insidious. It is extremely
important that the orthopaedic surgeon carry out a detailed
neurological evaluation at each visit. Any change in neurological
function may be the first sign that there are problems. Also, any
change in neurological function should generate referral to the
neurosurgeon for appropriate evaluation.
which require treatment by other medical specialists (neurosurgeons,
urologists, developmental pediatricians, and therapists). The mental
development of the child, and the formation of parent-child bonding,
may take precedence over the correction of skeletal deformity,
especially during early infancy. Very few orthopaedic deformities
cannot delay treatment until the child is one year old. Most of the
problems of infancy such as shunt malfunction, feeding difficulties,
respiratory problems, and developmental delay have been addressed.
Orthopaedic treatment should be coordinated with the overall treatment
plan for the child and also considered in light of orthopaedic
deformities of the spine, hips, knees, and feet. For instance, severe
congenital foot deformity in a patient with thoracic level paralysis
(nonambulatory patient) takes precedence over the possible presence of
a dislocated hip. On the other hand, severe kyphosis occasionally seen
in thoracic level patients should be corrected prior to foot
deformities. Correction of the spinal deformity provides more
significant overall patient improvement. The following centers
discussion on anatomic area of involvement in patients with
myelodysplasia.
anatomic features of the spine make treatment a challenge. Issues
pertinent to the immature spine with myelomeningocele include failure
of the neural arch to close, resulting in deficiency in the posterior
elements. The lack of posterior elements increases the difficulty of
obtaining surgical fixation and also decreases the bone available for
fusion. As such, individuals with severe spinal deformity and
myelomeningocele require anterior and posterior spine fusions to obtain
the fusion and improve fixation. In addition, the open posterior canal
increases the incidence of inadvertent injury to the thecal sac.
Sagittal plane abnormalities tend to be more extreme in spina bifida
than in idiopathic scoliosis with extreme lumbar lordosis and risk for
increased kyphosis in the thoracic spine. The posterior musculature of
the spinal erector muscles are anterior to the axis of the spine and
may lead to further kyphosis. In addition, a higher incidence of
congenital anomalies of the spine exists (hemivertebrae, unsegmented
bars, diastematomyelia, lipomas, and dermoid cysts). Patients with
myelomeningocele have high rates of urinary tract infections, which
lead to a higher rate of secondary infection of the spine following
surgery.
myelomeningocele. Most patients with thoracic level paraplegia have
spinal deformity; the incidence falls to 50 to 60% in patients with low
lumbar level paraplegia. In general, the curves tend to be greater at
an earlier age, therefore, increasing the potential for significant
curves with time. If the spinal deformity rapidly progresses, MRI scan
is needed to rule out syrinx in the cervical and thoracic spine. The
lumbar spine is also scanned for dermoid cysts, lipomas, and tethering
of the cord.
than 30°. Bracing may be considered in selected individuals with curves
greater than 30° who are less than 7 years of age. Unfortunately,
bracing is not as efficacious as in idiopathic scoliosis, and
progression of the spinal deformity can be expected. In addition,
problems with skin sores and rib deformities may result from the use of
bracing. Surgical stabilization is indicated in patients with
progressive curves or curves greater than 45°. The goals of surgical
treatment include obtaining a level pelvis, restoring normal sagittal
and coronal balance, and preventing further deformity. In general,
fusion levels are considered to extend from the neutral vertebral body
proximally into the distal stable zone. If questions arise, it is more
advisable to fuse longer than shorter. In general, instrumentation is
planned to avoid ending the fusion in the middle of a sagittal curve.
with pelvic obliquity greater than 15° or in patients with thoracic or
upper lumbar level paraplegia. Unfortunately, fusion to the pelvis may
increase secondary problems, such as pressure sore development.
Importantly, female patients should be informed of the possibility of
increased difficulty of self-catheterization due to stabilization of
the lumbar spine. Anterior interbody fusion is needed to augment the
fusion rates, particularly in the regions of deficient posterior
elements. Fixation of the posterior elements is possible with the use
of pedicle screws; sublaminar wires and cables are placed in the intact
lamina proximally and transverse connectors will prevent collapse of
the spine. Contoured rods may be placed over the ala of the sacrum, or
into the first sacral foramen. Anterior rods and instrumentation may be
used in patients with extreme deformity and increased stabilization can
be expected. Anterior only instrumentation for isolated lumbar curves
without posterior surgery may be considered; however, posterior
instrumentation should be considered in addition to this technique in
patients with larger curves or high level of thoracic
deformity.
Instrumentation should be low profile, due to the poor skin and
problems that can result from deficient soft tissues in
myelomeningocele.
in patients with myelomeningocele and may be treated somewhat
differently than in those patients with scoliosis without congenital
anomalies. In general, anterior and posterior spine fusion over the
apices of the deformity is needed to prevent further progression.
Earlier surgery is indicated in those patients who have congenital
anomalies that are prone to rapid progression, such as contralateral
bars and hemivertebrae. Anterior fusion is done through an open
anterior approach; however, transpedicular approaches can be used to
obtain anterior fusion. Rarely, osteotomies are needed to correct
significant deformities.
severe kyphosis that merits treatment. Children with significant
kyphosis will have difficulty with independent sitting without using
their upper extremities for balancing. Cephalad displacement of the
abdominal contents will restrict breathing, prevent normal nutrition,
and may lead to failure to thrive for these individuals. Try to correct
the kyphosis early in order to improve the ability of the child to sit
independent without the necessary use of the upper extremities. Bracing
is usually ineffectual. In general, kyphectomy is performed by
decancellation of the vertebrae above and below the apical vertebrae
and is done to avoid violation of the endplates and, therefore, the
growth centers of this deformity. Instrumentation is placed
extraperiosteally to allow for future growth. Usually a Luque rod is
contoured and placed over the ala of the sacrum or into the first
sacral foramen. Proximal fixation with pairs of sublaminar wires placed
in an extraperiosteal fashion will allow for continued growth. At
closure, the paraspinal muscles are closed posteriorly over the
instrumentation, reducing them to an anatomic position and decreasing
the propensity for progressive kyphosis. Rigid S-shaped kyphosis may be
treated with cordectomy and resection of the vertebral bodies and
fixation with segmental fixation.
have hip dislocation and dysplasia. The incidence of dislocation of the
hip is 20% compared to 0.1% in the general population. This is
presumably due to altered forces across the hip in utero and in the
postnatal period. Thoracic level patients may have contractures of
their hips due to positioning. Upper lumbar level patients will have
actively firing hip adductors and hip flexors, but will be deficient in
hip abductors and hip extensors. This muscle imbalance leads to higher
rates of dislocation and dysplasia than in thoracic level patients.
Compared to thoracic level and lower lumbar, sacral level patients will
usually have stronger hip extensors and abductors, in addition to
weight-bearing activities, which will prevent dislocations.
to avoid hip contractures, which will preclude comfortable sitting, as
well as standing. Thoracic level patients with dislocated hips or
progressively dislocating hips should not undergo surgery to reduce the
hips. Surgery is only performed in order to prevent significant hip
contractures that preclude sitting or standing. All children who
develop independent sitting by 18 to 24 months of age should be started
on a prone standing program in order to improve their upper extremity
function. Patients with the upper lumbar level function may be
candidates for a reciprocating gate orthosis. In these patients,
swaying of the hips in a side-by-side manner will allow for forward
propulsion. Unfortunately, in order to use this orthosis, patients must
be fairly contracture free.
of hip dislocation and dysplasia than in the thoracic level paraplegic
patients. Historically, efforts have been made to maintain the hip
position in these patients, including muscle transfers. Today,
treatment in these patients is similar to those in the thoracic level
paraplegic patients and surgery is only indicated to prevent muscle
contractures. In the past, was recommended reduction of hips in lower
lumbar patients with the addition of different tendon transfers
designed to augment weak hip extension and abduction. These reductions
include transfer of the psoas muscle to the greater trochanter;
posterior transfer of the origins of the adductor longus, brevis and
gracilis muscles; external oblique transfer to the greater trochanter;
and posterior tensor fascia lata transfer to the gluteal muscle sling.
Controversy surrounds whether these extensive muscle transfers prevent
later dysplasia and improve the function and gait of these patients.
Recently, surgeons have not been reducing dislocated hips in lumbar
level patients, unless patients are experiencing ongoing pain. The
function of these patients whether the hips are dislocated or located
does not seem to vary; and they seem to function whether the hips are
located or not.
should undergo hip reduction and stabilization procedures. Standard
open reduction through an anterior approach and femoral shortening in
older patients may be indicated. If weakness of the hip abductors is
noted, the transfer of the external oblique is probably appropriate.
several problems, including hyperextension deformity, flexion
contracture, and valgus deformities of the knee. The important knee
functions of motion and stability are to be reasonably maintained in
patients in order to allow sitting, standing, and gait.
patients with L3 level of motor function. These patients have active
quadriceps function with no hamstring function thus leading to
hyperextension deformities. These deformities are usually treated well
with physical therapy or casting in the early period. Surgery may be
performed in cases with persistent dislocation of the knee and
inability to obtain flexion. In these patients, the hamstring muscles
can subluxate anteriorly and provide a deforming knee extensor moment
verses the normal knee flexion. These patients are treated with an
anterior VY quadriceps plasty, thus allowing the hamstring muscles to
then drift posteriorly to the axis of the knee.
with myelodysplasia. Patients with deformities less than 20° may be
treated with an anterior floor reaction AFO and progressive stretching.
If contractures are greater than 30°, this degree of flexion will
preclude efficient gait and may lead to anterior knee pain, due to
increased stresses of the patellofemoral joint. Posterior release is
indicated in patients with a significant flexion contracture. In this
technique, the hamstring muscles are transferred proximally to the
insertion of the gastrocnemius muscle. The gastrocnemius muscle can be
released if it is a deforming force for flexion. Posterior capsulotomy
and partial sectioning of the cruciate ligaments may be needed in order
to obtain full extension. In the author’s experience, posterior
capsulotomy is a procedure that works better in younger patients. Older
patients tend to have higher rates of recurrence. Therefore, distal
femoral osteotomy with extension is more reproducible in older
patients. It is important to ensure that the patient has active knee
flexion greater than 100° of flexion, as distal femoral extension
osteotomy will decrease the flexion arc. Anterior physeal stapling may
be indicated in growing patients with knee flexion contracture,
avoiding the morbidity of distal femoral osteotomy.
progressive valgus deformities of the knee. This may be due to
tightness of the iliotibial band and increased valgus moment of the
knee as a result of the Trendelenburg gait. Patients with a valgus
deformity of the knee can be treated with a knee-ankle-foot orthosis.
Selective surgical procedures may include iliotibial band release,
osteotomy of the distal femoral joint and medial distal femoral physeal
stapling.
of foot pathology requires treatment of structural deformity and
balance of the extrinsic and intrinsic foot muscles as well. The
preservation, removal, or transfer of muscle activity must be carefully
considered in relationship to the function of the foot during walking.
For instance, a muscle that functions as a deforming force may not have
the power to fully correct the structural deformity after transfer.
Transfer of a tendon may result in muscle weakness and production of a
secondary problem. In thoracic level and upper lumbar level patients,
the feet should be kept contracture and deformity free to allow
appropriate orthosis wear and prevention of foot sores. This is
important for shoeing and to start standing protocols, which may
promote future independence with activities of daily living. Lower
level lumbar patients have good long-term ambulatory potential and may
require removal of deforming forces in the foot to maximize ambulatory
potential and to prevent areas of high pressure and subsequent pressure
sores. The motion of the joints in the foot should be preserved as much
as possible. This will allow preservation of the shock absorptive
capacity of the foot, and lessen the possibility of ulceration and
joint degeneration. Sacral level patients have at least moderate
plantar flexion power in the triceps surae, yet intrinsic paralysis may
lead to foot deformities consisting of hind foot and forefoot
malalignment.
of foot deformities. The clubfoot deformity is significantly stiffer
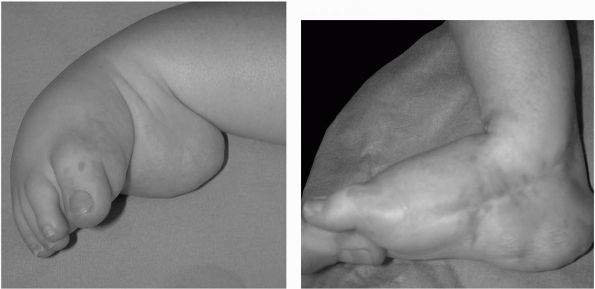
and more resistant to manipulate in comparison to idiopathic clubfeet (Figure 8-7).
As such, correction of the equinovarus deformity in MMC patients is
rarely accomplished by nonoperative means. An attempt to manipulate and
cast the deformity during the newborn period may be worthwhile in the
extremely rare instance when the foot is supple and can be manipulated
into a satisfactory position prior to the application of plaster. If
the foot has not achieved satisfactory correction by the time the
infant is 3 months old, make no further attempts at conservative
treatment; surgical correction is recommended when the child is 1 year
old. Performing the surgery at an age when the child is able to stand
utilizes weight bearing as well as an orthosis to maintain correction.
presents a major potential problem during surgical correction of the
severe hind foot deformity common in the MMC clubfoot. Many different
incisions have been tried, but no approach is completely devoid of
limitations. After skin incision, all 1 to 2 cm of the paralyzed and
spastic tendons should be resected rather than lengthened.
Additionally, in patients with L5 level paraplegia the functioning,
anterior tibialis and the peroneal tendons will often result in a
calcaneal valgus deformity. In these cases, consider resection of these
tendons, as it is better for the patient to have a flaccid braceable
foot than a deformed foot due to muscle activity that is inappropriate
for standing. Limited surgery rarely corrects the clubfoot. Complete
circumferential subtalar release is necessary in order to allow the
calcaneus to rotate sufficiently underneath the talus to reduce varus
and align the axis of the foot with the axis of the ankle and knee.
Postoperatively, the patient is placed into a long leg cast and the
pins are removed at 6 weeks. The child is then fitted for a night
splint and AFOs are worn during the day.
 |
|
FIGURE 8-7.
Preoperative and postoperative photographs from a child with severe clubfoot treated with extensive posterior medial and lateral release. |
frequency in those patients with an MMC. The treatment of the recurrent
deformity depends on several factors including the age of the patient,
the functional level of the patient, as well as the previous treatment
of the clubfoot. In toddlers treat recurrent clubfoot with a repeat
posteromedial release. Recurrent clubfoot deformity may require
talectomy; however, the resultant correction is rarely satisfactory due
to possible recurrence. Less severe deformities may be managed with a
combination of osteotomies such as a tarsal or metatarsal osteotomy to
correct the midfoot deformity and a calcaneal osteotomy to correct the
fixed varus deformity of the heel.
unopposed voluntary or spastic pull of the anterior tibialis muscle,
the toe extensor muscles, or the peroneal muscles. With time, the
calcaneus eventually becomes vertical underneath the talus, promoting
excessive pressure under the calcaneus and preventing the forefoot from
contacting the floor. Nonoperative treatment of calcaneal deformity is
rarely successful in the long term. Simple resection of the offending
tendons at an early age (in the child under 5 years of age) allows the
foot to be brought into satisfactory position for bracing. However, in
cases of more severe deformity with vertical alignment of the
calcaneus, the anterior tibialis tendon is transferred through the
interosseous membrane and attached to the os calcis. The remaining
tight anterior structures are released so that the foot can be brought
into a satisfactory plantarflexed position for bracing. The transferred
tendon rarely provides enough power for braceless ambulation and is
designed to make the foot braceable and ulcer free, not to eliminate
brace use.
contraction of the lateral musculature of the foot with equinus
deformity of the calcaneus; severe cases present with lateral
displacement of the calcaneus from beneath the talus. The deformity may
be flexible or rigid; in the former, a trial of manipulation and cast
treatment followed by an orthosis designed to hold the foot in the
corrected position may be successful. In the rigid form of congenital
vertical talus, an aggressive surgical approach is needed. As in
1-year-old patients, surgery consists of an extensive subtalar release,
resection of deforming tendons, and postoperative ankle release. The
anterior tibialis tendon can either be resected or transferred back to
the neck of the talus. After postoperative cast immobilization, it is
important to protect the foot and ankle with a rigid ankle foot
orthosis to prevent neuropathic degeneration of the ankle joint.
deformity seen in older children. The valgus deformity may occur in the
subtalar joint, in the ankle, or in both sites. Ankle valgus is noted
on radiographs when the fibular physis is above the level of the
plafond, if there is wedging of the distal tibia epiphysis and if the
longitudinal axis of the tibia is not perpendicular to the dome of the
talus. In many cases, the deformity is mild and requires only custom
inserts such as a UCBL. In rare cases, the ankle valgus deformity will
be excessive and lead to decreased knee extension moment in stance
phase. In such instances, correction may be obtained with a
supramalleolar osteotomy that corrects both the valgus and rotation or,
alternatively, a medial tibia epiphysiodesis may be used in cases of
valgus deformity without external rotation of the ankle.
implies that valgus is present in the subtalar joint. Several different
options exist when the valgus is due to the subtalar joint. These
options include medial calcaneal osteotomy, calcaneal lengthening or
subtalar arthrodesis. The use of subtalar arthrodesis will usually
restore normal hind foot alignment but does remove motion from the
subtalar joint. This increases the risk of sores and Charcot-like
changes in the ankle joint as the child matures.
most commonly seen in sacral level MMC. Muscle weakness leads to
dropped first metatarsal and claw toes. The muscle imbalance (weak
peroneals) and plantar flexed metatarsal leads the hind foot into
varus. Children with a cavus deformity have sufficient sensation and
voluntary muscle control to walk without an orthosis. Deformity will
frequently be progressive, resulting in ulcerations on their toes and
over their metatarsal heads. Failure of conservative treatment with
severe calluses or ulcerations and ankle instability due to hind foot
varus is an indication for surgical correction. Cavus is corrected via
plantar fascia release and osteotomy of the metatarsals or midfoot.
Balancing of muscles may include transfer of the IP joints, transfer at
toe flexors to the extensors, and occasionally extensor hallicus longus
recession will be used to increase foot dorsiflexion power. A calcaneal
osteotomy is necessary when a dorsiflexion attitude of the calcaneus is
present and accentuates the cavus deformity. Following osteotomy the
calcaneus is moved backward and laterally as needed to correct cavus or
cavovarus deformities.
degeneration due to lack of protective sensation. This is primarily a
problem in the ambulatory young
adult
who has decreased sensation of the knee, ankle, and foot. The L4-L5
patient appears to be the most vulnerable; however, patients with
paralysis at the S-1 level of paraplegia may also have Charcot changes.
The pathologic process follows a traumatic episode that may be quite
mild. Progressive destruction results after the patient continues to
walk leading to further microfractures and joint destruction. Following
initial trauma there is usually a considerable amount of swelling and
redness around the joint and the appearance of the foot resembles an
infection and cellulitis. Because the initial radiographs are often
unremarkable, the patient may be given antibiotics for the mistaken
diagnosis of infection.
protection following the initial episode and before additional injury
occurs. Immobilization may be accomplished by splinting or casting and
non weight bearing. Typically, the swelling and erythema will subside
after 1 or 2 weeks and complete healing will take approximately 6 to 8
weeks. If the early treatment is successful, then radiographic changes
may never be identified. If redness and swelling recurs after the onset
of weight bearing, then protection must be resumed for a longer period
of time. In some cases diagnosis and treatment are delayed and the
radiograph becomes positive for joint deformity or degeneration.
Prolonged immobilization and protection must be provided until the
process has run its course. With this plan, joint instability and the
development of bony prominences are hopefully avoided. Such treatment
may take up to 6 to 8 months and joint protection should be maintained
until there is radiographic evidence of healing of the joint, and all
swelling and erythema has disappeared.
occasionally present with fractures of the lower extremity following
minimal trauma. These fractures occur in patients who are between 3 and
7 years of age and typically occur in the distal femur and proximal
tibia. Fractures occur at the epiphyseal plate or in the metaphysis.
Presenting signs and symptoms include erythema, swelling, and fever;
moderate elevations in sedimentation rate may be noted. Differential
diagnosis includes the possibility of cellulitis and osteomyelitis.
Fractures should be considered the primary cause, unless other signs
and symptoms and positive cultures are consistent with infection. These
fractures are treated with a bulky soft dressing with splinting for 3
weeks until callus formation is noted and then progressive weight
bearing is started to prevent further osteopenia.
problems and issues regarding the hip, knee, and foot. Treatment is
indicated and individualized to the patient according to the general
medical health of the child, the level of paraplegia and the functional
deficits inherit to each patient.
anterior horn cells of the spinal cord, resulting in distal weakness.
The inheritance is autosomal recessive in nature and consists of
different clinical pictures, based on differences in severity. Spinal
muscular atrophy is classified clinically into three types. Type I is
termed Werdnig-Hoffmann disease. Type II is a chronic form of
Werdnig-Hoffmann disease and Type III is the milder form of Spinal
Muscular Atrophy, termed Kugelberg-Welander disease. No clear
demarcation in the extent of disease, onset of disease and progression
of deformity exists between the three types. However, patients are
often classified into these three categories, even though clinical
presentation may not distinctly place a child into each of the
different groups. In general, the earlier onset of the disease results
in more severe clinical effects and poor outcome.
prior to 6 months of age. These patients present with severe pulmonary
restriction and complications resulting in early death. As such,
orthopaedic treatment is rarely needed except occasionally to provide
immobilization for pathologic fractures in the postnatal period. Type
II or chronic Werdnig-Hoffmann disease is diagnosed after 6 months of
age. These patients never become ambulatory and may live into the
middle decades of life. Type III or Kugelberg-Welander disease is
usually diagnosed after 2 years of age. These patients are ambulatory,
with decreasing ambulation over time, as weakness increases. These
patients have proximal muscle weakness and may have some similar
physical exam features as those seen in muscular dystrophy.
is needed for resultant muscle weakness, which leads to contractures of
the soft tissues, hip instability, and progressive spinal deformity.
Due
to the muscle weakness, muscle contractures become common, and muscle
is replaced with fat and fibrosis. Decreased function and an inability
to ambulate predispose individuals to develop hip and knee flexion
contractures. Physical therapy and orthoses may be of some benefit in
maintaining motion. As these individuals are usually wheelchair
dependent, muscle contractures are rarely severe enough to require
surgical intervention.
and pain, it is desirable to maintain reduction of the hips, even in
nonambulatory patients. Many patients have a life expectancy into the
fourth and fifth decade. Treatment to prevent hip displacement includes
surgical release of those muscle contractures that lead toward
displacement. These treatments include surgical releases of the
adductors and hamstrings. In patients with rapid subluxation, femoral
osteotomy may be of benefit. In patients with dislocated hips, an open
reduction, femoral osteotomy and pelvic acetabuloplasty osteotomy may
be required. Salvage operations, such as Chiari and shelf operations
will maintain posterior coverage, while increasing lateral coverage.
muscular atrophy. All type II patients will develop scoliosis within
the first decade of life. Individuals with type III spinal muscular
atrophy will have a more variable incidence of scoliosis. In general,
scoliosis is characterized as a long, C-shaped scoliosis involving
pelvic obliquity. Occasionally, isolated thoracic curves will be noted.
It is expected that all type II patients and most type III patients
will have progression of the deformity and will require surgical
intervention. Be that as it may, it is a benefit to delay surgery as
long as possible. Therefore, bracing with use of a TLSO, (Thoracic,
Lumbar, Sacral, Orthosis) may be indicated in patients younger than 8
or 9 years with minor curves. Bracing will allow patients to have
improved sitting balance. The main benefit allows increased spinal
growth and obviates the need to consider anterior and posterior spinal
fusion. Surgery is uniformly indicated in patients with curves over 40°
and who maintain good pulmonary function. Pulmonary function is
affected in response to weakness of the intercostal muscles. Pulmonary
function may be further affected by cephalad displacement of the
abdominal contents from severe scoliosis. Decreased complications and
improved outcome can be anticipated in patients whose expected forced
vital capacity is greater than 30 to 40%.
balance, an improvement in self-image, and cosmesis. Similar to spine
fusion in other neuromuscular disorders, posterior spine fusion
instrumentation improves the ability of the caregiver to provide for
the patients. Posterior spine fusion may also decrease or remove the
aching back pain that may occasionally be seen in individuals with
severe deformity. Scoliosis is corrected with posterior spine fusion
and instrumentation to the pelvis in order to reduce pelvic obliquity,
if present. Pelvic fixation is obtained with Galveston instrumentation
or multiple screw placements into the lower lumbar vertebra, sacrum,
and the iliac wing. Due to the significant osteoporosis seen in these
individuals, sublaminar wires at every level are preferred to segmental
instrumentation via hooks. Anterior release and instrumentation is
rarely indicated in these patients, due to the potential for
exacerbating poor pulmonary function. Increased blood loss and need for
transfusion is noted, as well as increased risk of infection, due to
complexity and length of these procedures. Spinal cord monitoring is
utilized. In general, spinal cord monitoring benefits patients with
muscle disease in order to maintain any residual bowel and bladder
function, as well as to maintain the protective sensation, thus
preventing decubitus ulcers.
aggressive pulmonary toilet Continuous positive airway pressure (CPAP)
is often beneficial in maintaining lung ventilation and decreases
atelectasis. Epidural pain control is of further benefit in order to
prevent the splinting of respiration as a result of postoperative pain.
cerebellar degenerative disorders and has an autosomal recessive
inheritance. These individuals present with increased ataxia as the
first presenting symptom. On physical exam, there is a loss of the knee
and ankle reflexes, and a positive Babinski sign. In addition, there is
a loss of proprioceptive and vibratory sense. The average age at
clinical onset is from 7 to 15 years of age, with rare cases of delayed
onset up to 25 years of age. Ataxia and symmetric weakness leads to
progressive decrease in walking and patients often become wheelchair
bound by the second to third decade. Weakness is usually symmetric,
involving the proximal musculature (gluteus maximus), more than in the
distal musculature. Premature death occurs in the fourth or fifth
decade as a result of complications from cardiomyopathy or
pulmonary
dysfunction following complicating events, such as aspiration
pneumonia. The orthopaedic manifestations of Friedreich’s ataxia
include foot disorders and spinal deformity.
have a cavovarus foot deformity as a presenting symptom. Ambulation
becomes difficult as a result of the foot deformity, combined with
ataxia. In order to produce a plantar grade foot, surgical treatment of
the cavovarus deformity requires muscle rebalancing, bony osteotomies,
or fusion. Soft tissue releases include plantar fascia release,
tendoachilles lengthening, transfer of the posterior tibialis tendon,
or lengthening and transfer of the anterior tibialis tendon. When a
transfer of the anterior or posterior tendon in entertained, it is
usually placed into the dorsum of the third cuneiform in the center of
the foot. Residual skeletal deformity of the foot may be managed with
calcaneal and midfoot osteotomies versus a triple arthrodesis.
Friedreich’s ataxia. Several distinct differences in spinal deformity
exist between patients with Friedreich’s ataxia and patients with
spinal deformity as a result of muscle weakness. Patients with
Friedreich’s ataxia develop scoliosis not as a function of muscle
weakness, but as a result of abnormalities in balance. It is theorized
that the spinal deformity in Friedreich’s ataxia is a result of the
perturbations in the proprioceptive and balance systems. This theory is
similar to the theories for the cause of curves in patients with
idiopathic scoliosis. Because of the similarities, the progression,
curve pattern, and treatment are more similar to patients with
adolescent idiopathic scoliosis. Curve onset may occur while
individuals are walking, as opposed to those patients with muscular
dystrophy. In the latter case, curve progression usually develops when
patients become wheelchair dependent. Similar to idiopathic scoliosis,
progression of scoliosis is age dependent; younger patients will have
rapid progression and larger final magnitudes than patients with later
onset of scoliosis. The curve pattern in Friedreich’s ataxia is also
similar to idiopathic scoliosis with greater incidences of double major
curve patterns, single thoracic and thoracolumbar curves. This is in
contrast to scoliosis from muscular dystrophy, which is characterized
by long C-shaped curves and pelvic obliquity.
similar to that in patients with adolescent idiopathic scoliosis.
Bracing may be utilized in younger, immature patients with mild to
moderate curves. Bracing may provide some decrease in progression of
the curve; however, the use of the TLSO may limit the patient’s ability
to walk. Surgery is generally indicated in patients who have curves
greater than 50°. Surgical planning and treatment is similar to
adolescent idiopathic scoliosis, with the bulk of patients requiring
posterior spine fusion with instrumentation. Because the curves rarely
result in pelvic obliquity, fusion to the pelvis is not indicated in
most cases. posterior instrumentation with segmental fixation from the
upper thoracic to the low lumbar region, maintaining curve balance in
both the sagittal and coronal planes is recommended. Unnecessary fusion
to the sacrum with screw fixation or Galveston fixation in an
ambulatory patient may significantly decrease their ability to walk.
Anterior release is indicated in those curves with severe rigidity and
with poor sitting balance.
with noted decreases in ambulation, often heralded with new onset of
toe walking. On initial physical exam, patients have loss in reflexes
and pseudohypertrophy of the calves. Delayed onset of toe walking due
to DMD is uniquely different from that seen in idiopathic toe walking.
These individuals walk on their toes from the very beginning of
independent ambulation. Patients with DMD may also present with
increased tripping, difficulty running, and climbing stairs. These
patients have difficulties as a result of proximal muscle weakness in
the shoulder and hip musculature. Weakness in the lower extremity
begins in the gluteus maximus, quadriceps, and anterior tibialis
muscle. In the upper extremity, weakness is noted in the deltoid,
pectoralis, and trapezius muscles. Pelvic weakness results in an
abnormal gait that is typical for muscular dystrophy. These individuals
have weak hip extensors and, therefore, develop compensatory flexion of
the hips and increased lordosis of the lumbar spine. Muscle
contractures in the tensor fascia lata promote development of a
wide-based gait. Static examination will further reveal contractures of
the tensor fascia lata, with a positive Ober test. In addition,
Trendelenburg lurch results from weak hip abductors. These patients
have further difficulty compensating for the abnormal gait due to
concurrent weakness in the upper extremity that precludes crutch use.
They will present with a positive Gower’s sign, which is
decreased ability to rise from a sitting position without use of the upper extremities.
prevention of contractures which preclude sitting in nonambulatory
patients and prevention of significant spinal deformity. In ambulatory
patients, judicious release of these contractures, in combination with
aggressive physical therapy in the motivated patients can preserve an
ambulatory ability for 2 to 3 more years. Such surgical procedures are
minimally useful in wheelchair-dependent patients. Other treatment
indications include pain from severe muscle contractures.
equinovarus foot deformities. The latter results from a combination of
the hypertrophy of the gastrocnemius muscle weakness of the anterior
tibialis and over-pull of the posterior tibialis muscle. These patients
are treated with percutaneous tendoachilles Z-lengthening and posterior
tibialis transfer through the interosseous membrane. Postoperative use
of an AFO will prevent recurrence. Ambulatory patients with abduction
contractures may benefit from selective release of the iliotibial band
and the fascia lata. Nonambulatory individuals with DMD may also
develop hip flexion contractures, which result in increased lordosis
and occasional back pain. Selective release of the sartorius, rectus
femoris, and tensor fascia lata may improve the hip flexion
contractures in nonambulatory patients. Knee flexion contractures may
occasionally be painful in nonambulatory patients and hamstring
releases can reduce the pain and spasms.
scoliosis. The onset of scoliosis is concurrent with loss of ambulation
in most patients. Rapid progression of the curve can be expected as
soon as a child becomes wheelchair dependent. Most spinal deformity is
a long thoracolumbar curve, with concurrent pelvic obliquity.
Nonoperative treatment for scoliosis may be indicated in selective
patients who have poor sitting balance and who have contraindications
for surgery. Use of an orthosis will not prevent curve progression in
affected individuals, but may help sitting balance in those patients
who are poor candidates for surgical treatment.
for a child with a 25° curve. There is no benefit to waiting as
relentless progression of scoliosis and concurrent decrease in lung
function may be expected. Standard surgical technique includes
posterior instrumentation from T2 to L5 or to the pelvis. Most utilize
instrumentation to the pelvis, especially if the curves are greater
than 40° with attendant pelvic obliquity. Posterior instrumentation
with Luque wiring provides solid fixation. Fixation to the pelvis can
be accomplished with pedicle or sacral screws into the sacrum or the
ilium. Alternatively, a unit rod may be placed into the pelvis via the
Galveston method. Spinal cord monitoring is used throughout surgery to
detect spinal cord injury. Although patients are nonambulatory,
maintenance of bowel and bladder function and protective sensation is
important.
comparison to those with idiopathic scoliosis. Patients with DMD may
have cardiomyopathy; therefore, a preoperative cardiology evaluation is
mandatory. All patients with DMD have a decrease in pulmonary function.
Once the child becomes wheelchair dependant, further loss of pulmonary
function is inevitable. Spinal fusion is reasonably safe in patients
who have a forced vital capacity greater than 30 to 40% of the expected
values. Increased complications from surgery may result when the forced
vital capacity is lower than 30% of expected values. With aggressive
postoperative pulmonary toilet, surgical intervention may still be
possible in patients with forced vital capacity of 20%. However,
families of these patients should be informed of the significant risk
and need for postoperative intubation. Posterior spine fusion with
instrumentation has also been noted to stabilize the loss of pulmonary
function. As such, increased life expectancy can be expected as a
result of posterior spine fusion in patients who have an uncomplicated
postoperative course.
need for transfusion. Higher rates of infection are also present as a
result of the long duration of surgery and as a result of the
significant blood loss. Surgical complications such as hardware failure
have also been reported. The benefit of surgical treatment for spinal
deformity is a clear improvement in the quality of life for affected
individuals by maintaining an erect sitting position and improving
their self image and cosmesis. On the other hand, some patients may
have increased difficulty in feeding, as it is harder to get food to
their mouth with their posture restored to near normal.
mild form of DMD. Although the anatomic distribution and problems are
similar to those seen
in
DMD, the onset of symptoms is later, at approximately 7 years of age or
older. Fortunately, the severity of the problems and the extent of
disability are much less and these patients can expect to be ambulatory
into the teen and young adult years.
Treatment for these deformities is individualized, using some of the
same surgical and nonoperative treatment strategies seen in DMD. For
instance, AFO use may prevent contracture and improve ambulatory
potential. If this fails, tendoachilles lengthening, plantar fascia
release, and posterior tibialis transfer may be needed. Affected
individuals with scoliosis need their treatment individualized
according to the location of the curve and extent of the curve size.
recessive muscular dystrophy. Clinical onset is noted by early muscle
contractures, such as equinus contractures. Paraspinal weakness, spasm,
and tightness of these muscles may lead to cervical spine contractures.
Affected individuals will also have elbow flexion contractures. These
patients can walk for many decades and treatment is individualized to
release any formal contractures of the foot. In rare cases, patients
with this disorder require stabilization of the spine when curves are
greater than 40°.
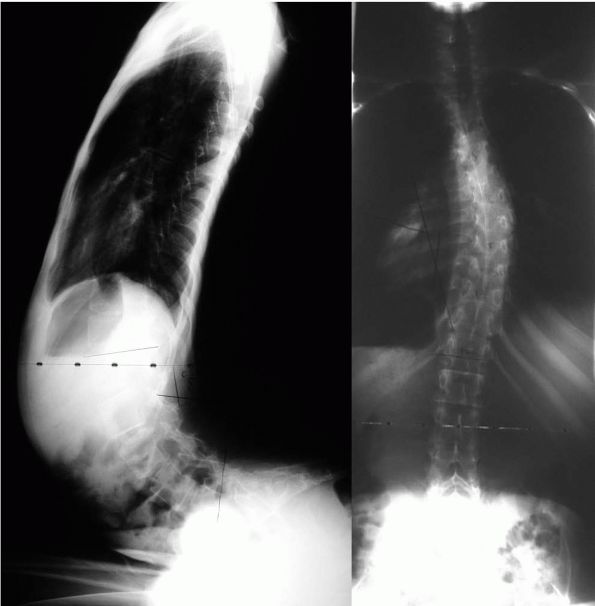 |
|
FIGURE 8-8. Lateral and AP of a 16-year-old boy with Becker muscular dystrophy. Extreme lordosis and minimal scoliosis is present.
|
dystrophy are differentiated according to differences in genetic
pattern. Patients with infantile Facioscapulohumeral muscular dystrophy
have an autosomal recessive inheritance. This is a very severe form of
muscular dystrophy, with onset of facial weakness in the first decade
of life. Patients become wheelchair dependent by the second decade and
will develop pulmonary complications resulting in death in the second
to third decade. Patients have very severe weakness of the hip
extensors, resulting in extreme lumbar lordosis and hip flexion
contractures. In these patients, orthopaedic surgery is rarely helpful in improving function.
variety of Facioscapulohumeral muscular dystrophy has a variable
expression with a less malignant course. The onset of clinical symptoms
is in late childhood and early adolescence. Patients present with
facial weakness and lack of facial mobility; decreased scapulothoracic
motion, and scapular winging is noted. Following onset of facial
abnormalities and scapulohumeral changes, weakness of the pelvic
muscles and anterior tibialis is noted (Figure 8-9).
Patients rarely present with scoliosis. The major orthopaedic problem
is winging of the scapula, resulting in decreased shoulder flexion and
abduction. In these patients, posterior stabilization of the scapula to
the thorax may benefit by improving shoulder flexion and abduction.
These surgical results are reliably maintained over time.
 |
|
FIGURE 8-9.
A 10-year-old female with fascioscapulohumeral muscular dystrophy. Note hyperlordosis and knee flexion secondary to weakness in the hip extensors and triceps surae. |
recessive muscular dystrophy. It is relatively benign in its clinical
course in comparison to patients with Duchenne muscular dystrophy. The
anatomic distribution is similar to that seen in Facioscapulohumeral
muscular dystrophy, except no involvement of the face is noted.
Clinical symptoms start in the second and third decade with weakness in
hip extensors, hip flexors, and quadriceps. The clinical exam is
similar to that seen in Becker and Duchenne muscular dystrophy, except
more accentuated effects may be noted in the shoulder girdle region.
Treatment is initiated in order to prevent and control muscle
contractures, which are similar to those in Duchenne and Becker
muscular dystrophy.
relax after spontaneous or reflexive muscle contracture; and it is this
early feature of myotonic dystrophy that is noted on physical exam.
Other clinical signs of myotonic dystrophy such as gradual atrophy of
the muscle, frontal baldness, and cataracts may be present in older
individuals. Cardiac problems such as arrhythmias or mitral valve
prolapse may be common in patients with myotonic dystrophy. Myotonia
tends to decrease with disease progression as a result of the muscle
weakness. Myotonic dystrophy is inherited as an autosomal dominant
disorder. Succeeding generations may have increased clinical effects
than preceding generations. Clinical onset of functional muscle
abnormalities begins in adolescence to early adulthood. As opposed to
muscular dystrophies, myotonic dystrophy results in more marked
abnormalities in the distal musculature. Early decreased function in
the hands and decreased grip strength is followed by more proximal
abnormalities. As the disease progresses, individuals will have
impaired walking, due to weakness of the quadriceps, hamstring, and hip
extensor muscles. Patients tend to lose their ability to
ambulate
within 15 to 20 years of disease onset. Very few orthopaedic
deformities of myotonic dystrophy merit surgical treatment. Foot
deformities, such as hind foot varus may be amenable to orthotic use.
diverse group of peripheral neuropathies, which have variable
inheritance patterns. The first three HMSNs have a predominant onset in
childhood and adolescence. Hereditary motor sensory neuropathies
IV-VII, typically present in the adult population. Charcot-Marie-Tooth
disease is considered the prototypical hereditary motor sensory
neuropathy. Charcot-Marie-Tooth disease is classified as HMSN I if it
is of the hypertrophic form. The less affected neuronal form of
Charcot-Marie-Tooth disease is considered HMSN II.
deep tendon reflexes and more weakness in the distal extremities, as
opposed to the proximal muscle weakness. The more severe form of
Charcot-Marie-Tooth disease has marked decreased nerve conduction
velocity studies. The milder form of Charcot-Marie-Tooth disease (HMSN
II) has clinically normal deep tendon reflexes with only mild
aberrations noted in the nerve conduction velocities. These individuals
have less weakness and, therefore, lead to later disease onset and less
significant deformity. The diagnosis of HMSN can now be reliably made
via genetic testing.
muscle imbalances; usually resulting in cavovarus foot deformities, hip
dysplasia, spinal deformity, and decreased upper extremity function.
The predominant cavovarus foot deformity results from weakness of the
peroneal muscles and relatively normal power of the posterior tibialis
tendon. This deformity leads to foot inversion, abduction, and a varus
hind foot. Cavus results from contracture of the plantar fascia and
intrinsic muscle weakness. Preferential loss in strength in the
peroneus brevis versus the peroneus longus will also contribute to a
plantar flexed first ray and the cavus deformity. The cavus deformity
and hind foot muscle imbalance (increased tone in the posterior
tibialis tendon, compared to the peroneals) results in a varus hind
foot deformity. This deformity is initially flexible, but may become
fixed with time. Anterior tibialis weakness results in over-activity of
the extensor tendons to the toes. This weakness combined with intrinsic
contracture leads to claw toe deformity.
observation, orthotic use, and surgical treatment. The nonoperative
methods are reasonable in patients with mild or nonprogressive
deformities. Surgical indications include progressive deformity, ankle
instability, and pain due to lateral column overload. Surgical
treatment involves muscle balancing by removing or weakening the
deforming forces and augmenting the deficient forces. The need for bony
surgery in the hind foot is based on whether the hind foot varus is
fixed or flexible. The Coleman Block Test is useful in determining
whether the hind foot varus is fixed or flexible. With this maneuver, a
1½-¾-inch wooden block is placed under the lateral aspect of the foot
and fifth metatarsal. Soft tissue only procedures would be appropriate
if the hind foot can correct to a valgus or neutral position with this
maneuver. Failure to correct the varus deformity implies a fixed
skeletal deformity. In these cases, forefoot balancing procedures are
combined with hind foot osteotomy or fusion.
release and in fixed deformities will require plantar based osteotomy,
either through the metatarsals or the first cuneiform. The peroneus
longus is usually transferred to the peroneus brevis tendon. Supination
and hind foot varus is treated with anterior tibialis transfer to the
third cuneiform if good power is present in the anterior tibialis
tendon. If the anterior tibialis muscle is weak, posterior tibialis
tendon transfer through the interosseous membrane will assist in
balancing the foot. If the individual has weak dorsiflexion, a transfer
of the extensor hallucis longus to the first metatarsal may increase
dorsiflexion power with the added advantage of diminishing the tendency
for clawing at the first ray. Fixed hind foot varus may be treated with
calcaneal sliding osteotomy, closing wedge osteotomy, or hind foot
triple arthrodesis. Tendoachilles release is rarely indicated as most
of the equinus deformity is due to forefoot plantar flexion. Triple
arthrodesis may be indicated in the severely deformed foot. However,
complications with this operation include overcorrection,
undercorrection, and pseudoarthrosis. Adjacent arthrosis in the ankle
and midfoot may occur with longer term follow-up. Toe deformities in
Charcot-Marie-Tooth disease may be treated with tenotomy of the long
toe flexor in flexible deformities. Dorsal transfer of the long toe
flexors may be indicated in more moderate deformities. The latter
operation,
however, has risks of complications to the neurovascular structure.
Other treatment options for toe deformities include a resection
arthroplasty of the Interphalangeal joints.
patients with hereditary sensory motor neuropathy type I and II; a
higher incidence is noted in patients with more severe HMSN I. The
spectrum of hip abnormalities may include dislocation at birth, as well
as progressive acetabular dysplasia, coxa valga, or hip subluxation. In
general, radiographic dysplasia is asymptomatic. On occasion, patients
may present with hip pain and the above anatomic deformities are
subsequently diagnosed. Treatment for progressive or symptomatic
acetabular dysplasia and subluxation involve balancing of deforming
muscle forces across the hip. Femoral and acetabular osteotomies may be
utilized in the rare patients with progressive hip subluxation or
dislocation.
of those in patients with HMSN I or II. Females seem to have a higher
incidence than males, and the deformity is more likely in those
individuals with more severe HMSN I. The spinal deformity is similar to
that seen in adolescent idiopathic scoliosis with usual age of onset at
10 years of age and with development of similar curve patterns, to 0.
In the unusual instance of large curves (35 to 40° in rapidly growing
individuals) orthotic treatment with a brace may be indicated.
Posterior spine fusion with segmental instrumentation and fusion is
indicated in those rare patients with progressive deformities greater
than 50 to 60°. Use of spinal cord monitoring during surgery may be
limited due to the poor nerve conduction inherent to the
demyelinization of the patients with hereditary sensory motor
neuropathies.
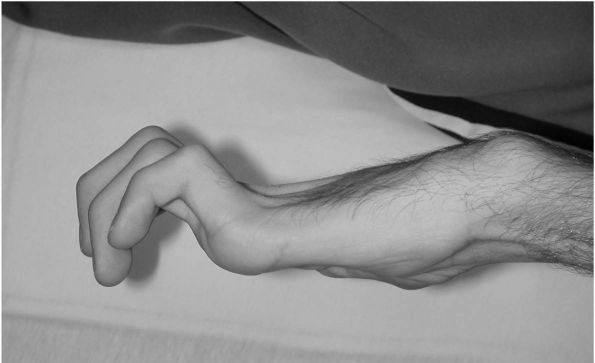 |
|
FIGURE 8-10. Intrinsic minus hand seen in a 20-year-old with HMSN I.
|
deformities may be present in severely involved patients with HMSN I.
These patients have intrinsic weakness, resulting in claw hands of the
second through fifth digits (Figure 8-10). In
addition, decrease in the intrinsic muscles will result in decrease
pinch and opponens function of the thumb. Surgical treatment may be
considered in those patients with severe clawing and would consist of
flexor digitorum superficialis tenodesis.
condition with joint contractures, atrophy of muscles noted on physical
examination. In general, there are over 60 identifiable causes for
arthrogryposis, and all patients have joint contractures detected at
birth. No unifying theory as to the origin of these disorders is
currently identified. Some may be due to in utero infection of some
variety that affects the anterior horn cells; some may have a strong
genetic component such as in Larsen’s syndrome. Yet all have the
characteristic of limited movement of the limb in utero that probably
leads to the fibrosis and stiffness seen at birth. A diagnosis of
arthrogryposis may be suspected in a fetus that has limited movement
and with characteristic deformities such as clubfeet noted on
ultrasound. Once delivered, these infants may have their arthrogryposis
grossly classified according to distribution of the deformities. These
contracture syndromes may involve all four extremities, as seen in
patients with Larsen’s syndrome or arthrogryposis multiplex congenita.
Contractures may involve just the hands and feet and be considered
distal arthrogryposis. Pterygium syndromes include those patients
with
severe pterygia of their major joints. Initial work-up involves
consultations with a pediatric geneticist and neurologist and may
require evaluation with MRI scans and skin or muscle biopsies.
characteristic appearance of the limbs. Limbs appear tubular without
normal contours of the muscles and joints and the overlying skin is
without wrinkles; however, there may be occasional dimples over joints
or muscles. The shoulder is typically adducted and internally rotated;
and the elbow is usually fixed in extension. At the wrist, the hand is
invariably flexed and ulnarly deviated and there may be a thumb and
palm deformity with severe finger flexion. More severe finger deformity
is noted on the ulnar side of the hand as opposed to the radial side.
Hips are usually flexed, abducted, and externally rotated; we have
occasionally seen children with hips and knees in frank breech
position. Knees may be fixed in the extension and the feet usually have
severe, stiff clubfeet. In general, examination of the joints
demonstrates a rigid block to motion with firm nonelastic endpoints.
with large amounts of fibrous and fatty deposition between the muscle
fibers. Myopathic and neuropathic features can be noted in the muscles.
The joints have severe ankylosis with dysplastic and contracted joint
capsules with thickening and fibrosis of the ligaments and joint
capsules. Intra-articular deposits of fibrocartilagenous material may
accompany the contracted joint capsules and be a mechanical block to
reduction in the hips (pulvinar) and the knees. As time progresses,
joint motion is not likely to worsen. Eventually, 25% of patients will
fail to develop ambulation due to stiffness of the joints and with
variable weakness in the extremities.
the lower limb alignment and allow ambulation. Upper extremity
treatment may be geared with the goal to improve the ability of the
hand to feed and assist with perineal care and with transfers. Physical
therapy is important for increasing range of motion, which is started
at an earlier age. Orthotics can be used to maintain a standing
position and also to brace the upper and lower extremity into positions
of functions. In general, surgical release of soft tissues is done at
an earlier age in order to improve joint motion and correct deformity.
It is not uncommon for initial surgical correction to be lost and then
to require further surgical treatment to correct residual or recurrent
deformity. However, bony operations such as osteotomy or fusion are
usually done at a later age to correct residual deformity.
may be noted in 60% of patients. In general, the goals of hip treatment
are to improve motion and allow the hips to be in a functional position
for standing and sitting. Maintaining a located hip based on
radiographic presentation is less important; it is reasonable to accept
bilaterally dislocated hips that are freely mobile without pain. On the
other hand, patients with unilaterally dislocated hips generally do not
function as well as in patients with bilaterally dislocated hips.
Therefore, early efforts to reduce unilaterally displaced hips may be
indicated by some surgeons. Open reduction via an anterior approach is
used; femoral shortening may assist in reducing the head and in
preventing avascular necrosis from surgery. Postoperative reduction is
maintained with spica casting for a short period of time and in a
position of extension to assist functional standing in the future.
have variable deformities of the knee, including flexion deformities,
hyperextension, and dislocated knees. In general, patients with
extension deformities of the knees do better than fixed flexion
deformities, as the extended knee allows for reasonably good standing.
Physical therapy and serial casting may be of some benefit in obtaining
further knee flexion in the cases of fixed extension or hyperextension
of the knee. It can be a challenge to determine the proper plane of
knee motion for appropriate therapy and manipulation and casting.
Gentle attempts of physiotherapy and casting are encouraged to avoid
risks of fracture. Once 90° of knee motion is obtained a Pavlick
harness may be used to maintain reduction. An organized strategy is
needed in those patients with combinations of dislocated hips and
hyperextension deformities of the knees with and without clubfoot
deformities. In the short term, serial long leg casting may be of some
benefit in reducing the clubfoot deformity and increasing knee flexion.
After patients reach 1 year of age, consider simultaneous clubfoot
release and quadriceps VY plasty to obtain final clubfoot correction
and further knee flexion to 100°. Short-term immobilization (5 to 6
weeks) in a long leg cast is utilized and then converted to a short leg
cast so that foot correction can be maintained and knee motion can be
obtained with therapy. Once knee motion is stabilized, an anterior
approach and open reduction with femoral shortening will be considered
in patients with a unilaterally dislocated hip.
tend to limit ambulatory ability. Therefore, soft tissue releases,
including posterior release of the hamstring muscles and joint capsule
are indicated. Many of these patients will have anterior joint fibrosis
and, therefore an anterior release and removal of the pulvinar from the
anterior aspect of the knee is needed to obtain full knee extension. In
older patients with flexion deformities, a distal femoral extension
osteotomy is reasonable. Other surgical options would include anterior
hemiepiphyseal stapling in actively growing individuals or
consideration for external fixation and gradual joint mobilization.
Progressive stretching of the knee with external fixation will improve
the extension of the knee but it must be done in a gradual fashion to
avoid physeal fractures; unfortunately high recurrence rates are often
noted.
present with a very severe clubfoot deformity that is rigid. Treatment
goals for foot deformity are to have the foot flat to allow for weight
bearing and ambulation. In general, nonoperative methods for complete
treatment of this deformity are met with very little success. Other
foot deformities are more likely seen in patients with distal
arthrogryposis (such as congenital vertical talus). The standard
clubfoot release that is needed differs from than used for the
idiopathic clubfoot. For example, extensive capsular releases are
always needed in arthrogryposis, and it is wise to resect portions of
the extrinsic tendons to prevent recurrence of deformity. After surgery
and 4 months of casting, patients should be immobilized in AFOs for
several years to prevent recurrence. Patients with residual deformity
may be candidates for a talectomy at a later age. In rare cases, the
posterior medial lateral release and second stage talectomy does not
completely correct the deformities. In these cases, distal tibia
extension osteotomy may be useful with residual, stiff equinus
deformity.
is usually limited due to the shoulder contracture with internal
rotation and adducted; further upper extremity limitation is due to
extension contractures of the elbows. In order to improve the ability
to feed oneself, it is beneficial to obtain active elbow flexion of one
elbow. Extensive release of the triceps contracture and improved elbow
flexion is obtainable; however ultimate active elbow flexion may not be
possible due to deficient motor muscle power. Treatment may be
indicated if the patient has bilateral elbow extension contractures and
good triceps or pectoralis motor function. In these patients, efforts
are made to improve joint range of motion by doing posterior
capsulotomy, followed by muscle transfer of the triceps or pectoralis.
In general, these surgeries work better in children who are greater
than 4 to 5 years of age with 4+ power and who may be compliant with
the occupational therapy. As in the lower extremity, humeral
osteotomies can be used at a later date in order to improve the
positioning of the arm for activities of daily living.
will have scoliosis. They respond poorly to brace treatment and, in
general, surgery is indicated for curves that are greater than 50°.
the same physical exam features as patients with arthrogryposis.
Larsen’s syndrome may be a sporadically occurring disorder or may be
autosomal dominant or autosomal recessive in inheritance. The autosomal
recessive form seems to be more severe than that found in the patients
with autosomal dominant inheritance. Clinical features of patients with
Larsen’s syndrome include multiple large joint dislocations,
ligamentous laxity, and flat facies. The joints that tend to be
dislocated are the knees (Figure 8-11), elbows,
and hips; in addition, many patients have clubfoot deformities.
Importantly, patients have very severe cervical spine kyphosis. All
patients with Larsen’s syndrome should have regularly scheduled
cervical spine screening radiographs. A certain portion of these
patients will also develop scoliosis.
dislocation of each joint. In general, upper extremity deformities and
dislocation are not treated. With time and development, affected
patients adapt and learn to function well with the current disability.
In the lower extremity, efforts are made to improve knee range of
motion in order to stand and sit. This may involve serial casting to
obtain knee flexion in those patients with extension contractures. Some
patients will have normal motion but will have some instability and are
best treated with a knee-ankle-foot orthosis (KAFO). Surgical
reconstruction for ligamentous exam may be considered. Patients with
Larsen’s syndrome may also have hips affected with acetabular
dysplasia, subluxation, or dislocation. Efforts to reduce hips at an
early age through an anterior or anterior medial approach helps
patients who are highly
functioning
with a normal life span. Consideration for pelvic and femoral
osteotomies may be needed in dislocated hips with severe acetabular
dysplasia.
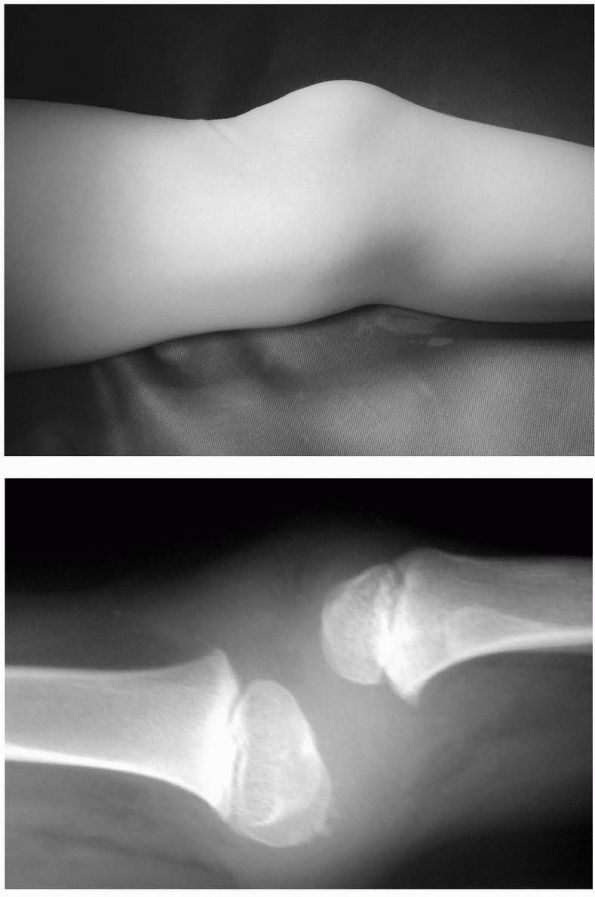 |
|
FIGURE 8-11. A 6-year-old boy with Larsen’s syndrome and a dislocated knee.
|
that in arthrogryposis multiplex congenita and is more amenable to
nonoperative treatment with the Ponseti approach. Although initial
improvement is probable with casting and manipulation, there may be
some residual deformity, which will require later foot reconstruction
or release. Alternatively, initial extensive posterior medial lateral
release may be preferred in these individuals. Try to obtain as much
correction with casting before considering surgical release. Following
surgery, an AFO may be beneficial for prevention of recurrent deformity.
screening lateral radiographs to determine whether there is cervical
kyphosis, which is apparent by radiographic midcervical kyphosis and
hypoplasia of the vertebral bodies. This spinal deformity may be
myelopathic and lead to complications, such as cord compromise or early
death. In
general,
patients should be monitored for myelopathy and curve progression. In
these instances, a posterior arthrodesis is indicated. With time,
posterior tethering via a bone fusion will promote the cervical spine
of the patient to grow into a more normal sagittal alignment.
of the hand and feet; the large joint dislocations and contractures
seen in arthrogryposis multiplex congenita are usually not present.
Patients may also have the facial flattening as seen in Larsen’s
syndrome. In the upper extremity, patients with distal arthrogryposis
will have ulnar deviation of the fingers and flexion deformities of the
IP joints. The hand has been described as a cup-like palm. Individuals
with distal arthrogryposis may have severe foot deformities, including
metatarsus adductus, congenital vertical talus, or clubfoot
deformities. Surgery in the upper extremity is useful to improve hand
function by lengthening the flexor tendons and rebalancing the extensor
tendons. In general, patients with distal arthrogryposis have more
severe foot deformities than hand deformities. Foot deformities in
distal arthrogryposis are slightly more amenable to conservative
management then in arthrogryposis multiplex congenita. Treatment of
clubfeet starts with a series of clubfoot casting to try to obtain as
much correction as possible. Recurrence rates seem to be less in
patients with distal arthrogryposis than those in the standard
arthrogryposis multiplex congenita.
O, Lonstein JE, Winter RB et al. Management of neuromuscular spinal
deformities with Luque segmental instrumentation. J Bone Joint Surg
1989;71A:548. Average 3-year follow-up of 46
patients with neuromuscular scoliosis. Twenty-two of the 46 patients
with neuromuscular scoliosis. Twenty-two of the 46 patients (48%) had
cerebral palsy. The authors discuss the indication for surgery, need
for pelvic extension of the instrumentation and fusion, role of
postoperative immobilization, role of two-stage procedures, and
complications in this most difficult group of patients.
MJ, Banta JV, Renshaw TS. Spinal fusion augmented by Luque-rod
segmental instrumentation for neuromuscular scoliosis. J Bone Joint
Surg 1989;71A:32. Although less than half of the
patients in the series had cerebral palsy, this diagnosis accounted for
the largest group. With a mean follow-up of 42 months, the authors
demonstrate that Luque rod segmental instrumentation with posterior
spinal instrumentation is an effective treatment for patients with
neuromuscular scoliosis. Failure rates were higher when 3/16-inch rods
were used, and functional kyphosis occurred above the fusion when it
did not extend to the upper thoracic spine.
simple procedure to relieve pain and maintain sitting position in
severely involved patients with dislocated hips is described.
RL, Allen BL. Considerations in the treatment of cerebral palsy
patients with spinal deformities. Orthop Clin North Am 1999;19:419. Review article covering all aspects of decision making with regard to spinal deformity in patients with cerebral palsy.
JR, Fabian D, Hicks R, Tashman S. Pre- and postoperative gait analysis
in patients with spastic diplegia: a preliminary report. J Pediatr
Orthop 1984;4:715. A study of 20 patients with
diplegia preoperatively and 6 to 18 months postoperatively. This
article shows the potential benefits of gait analysis in a group of
patients with complex gait abnormalities. By gait analysis criteria, 13
patients were improved, 6 were unchanged, and 1 was worsened by surgery.
WK, Renshaw TS. The treatment of scoliosis in cerebral palsy by
posterior spinal fusion with Luque-rod segmental instrumentation. J
Bone Joint Surg 1988;70A:41. Review of 33
patients with cerebral palsy who underwent posterior spinal fusion from
Luque rod instrumentation at a mean follow-up of 40 months. Luque
rodding accompanying posterior spinal fusion allows safe correction of
the deformity, maintenance of correction, and achievement of a solid
spinal fusion with minimal complications.
article explicates the surgeon’s role in treating the infant and young
child. The comments on physical therapy are enlightening.
MM, Stein GA, Koffman M et al. Femoral varus derotational osteotomy in
spastic cerebral palsy. J Bone Joint Surg 1985;67A:1229. The
indications for and outcomes of this commonly performed procedure in 20
patients are presented. Good results, including decreased pain and
maintained walking ability were found.
T, Schoenberg BS, Kurland LT, Groover RV. Cerebral palsy: survival
rates, associated handicaps and distribution by clinical subtype
(Rochester, MN, 1950-1976). Neurology 1985;35:900. This
review of a cohort of cerebral palsy children in Rochester, Minnesota,
emphasizes the correlation between intelligence and survival and the
common resolution of mild motor deficits.
article discusses incidence of scoliosis in cerebral palsy and methods
of management, including seating orthoses, bracing, and surgery.
article discusses the magnitude of the problem of scoliosis mainly in
spastic quadriplegics and emphasizes the pattern of curve, the
association of severe curves with severe motor involvement, and the
lack of correlation of curve with hip dislocation.
excellent review of factors associated with cerebral palsy derived from
the study of 54,000 consecutive births at several institutions. The
major predictive factors were maternal mental retardation, birth weight
below 2,001 g, and fetal malformation. Breech presentation was a
predictor, whereas breech delivery was not.
J, Hoffer MM. Pre- and postoperative dynamic electromyography as an aid
in planning tendon transfers in children with cerebral palsy. J Bone
Joint Surg 1977;59A:531. A classic study
demonstrating the use of dynamic EMG in improving outcomes of surgery
especially about the foot and ankle for the common equinovarus
deformity as well as other deformities.
is a rare long-term follow-up in cerebral palsy of 242 patients who had
500 operations. This article is part of the basis of pessimism in
orthopaedic surgical treatment of this disorder. A high rate of failure
of soft tissue surgery was found. Bone surgery was recommended as more
predictable and lasting. The difficulty in effecting improvement in
athetoids is emphasized.
brief review of this difficult area. This article emphasizes patient
selection criteria and offers specific treatment recommendations.
superb investigation into the cause of crouch gait emphasizing the
primacy of weakness of the triceps surae resulting from over
lengthening of the Achilles tendon in causing this problem.
DH, Schottstaedt ER, Larsen LJ et al. Clinical and electromyographic
study of seven spastic children with internal rotation gait. J Bone
Joint Surg 1969;51A:1070. This article
thoughtfully, carefully, and with explicit assumptions evaluates the
clinical outcomes of treatment in this difficult patient population.
Although derotational osteotomy is commonly done for internal rotation
deformity, this is an excellent article exemplifying an approach to
treatment and documentation of results.
JG, Simon SR. Progression of scoliosis after skeletal maturity in
institutionalized adults who have cerebral palsy. J Bone Joint Surg
1988;70A:1290. This study discusses the natural
history after maturity of scolitic curves in cerebral palsy. More rapid
curve progression was found in those patients with greater than 50°
curves at maturity.
M, Olson J. Factors affecting the ambulatory status of patients with
spina bifida cystica. J Bone Joint Surg 1983;65A:350. A
careful assessment of factors affecting ambulatory status in 98
patients is presented. Overall, all sacral and L5-level patients
walked; thoracic through L3-level patients were general not functional
ambulators.
series investigated the effect of hip stability on function in 24
patients. Of those undergoing surgery to stabilize the hip, 45% failed.
No functional improvement was found because of hips being located. The
worst function by level were those having complications following hip
surgery, mainly stiffness.
DG, Menelaus MD. The results of transfer of the tibialis anterior to
the heel in patients who have a myelomeningocele. J Bone Joint Surg
1986;68A:1258. A long-term follow-up of 46
transfers to avoid or treat the disabling calcaneus deformity resulting
from triceps surae weakness. Several overcorrections requiring further
surgery occurred, prompting the recommendation that spastic anterior
tibialis muscles not be transferred to the calcaneus.
article describes the results of iliopsoas transfer to the greater
trochanter with adductor longus weakening. Good results with few
complications are presented. The authors recommended this surgical
procedure to balance muscle forces about the hip and maintain hip
reduction.
excellent review of the treatment and complications of the common
rotational deformities seen in ambulatory patients. Fifty children are
described with 66 limbs treated. A significant number of complications
occurred in tibia derotational osteotomies.
E. Saker D, Blatt T. The effect of hip reduction on function in
patients with myelomeningocele. J Bone Joint Surg 1978;60A:169. A
study of 76 patients was done with 41 patients having had no surgery on
the hip and 35 having had attempts to surgically stabilize the hip. No
difference in functional abilities or pain was found regardless of
whether the hip was located, subluxated, or dislocated. Complications
from surgery occurred in 40% of patients. A level pelvis and free hip
motion were important for function.
P, Lindseth R, Campbell R et al. Scoliosis and hydrocephalus in
myelocele patients: the effects of ventricular shunting. J Neurosurg
1979;50:174. Progressive scoliosis may be due to shunt malformation. The importance of neurologic examinations at each visit is stressed.
review of 56 patients revealed no functional ambulators at the thoracic
level and all functional ambulators at the sacral level. Lumbar level
patient’s walking abilities were affected by level (high, versus low),
age, mental retardation, lower extremity deformities, and social
situations.
JS, Gillespie R. Management of myelomeningocele kyphosis in the older
child by kyphectomy and segmental spine instrumentation. Spine
1986;12:37. Short-term review of 12 patients
treated by kyphectomy and segmental spinal instrumentation. The authors
present a method of anterior fixation of the pelvis.
RE. Treatment of the lower extremity in children paralyzed by
myelomeningocele (birth to 18 months). AAOS Instruct Course Lect
1976;25:76. A review of the early treatment of
MMC with descriptive data on a large population of affected children. A
simplified scheme for classifying the functional neurologic level is
presented.
review of valgus deformity of the hind foot that emphasizes that the
valgus can exist in the ankle joint, subtalar joint, or both.
J, Menelaus MB, Dickens DRV et al. Efficacy of surgical management for
scoliosis in myelomeningocele: correction of deformity and alteration
of functional status. J Pediatr Orthop 1986;6:568. The
authors evaluated the effect of spinal fusion in 49 patients with MMC.
Combined anterior interbody fusion and posterior fusion and
instrumentation to the sacrum gave the best results with respect to
correction of the deformity; however, the ability to ambulate was
adversely affected.
MJ. Anterior and posterior instrumentation and fusion of thoracolumbar
scoliosis due to myelomeningocele. J Bone Joint Surg 1987;69B:20. The
author demonstrates improved posture and function in 21 of 23 patients
with MMC scoliosis treated by anterior and posterior spinal
instrumentation and fusion. Indications, operative techniques, and
complications are discussed.
small series of 10 patients treated by kyphectomy and various forms of
instrumentation for MMC kyphosis and followed for a mean of 7 years is
presented. A long fusion from the midthoracic region to the sacrum was
necessary to provide long-term stability and to prevent the development
of thoracic lordosis. Operative techniques, risks, and complications
are discussed.
excellent single source for all aspects of the aggressive treatment of
this disorder derived from a large clinical experience in Melbourne,
Australia.
study followed 41 operations in 23 children an average of 2.5 years.
Principles, indications, and techniques are discussed in this short
follow-up that recommends talectomy for rigid neuromuscular clubfeet.
WR, Mayfield JK, Winter RB, Moe JH. Surgical treatment of paralytic
scoliosis associated with myelomeningocele. J Bone Joint Surg
1982;64A:841. Series discussing results and evolution of treatment of scoliosis in meningomyelocele.
of the prevalence, type, and magnitude of scoliosis in 163 patients
with MMC. Of the 163 patients 143 developed scoliosis, 21 (15%) of
which were congenital in origin. Scoliosis severity increased with
higher neurologic level (particularly above L3) and increasing age.
Curve direction correlated with pelvic obliquity but not with hip
dislocation.
general review of evaluation and treatment of MMC by an author with a
large experience. Not all procedures described are popular now, but the
approach is current.
of high lumbar and thoracic level patients were community ambulators in
this study. This is the only report with a large number of high level
patients maintaining functional walking.
patients had 17 feet corrected by talectomy. Follow-up averaged 7.4
years. Nearly all had good hind foot correction, but one-half had poor
forefoot correction.
DA, Lindseth RE. Effectiveness of muscle transfers in myelomeningocele
hips measured by radiographic images. J Pediatr Orthop 1982;2:121. These
authors recommend external oblique transfer to the greater trochanter
and adductor transfer to the ischium to balance muscle forces about the
hip. Good results are reported. The results of other procedures are
desired.
YH, Lonstein JE, Winter RB, Moe JH. Spinal deformities in patients with
arthrogryposis: a review of 16 patients. Spine 1985;10:609. Literature review and results of management of a small group of patients with arthrogryposis.
DS, Cruess RL. The management of the foot and ankle in arthrogryposis
multiplex congenita. J Bone Joint Surg 1978;60B:96. The
range of treatment options is discussed. The use of primary talectomy
in severe, rigid clubfoot deformities is emphasized. Follow-up to near
skeletal maturity in most patients was available.
14 patients with scoliosis, both congenital scoliosis and long C-shaped
curves were observed. Progression to severe and rigid curves was
typical.
excellent review of 114 patients treated over a 15-year period.
Multiple tables help organize the complex problems and treatment of
these patients.
in 18 of 88 patients with AMC was found. The curves were commonly
thoracolumbar and tended to progress early despite brace or cast
treatment.
article outlines criteria for identifying nonambulators, which is
important in developing a treatment plan for severely involved patients.
review of treatment and results in 52 patients. Treatment to gain
functional range in the first year is emphasized. Soft tissue rather
than bone procedures are recommended in the young child.
in-depth discussion of the indications, technique, and complications of
talectomy. The results of 41 operations are reviewed, but follow-up is
short (average of 2.5 years).
R, Williams PF, O’Connor JCB. The 1960’s epidemic of arthrogryposis
multiplex congenita. J Bone Joint Surg 1981;63B:76. This
article describes the epidemiology of this disorder. Specifically, the
high number of new cases presenting in the 1960s is documented. This
suggests an environmental factor as part or all of the etiology.
FR, Hoffman EP, Amos JA. Dystrophin analysis in duchenne muscular
dystrophy: use in fetal diagnosis and in genetic counseling. Am J Hum
Genet 1989;45:362. The use of dystrophin assay to
identify an affected fetus and establish carrier status in the mother
is presented. A discussion of the general use of dystrophin analysis is
presented.
superb clinician’s extensive experience with the muscular dystrophies
and other neuromuscular disorders is detailed in this relatively short
book.
MH, Fenichel GM, Griggs RC et al. Duchenne Muscular dystrophy: patterns
of clinical progression and effects of supportive therapy. Neurology
1989;39:475. A detailed analysis of 283 boys
enrolled in a prospective study. Best available data on natural history
and effects of intervention except surgery of lower extremities, which
was not included in the analysis.
DH, Fischbeck KH. Molecular biology of Duchenne and Becker’s muscular
dystrophy: clinical applications. Ann Neurol 1989;26:189. A
review of the research that lead to the identification of the defective
gene product in Duchenne and Becker muscular dystrophy. Clinical
implications are discussed.
JD. The natural history of spine curvature progression in the
nonambulatory Duchenne muscular dystrophy patient. Spine 1983;8:771. When
the curve progresses beyond 40°, patients experience loss of sitting
balance, decreased sitting tolerance, pain, decreased vital capacity,
and need to use hands and arms to prop the body. Surgery is indicated
to prevent these problems.
LT, Mubarak SJ, Schultz P et al. Correlation of scoliosis and pulmonary
function in Duchenne muscular dystrophy. J Pediatr Orthop 1983;3:347. Forced
vital capacity peaks at the age when standing ceases. It decreased by
4% each year and with each 10-degree increase in curvature. Early
spinal instrumentation is indicated to slow rate of decline of forced
vital capacity.
function (forced vital capacity) declines most rapidly during
adolescent growth spurt. Surgery does not influence the rate of
pulmonary function deterioration.
study of 51 boys with Duchenne muscular dystrophy followed until death
without surgical treatment. All patients developed scoliosis and severe
curves led to difficulty sitting, skin breakdown, and pain. When the
curve exceeded 35°, the vital capacity usually was less than 40% of
predicted. Spinal arthrodesis should be considered when walking becomes
impossible.
KF, Smith SA, Swaiman AF, eds. Progressive Muscular Dystrophies in
Pediatric Neurology: Principles and Practice. St. Louis: CV Mosby
1989:1105. A concise, readable review of clinical characteristics and diagnosis of these disorders.
PJ, Wagner MB, Kaplan JS et al. Predicting the success of reambulation
in patients with Duchenne muscular dystrophy. J Bone Joint Surg
1983;65A:719. A detailed approach to the selection of patients for bracing and surgery is presented.
in 24 patients with Duchenne muscular dystrophy. Discussion of natural
history, curve progression and results of early surgery.
report of 22 surgical fusions. Bracing was ineffective in halting
progression preoperatively. Several techniques were employed. A high
rate of complications occurred.
GA, Drennan JC, Russman BS. Functional classification and orthopaedic
management of spinal muscular atrophy. J Bone Joint Surg 1981;63B:516. Fifty-four
patients formed the basis of a functional grouping with differences in
natural history and orthopaedic treatment among the groups. An
explicit, detailed approach to the management of spine and lower
extremity deformities is presented.
review of orthopaedic problems in 50 patients with a mean age of 11.5
years. Scoliosis is the major problem with timing of surgery and
complications discussed in detail. Loss of function is common after
spinal stabilization. Release of lower extremity contractures is
discussed.
KF. Anterior horn cell and cranial motor neuron disease. Pediatr
Neurology: Principles and Practice. St Louis: CV Mosby, 1989:1086. A brief review of the clinical characteristics, diagnosis and pathology of these disorders.
RB, Bobechko WP. Incidence, natural history and treatment of scoliosis
in Friedreich’s ataxia. J Pediatr Orthop 1984;4:673. This
report on 42 patients found relentless progression even after skeletal
maturity, suggesting a more typical neuromuscular scoliosis that
suggested in the reference by Labelle and associates in patients with
this disorder.
S, Shaw J, Wallis J et al. Genetic homogeneity at the Friedreich ataxia
locus on chromosome 9. Am J Hum Genet 1989;44:518. Two
populations with clinically different Friedreich ataxia (French
Canadians and Acadians from Louisiana) were investigated by genetic
linkage and found to have the same or nearly the same locus of gene
abnormality on chromosome 9. This type of investigation will doubtless
refine classification and improve prognostication in the future.
follow-up of 78 patients showing similar curve pattern and progression
to that found in idiopathic scoliosis. No correlation of curve
progression with weakness was found. Early age of disease onset and
presence of scoliosis before puberty correlated with progressive
curves. Virtually all patients had curves of 10° or more.
RL, Canale ST, Cooke AJ Jr et al. The role of foot surgery in
progressive neuromuscular disorders in children. J Bone Joint Surg
1973;55A:1396. This article reemphasizes the need for triple arthrodesis to obtain and maintain a stable foot in this disorder.
review of 34 patients undergoing foot surgery with an average follow-up
of 7.2 years. Good results are reported using subtalar or triple
arthrodesis to stabilize the foot. Tendon transfers were used as needed
for foot muscle balance and toe deformities.
thoughtful and rational approach to treatment of cavo varus. It
emphasizes the different treatment options available depending on
whether the hind foot is correctable or fixed in varus.
JE, Carr CR. Progressive muscular atrophy of the peroneal type
(Charcot-Marie-Tooth disease). J Bone Joint Surg 1950;32A:27. Forty-five
patients having had foot surgery are reviewed. Sixty-six good results
and 23 unsatisfactory results were obtained using a combination of soft
tissue and bony procedures.
RL, Canale ST, Cooke AJ et al. The role of foot surgery in progressive
neuromuscular disorders in children. J Bone Joint Surg 1973;55A:1396. Fifteen
patients with Charcot-Marie-Tooth disease or Friedriech’s ataxia with
foot deformity were reviewed. Soft tissue and bone procedures other
than triple arthrodesis failed to maintain correction.
SA. Peripheral neuropathies in children. In: Swaiman KF, ed. Pediatric
Neurology: Principles and Practice. St Louis: CV Mosby, 1989:1110. A brief review of the present classification of these disorders.
triple arthrodesis were followed an average of 21 years. Fourteen (47%)
were considered poor results. Some bad results seem secondary to
disease progression, but arthritis in adjacent joints and recurrent
deformity were common. Attempts to maintain foot position without
fusion seemed warranted in light of these results.