
General Disorders of the Musculoskeletal System > 6 – Rheumatic
Diseases: Diagnosis and Management
transudate from synovial capillaries, modified by the secretory
activities of the type B synovial lining cells. They secrete the
hyaluronic acid-protein complex (mucin) that gives synovial fluid its
viscosity and lubricating properties. Glucose and electrolytes are in
equilibrium between synovial fluid and serum. The nutrition of hyaline
cartilage depends on synovial fluid exchanged by diffusion plus
compression and decompression of the cartilage during motion and weight
bearing.
viscous that droplets expelled from a needle tip fall in a long string.
Normal synovial fluid does not clot because it lacks fibrinogen. The
normal volume of fluid in the largest synovial cavity, the knee, is 0.2
to 4 mL. Thus, aspiration of the normal joint may yield only enough
fluid to wet the needle.
culture; (2) less than 100 leukocytes per cubic millimeter, mostly
lymphocytes and monocytes; (3) specific gravity of 1.008 to 1.015; (4)
protein content of 20 to 200 mg/mL, with an albumin/globulin ratio of
1.5:1; (5) mucin content that varies widely and is the main determinant
of the high viscosity; (6) glucose that is normally more than 75% of
the concentration in serum; and (7) no crystals.
maneuver whenever the cause of arthritis is in doubt but especially
when the inflammation is concentrated in a single joint or has an acute
onset. Ruling out infection or crystal deposition as a cause of
arthritis is the strongest indication for synovial fluid analysis
because the treatment of these disorders is substantially different
from that of other causes of joint inflammation. In the case of
bacterial infection, the results of misdiagnosis can be catastrophic.
fluid includes (1) inspection for color, transparency, and viscosity;
(2) cell count; (3) crystal examination; (4) bacterial culture; and, if
bacterial infection is suspected clinically, (5) Gram’s stain. Samples
for both cell count and crystals can be collected in sodium heparin
(green-top) tubes. All of these tests are most reliable when performed
on fresh specimens. Culture of the fastidious gonococcus requires
employing specialized culture techniques within a few minutes of
collection.
the diagnosis of gout, pseudogout, and basic calcium phosphate (BCP)
crystal disease. Examination of fresh synovial fluid on a polarizing
microscope with a first-order red compensating filter permits the
recognition of birefringence in addition to crystal morphology. Sodium urate
crystals are thin, pointed, and strongly negatively birefringent
(yellow if their axis is parallel to that of the compensator). Calcium pyrophosphate dihydrate
(CPPD) crystals are short, thick, and weakly positively birefringent.
Diagnosis is more certain if crystals are seen within phagocytes. BCP
(including hydroxyapatite) crystals may sometimes be identified under
ordinary light microscopy as small, chunky Alizarin red-staining
crystals in clumps.
steroid crystals (flat parallelograms occurring in joints recently
injected with depot steroid preparations).
of synovial fluid analysis, with usual diagnostic implications. Typical
cell counts and fluid characteristics overlap among the
classifications, and only in the case of a positive crystal examination
or culture can a definitive diagnosis be made from fluid alone. Because
septic joints (especially if partially treated) may have low leukocyte
counts, culture should be performed regardless of the fluid’s
appearance.
Furthermore, more than one diagnosis may be present, as in the gout patient with a septic joint.
|
TABLE 6-1. Results of Synovial Fluid Analysis
|
||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
hemophilia. A dark brown fluid full of hemosiderin is seen in
hemophilia and pigmented villonodular synovitis. Fat floating on a
bloody fluid indicates fracture or tumor exposing the marrow cavity to
the synovial space. Floating fragments resembling ground pepper
represent pigmented cartilage fragments in ochronosis. Small white
clots (“rice bodies”) represent fibrin bits seen in some inflammatory
fluids. Phagocytic mononuclear cells with vacuoles may appear in
rheumatoid arthritis (RA) and in reactive arthritis.
fluid analysis, direct visualization by arthroscopy, or arthrography
usually provides most of the information required for diagnosis. The
suspicion of tumor, pigmented villonodular synovitis, tuberculous or
fungal arthritis, sarcoidosis, amyloidosis, or synovial chondromatosis
may be confirmed by synovial biopsy.
tissue is usually formalin fixed for hematoxylin and eosin staining,
but preservation of urate crystals requires absolute ethanol, and
cultures require unfixed sterile samples. The yield of positive
cultures from synovium in tuberculous and fungal arthritis is superior
to that of fluid cultures. Other special staining procedures include
iron stains for hemosiderin in pigmented villonodular synovitis or
hemochromatosis and Congo red stains for amyloid.
cartilage metaplasia of synovial chondromatosis, malignant synovial
sarcoma and chondrosarcoma, metastatic tumors or leukemias, caseating
granulomas (tuberculosis), noncaseating granulomas (sarcoidosis), the
histiocytes and giant cells of multicentric reticulohistiocytosis, and
the periodic acid Schiff-positive macrophages of Whipple disease. Of
course, inflammation is a far more common finding, but unfortunately,
the histology of inflamed synovium is similar in the major rheumatic
diseases. Early in inflammatory arthritis, the synovial lining cells
accumulate, capillaries proliferate, and then mononuclear cells (mostly
monocytes and T lymphocytes) infiltrate the tissue. Finally, in the
chronic phase, complete germinal centers resembling those of lymph
nodes may appear. The exuberant thickening of this hypercellular
synovium resembles granulation tissue and forms long villous fronds.
age related, affecting more than 80% of people over the age of 55. It
is more common in women, especially after menopause. OA in
weight-bearing joints is strongly linked to body mass index. As life
expectancy increases, and the rate of obesity reaches epidemic
proportions, it is no surprise that OA is increasingly common. OA
pathogenesis involves an imbalance between normal cartilage degradative
and repair mechanisms, which results in net loss of cartilage,
hypertrophy of bone, and generation of osseous outgrowths called
osteophytes. OA has a predilection for finger joints, knees, hips,
shoulders, and the spine. Occurrence in an atypical joint, such as an
elbow, can usually be traced to prior trauma, a congenital joint
abnormality, underlying systemic disease, or a chronic crystalline
arthropathy. The heterogeneity of OA arises from the many factors that
can contribute to cartilage damage.
with activity, and relieved by rest, though rest pain occurs in
advanced disease. “Gelling”— stiffness that occurs after any period of
rest—is also common. Morning stiffness, when present, rarely lasts more
than 30 minutes. Symptom severity ranges from asymptomatic disease
diagnosed radiographically to significant pain with functional
limitation. Physical exam findings of the affected joints include
tenderness on palpation, crepitus (palpable friction) with movement,
bony enlargement, abnormal alignment, decreased range of motion, and
sometimes joint effusion.
|
TABLE 6-2. Radiographic Classification of Degenerative Joint Disease
|
||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||||||||||||||||||
osteophytes, asymmetric joint space narrowing, subchondral bone
sclerosis, and subchondral cysts. The severity of OA can be scored
based on these four common features (Table 6-2). Osteophytes,
commonly called “bone spurs,” are osseous outgrowths that most commonly
arise at the joint margins, and are the single most common feature of
OA. Joint space narrowing results from
loss of hyaline cartilage and should be present to some degree in all
joints that exhibit osteophytes. Because cartilage loss is
greatest
where the stress on the joint is greatest, radiographic joint space
narrowing in OA is usually asymmetric, in contrast to the more uniform
pattern that occurs in chronic inflammatory arthritides such as
rheumatoid arthritis. Subchondral bony sclerosis
also occurs in the most stressed areas of the joint, where cartilage is
most thin. Once cartilage is lost, the bone surfaces rub and polish
each other, a process termed eburnation. Eventually, in severe disease, cysts may form in the subchondral bone beneath eburnated or sclerotic surfaces.
interphalangeal (DIP), proximal interphalangeal (PIP), and the first
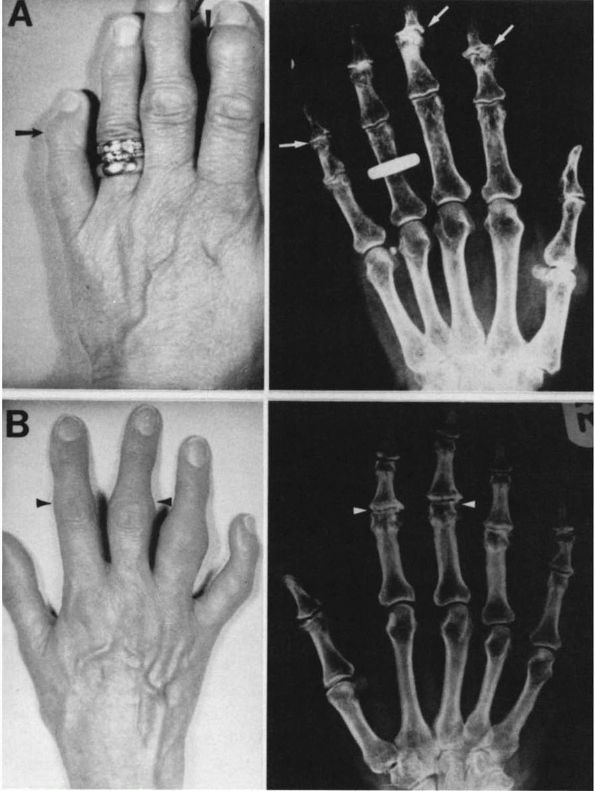
carpometacarpal (CMC) joints. Osteophytes of the DIP joints (called Heberden’s nodes), and of the PIP joints (called Bouchard’s nodes) may be prominent and are sometimes tender (Figure 6-1).
In severe cases, angular deformity, dislocation, or ankylosis may
occur. OA of the first CMC (trapeziometacarpal) joint causes pain with
grasping or pinching and crepitus on movement. Occasionally, the
metacarpophalangeal (MCP) joints are involved but never without
concomitant disease in the DIP and PIP joints. Isolated MCP
degeneration is usually secondary to some other disease process, such
as a recurrent crystal arthritis. Radiographs of the hand and wrist
joints will show uniform or nonuniform joint space narrowing,
osteophytes at the joint margin, bony sclerosis, and sometimes
subchondral cysts. At the DIP and PIP joints, osteophytes have been
described as “seagull wings” because the pattern of the marginal
osteophytes with irregular joint space narrowing resembles a seagull
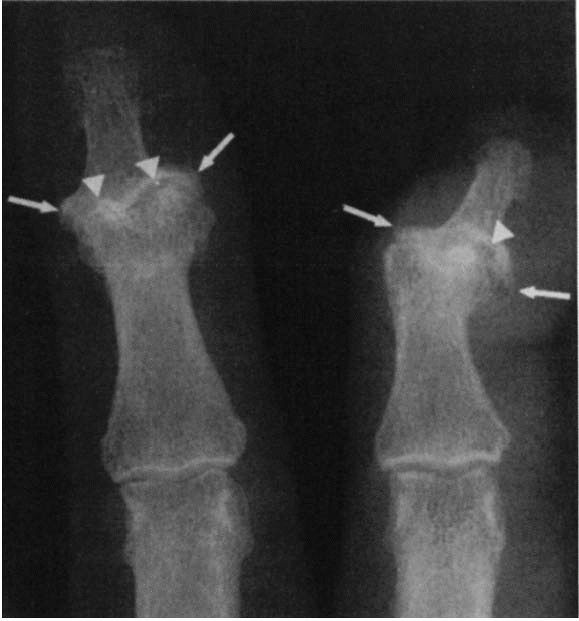
flying (Figure 6-2). Erosive osteoarthritis
of the DIP and PIP joints is a particularly painful disease primarily
of middle-aged and older women, characterized by degenerative changes
plus inflammation. Bone erosions occur centrally, but can also extend
laterally along the joint margin, and coupled with marginal
osteophytes, give a scalloped appearance to the joint on plain
radiographs.
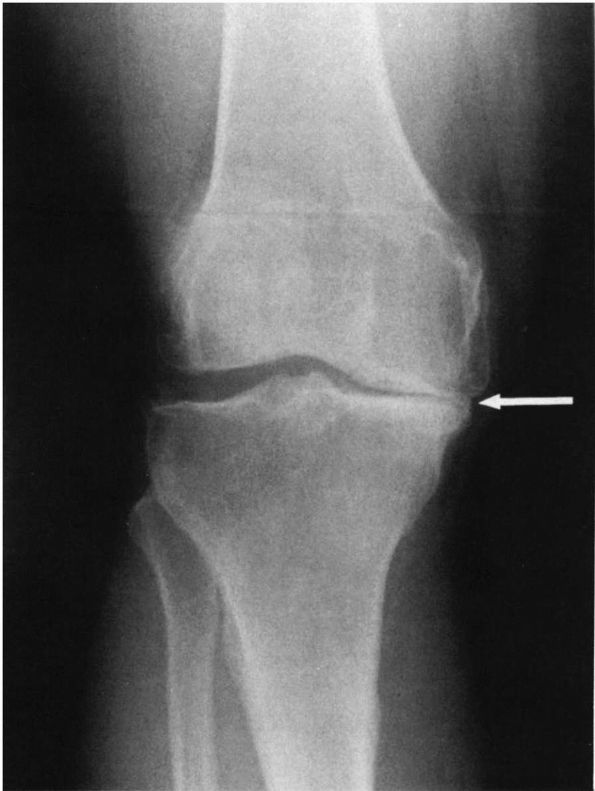
to body mass index. Symptoms include pain with walking, standing up
from a chair, climbing or descending stairs; and stiffness after
periods of rest. Exam findings may include joint effusion, crepitus
with movement, and with more advanced disease, loss of full knee
extension and a palpable osteophytic ridge. Because it bears the most
weight, the medial aspect of the joint tends to degenerate more rapidly
than the lateral aspect, resulting in varus (“bowlegged”) angulation of
the knee, and asymmetric joint space narrowing on weight-bearing
radiographs (Figure 6-3). Although less common,
the opposite pattern of lateral joint space loss and valgus
(“knock-kneed”) deformity does occur. Plain films also typically reveal
sclerosis, with or without subchoncral bone cysts, and osteophytes
arising from the tibial spines, intercondylar notch, and from the joint
margins of the tibia and femur. In patellofemoral disease, merchant and
lateral radiographs of the knees will reveal narrowing of the
patellofemoral space, often with the patellae displaced laterally, and
osteophytes at the superior and inferior poles of the patella. Uniform
tri-compartmental (medial, patellar, and lateral) joint space narrowing
without osteophytes usually indicates the aftermath of an inflammatory
arthritis, such as rheumatoid arthritis.
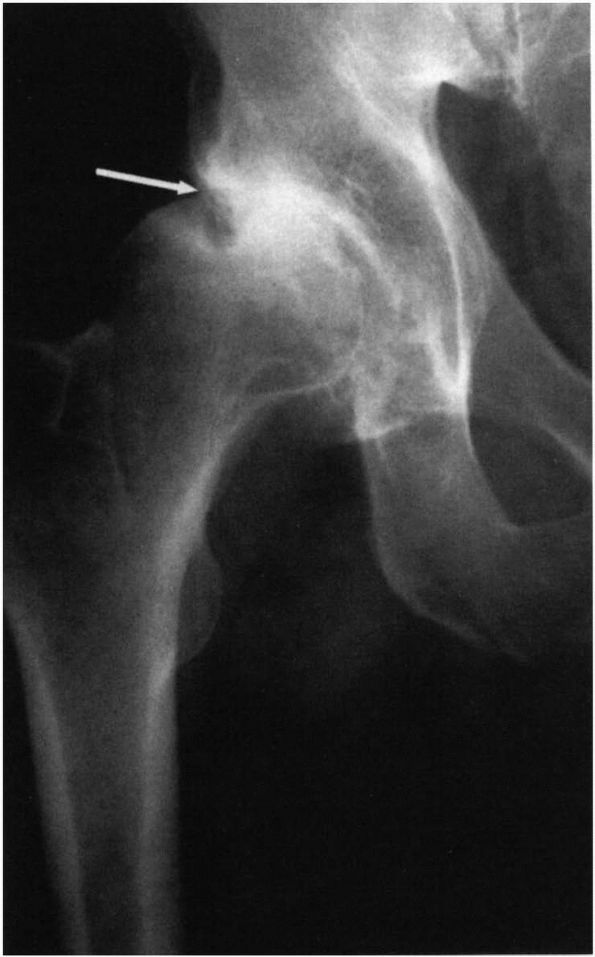
buttock pain with walking, but sometimes experience pain that radiates
toward the knees. The exam may reveal decreased range of motion,
particularly internal hip rotation, sometimes with pain at the limit of
rotation. Radiographs of early disease show changes concentrated in the
superolateral aspect of the joint, the area under most mechanical
stress. Joint space narrowing is followed by osteophyte formation,
sclerosis, and cyst formation. Remodeling of the medial and lateral
femoral head occurs and can lead to its collapse and flattening. The
medial acetabulum space fills in with osteophytes, resulting in gradual
superolateral migration of the femoral head (Figure 6-4). The cortex may thicken with new bone formation along the medial aspect of the femoral head (buttressing).
In contrast, chronic inflammatory arthritis, such as rheumatoid
arthritis, will typically lead to diffuse loss of joint space and
medial migration along the axis of the femoral head, or protrusio acetabuli.
occurs at C5, T8, and L3, the areas of greatest spinal flexibility.
Joints of the spine include cartilaginous joints, nucleus pulposus of the intervertebral discs; synovial lined apophyseal joints; cervical pseudoarthroses called uncovertebral joints; and fibrous articulation, the annulus fibrosus of the
intervertebral discs (Figure 6-5).
OA will typically affect all spinal joints in the same area, but
features of disease at each joint type are described separately for
clarity.
 |
|
FIGURE 6-1. (A and B) Heberden’s and Bouchard’s nodes. (A) Enlargement of the distal interphalangeal joint (arrow) due to joint osteophytes is called a Heberden’s node; (B) similar enlargement at the proximal interphalangeal joint is called a Bouchard’s node (arrowhead). (see color image)
|
 |
|
FIGURE 6-2. Osteoarthritis of the DIP joint: the combination of osteophytes at the margin (arrow) and irregular joint space narrowing (arrowhead) resembles a seagull flying.
|
is primary degeneration of the nucleus pulposus of the intervertebral
discs. Radiographs reveal progressive uniform or nonuniform narrowing
of the disc space and reactive subchondral sclerosis at the vertebral
endplate with osteophytes extending from the anterolateral vertebral
margins. Progressive desiccation or rupture of the disc will sometimes
allow gas to appear in the disc substance, which on radiographs will
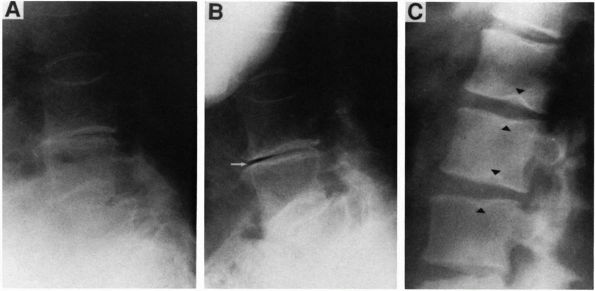
appear as a thin linear lucency within the disc (vacuum sign) seen on extension and often disappearing on flexion (Figure 6-6, A and B). The vacuum sign helps to exclude infection as a cause of disc space loss. Discogenic
low back pain caused by ruptured discs is typically worsened by flexing
the spine, which increases pressure on the disc, and likewise decreased
by extension of the spine, or by lying supine. The degenerating disc
may herniate into the adjacent vertebral body, producing a Schmorl node (Figure 6-6C), or impinge on the spinal cord, causing spinal stenosis.
 |
|
FIGURE 6-3. Degenerative joint disease of the knee is manifested by asymmetric joint space narrowing. The medial compartment (arrow) that bears the most weight is more affected than the lateral compartment.
|
 |
|
FIGURE 6-4. Osteoarthritis of the hip. An AP radiograph of the hip joint illustrates superolateral migration of the femoral head (arrow) with asymmetric joint space narrowing.
|
 |
|
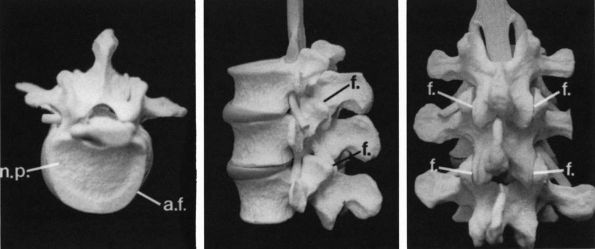
FIGURE 6-5.
The vertebral bodies and intervertebral discs contain three structures that degenerate in OA. The intervertebral facet joints (f) show classic features of osteoarthritis. The intervertebral disc is composed of the central nucleus pulposus (n.p.), where disease is termed intervertebral osteochondrosis, surrounded by the annulus fibrosis (a.f.), where disease is termed spondylosis deformans. |
 |
|
FIGURE 6-6. Lateral views of the lumbosacral spine show a horizontal linear lucency (arrow), called a vacuum sign, created when gas forms in the degenerated disc due to negative pressure. It is seen in extension (B) when disc pressure is lowest, and vanishes in flexion (A), which increases disc pressure. (C) A Schmorl node (arrowhead) forms when the nucleus pulposus herniates into an adjacent vertebral body.
|
characterized by joint space narrowing, marginal osteophytes, and bony
sclerosis. Lumbar facet disease may produce central low back pain that
worsens on extension, which loads the facet joints and decreases with
flexion. Osteophytes and hypertrophy of the joint capsule may cause
spinal stenosis by encroaching onto the spinal cord or the nerve roots
at the intervertebral foramina. Pain due to stenosis may radiate below
the knee, worsen with exertion and extension, and resolve with rest or
by bending forward, a pattern known as neurogenic claudication or pseudoclaudication.
Concurrent intervertebral disc degeneration worsens intersegmental
instability and increases the load on the lumbar facet joints, which
can lead to subluxation of the joints, allowing the forward movement of
one vertebral body over another (spondylolisthesis).
vertebral bodies project slightly beyond the disc to form
pseudoarthroses called uncovertebral joints. Osteophytes at both the
uncovertebral and facet joints can cause cervical spinal stenosis.
to degenerative disease of the annulus fibrosus of the intervertebral
disc and is characterized by formation of large osteophytes along the
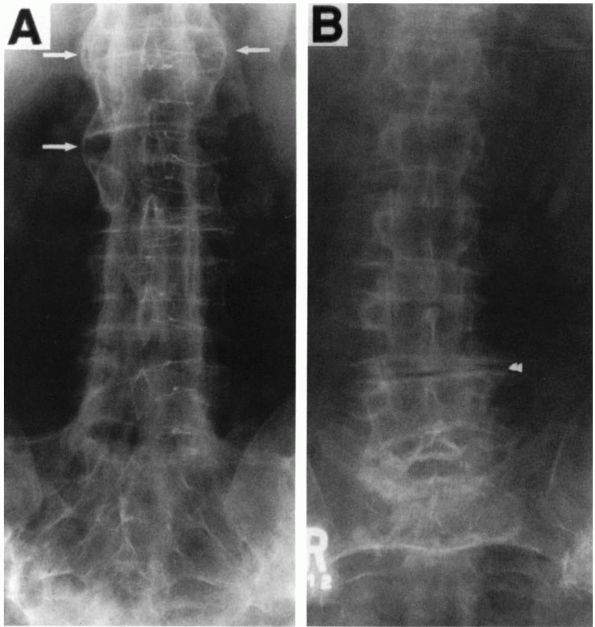
anterior and lateral aspects of the spine. Osteophytes of axial OA are
oriented horizontal to their point of origin, which distinguishes them
from the fine, vertically oriented syndesmophytes of the inflammatory spondyloarthropathies (Figure 6-7). Axial osteophytes of OA also differ from the paravertebral hyperostoses of diffuse idiopathic skeletal hyperostosis
or DISH. In DISH, there is ossification of the paravertebral ligaments,
particularly at the anterolateral aspects of the vertebral bodies,
producing upward or downward pointing hyperostoses, which are sometimes
bridging, appearing to flow like candle wax from one vertebra to the
next.
lumbosacral spot radiographs are adequate to diagnose OA of the spine,
and oblique views of the lumbar spine to show the facet joints are not
usually necessary. Magnetic resonance imaging (MRI), or alternatively,
computed axial tomography (CT), are necessary to diagnose herniated
vertebral discs and/or lumbar spinal stenosis. It is important to make
a specific diagnosis when these problems are suspected, as physical
therapy designed to alleviate one disorder may aggravate the other.
 |
|
FIGURE 6-7.
Osteophytes versus syndesmophytes. Syndesmophytes of spondyloarthropathy are fine, project vertically, and seem to flow from one vertebral body to the next (arrows in A), whereas osteophytes of OA are course and project more horizontally (arrowheads in B). |
individuals and presents on radiographs as asymmetric joint space
narrowing, osteophytes at the inferior aspect of the joint, and
distinct sclerotic joint margins. The finding of pseudowidened joint
spaces, indistinct joint margins, and erosions or fusion of the joint,
particularly in a young person, suggests the presence or aftermath of
an inflammatory sacroiliitis, rather than OA.
prone to develop OA, with the usual radiographic findings of joint
space narrowing, osteophyte formation, sclerosis, and subchondral
cysts. It often presents with pain on walking, and limited dorsiflexion
or plantar flexion of the MTP, or hallux rigidus. Asymmetric degeneration may lead to abduction of the great toe with lateral angulation of the joint, or hallux valgus (bunion) deformity.
inversion and eversion of the foot. Tibiotalar joint degeneration
rarely occurs without prior trauma or an inciting anatomic abnormality.
Rapid and destructive degeneration of the foot and ankle joints usually
indicates a neuroarthropathy (Charcot
joint), in which loss of sensation and proprioception leads to improper
joint loading and repetitive trauma. The neuropathy associated with
diabetes mellitus is the most common cause.
arthritis, and none are needed to confirm the diagnosis. Studies are
sometimes done, however, to rule out diseases that can be associated
with or mimic OA. Assessment of serum creatinine and liver
transaminases may be necessary in order to safely prescribe analgesics
for pain. Analysis of synovial fluid from joints with OA typically
reveals normal to mildly decreased viscosity and a cell count that is
frequently less than 100 cells/µL, and rarely higher than 8,000
cells/µL. Cell counts at the high end of this range are more common in
long-standing joint disease, and very high cell counts should prompt
further investigation for infection, crystal arthritis, or other
superimposed inflammatory joint disease. When new joint effusions
occur, fluid should also be analyzed by polarized light microscopy for
calcium pyrophosphate dihydrate (CPPD) crystals, as
the
incidence of CPPD is increased in degenerative arthritis. Basic calcium
phosphate crystals are also increased in degenerative arthritis, but
are too small to detect by light microscopy.
reduce pain, but to slow or prevent further decline in functional
status. The natural history of OA may be one of slow, chronic
progression, or of stable periods with intermittent worsening. With new
or increased pain there is a natural tendency to reduce activity. As
activity decreases over time, so, too, does muscle bulk and strength,
which may lead to decreased joint stability, worsening of joint
degeneration, and further decline in functional status. Even though OA
is not a systemic disease, the related decline in functional status can
have major systemic consequences, affecting cardiovascular health,
emotional health, and sense of well-being. Breaking this cycle may
require a team approach targeted toward educating the patient and
family, evaluating and sometimes altering the patient’s lifestyle,
offering assistive devices, and prescribing both physical and
pharmacotherapy.
where intervention is most likely to be effective. Obesity is the
number one modifiable risk factor for OA.
Therefore, obese patients should be counseled by dietitians and be
continually encouraged by their physicians to adhere to a diet program
that safely promotes and maintains weight
loss. Patients may alter the course of their disease and improve their
level of safety and functioning, simply by losing weight and increasing
or at least maintaining muscle strength. They need to understand the
benefits and limitations of their medications, which are prescribed to
alleviate pain and reduce inflammation, but by themselves may not alter
the disease process or slow its progression.
especially in acute disease exacerbations, but excessive rest or
reduction in activity will begin the cycle of atrophy, weakness, and
further functional decline, which may actually worsen pain over time.
Exercise is required to strengthen muscles, and many studies have shown
that regular, moderate exercise can both reduce pain, and improve the
functional status of patients with mild to moderate OA. Finally,
exercise is often required, in addition to dietary modifications, for
effective weight loss. The type and intensity of exercise should be
tailored to the individual needs of the patient. For instance, for
patients with OA of the knees who have not yet developed significantly
abnormal joint angulation, a twice daily set of supine 10-second
isometric quadriceps contractions can improve strength, reduce pain,
and reduce the risk of falling. This exercise is generally well
tolerated even by patients with moderately severe knee pain. In
general, low impact exercise, such as walking, is preferred to high
impact exercise, such as running or jogging. Exercise in water, which
helps to unload the weight-bearing joints, can be especially beneficial
when weight-bearing pain in the knees, back, or hips limits tolerance
for land exercise.
reasons that physical therapy is an integral part of the management of
OA. Physical therapists can instruct patients on the proper use of
canes and walkers, to decrease weight-bearing stress on knees and hips,
reduce the risk of injurious falls, and reduce the fear of falling that
by itself can greatly limit patient mobility. Therapists can also
instruct patients on the proper use of transcutaneous electrical nerve
stimulation (TENS) units to reduce pain in specific areas. Insoles,
braces, and orthopaedic shoes can benefit OA of the knees, ankles, and
feet. Lateral wedged shoe inserts can sometimes reduce pain of medial
knee joint OA by shifting weight to the less affected lateral
compartment. Medial patellar taping may reduce the lateral compartment
pain of patellofemoral syndrome. Patients with chronic back pain due to
lumbar spinal stenosis will benefit from education about appropriate
spinal biomechanics used in daily activities and about those positions
of the spine that exacerbate back and leg pain.
with hand and wrist OA. Therapists can fit patients with finger or
first CMC splints to stabilize affected joints, and instruct them on
how to reduce joint stress during daily activities. Small sleeves of
silicone can pad tender Heberden’s and Bouchard’s nodes. Careful use of
heat or cold may also alleviate pain. Paraffin baths are particularly
soothing for the pain of erosive OA.
(rubifacient) therapies, intra-articular therapies, oral analgesic
therapies, and dietary supplements.
have a transient, soothing effect on joint pain, and are generally
safe. Topical capsaicin, if applied frequently and consistently to a
region, can selectively reduce pain sensation in that region by
depleting substance P from type C unmyelinated pain neurons. However,
not everyone can tolerate the initial burning sensation caused by
capsaicin, and patients must be well informed in order to use this
agent properly.
particularly helpful for OA of the knees, especially when there is
inflammation and joint effusion, and can be safely repeated up to four
times per year. The injection may work best if the knee joint is first
aspirated to remove excess synovial fluid. If the fluid appears at all
turbid, it should be sent for cell counts and for culture, and the
corticosteroid injection should be postponed until it is clear that the
joint is not infected. Epidural corticosteroid injections, performed
under fluoroscopic guidance, can sometimes reduce pain of lumbar
stenosis. Viscosupplementation,
intra-articular injection of hyaluronic acid derivatives, may also be
beneficial for mild to moderate OA of the knee, particularly for
patients who cannot take analgesics, or who are not candidates for
joint replacement.
of OA pain, but choice of agent must be guided by knowledge of the
patient’s other medical conditions and concurrent treatment. Oral
analgesics include acetaminophen, nonsteroidal anti-inflammatory drugs
(NSAIDs), selective cyclooxygenase-2 (COX-2) inhibitors, nonacetylated
salicylates, synthetic opioid agonists, and narcotics.
patients will provide adequate pain relief when used at a full dose of
1 g three to four times per day. Though long thought to be very safe, a
recent meta-analysis suggests that acetaminophen may cause more
gastrointestinal and renal toxicity then originally believed,
especially when used chronically. Impaired liver function is a
contraindication for high dose acetaminophen.
inhibitor will be more effective than acetaminophen for relieving
OA-related pain. However, the benefit of added pain relief must be
balanced with the potential for significant toxicity, especially in the
elderly, who are the majority of patients needing analgesics for OA.
NSAIDs inhibit gastric COX-1, thus blocking production of
gastroprotective prostaglandins, and are known to cause significant
gastrointestinal (GI) side effects and toxicity, including pain, acid
reflux, gastric ulcers, and erosive esophagitis. Therefore, many
patients who require chronic use of NSAIDs will also require concurrent
use of gastroprotective agents such as misoprostal, or proton-pump,
inhibitors. Selective COX-2 inhibitors are as effective as NSAIDs for
relieving OA-related pain, and cause fewer GI side effects and ulcers,
however, they may be associated with an increased risk of heart attack
and stroke. Both NSAIDs and COX-2 inhibitors can reduce glomerular
filtration, and thus increase sodium and fluid retention. Therefore,
patients who have uncontrolled hypertension, renal insufficiency, or
congestive heart failure should, in most cases, avoid these agents
altogether and should certainly be monitored very closely when these
agents are prescribed. The nonacetylated salicylates, which do not
inhibit COX-1 or COX-2, may be safer choices for elderly patients with
hypertension or mild renal insufficiency. However, ototoxicity and CNS
side effects may limit tolerance to these medications.
agonist, tramadol, has a role in management of OA-related pain.
Short-term use for acute exacerbation of pain is safe and effective in
most circumstances. Chronic use should generally be avoided, but may be
appropriate in selective cases where pain and functional limitation are
significant, surgery is not possible, and other medical conditions
prohibit use of NSAIDs or COX-2 inhibitors. Narcotics and tramadol are
often prescribed as combination pills containing acetaminophen, which
may improve efficacy but also creates a risk for acetaminophen overdose
if additional acetaminophen is taken along with them. Patients must be
educated to avoid acetaminophen overdose.
hepatic toxicity and should be used with caution in patients with liver
disease. Physicians should obtain baseline creatinine and liver
transaminases, and later repeat these tests, in all patients who are
starting a course of chronic analgesic use for OA.
gained great popularity and are now widely used. Studies in animal
models suggest that glucosamine may slow cartilage breakdown. Human
studies do show at least a modest benefit of reduced pain, or reduced
need for other oral analgesics, such as acetaminophen. Human studies
have also purported to show that glucosamine use reduces loss of knee
cartilage, by showing differences in radiographic joint space between
glucosamine versus placebo users. However, these studies have been
criticized
for not accounting for the possibility that pain reduction in the
glucosamine users may have altered their stance and thus increased the
measured joint space. Glucosamine is generally safe, but should be
avoided by patients who are allergic to shellfish. Many glucosamine
preparations also contain chondroitin sulfate, and small controlled
trials suggest that this agent may also reduce pain of OA. However,
chondroitin is derived from animal cartilage, which raises issues of
safety, and there is little evidence that chondroitin will provide any
additional benefit beyond that derived from glucosamine alone. The
American College of Rheumatology Subcommittee on Osteoarthritis
Guidelines does not recommend use of these agents at this time.
or deformity that limits function and is discussed separately
elsewhere. It does not obviate the need for continued medical
management.
polyarticular inflammation that leads to joint swelling, joint
deformity, loss of joint function, and early death. Advances in the
underlying immunobiology, earlier diagnostic possibilities, and major
therapeutic approaches could well limit the previous inexorable decline
in function. RA occurs in 1 to 3% of the white adult population, but
prevalence varies depending on age, race, and classification criteria
used. Women are slightly more affected than men (3:2), but the disease
is seen in all races, teenagers, and the elderly, and has a worldwide
distribution.
major histocompatibility complex control both immune responses and
susceptibility to rheumatoid arthritis (Color Figure 6-1).
People who are HLA DRB4 positive are more likely to develop erosive,
disabling disease, but only one-third of RA patients are DRB4 positive.
Infection is suspected to play a role in RA onset, although no specific
bacterial or viral causes have been proven. Perhaps RA is a final
common pathway for several infections.
and T-lymphocyte infiltration occur in the subsynovial tissue, followed
by synovial lining cell proliferation. Increased cellularity includes
synovial infiltration by B-cells, macrophages, and fibroblasts. B-cells
develop into plasma cells and reside in the synovium chronically,
producing rheumatoid factor (an immunoglobulin), leading to complement
activation. Fibroblasts migrate to the synovial surface, with
granulation tissue development, entailing further proliferation of
fibroblasts, synovial lining cells, and enhanced vascular infiltration.
Responding to chemotactic factors including complement byproducts,
granulocytes migrate through capillary walls and synovial tissue into
the joint space. These cells then permanently reside in the joint space
and discharge enzymes. These hydrolases, DNAase, proteinases (elastase
and collagenase) accumulate in the synovial fluid, articular cartilage,
and bone-destroying structural proteins and other cells. Soluble
pro-inflammatory substances produced by activated lymphocytes,
monocytes, and macrophages (TNF-α, interleukin 1 and other cytokines, E
series prostaglandins, leukotriene B4) are generated in the joint.
These increase vascular permeability and further activate granulocytes,
lymphocytes, and monocyte-derived macrophages, synovial cells,
osteoclasts, and fibroblasts. Soluble mediators augment and perpetuate
the inflammatory response.
called pannus, advances across the joint surface, destroying marginal
articular cartilage and invading subchondral bone. Pannus attached to
the joint capsule, ligament, and tendons results in joint deformity.
Active inflammation is accompanied by attempted repair, and collagen
production may become dominant, leading to fibrosis and joint
contractures.
with RA are stiff and sore in the morning, lasting 1 to or more hours,
improving with low-grade activity.
Visible
joint swelling, especially that observed by an experienced health care
provider, is highly specific for an inflammatory arthritis. Rheumatoid
arthritis has a predilection for small joints of the hands (PIP and
MCP), the wrists, the knees and the feet. Population-based studies
suggest pain elicited by squeezing the MTPs or the MCPs in the presence
of two or more swollen joints is highly specific for RA. Symmetrical
involvement is typical of RA, and it can be associated with fatigue,
malaise, fever, weight loss, and lymphadenopathy. Nodules may occur,
typically along the olecranon border, Achilles tendon, or extensor
surfaces of the hands and feet, but are often a later finding.
polyarthritis. MRI reveals bone edema and cartilage degradation within
weeks of symptom onset. In time almost all synovial joints may become
involved, including TMJ, shoulders, elbows, wrists, MCP, PIP, knees,
ankles, feet, and cervical spine. The hips may be spared early.
Involvement of the DIPs of the hands and inflammatory spinal
involvement are extremely rare. Tenosynovitis is common, especially in
the hands and feet and can lead to nerve root entrapment (carpal and
tarsal tunnel) and tendon rupture. Documentable joint inflammation is
detected as palpable synovial swelling or joint tenderness. Although
swelling is often visible and palpable, joint tenderness is elicited by
applying direct pressure to the joint or at the end range of passive
joint motion. Tenosynovitis is diagnosed as swelling along the tendon,
pain with passive stretching of the tendon, and pain on resisted
movement.
diagnostic criteria, which require the presence of four or more of the
following seven items:
-
Morning stiffness for at least one hour and present for at least 6 weeks
-
Swelling of three or more joints for at least 6 weeks
-
Swelling of wrist, PIP, or MCP joints for at least 6 weeks
-
Symmetrical joint swelling
-
Hand radiograph changes typical of RA, including erosions or unequivocal bony periarticular decalcification
-
Subcutaneous nodules
-
Rheumatoid factor
polyarthritis, such as rubella or parvovirus, SLE, Sjögren’s syndrome,
sarcoidosis, systemic immune complex reactions, reactive arthritis,
psoriatic arthritis, polyarticular CPPD (pseudogout), and erosive
osteoarthritis involving mainly the PIPs. All can have polyarthritis,
but historical features of infection, cutaneous abnormalities, and
patterns of joint involvement all suggest these alternate diagnoses.
years, in as many as 10% of patients. Risk factors for progression
include rheumatoid factor positivity, DRB4 positivity, nodules,
persistent elevation of the CRP, and a progressive, additive onset.
Intermittent disease flares with increased systemic symptoms and
increased numbers of swollen joints is the rule. Joint damage
correlates with persistent joint inflammation.
6 months and in 80% over time. Other conditions with positive
rheumatoid factors include cryoglobulinemia, parvovirus 19 infection,
hepatitis C, Sjögren’s syndrome, SLE and occurs in 5% of normal,
healthy individuals. A positive ANA occurs in 40%. The recently
described anti-citrullinated cyclic peptide (anti-CCP) antibody can be
positive early in RA when the rheumatoid factor is negative. It appears
to have prognostic importance. Persistent elevation of the ESR or CRP
and anemia of chronic disease are common in undertreated RA.
affects the MCP, PIP, MTP, and ulnar-carpal joints, and are present in
up to 30% of patients in the first year with 90% having erosions after
2 years. MRI is a far more sensitive test for erosions than is plain
radiography.
deformities. Rotation of the carpometacarpal complex on the radius, and
volar subluxation of the carpus on the radius leads to reduced grip.
Disruption of the distal radio-ulnar ligament leads to dorsal
subluxation of the distal ulna, appreciated clinically as the “piano
key sign.” Damage to the joint capsule and collateral ligaments of the
MCP
joints
combined with rotational deformity of the metacarpal complex leads to
ulnar deviation of the fingers, often accompanied by volar or ulnar
subluxation of the PIP joints. Swan neck deformity is fixed
hyperextension of the PIP joint and accompanying flexion of the DIP
joint. Boutonniere deformity is flexion contracture of the PIP joint
with hyperextension of the DIP, resulting from damage to the central
portion of the extensor tendon overlying the PIP joint. A Baker’s cyst
develops in the popliteal space from increased intra-articular pressure
and a gradual weakening of the posterior capsule. Baker’s cysts are
prone to rupture, with acute pain, swelling, and heat in the calf,
mimicking deep venous thrombosis or cellulitis. Ultrasonography can
differentiate a cyst from venous thrombosis. Forefoot involvement
includes widening of the forefoot, hallux valgus, hallux rigidus, and
cock-up toe deformities. Atrophy of the soft tissue pads on the plantar
surface of MTP results in weight-bearing pain and ulceration. Hindfoot
involvement includes contracture of the subtalar joints or excessive
laxity associated with abduction and pronation of the midfoot and
forefoot.
hand involvement. Particularly concerning is weakening of ligaments
attaching the odontoid process to C2 and to the lateral masses of C1.
Anterior subluxation of C1 relative to C2 and the odontoid process
leads to atlanto-axial subluxation. Progressive subluxation in concert
with degenerative and inflammatory changes at levels below C2 may lead
to neurological impingement and motor deficits. Cranial settling
results when erosion of the occipital condyles or the lateral masses of
C1 cause settling of the skull relative to the odontoid process. The
odontoid may protrude into the foramen magnum where it compresses the
medulla or pons.
serositis, and vasculitis. Nodules occur in 25% of patients and are
often a late feature. They have a central area of fibrinoid necrosis
surrounded by histiocytes and inflammatory cells. Common locations are
elbows, hands, feet, although they can occur in all locations including
internal organs. Repeated pressure may encourage their development.
Sjögren’s syndrome occurs in 10% of RA patients, involving chronic
inflammation of exocrine glands, most commonly the lacrimal and
salivary glands. Clinically it presents as dryness of the eyes and
mouth, leading to corneal ulceration and accelerated dental carries.
Serositis is relatively uncommon and presents with recurring,
moderate-sized exudative pleural effusions with a low glucose. Diffuse
interstitial pulmonary fibrosis is bilateral, principally affecting the
lower lung zones. Felty’s syndrome is the co-occurrence of RA,
splenomegaly, cytopenias (white cells or platelets), and leg ulcers.
Systemic vasculitis can lead to ischemia of the skin (purpura), vasa
nervorum (peripheral neuropathies), and rarely internal viscera. Small
1 to 2 mm hemorrhagic cutaneous infarcts in the periungal region of the
digits are the result of an obliterative vasculopathy and do not
indicate a systemic vasculitis.
include reducing pain, reducing inflammation, and preventing disability
(Figure 6-8). Other goals include minimizing
drug toxicity and management of extra-articular features. These goals
are best prioritized through careful history taking, physical
examination, and selected laboratory and radiographic studies. Several
well-validated patientcompleted questionnaires of disability, such as
the health assessment questionnaire (HAQ), predict short- and long-term
functioning and should be a routine component of RA care.
control, although they may be performed in concert. Analgesia with
acetaminophen, nonsteroidal anti-inflammatory drugs, weak opiates
(codeine and tramadol), use of local depot steroid injections, physical
therapy, joint splinting, and education (Arthritis Foundation) are
appropriate for pain management. NSAIDs and injections have only a
minor role in inflammation management. Pain can result from several
causes including inflammation, but also from joint and tendon damage,
muscle pain, and fatigue and should not be assumed to be inflammation
related. Joint inflammation is diagnosed by asking about inflammatory
symptoms (morning stiffness and improvement with activity) and the
presence of joint swelling. Laboratory tests (ESR/CRP) can be a helpful
adjunct to the history and examination.
 |
|
FIGURE 6-8. Managing rheumatoid arthritis.
|
which do not have immediate onset. While oral corticosteroids
(prednisone) and joint injections may provide temporary relief, their
long-term toxicity and the availability of more effective drugs argue
against their long-term use. Corticosteroids should be used
temporarily, for 1 or 2 weeks with acute flares and while waiting for
long-term inflammation control with disease-modifying agents. Low
potency, low toxicity DMARDs include hydroxychloroquine, sulfasalazine,
and minocycline. These drugs should be used early in patients with mild
disease, and in combinations with more potent DMARDs as disease
progresses. One of these drugs should be started as soon as the RA
diagnosis is suspected.
of inflammatory control for the last 20 years, with two other
antimetabolites, azathioprine and leflunomide, as alternatives. For
persistent uncontrolled inflammation, research has shown that
combinations of DMARDs (two or three of the above listed agents) or the
addition of TNF-α inhibitors are required. Recent large, long-term,
randomized controlled trials with TNF-α inhibitors have shown
improvements in joint inflammation and joint damage, with prevention of
radiographic erosions. Use of antimetabolites and TNF-α inhibitors
require expert rheumatologist advice. The antimetabolites have
important toxicity in the liver and bone marrow and are associated with
infection risk. Recent data shows methotrexate does not impair wound
healing. The TNF-α antagonists also have increased infection risk and
should be stopped 2 to 4 weeks before surgery.
goal and requires optimal pain control, control of joint inflammation,
and attention to personal, social and occupational factors that
contribute to disability burden. Depression, anxiety, low educational
attainment, a physically demanding job, and persistent unchecked
inflammation all predict job loss. Involvement of the primary care
provider, social workers, physical therapists, occupational therapists,
and the work site may be required to match job requirements with the
patient’s abilities.
management. Sicca complex (Sjögren’s syndrome) may be improved with
eyedrops, oral lozenges, and oral pilocarpine or oral cholinergic
agonists. Nodules are best left alone unless they are causing pain and
functional loss. Local corticosteroid injection may cause nodules to
regress. They often recur after surgical removal. Entrapment
neuropathies require surgical release. Tendon ruptures (especially
wrist and finger extensors) require surgical repair. Recurrent Baker’s
cysts may respond to surgical excision.
for surgical stabilization. Cervical instability requires cooperation
between the orthopedist and the rheumatologist. Advanced degenerative
disease responds well to total joint replacement, particularly knees
and hips, although shoulder, wrist, MCP, and PIP arthroplasty may all
be appropriate in some instances. The need for surgical intervention is
evidence of unchecked inflammation in the past, and surgery is not an
alternative to optimal medical management.
cytopenias, and drug toxicity mandate consultation with a
rheumatologist and other appropriate medical specialists, with a
rheumatologist coordinating care.
fundamentally alter the natural history and subsequent disability
associated with rheumatoid arthritis. RA need not be a progressive,
disabling condition, when expertly managed.
chronic autoimmune disease that is distinguished by characteristic
organ manifestations. It most commonly involves the musculoskeletal,
cutaneous, and renal systems. Its cause is unknown but likely involves
hereditary and environmental susceptibility factors. Autoantibodies are
the hallmark of this condition and are directed primarily to cell
nuclei and their constituents, for example, antinuclear antibodies
(ANA), and anti-double stranded DNA (anti-dsDNA). They mediate tissue
injury by forming immune complexes, which promote inflammation. They
also promote cell destruction by the reticuloendothelial system and
perhaps exert direct toxic effects on cell function.
peak incidence in the reproductive years. It affects individuals of all
races and ethnicities with a prevalence of 15 to 52 cases/100,000
persons. Diagnosis is made on the basis of characteristic clinical and
laboratory features (for formal criteria used in making the diagnosis,
see Tan 1982). The course is variable, and is
related to race, type, and severity of organ involvement. Prognosis has
improved dramatically over the last 50 years secondary to earlier
recognition and more effective management.
due to the multitude of clinical manifestations. Moreover, these
clinical manifestations may change over time. Joint and skin complaints
followed by constitutional symptoms (fever, fatigue, malaise, weight
loss) are the most common presenting complaints. The majority of
patients have arthralgias (95%), arthritis (90%), fever (90%), fatigue
(81%), skin rashes (malar rash, discoid lupus, photosensitivity, 74%),
or glomerulonephritis (50%) at some time in their illness. Myalgias and
myositis occur less frequently. Other clinical features used in making
the diagnosis of SLE include serositis, seizures, psychosis, and oral
ulcers. The classic presentation of a butterfly (malar) rash (erythema
over the cheekbones and nose) with simultaneous arthritis occurs in a
minority of patients.
and swelling (arthritis). Symptoms are inflammatory in nature, and may
be evanescent and migratory, persistent, or progressive. Any peripheral
joint may be involved, although the metacarpophalangeal and proximal
interphalangeal joints of the hands, wrists, and knees are most
frequently affected. Symmetric polyarthritis affecting the hands and
wrists is the most common arthritis presentation. Pain and palpable
tenderness is often more prominent than swelling. This joint
distribution resembles the pattern in rheumatoid arthritis (RA), and
often, patients are initially diagnosed with RA. The diagnosis of SLE
becomes obvious when other characteristic clinical and laboratory
features develop. Usually the joint complaints in SLE are milder than
in RA, and destruction of cartilage and bone does not occur.
Deformities of the joints can occur in SLE, and in the hand include
ulnar deviation and subluxation, swan neck deformities, and
hyperextension of the thumb interphalangeal joints. This usually
reducible subluxation is called
Jaccoud’s
arthropathy, and occurs in 5 to 40% of patients. It results from
stretching and laxity of ligaments and tendons rather than from
destructive changes typical of RA. Jaccoud’s arthropathy can occur in
other joints as well. Symptomatic axial skeleton involvement is
unusual, although radiographic evidence of sacroiliitis is frequently
present.
are susceptible to a variety of musculoskeletal complications. Steroid
myopathy can cause progressive weakness. Glucocorticoid-induced
osteoporotic compression fractures are a potential cause of acute back
pain. Stress fractures near a joint can cause joint pain and swelling,
and should also be considered in those patients on chronic
corticosteroids. Septic arthritis and osteonecrosis are other potential
causes of joint pain. Immunosuppressives, glucocorticoids, and
intrinsic susceptibility to infection predispose these patients to
septic arthritis. Clinically apparent osteonecrosis occurs in 4 to 15%
of these patients. It affects the humeral head in 80% of cases,
followed by the knees and shoulders. Multiple joint osteonecrosis is
common. Pain may be mild to severe and develops insidiously to
abruptly. Thus, acute mono- or oligoarthritis needs to be further
evaluated with arthrocentesis and MRI to eliminate septic arthritis and
osteonecrosis, respectively.
resulting in Jaccoud’s arthropathy, tenosynovitis, and tendon rupture.
Tenosynovitis often involves the extensor tendons of the fingers and
toes. Tendon rupture frequently affects the patellar tendons, long head
of the biceps and triceps, and extensor tendons of the hands. Trauma
and corticosteroid use are associated risk factors.
those observed in RA although they are milder in intensity. Typical
features include perivascular inflammation, synovial cell
proliferation, and fibrin deposition at the synovial membrane. Bone and
cartilage destruction rarely occur, in contrast to what is observed in
RA. Histologic examination of ruptured tendons may show inflammatory
changes.
to 50% of patients may have mild abnormalities. These abnormalities
include soft tissue swelling, periarticular osteopenia, osteoporosis,
and subluxation. Joint space narrowing and marginal erosions, which are
common in RA, are rarely if ever observed in SLE.
laboratory evidence of SLE. ANAs are present in greater than 98% of
patients with SLE. However, the specificity of a positive ANA is low.
Positive ANAs are observed frequently in other autoimmune diseases, as
well as in healthy individuals and particularly in the elderly.
Consequently, their utility is the greatest when used to support the
clinical and laboratory impression of SLE. Anti-dsDNA and anti-Sm
autoantibodies are more specific for SLE. Unexplained leukopenia,
lymphopenia, hemolytic anemia, and thrombocytopenia are hematologic
manifestations of SLE. Proteinuria, hematuria, and red blood cell casts
suggest renal involvement by SLE.
inflammatory. Fluid is clear with normal viscosity. White cell counts
typically range from 2,000 to 15,000, although values up to 40,000 have
been detected. There is often a lymphocyte predominance.
pattern and severity of organ involvement. If musculoskeletal
complaints are accompanied by major organ involvement (kidneys, central
nervous system, blood cells), then aggressive therapy with high dose
glucocorticoids with or without immunosuppressive therapy is used.
Cyclophosphamide, mycophenolate mofetil, and azathioprine are commonly
used immunosuppressive agents. If musculoskeletal complaints accompany
minor organ involvement (skin, oral mucosa, pleura), then antimalarials
are often used. Isolated mild musculoskeletal complaints are initially
treated with NSAIDs. If they are refractory or more severe,
antimalarials are added. Methotrexate is used for arthritis that has
failed antimalarial treatment. Low dose glucocorticoids (usually ≤ 10
mg prednisone each day) are used in arthritis that fails to respond to
NSAIDs while waiting for antimalarials and/or methotrexate to become
effective. More aggressive therapy of SLE arthritis with combination
immunosuppressive therapy, such as that used for RA, is rarely
indicated. TNF-α blocking agents are not used in SLE, and may worsen it.
musculoskeletal complaints. Physical and occupational therapy are
important adjuncts to pharmacologic therapy. Splints may be effective
in limiting deformities and, with appropriate exercise, may preserve
function. Surgical interventions may be necessary to correct
deformities, restore joint function, and manage tendon rupture and
osteonecrosis.
disorders which include ankylosing spondylitis, inflammatory bowel
disease arthritis, reactive arthritis (Reiter’s syndrome), and
psoriatic arthritis. They are clinically, radiographically,
pathologically and genetically related to ankylosing spondylitis. The
absence of rheumatoid factor (leading to the name “seronegative”
spondyloarthropathy) is not an essential diagnostic feature. The
predilection of these disorders to axial, spinal inflammation is the
dominant clinical feature leading to the preferred term spondyloarthropathy.
A number of clinical distinctions permit differentiation from
rheumatoid arthritis, including important differences in articular and
extra-articular features as well as management and prognosis.
spondyloarthropathies is the presence of “enthesopathy.” An enthesis is
the insertion point of tendons, ligaments, or joint capsule on bone and
the term enthesopathy refers to a physical alteration at the site of
such attachments. Inflammatory enthesitis
is the clinical hallmark of all the spondyloarthropathies and is an
important feature shared by all the members of this family. The
presence of widespread enthesitis leads to multiple spinal and
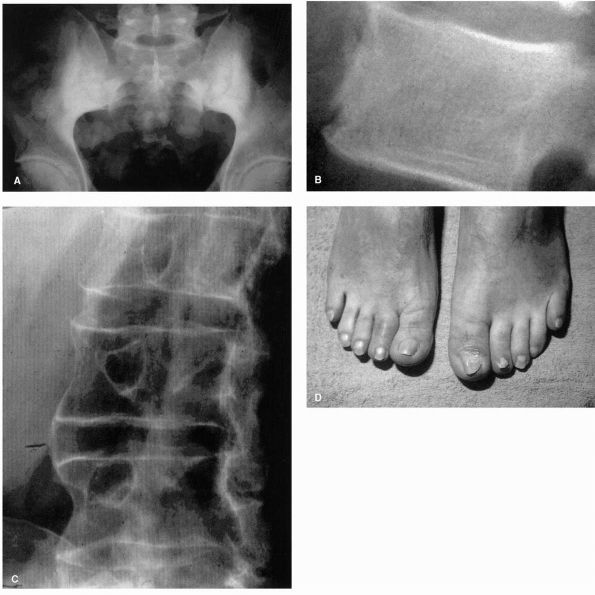
peripheral manifestations so characteristic of these disorders (Figure 6-9).
Inflammation of spinal entheses at paraspinous ligaments leads to
spondylitis (inflammatory involvement of the spine). Inflammation at
axial cartilaginous joints contributes to the arthritis at the SI
joints, intervertebral discs, symphysis pubis, manubriosternal joint,
and sternoclavicular joints.
Axial “root” joint synovitis in shoulders and hips is most commonly
seen in ankylosing spondylitis. An asymmetric lower extremity
oligoarthritis (2 to 4 joints), especially involving the knee and ankle
joints, is seen in patients with inflammatory bowel disease arthritis.
A similar, predominantly large joint lower extremity oligoarthritis may
be seen in reactive arthritis or Reiter’s syndrome with the addition of
small joint synovitis, particularly with distal interphalangeal (DIP)
involvement of toes and fingers (DIP involvement is not usually seen
clinically in RA). In addition, a particularly distinctive feature of
the spondyloarthropathies (especially seen in reactive and psoriatic
arthritis) is the presence of “sausage digits” (Figure 6-9D).
This distinctive pattern of swelling represents the combination of
synovitis of small synovial joints combined with enthesitis of tendon
sheaths, tendon insertions, joint capsules and supporting ligaments,
giving rise to sausage-like swelling of the entire digit. A spectrum of
progressively increasing peripheral joint involvement is seen when
comparing ankylosing spondylitis (least peripheral) to inflammatory
bowel disease arthritis to reactive arthritis to psoriatic arthritis
(most peripheral).
frequently present in this family of disorders: ocular (conjunctivitis
and uveitis), GU (urethritis), GI (diarrhea and dysentery), and
cutaneous (psoriasis) manifestations.
1% of the population. It begins most frequently before age 40, usually
in the third or fourth decades of life. Symptoms and signs of
inflammatory spinal disease predominate. Inflammatory back pain,
suggesting sacroiliitis and spondylitis, has five important historical
features that help to
distinguish
it from more common mechanical low back pain: age less than 40,
insidious onset, duration of less than 3 months, significant morning
stiffness, and improvement with exercise. Patients with inflammatory
back pain give a history that frequently sounds quite vague, the
significance of which can easily be missed. Discriminating questions
focusing on sleep, mornings, and the effect of rest and exercise can be
extremely helpful in suspecting the correct diagnosis. Family histories
of patients with ankylosing spondylitis frequently reveal other
individuals with early onset low back pain, uveitis or iritis,
inflammatory bowel disease, or psoriasis.
 |
|
FIGURE 6-9. (A) Bilateral sacroiliitis (ankylosing spondylitis). (B) Syndesmophyte in upper lumbar spine. (C) Exuberant periosteal new bone formation in lower thoracic spine. (D) Sausage digits of right 2nd and left 1st toe (psoriatic arthritis).
|
Sacroiliac joints may be tender to direct percussion. Specific
maneuvers applying mechanical stress to the pelvis frequently result in
discomfort felt directly at the SI joint (upper, inner buttock region).
Visual inspection of the lumbar spine during
flexion
is very important. Due to extensive inflammation in spinal entheses,
the lumbar lordotic curvature frequently does not reverse (as it
should) during spinal flexion. Chest expansion may also be limited due
to inflammatory changes at costovertebral joints in the thoracic spine.
The cervical spine may also be involved, demonstrating restriction in
all planes of motion (especially extension). Early loss of spinal range
of motion in patients with ankylosing spondylitis may be due to
inflammatory pseudofusion of spinal ligaments, and may be reversible
with aggressive anti-inflammatory therapy combined with range of motion
exercises. Later, more fixed reductions in range of motion, due to bony
fusion, are not reversible.
symmetric sacroiliitis characterized by sclerosis, articular erosions,
and the later development of bony fusion across the joint. A single AP
pelvis radiograph (Ferguson view) is the most helpful to confirm
suspected sacroiliitis. Additional radiographic features of ankylosing
spondylitis include progressive vertebral squaring and marginal
sclerosis due to remodeling of the vertebral bodies and new bone
formation. Syndesmophytes (bony bridging across the annulus of the
intervertebral discs) may develop and, years later, result in the
development of a bamboo” spine (multiple symmetrical syndesmophytes
giving the radiographic appearance of bamboo).
Crohn’s disease or ulcerative colitis, and may involve either the
sacroiliac joints or peripheral joints of the lower extremities.
to sacroiliitis with spondylitic symptoms characteristic of ankylosing
spondylitis. Radiographically, these patients have symmetric
sacroiliitis indistinguishable from ankylosing spondylitis.
Inflammatory spondylitis may precede, occur simultaneously with, or
follow inflammatory bowel disease (IBD). The clinical course of
spondylitis is independent of the clinical activity of the bowel
inflammation.
extremity, large joint oligoarthritis predominantly involving knees and
ankles. The course of the peripheral arthritis tends to be episodic,
but parallels the activity of the bowel disease.
arthritis and extra-articular features sometimes seen in susceptible
individuals following a genitourinary or gastrointestinal infection.
Such patients characteristically have a seronegative (rheumatoid factor
negative) arthritis lasting greater than 1 month associated with
mucocutaneous, ocular, gastrointestinal (GI) or genitourinary (GU)
manifestations. Mucocutaneous features include painless oral ulcers,
balanitis (scaly rash on the glans penis), and keratoderma
blennorrhagicum (scaly rash on palms and soles). Ocular features
include conjunctivitis (which may be completely asymptomatic) or
uveitis with ocular redness and photophobia. GI involvement is
typically a dysenteric or diarrheal illness. GU involvement consists of
urethritis or cervicitis.
species. These agents have not been cultured from synovial tissues
despite the presence of significant joint swelling and synovitis, hence
the term reactive arthritis.
involvement in reactive arthritis is usually an asymmetric lower
extremity oligoarthritis. However, small joint involvement is also seen
in toes and fingers, particularly with the presence of sausage digits.
Furthermore, the arthritis and extra-articular manifestations may occur
at different times.
affecting at least 2% of the population. Psoriatic arthritis, however,
develops in only about 5% of patients with cutaneous psoriasis.
Psoriatic arthritis usually begins in young adulthood but may occur at
any age. A number of patterns of clinical involvement can be seen with
psoriatic arthritis, but the predominant peripheral expression of
arthritis is an asymmetric oligoarthritis, so typical of the
spondyloarthropathies. Approximately 50% of patients with psoriatic
arthritis have a lower-upper extremity oligoarticularthritis.
Approximately 30% will have
an
asymmetric to nearly symmetric polyarthritis, which may resemble
rheumatoid arthritis. However, careful examination revealing persistent
asymmetry, DIP joint involvement, sausage digits, and peripheral
enthesopathy (Achilles tendonitis or plantar fasciitis) strongly
suggests a spondyloarthropathy rather than rheumatoid arthritis. Less
commonly, patients with psoriatic arthritis have sacroiliitis and
spinal inflammation (approximately 20%), exclusive DIP involvement in
hands and feet (approximately 10%) and, rarely, severe destruction of
the finger joints called arthritis mutilans (uncommonly seen today).
Sausage digits are frequently seen in psoriatic arthritis and may be
very symptomatic or completely asymptomatic (thus, easily missed if
both shoes and socks are not removed for a careful joint exam).
essentially confined to the skin and nails. The five most common site
of cutaneous involvement include the elbows, knees, temporoparietal
scalp, umbilicus, and intergluteal cleft (these last three sites can
easily be missed if not inspected carefully).
development of fluffy periosteal new bone at sites of entheseal
inflammation, very characteristic of the spondyloarthropathies.
Extensive and aggressive inflammation at the distal interphalangeal
joints may result in a classic “pencil in cup” deformity at the DIP
joints of the hands or the IP joint of the great toe (a favorite site
of inflammation in psoriatic arthritis).
are clearly recognized and distinguished, patients may present with
clinical features suggesting a spondyloarthropathy (seronegative
oligoarthritis and peripheral enthesitis) without additional
abnormalities allowing a proper name diagnosis. Patients with such
findings are best diagnosed as having an undifferentiated
spondyloarthropathy, with treatment focused on dominant clinical
features.
enthesitis is best managed with a combination of patient education
(increasing understanding and reducing fear), nonsteroidal
anti-inflammatory drugs (NSAIDs) to reduce pain and stiffness,
development of a lifelong daily exercise program (to reduce the
tendency toward spinal fusion) and, more recently, the addition of
anti-tumor necrosis factor (anti-TNF) therapy including etanercept,
infliximab and adalimumab, and others.
plantar fascia and other sites) can best be managed through patient
education, NSAIDs, and orthotics (heel cushions, arch supports, and
splints to reduce physical stress on inflamed entheses). Selective,
local corticosteroid injections may be helpful at reducing inflammation
at painful entheses (with the exception of the Achilles tendon and its
insertion, which should not be injected because of the danger of
rupture).
managed with patient education, NSAIDs, joint aspiration, and
corticosteroid injection, and the addition of systemic medication to
reduce the inflammatory process throughout the body. Sulfasalazine
(entericcoated preparations) can be an especially helpful for patients
with peripheral arthritis. Methotrexate is perhaps the most widely
used, potent anti-inflammatory and is usually given in weekly oral or
parenteral pulses. More recently, anti-TNF therapy (etanercept,
infliximab, and adalimumab) has been used in patients with
spondyloarthropathy and severe axial and peripheral inflammation with
significant disease-modifying effects. Long-term follow-up of patients
given these newer biologic therapies (anti-TNF and other anticytokine
therapy) will be required to firmly establish both their efficacy and
toxicity. At present, the potential for serious infections appears to
be the most significant adverse effect of these agents.
the spondyloarthropathies should be directed at the organs involved.
Ocular involvement with uveitis frequently responds to topical and
systemic corticosteroids. GI involvement with inflammatory bowel
disease requires treatment of the underlying Crohn’s disease or
ulcerative colitis. GU involvement with urethritis may be treated with
a tetracycline or erythromycin during acute episodes of reactive
arthritis (which appears to have no beneficial effect on the duration
and severity of subsequent arthritis). Cutaneous psoriasis frequently
responds to topical preparations and especially well to weekly pulse
methotrexate.
has recently replaced the American term juvenile rheumatoid arthritis
(JRA) and European term juvenile chronic arthritis (JCA). Although the
nomenclature continues to evolve, the rationale for new ILAR
(International League of Associations for Rheumatology) classification
is to recognize subgroups with enhanced homogeneity and to provide
internationally standardized terminology in order to facilitate basic
and clinical research.
systemic arthritis, oligoarthritis, extended oligoarthritis,
polyarthritis (RF negative), polyarthritis (RF positive), psoriatic
arthritis, enthesitis related arthritis, and an “other” arthritis
category. It is important to correctly classify the subtype of
arthritis as the pertinent differential diagnoses, the prognoses, and
the complications vary with the mode of JIA onset. Common to all forms
of JIA are the challenges involved in therapy of a chronic inflammatory
condition occurring in growing, developing individuals. A
multidisciplinary approach, including physical therapist, occupational
therapist, medical social worker, orthopaedist, ophthalmologist, and
rheumatologist, is necessary to ensure the best possible outcome.
unknown. However, evidence suggests that both genetics and environment
likely play a role. The strongest evidence is for linkage with certain
HLA alleles, but increasing evidence shows non-HLA genes may be
important as well.
that present with arthralgia or apparent arthritis) have multiple
causes. Essential to the diagnosis of JIA is the presence of chronic
arthritis (longer than 6 weeks duration), onset before the 16th
birthday and the exclusion of other conditions by history, physical
examination, and appropriate laboratory testing. The possibility of
malignancy must always be considered in the evaluation of a child with
joint pain and ruled out with appropriate studies prior to instituting
therapy for the arthritis. Pain out of proportion to physical findings,
cytopenia, elevated acute phase response with a normal or low platelet
count, elevations in uric acid and/or LDH, and abnormalities on
radiographs are all clues that malignancy may be the underlying cause
of the joint symptoms. Giving corticosteroids to a child with occult
malignancy can dramatically worsen outcome and should absolutely be
avoided. Neuroblastoma and lymphoid malignancies are the most common
neoplasms that present as arthritis in children.
arthralgia or arthritis. Analysis of synovial fluid is mandatory in
cases in which septic arthritis is a diagnostic possibility. Other
infectious causes of arthritis include Lyme disease and tuberculosis.
Beyond malignancy and infection, the differential diagnosis is still
extensive, including trauma, reactive arthritis, acute rheumatic fever,
transient synovitis of the hip, hemophilia, inflammatory bowel disease,
bacterial endocarditis, viral infections, serum sickness, lupus,
dermatomyositis, metabolic disorders, among others. Growing pains, or
benign limb pains, are common in school-aged children. These poorly
localized pains usually occur in the lower extremities in the evening
or at night and normally last a few days to weeks. Severe pain, altered
gait, morning stiffness, and abnormalities on physical examination such
as joint swelling suggest consideration of alternative diagnoses,
including JIA.
Inflammatory arthritis in children (particularly younger children) is
much less likely to present with complaints of pain, even when there is
easily demonstrable inflammatory arthritis on examination. Like adults,
children with chronic arthritis may develop destructive bony changes
and soft tissue flexion contractures. Children, however, are much more
prone to develop ankylosis of peripheral joints and the cervical spine.
Growth disturbances can result from suppression of linear growth by
inflammatory disease and premature epiphyseal closure. Alternatively,
chronic inflammation can cause boney
overgrowth
due to enhanced blood supply to an open epiphysis. Only a minority of
JIA patients are RF positive, and virtually all of these have
polyarticular involvement at presentation.
most common form of JIA. This designation is utilized for children with
up to four joints affected within the first 6 months of disease onset.
Typically, the large joints are affected. In about half of patients,
the disease is limited to a single joint, most often the knee. Elbows
and ankles are also commonly involved. This presentation often afflicts
young girls, with a peak age of onset at about 2 years of age. These
girls have a high incidence of concomitant inflammatory eye disease
(uveitis) as well as serum ANA. Uveitis is typically asymptomatic in
oligoarthritis.
and of insidious onset. The patient may present to the pediatrician for
evaluation of abnormal gait or a reluctance to walk or play. These
patients do not appear systemically ill. Undiagnosed or untreated
disease may result in (and present with) muscular atrophy and joint
contractures, particularly of the knee. As with other subtypes of JIA,
growth disturbances of variable degree occur, depending on severity of
disease, age of affliction, and duration of joint inflammation. Most
patients persist with an oligoarticular course and are classified as
“persistent oligoarthritis.” However, a subset may go on to develop
additional joint involvement over time. Those patients that develop a
polyarticular course after the initial 6 months of oligoarthritis are
classified as “extended oligoarthritis.”
first 6 months of their illness and do not have circulating serum
rheumatoid factor. Patients often present with a gradual onset of
symptoms: decreased activity, morning stiffness, joint swelling, and
occasionally joint pain. Girls are most commonly affected. Both large
and small joint involvement may be seen. Systemic symptoms occur, but
are generally mild. Low-grade fever, fatigue, and poor appetite may
occur for weeks or months before diagnosis. Examination reveals
proliferative synovitis and effusions and often loss of range of
motion. Mild adenopathy or hepatosplenomegaly is sometimes present.
Chronic uveitis occurs less frequently than in the oligoarthritis
category. JIA polyarthritis patients demonstrate a striking tendency
for ankylosis of joints, particularly of the cervical spine.
symptoms but many present with acute polyarthritis. Girls are most
commonly affected, and both large and small joint involvement may be
seen. Low-grade fever, fatigue, and poor appetite are often present.
Examination reveals proliferative synovitis and joint effusions often
with decreased range of motion. Mild adenopathy or hepatosplenomegaly
may be present. Chronic uveitis is rare. These patients often have a
persistent destructive arthropathy and associated subcutaneous
rheumatoid nodules. This small group of patients represents the onset
in childhood of classic adult RA.
approximately 10% of cases. This diagnosis can be quite difficult to
make, as the inflammatory arthritis is not always present on initial
evaluation. These children present with high fever, malaise, and rash.
The fever pattern in Still’s disease is classically quotidian, with one
or more daily spikes to the 38.8°C to 40.5°C (102°F to 105°F) range,
followed by a return to normal or occasionally subnormal temperatures.
The rash is often present only during fever spikes or after a hot bath,
when it transiently appears as a fine, salmon-colored, macular eruption
of the trunk, proximal extremities, and skin overlying affected joints.
Most patients have adenopathy and hepatosplenomegaly and are found to
have moderate to severe anemia and a striking neutrophilic
leukocytosis. Other manifestations of systemic onset disease may
include pericarditis, myocarditis, pleural effusion, and interstitial
lung disease. Renal disease is rare. Other important considerations in
the differential diagnosis include infections (particularly
osteomyelitis and abdominal abscesses), inflammatory bowel disease, and
malignancy. RF and ANA are usually absent, and diagnosis is made on the
basis of clinical findings.
psoriasis and arthritis, or arthritis plus two of the following:
dactylitis, nail changes (pitting or
onycholysis),
or dermatologist-confirmed psoriasis in a first-degree relative.
Generally, there are no associated systemic symptoms and the number of
joints involved is variable. Rheumatoid factor is absent by definition.
Psoriatic arthritis has a variable course but can be quite destructive.
years and often have a family history of HLA-B27 spondyloarthropathy in
a 1st or 2nd degree relative.
arthritis. Or alternatively, they have only arthritis or only
enthesitis plus two of the following: sacroiliac joint tenderness,
presence of HLA-B27, a close relative with a spondyloarthropathy,
symptomatic anterior uveitis, or, if male, onset of arthritis before 8
years.
features of two or more of the categories, make precise classification
impossible. Future revisions of the ILAR criteria and further advances
in establishing etiology will likely occur.
prognosis, and the responsiveness to therapy of JIA. Patients with
oligoarthritis (pauciarticular disease) generally have milder synovitis
and a better prognosis. However, 10 to 15% of patients with
oligoarthritis later progress to polyarticular disease. Children with
oligoarthritis may develop significant disability due to joint
contractures and muscular atrophy, even with control of inflammatory
arthritis activity. One cannot overemphasize the importance of physical
therapy in the management of these patients.
with RF often develop progressive disease extending into the adult
years, and develop significant disability in the absence of effective
therapy. New anticytokine therapies have brightened the long-term
outcome for patients with polyarticular (both RF positive and RF
negative) disease.
acute systemic illness without major sequelae, but synovitis with
either oligoarticular or polyarticular pattern may then persist. A
subset develop persistent treatment-resistant arthritis. Mortality from
JIA is rare but does occur. The vast majority of the deaths occur in
children with systemic onset disease. Macrophage activation syndrome
(MAS) is a rare form of the hemophagocytic lymphohistiocytosis
syndrome, and when it occurs it is a life-threatening complication of
the disease. Viral infections and drugs have been implicated in the
onset of MAS. These children become rapidly ill with fever, worsening
adenopathy, organomegaly, petechiae, and bleeding. ESR rapidly falls
and pancytopenia is present. The ferritin level is often markedly
elevated. This complication needs prompt recognition and treatment.
with JIA and is typically asymptomatic at onset. This is most common in
oligoarthritis patients but may be seen in up to 10% of rheumatoid
factor negative polyarthritis patients, and rarely in systemic-onset
patients. A chronic anterior uveitis occurs most commonly in young
girls who are ANA positive. Insidiously progressive disease may result
in posterior synechiae with resultant pupillary abnormalities.
Occasionally band keratopathy may be seen. Loss of vision results from
the development of secondary glaucoma, cataracts, and keratopathy.
Early detection and treatment are essential to improving the outcome of
JIA-associated uveitis. The eye disease is usually asymptomatic at
onset. Patients with oligoarthritis should be seen by an
ophthalmologist at least every 3 months during the first 2 to 3 years
of disease, and then every 6 months for several more years. There is no
correlation between the severity of arthritis and risk for development
of uveitis. In addition, children with enthesitis-related arthritis may
develop anterior uveitis, but it is typically symptomatic, presenting
as a red, painful eye with or without photophobia.
may have generalized inhibition of growth and subsequent short stature.
Occasionally, periarticular hyperemia results in premature epiphyseal
fusion with resultant shortening of the affected extremity. Prolonged
hyperemia may at times cause accelerated bone growth. Leg length
discrepancies are the most common orthopaedic sequelae of JIA and can
be severe. Temporomandibular Joint (TMJ)
involvement
may result in a shortened mandible and micrognathia. This may result in
disturbances of speech and chewing, in addition to the cosmetic
alteration. Surgery may be complicated by difficulties with
endotracheal intubation particularly in patients with associated
cervical spine fusion.
seen later in patients with polyarticular disease. Muscle spasm and
disuse may result in flexion contracture. Hip involvement in early
childhood may contribute to valgus deformity of the femoral neck,
persistent femoral anteversion, and dysplasia of both femoral head and
acetabulum. Postoperative ectopic bone formation occasionally
complicates surgical management of severe disease. Nonetheless, joint
replacement can be successful in patients with disabling, end-stage
disease. Knee involvement typically results in flexion contracture.
Associated leg length discrepancy and hip disease may also contribute
to development of contracture as well as genu valgus. Secondary
scoliosis may occur. In the hand and wrist, deformities similar to
adult rheumatoid arthritis are seen, although ulnar deviation at the
wrist and radial deviation at the MCP joints is common. Additionally,
some patients develop extensive fusion of carpal bones. Ankles and feet
are similarly prone to fusion, particularly at the subtalar joint.
Complex foot deformities may be seen as a result of soft tissue damage
and growth disturbances.
diagnosis and prompt institution of treatment. As our arsenal of
therapeutic agents grows and is utilized appropriately, we are likely
to see long-term outcomes continue to improve.
of JIA. They control pain, swelling, and stiffness, but have no effect
on the long-term outcome. NSAIDs approved by the U.S. Food and Drug
Administration for use in childhood include aspirin, ibuprofen,
naproxen, and tolmetin sodium. Other NSAIDs are used for patients who
cannot tolerate the FDA-approved NSAIDs. Ibuprofen, naproxen, and
several salicylate preparations are available in liquid form. Aspirin
has a long historical record of use in JIA but its use has decreased
markedly due to the risk (albeit very slight) of Reye syndrome, the
potential for drug toxicity, and the availability of other effective
drugs. Salicylates (if used) should be promptly discontinued in JIA
patients with concomitant influenza or varicella infections. Most
children with JIA do not do well on an NSAID alone and require
additional therapeutic intervention for maximum control of their
disease.
corticosteroids and/or anti-malarial drugs are frequently utilized
along with regular physical therapy. Systemic corticosteroids are
reserved for refractory cases of JIA and are particularly useful in the
treatment of severe systemic JIA. The lowest possible dose should be
used.
joints involved. Increasingly, biologic agents are being utilized that
block a particular proinflammatory cytokine. Children with severe
polyarticular arthritis who fail or have an incomplete response to
methotrexate are candidates for treatment with this class of agents.
These drugs must still be used with caution, as their long-term safety
has not yet been demonstrated.
rheumatology center require surgical intervention, and the specific
procedures used are reviewed more extensively in the references.
Children with severe JIA have significant osteoporosis and can
experience spontaneous fractures. JIA patients require surgical
intervention when medical and physical therapy (e.g., splinting,
casting) are not sufficient to control pain, improve contracture, or
correct deformity. The most common procedure in JIA patients is soft
tissue release, particularly of the knee or hip. Occasionally, synovial
biopsy may be necessary for diagnostic reasons. Synovectomy can be
helpful for the child with severe pain or loss of function and is most
commonly required for the knee in oligoarthritis patients. Arthroscopy
can be performed, but the presence of severe pericapsular contracture
may decrease distensibility and visibility of the joint. Severe bone
ankylosis may require a corrective osteotomy to improve joint position.
Joint replacement is a well-established treatment of end-stage JIA.
Children with severe JIA are often smaller and lighter than other
children of the same age, and the joint prosthesis may need to be
custom made.
is necessary in all children. Children on systemic corticosteroid
therapy need intravenous stress corticosteroid coverage and are at
greater risk of infection. Involvement of the cervical spine, TMJs, and
cricoarytenoid joints can make airway management difficult.
etiologic role in the initiation and propagation of some acute and
chronic arthritides. Symptoms suggesting a possible infectious
arthritis need to be evaluated promptly and treated appropriately,
because failure to recognize infectious arthritis may result in
avoidable catastrophic joint destruction. However, in reactive
arthritis, the interaction of a genetic susceptible background,
especially HLA-B27, with certain infectious agents (e.g.,
Campylobacter, Chlamydia, Clostridium, Salmonella, Shigella, or
Yersinia) may result in inflammatory reactive arthritis. In these
instances bacterial DNA may be found in joint fluid or tissue using
PCR, but cultures remain negative. This section discusses infectious
arthritis based on the different causes: bacterial, fungal, and viral.
to a destructive acute arthritis. Direct invasion of the joint by
pyogenic bacteria may occur through inoculation, or by contiguous
spread from soft tissue infection or from osteomyelitis. In the primary
care setting, the majority of cases arise from hematogenous spread to
the joint. Common predisposing factors include injection drug use,
in-dwelling catheters, and underlying immunocompromised states such as
HIV infection, alcoholism, diabetes mellitus, malignancy, allogeneic
transplants, chronic inflammatory arthritis, or concomitant use of
immunosuppressive medications. Immuno-compromised patients may display
few symptoms of infection, making the clinical diagnosis of septic
arthritis difficult. Orthopedic surgeons are more likely to be
confronted with joint infections as a result of trauma or surgical
procedures. Examples include penetrating injury, introduction of a
foreign body into a joint, arthroplasties, or total joint replacements.
Late infections of prosthetic joints, albeit rare, may be the result of
contamination at the time of surgery, or due to bacterial seeding
during a bacteremic episode.
native joints are Gram-positive cocci (in 75% of all cases), whereas
Gram-negative organisms are the second most common (15 to 20%). Staphylococcus aureus
is the most common cause, both in native and prosthetic joints.
Streptococci, including pneumococci, are the second most common
Gram-positive bacteria isolated. Staphylococcus epidermidis
is commonly encountered in prosthetic joints but rarely seen in native
joint infections. In intravenous drug abusers or patients with
comorbidities, such as in the elderly, Gram-negative organisms
including Escherichia coli, Proteus spp., and Serratia spp., may be more commonly encountered.
initiating therapy may lead to joint destruction. Acute septic
arthritis usually presents with joint swelling, warmth, pain, erythema,
and loss of function. Although usually a single joint is affected (in
about 80% of the cases), multiple joints can be infected, especially in
immuno-compromised patients. Sudden onset of monoarticular arthritis
should be considered septic until proven otherwise. The large
weight-bearing joints, especially the knees, are most commonly
involved. Ankles, shoulders, elbows, and wrists are other commonly
affected sites. In a patient with known inflammatory polyarthritis,
such as rheumatoid arthritis, the sudden worsening of a single joint
out of proportion to disease activity in the other joints should
suggest the possibility of septic arthritis. Nonarticular infection,
such as cellulitis, pneumonia, dental abscess, or urinary tract
infections, are often the distant source for bacterial seeding of
joints.
helpful in suggesting an infection but in themselves are nonspecific,
as are general laboratory studies. An elevated ESR is common.
Leukocytosis occurs in up to two-thirds of patients. Plain radiographs
are usually normal early in infectious arthritis, except perhaps for
evidence of soft tissue swelling or joint effusion. Nevertheless,
joints with suspected infection should be radiographed at presentation,
since a baseline study is useful in interpretation of subsequent
examinations. Periarticular osteopenia may be seen but is nonspecific.
Joint space narrowing due to cartilage destruction may occur in days to
weeks without appropriate treatment. Subchondral bone destruction is a
late
finding.
Contiguous osteomyelitis is a late but grave complication. Radionuclide
imaging techniques, such as technetium gallium, or indium scans, may
aid diagnosis of septic arthritis in joints difficult to aspirate, such
as intervertebral, sacroiliac, or hip joints.
confirmed by arthrocentesis with synovial fluid analysis and culture.
Synovial fluid is usually purulent, with greater than 50,000 cells/mm3
and over 80% polymorphonuclear leukocytes. Initial leukocyte counts,
however, may be only minimally elevated. In immuno-compromised hosts,
leukocyte counts in synovial fluid may remain low or even normal. In
cases with low or normal leukocyte counts, repeat arthrocentesis in 12
to 24 hours may demonstrate rising counts. A high leukocyte count in
itself is not pathognomonic, since high counts may be seen in nonseptic
inflammatory arthritis, especially crystal-induced arthritis or
Reiter’s syndrome. Identification of crystals, however, does not rule
out septic arthritis, because both entities may occur simultaneously.
The definitive diagnosis requires demonstration of the causative
bacteria on Gram’s stain, by culture, or both. The main reason for
false-negative culture results are prior use of antibiotics and the
special growth requirements of some fastidious microorganisms. In those
cases the use of PCR may be extremely helpful. Although the utility of
this sensitive technique is still being defined, DNA detection of Neisseria gonorrhoeae and Mycoplasma spp. has been shown to be useful when cultures remained negative.
recovery, and reduce morbidity. Antibiotic treatment based on a
presumptive diagnosis of septic arthritis should be initiated after
arthrocentesis and collection of samples for microbiologic studies. The
choice of antibiotic is guided by the history, clinical presentation,
and results of the Gram’s stain. If the Gram’s stain of the synovial
fluid reveals Gram-positive cocci, cefazolin or vancomycin are
preferred choices. If there are Gram-negative organisms, a third
generation cephalosporin is indicated. If Pseudomonas aeruginosa
is suspected (in patients with injection drug use), ceftazidime with an
aminoglycoside is the first choice. If a young, otherwise healthy,
sexually active person presents with tenosynovitis and migratory joint
pain, without any visible bacteria on Gram’s stain, therapy against
presumptive gonococcal infection may be appropriate (see next section).
hasten eradication of the infection. Surgical drainage may be indicated
if repeated aspirations are technically difficult. Although
antimicrobial therapy needs to be tailored to the individual patient,
typically, intravenous antibiotic therapy is given for 2 to 4 weeks
followed by oral therapy for a total course of treatment of 4 to 6
weeks, depending on the severity of the infection. In prosthetic
joints, therapy may be even more protracted.
typical patient who develops gonococcal arthritis is frequently young
and healthy. Infectious arthritis due to N. gonorrhoeae
follows dissemination from a primary site, such as urethra, cervix,
rectum, or pharynx. The clinical course is classically biphasic. The
first stage is characterized by migratory polyarthralgias,
polyarthritis, or tenosynovitis. Multiple vesiculopustular skin lesions
that develop necrotic centers may be seen. Cultures from these skin
lesions and from blood are often positive for N. gonorrhoeae
during this stage. Untreated, the patient may develop the second stage,
in which infection settles into one or a few joints, which become
purulent. Cultures from purulent joints are positive only one-quarter
of the time because N. gonorrhoeae is a
fastidious organism and difficult to grow. Patients with suspected
gonococcal infection should have all possible sites examined and
cultured, including pharynx, rectum, blood, cervix in women, and
urethra in men. Special transport media should be used if samples will
be delayed in reaching the microbiology laboratory.
resolution of symptoms followed by a 2 weeks of oral cefuroxime or a
quinolone. However, quinoloneresistant strains are common in Asia and
the Pacific and are also increasingly noted in Hawaii and California,
so quinolones are no longer appropriate in these areas. Concurrent
treatment for Chlamydia trachomatis with doxycycline for 7 days is mandatory. Sexual partners should be traced and offered treatment as well.
differential diagnosis of chronic monoarticular and pauciarticular
arthritis at any age. The arthritis is frequently insidious in onset.
It tends to appear “cold,” lacking the usual signs of active
inflammation, especially erythema and heat. Pott’s disease, tuberculous
involvement of the spine, classically involves the thoracolumbar
junction. Anterior destruction of vertebral bodies and disks eventually
leads to angulation of the spine and kyphosis (gibbous deformity).
Although constitutional signs of tuberculosis (e.g., fever, malaise,
and weight loss) may be present, active pulmonary tuberculosis is rare.
A history of past infection may be absent. A positive skin test for
tuberculosis is helpful, although a negative test in the presence of
anergy does not rule out the diagnosis. Diagnosis is based on finding
acid-fast bacilli in synovium or synovial fluid or caseating granulomas
in biopsied synovium. A CT-guided biopsy of inaccessible sites may be
required. Synovial fluid or tissue cultures are positive 90% of the
time. Tuberculosis and its complications should be considered in
patients with AIDS or with a history of immigration from an endemic
area. In addition, reactivation of latent tuberculosis may occur as a
result of treatment with corticosteroids or tumor necrosis factor-α
(TNF-α) antagonists. Tuberculous arthritis is usually due to
hematogenous spread, and thus active disease elsewhere needs to be
investigated. Poncet’s disease is defined as reactive arthritis during
active tuberculosis, most commonly seen as polyarticular arthritis of
the hands and feet, while cultures of synovial fluid remain negative.
Improvement ensues with antituberculous medication. Antimicrobial
treatment of tuberculous arthritis or osteomyelitis is the same as for
pulmonary tuberculosis.
Infections are indolent. Diagnosis is made by the demonstration of the
organism in the synovial fluid or tissue. Delay of diagnosis up to a
year or more is not uncommon. Most nontuberculous mycobacterial joint
infections are due to local trauma, surgery or intra-articular
injection. Treatment may require surgical debridement for both
diagnosis and therapy, especially for the closed spaces of the hand and
the wrist and for infections of long bones, in addition to specific
drug therapy.
more than 85% of patients are immigrants. The six countries with the
highest incidence of leprosy include Brazil, India, Madagascar,
Mozambique, Myanmar, and Nepal. Lepromatous leprosy may present with
polyarthralgia or polyarthritis. Erythema nodosum leprosum is an
associated finding and consists of nodules on the legs, arms, or trunk.
Malaise and fever may occur. Swollen hands syndrome is another
presentation of leprosy. Thickening of peripheral nerves and typical
skin changes suggest the diagnosis. Chronic erosive arthritis may
resemble rheumatoid arthritis and improves with treatment of the
leprosy. A classic finding in the late stages of leprosy is the Charcot
joint as a result of sensory neuropathy with repeated minor trauma. The
diagnosis is made by identifying M. leprae in aspirates of skin lesions or biopsy specimens. M. leprae may be found in synovial and periarticular tissues. Combination therapy of several drugs with activity against M. leprae has replaced monotherapy with dapsone.
syphilis can cause inflammatory polyarthritis that can be confused with
many other joint diseases such as rheumatoid arthritis, SLE, and
sarcoidosis. Syphilis is suspected if there is a concomitant
maculopapular rash on the palms and soles. Other characteristics may
include a systemic illness with fever, lymphadenopathy, sore throat,
and mucosal ulcers. The arthritis is symmetric and involves
predominantly the lower extremities. Treponema pallidum spirochetes cannot be grown from human specimens in vitro, and hence cultures of joint fluid are negative. The diagnosis relies on serology (RPR or VDRL) and a confirmatory FTA-ABS.
syphilis. The congenital form of syphilis is a severe, disabling, and
often life-threatening condition, and occurs when the mother is
infected and transmits the pathogen to her unborn infant. Perinatal
mortality is high. Infants develop early stage and late-stage symptoms
of syphilis if not treated. Early stage symptoms include irritability,
failure to thrive,
and
fever. Bony abnormalities include osteochondritis, osteomyelitis,
osteitis, and periosteitis. In older children, painless effusions,
especially of the knees, may occur. In acquired primary syphilis,
transient bone pain of a boring nature may be prominent. The tibia,
humerus, and cranium are most frequently involved, but radiographs are
normal.
overlying soft tissue swelling may be seen in superficial bones, such
as anterior tibia, sternum, ribs, and skull. Symptoms and signs are
variable but characteristically worse at night. Proliferative
periosteitis is the most common radiographic change. It is associated
with new bone formation that may be extensive, resulting in marked
cortical thickening. The tibia, sternum, ribs, and skull are most
significantly involved, but changes may also be seen in the femur,
fibula, clavicle, hands, and feet. Periosteitis in the adult that
involves both clavicles or tibiae is frequently syphilitic. Destructive
bony lesions suggest syphilitic osteomyelitis or osteitis, but these
are less common than periosteitis. Areas of lysis may be seen.
osseous lesions. The lesion pathologically resembles a tubercle with
necrosis of adjacent bone. Lytic and sclerotic areas of bone may reach
large size and may be associated with pathologic fracture. Periosteitis
adjacent to gummatous lesions is frequent. Nongummatous osseous lesions
that consist of periosteitis, osteitis, or osteomyelitis may occur in
conjunction with or in the absence of gummatous bony lesions. Charcot
joints, characteristically of the knees, result from loss of
proprioception due to tabes dorsalis in tertiary syphilis. Hip, ankle,
shoulder, elbow, spine, and other joints may be affected as well in
tabes dorsalis.
The geographic distribution of Lyme disease is dictated by the presence
of the vector. In the Northeast and upper Midwest, transmission occurs
by the tick Ixodes scapularis or deer
tick. Although Lyme disease has been reported in most states, more than
90% of all cases in the Unites States are seen in only eight states:
Massachusetts, Rhode Island, Connecticut, New York, New Jersey,
Pennsylvania, Minnesota, and Wisconsin. Transmission in western states
occurs by Ixodes pacificus (black-legged tick), while Ixodes ricinus
is responsible for transmission in Europe. Normally ticks require up to
4 to 5 days to feed to completion and transmission of the organism
occurs most efficiently after 48 hours. Ticks removed within 24 hours
of attachment will not have transmitted the spirochete. Discovery and
removal of an engorged tick on the day following outdoor activities in
an endemic area is sufficient to prevent Lyme disease in most cases,
provided that a “total body tick check” is performed. Less than 50% of
patients ever recall a tick bite.
includes the typical erythema migrans, or bull’s eye rash, which occurs
in about 80% of Lyme disease patients and within one month of the tick
bite. The rash (usually more than 3 to 5 cm) should not be confused
with the small local reaction (just a few mm), which occurs at the site
of any bite. The diagnosis of this stage is made by the combination of
the typical history and the rash.
occurs in several weeks to a few months and is characterized by
disseminated skin lesions at sites other than the tick bite, cardiac
disease, and neurologic involvement. This phase represents systemic
infection. Spirochetes may be present in the skin, in cardiac tissue
(leading to conduction block), and central nervous system (Bell’s
palsy).
after the initial infection. Most frequently it consists of a
monoarthritis of the knee, but other large joints can be involved.
Patients with Lyme arthritis are usually strongly seropositive.
Episodes of joint inflammation may recur several times with
disease-free intervals, but the ultimate outcome is favorable with
eventual resolution of the inflammation over the course of several
years. Late neurologic symptoms are designated as tertiary
neuroborreliosis in analogy with tertiary neurosyphilis. Patients with
neuroborreliosis are strongly seropositive. Features include
encephalopathy, neurocognitive dysfunction, and peripheral neuropathy.
Neuroborreliosis is relatively rare, especially because most patients
are now correctly diagnosed and treated early within the disease course.
disease is obtained by ELISA. Positive or equivocal ELISA results are
confirmed by immunoblot, because significant false positive ELISA
results are due to other (spirochetal) infections (i.e., relapsing
fever, syphilis, Epstein-Barr virus) and other autoimmune diseases
(i.e., rheumatoid arthritis and SLE). A positive Lyme test includes a
positive
ELISA
and a positive immunoblot consisting of at least two of three bands for
IgM (23, 39 and 41 kD), or five of ten bands for IgG (18, 21, 28, 30,
39, 41, 45, 58, 68, 93 kD). Note that a positive IgM immunoblot can
only be used to support the diagnosis within 4 weeks of onset of
clinical disease. Laboratory testing in the setting of early disease is
not recommended because of a high rate of false-negative results, but
treatment should be initiated promptly.
weeks of 100 mg orally 2 times a day doxycycline, or amoxicillin 500 mg
4 times a day. Early disseminated or late Lyme disease requires 3 weeks
of a third generation cephalosporin intravenously.
from infected animals or through the ingestion of untreated milk or
milk products; raw meat and bone marrow have also been implicated.
Other portes d’entrée include skin abrasions and conjunctiva or by
inhalation during contact with animals, especially by slaughterhouse
workers, farmers, and veterinarians. Aerosolized B. melitensis is considered an agent of biological warfare.
brucellosis. However, it is suggested that even in developed nations,
the true incidence of brucellosis may be up to twentyfold higher than
official incidence numbers. In the United States, about 200 new cases
are reported every year, but only a fraction of cases are recognized
and reported. Consumption of imported cheese, travel abroad, and
occupational exposures are the most frequently identified sources of
infection. Brucellosis has many synonyms depending on geographical area
(e.g., Mediterranean fever, Malta fever, etc). Four species can cause
human brucellosis: Brucella melitensis (the most common cause of brucellosis) and is acquired primarily from goats and sheep; B. abortus from cattle; B. suis from hogs; and B. canis from dogs.
acute febrile illness and are not specific for brucellosis. Acute
infection is associated with bacteremia, fever, myalgias,
polyarthralgia, headache, and general malaise. Most commonly, brucella
arthritis is monoarticular and involves the large weight-bearing joints
of the lower extremities. However, 30 to 40% of patients have reactive
asymmetric polyarthritis involving the knees, hips, shoulders, and
sacroiliac and sternoclavicular joints. Infection with brucella
organisms commonly causes osteomyelitis of the lumbar vertebrae,
starting at the superior endplate and occasionally progressing to
involve the entire vertebra, disc space, and adjacent vertebrae.
Extraspinal brucella osteomyelitis is rare. In brucella septic
arthritis and osteomyelitis, the peripheral white cell count is
typically normal, while the erythrocyte sedimentation rate may be
either normal or elevated. Diagnosis is based on a positive culture or
on a rising or high brucella antibody titer. Cultures of synovial fluid
are positive in about 50% of cases. Radiologic investigations aimed at
detecting skeletal involvement include plain radiography, bone
scintigraphy, CT, and MRI. Bone scintigraphy is more sensitive than
conventional radiography in detecting areas of spinal and extraspinal
involvement, particularly in the early stage of infection. Plain
lateral radiography of the spine may reveal bone sclerosis, with
destruction and erosion of the superior end plate anteriorly. As the
disease progresses, healing with osteophyte formation and reduction of
disc space may take place.
combination therapy because of the high rates of failure and relapse
and the potential development of antibiotic resistance. Combination
therapy lasting less than 8 weeks has also been associated with high
rates of relapse. The combination of doxycycline and an aminoglycoside
for 4 weeks followed by the combination of doxycycline and rifampin for
4 to 8 weeks is the most effective regimen.
have an indolent course, and are associated with mild inflammation.
Diagnosis is often delayed and frequently the only laboratory finding
is a positive culture. Septic arthritis caused by Candida albicans is rare and is usually due to direct inoculation or hematogenous spread. C. albicans
is a rare cause of infection in prosthetic joints (< 1%). In adults
disseminated candida infections are usually related to IV drug abuse or
severe illness requiring intensive care and the use of multiple
broad-spectrum antibiotics, or associated with chemotherapy or
immunosuppressive medication. Treatment includes amphotericin B,
ketoconazole, and fluconazole.
arthritis. Osteomyelitis has a predilection for vertebral sites and
ribs, and can mimic tuberculous infection. Large joint arthritis such
as the knee can be acute, usually in the setting of pulmonary disease
or cutaneous infections. Diagnosis is confirmed by culture and
treatment options include amphotericin B, ketoconazole, and
itraconazole.
It may present with erythema nodosum, periarthritis, and bihilar
lymphadenopathy. This triad is also seen in sarcoidosis (known as
Lofgren syndrome), and therefore, the two entities must be
differentiated. In coccidioidomycosis, the triad is a hypersensitivity
reaction to primary infection and resolves spontaneously within weeks.
Persistent arthritis is uncommon, resulting from hematogenous spread or
extension from contiguous osteomyelitis. The most commonly involved
joints are knee, wrist and hand, ankle, elbow, and foot, in order of
decreasing frequency. Arthritis progression is slow and indolent.
Diagnosis is by synovial biopsy and culture.
typically occurs in agricultural or mine workers or in gardeners after
minor skin trauma. Disease usually presents as a painful erythematous
cutaneous nodule and spreads by local extension and via the lymphatic
system. Indolent arthritis occurs rarely and involves the knee, hand,
wrist, or ankle, in order of decreasing frequency. About half the
patients have a monoarthritis. Alcoholism or myeloproliferative disease
predisposes to infection. Diagnosis is by synovial histology and
culture.
spreads hematogenously and may seed the central nervous system and can
result in bone and joint infection similar to blastomycosis.
Demonstration of the organisms in joint fluid or biopsy specimens
confirms the diagnosis.
with amphotericin B but renal toxicity has been a major limitation.
Newer agents including liposomal amphotericin and caspofungin seem to
have less toxicity.
infections has long been recognized. Viral arthritis characteristically
is polyarticular and usually develops at the time of the rash or
shortly thereafter. In most cases pathogenesis seems to be due to
immune-complex deposition. It is important to remember that the
arthritis associated with viral infections never leads to joint
destruction or chronic joint inflammation.
as 12% of patients presenting with recent onset of polyarthralgia or
polyarthritis. B19, first described in 1975, is very common, and causes
the common childhood exanthem erythema infectiosum, or fifth disease,
characterized by “slapped cheeks” and a rash of the torso and
extremities. A majority of adults have serologic evidence of past
infection. About 10% of children with fifth disease have arthralgias,
and 5% have arthritis, usually short-lived. In contrast, 60% of
infected adults have arthralgias or frank polyarticular arthritis, in a
distribution that resembles rheumatoid arthritis. In fact, the history
of a viral syndrome and a B19 seroconversion may help differentiate
viral polyarthritis from rheumatoid arthritis in an early stage. B19
can also cause transient aplastic anemia in patients with underlying
hematologic disorders, and thus B19 infection may resemble SLE. A
recent B19 infection is confirmed if a B19 IgM is detected, which is
only present for several weeks (occasionally up to 2 to 3 months) after
infection. IgG persists for years and is not helpful unless a
seroconversion between paired samples is detected. Detection of B19 DNA
by PCR is useful but can remain positive for months after the initial
infection. The long-term outcome is good, though a few patients have
recurrent episodes of arthralgias over several years.
usually self-limiting, followed by the appearance of hepatitis B
surface antibodies. However, 5 to 10 % of patients may develop a
chronic infection, with continued antigen production (i.e., HBeAg,
HBcAg, HBsAg). In the prodromal phase, which may precede clinical
jaundice circulating immune complexes may cause symptoms ranging from
arthralgias to a sudden and severe polyarthritis. Symptoms usually last
several weeks, but in patients with chronic active hepatitis,
arthralgias and arthritis may be prolonged. Because patients may not be
icteric in chronic active hepatitis, laboratory testing for hepatitis
may be an essential to the diagnosis. Polyarteritis nodosa (PAN) is
frequently associated with chronic HBV viremia. PAN is characterized by
inflammation of small and medium-sized arteries and is a multisystem
disease: arteries in skin, kidneys, peripheral nerves (vasa nervorum),
intestines, and muscles can be affected.
hepatitis and cirrhosis. Mixed cryoglobulinemia is most frequently
associated with HCV, although a number of other viruses and bacteria
have also been
implicated.
Other disorders such as lymphoma and myeloma can also cause
cryoglobulinemia. Clinical features of mixed cryoglobulinemia include
palpable purpura, hepatitis, arthralgias/arthritis, renal involvement,
Raynaud’s and peripheral neuropathy. The cutaneous vasculitis and
glomerulonephritis is immune-complex mediated, and a similar
pathogenesis has been suggested for the arthritis. Treatment consists
of interferon 2b, preferably the pegylated form, combined with
ribavirin, with the objective of clearing the HCV. Glucocorticoids and
immunosuppressive agents such as methotrexate have been used, but
should probably be reserved for severe vasculitis due to the
cryoglobulinemia, because these drugs may increase viral replication,
and hence perpetuate the problem.
maculopapular rash, lymphadenopathy, and arthralgias or arthritis. The
joint symptoms are usually self-limited. Vaccination with live
attenuated strains can cause joint symptoms in up to 25% of vaccinated
individuals.
epidemics with associated polyarthritis, but to date, these have been
all outside North America. The viruses include Sindbis, Mayaro, Ross
River, Chikungunya, and O’nyong-nyong viruses, all of which are
mosquitoborne. The recent emergence of West Nile virus in the United
States demonstrates the potential that these viruses may be encountered
in this country in the future. The arthritis is polyarticular with a
preference for the small joint of the hands, wrists, elbows, knees, and
ankles. Most cases resolve within a week, although joint symptoms
persisting for up to a year have been reported.
musculoskeletal syndromes, only a few of which are specifically related
to HIV. Reiter’s syndrome, without eye inflammation, has probably an
increased frequency in HIV patients in the study populations. However,
extra-articular manifestations such as eye inflammation may be absent.
HIV increases the frequency of psoriatic arthritis in patients with
psoriasis. Diffuse infiltrative lymphocytosis syndrome (DILS) and
HIV-associated arthritis are seen specifically in HIV. These syndromes
occur in the setting of significant immunosuppression. Treatment with
combination antiviral therapy seems to reduce the incidence, suggesting
that HIV may be involved directly or indirectly in the pathogenesis.
of short duration.” “The cause of this disease is an immoderate use of
stimulating food and drinks. Plethoric persons are its most frequent
victims.”
as an acute gout attack. With its clinical description rooted in
antiquity, gout can be considered the prototypic acute monoarthritis as
well as crystal-induced arthritis. Gout is due to the presence of
monosodium urate (MSU) crystals in articular tissues.
supersaturation (generally, serum uric acid > 7.0 mg/dL) leads to
precipitation of MSU crystals in soft tissues. When sufficient MSU
accumulates within joints, several factors, such as trauma, abrupt
dietary changes, acute illness, surgery, dehydration, and
administration of serum urate-lowing medications, probably induce acute
MSU crystal precipitation within joints or shedding of crystals from
deposits in synovium into synovial fluid. MSU crystals recovered from
joints are coated with a variety of serum proteins, notably
immunoglobulins, which attract and enhance phagocytosis by
polymorphonuclear (PMN) leukocytes. Crystal contact with leukocytes
results in generation of mediators of inflammation and
chemoattractants, including interleukin-1 and a specific PMN-derived,
crystal-induced chemotactic factor. A massive influx of PMN into the
joints ensues. Partially due to the membranolytic properties of the MSU
crystals, PMN lysis occurs, releasing lysosomal contents that further
intensify the inflammatory response. Simultaneous influx of serum
factors into the joint, including lipoproteins and proteolytic enzyme
inhibitors, help make gout attacks self-limited.
hyperuricemia. Consequently, the epidemiology of gout closely parallels
the epidemiology of hyperuricemia. Because lower levels of serum uric
acid are found in the normal population of prepubertal men and
premenopausal women, gout develops mostly in postpubertal men or
postmenopausal women. Other factors that result in sustained elevations
of serum uric acid above the solubility product of MSU can lead to
increased total-body urate and predispose to gout. Such factors can be
divided into those that involve either uric acid underexcretion or
overproduction.
onset (usually over a few hours) of extreme pain, swelling, and
erythema in one joint. The inflammatory response is so intense that
patients describe an inability to tolerate anything touching the joint.
The first metatarsophalangeal (MTP) joint is the most frequently
affected joint (podagra). Other joints commonly affected early include
the midfoot (tarsalmetatarsal joints) and the ankle. The attacks are
usually self-limited and resolve completely in 1 to 2 weeks without
therapy. Recurrent episodes are common. The period of complete symptom
relief between episodes of intense joint inflammation, termed the
intercritical period, is a hallmark of crystal-induced arthritis.
intercritical period shortens leading to chronic polyarticular
arthritis. Polyarticular gout can affect knees, wrists, elbows,
metacarpophalangeal (MCP) joints and proximal interphalangeal (PIP)
joints. Hips, shoulders and the axial spine are rarely affected.
called tophi that can be seen in cartilage, tendons and soft tissues.
Extensor surfaces of the extremities and the Achilles tendon are common
sites of deposition, but tophi can be seen on the ear, on the skin, and
around the small joints of the fingers and toes. Tophi may drain
spontaneously and may be confused with draining abscesses. The drainage
from tophi usually includes particles of white, chalky material and may
even have an appearance suggestive of toothpaste. Patients who have
undergone organ transplant may develop gout that behaves differently
than idiopathic gout. These patients may present initially with
polyarticular, tophaceous gout.
monoarthritis should exclude cellulitis, bursitis, or tendonitis, which
can all be associated with warmth, pain, and swelling. Demonstration of
a joint effusion or pain on slight movement of the affected joint helps
to confirm true joint involvement. The finding of redness over a joint
with exquisite tenderness requires arthrocentesis to exclude an
infected joint. MSU crystals can be demonstrated under polarized
microscopy after aspirating a tophus or by touching a microscope slide
to the tophaceous drainage.
crystals within synovial fluid of an affected joint. Compensated,
polarized light microscopy will reveal negatively birefringent,
needle-shaped crystals found within PMNs. Other tests on synovial fluid
are of limited use in acute, crystal-induced arthritis. Synovial fluid
leukocyte counts should be clearly inflammatory, often in the 20,000 to
50,000 cells/µL range. In acute gout, synovial fluid leukocyte counts
can be as high as those seen in septic arthritis or higher. Coexistent
gout and septic arthritis is rare but can occur. If clinical suspicion
of a septic joint persists, even after crystals are identified,
synovial fluid Gram’s stain and culture should be obtained.
patients with suspected acute gout. Although a gout attack is preceded
by many months of sustained hyperuricemia, at the time of an acute gout
attack, serum uric acid may be low, normal or high. Serum uric acid
levels are not helpful in diagnosing acute gout; however, levels are
useful in guiding uric acidlowering therapy, later. Further laboratory
investigation to characterize the hyperuricemia may be indicated when
the acute gout attack resolves. In most cases, hyperuricemia is due to
renal underexcretion of uric acid. If the onset of gout occurs in a man
before the age of 20 years, in a menstruating woman, or in a patient
without an obvious reason for renal underexcretion, it is appropriate
to determine whether urate accumulation is a result of overproduction
or underexcretion. This is accomplished by measuring 24-hour urine uric
acid excretion. An excretion of greater than 800 mg of uric acid, on an
unrestricted diet is evidence of uric acid overproduction. In the
presence of normal renal function, a 24-hour urinary uric acid of less
than 800 mg in a hyperuricemic patient indicates underexcretion.
may only show nonspecific soft tissue swelling. With long-standing
disease and a history
of
several attacks, asymmetric periarticular swelling suggestive of a soft
tissue mass may indicate the existence of a tophus. Tophi may also
develop inside juxta-articular bone and appear on radiographs as
subchondral, cystic lucencies. Still later, bone erosions may be seen
that are either intra-articular or extra-articular (proximal to the
point of confluence of synovium, cartilage, and subchondral bone).
These erosions may exhibit a characteristic appearance in which
overhanging edges of the disrupted bone cortex are seen.
administration of a nonsteroidal anti-inflammatory drug (NSAID). An
example of an effective regimen is indomethacin, 50 mg taken every 6
hours for 1 to 2 days then tapered to 50 mg taken every 8 hours for 1
to 2 days then tapered to 25 mg every 8 hours until the attack abates.
There is little difference in efficacy among the NSAIDs and an
inexpensive, short-acting choice may be the most appropriate. Caution
should be exercised when using NSAIDs. Gastrointestinal ulceration is a
major problem with chronic use. Worsening renal insufficiency or
congestive heart failure can also occur. Decreased doses of NSAID and
more frequent monitoring of serum creatinine is indicated in a
high-risk patient. NSAIDs that inhibit cyclooxygenase 2 have a lower
incidence of gastrointestinal toxicity but are no more effective, have
the same potential renal toxicity and are more expensive. Concerns
about cardiovascular morbidity may limit the use of these agents.
Aspirin, other salicylates, allopurinol, and the uricosurics should be
avoided in gout attacks, because they are likely to alter serum uric
acid levels and lead to prolongation of the attack.
gout. The traditional oral regimen is 0.6 mg every 1 to 2 hours until
(1) improvement in the attack is seen; or (2) until the development of
nausea, vomiting, or diarrhea; or (3) until a maximum of 10 doses (<
6 mg in a 24-hour period) have been taken. Intravenous colchicine is
rarely used because of the potential for severe bone marrow toxicity.
The dose of colchicine should be reduced in those with impaired renal
or hepatic function and in the elderly.
20 mg daily for 3 days, 15 mg daily for 3 days, 10 mg daily for 3 days,
5 mg daily for 3 days, then stop) is frequently efficacious and well
tolerated. Intra-articular corticosteroids can also be beneficial
provided one is sure the joint is not infected. Intramuscular ACTH has
been shown to be effective. Finally, in a patient with multiple
comorbidities that limit therapy, simply controlling the pain while the
self-limited attack abates may the most appropriate course.
patients may suffer only a small number of gout attacks in their
lifetime and never develop detectable tophi or joint damage.
Intermittent use of NSAIDs in this case may be the most appropriate
course. Repeated attacks, the development of tophi, joint damage, or
renal damage may necessitate serum urate-lowering therapy. Allopurinol
inhibits the enzyme xanthine oxidase and prevents the formation of uric
acid. A usual dose is 300 mg orally, once a day. Smaller doses (i.e.,
100 mg) may be appropriate in the elderly or in a patient with renal
insufficiency. The dose is adjusted upward until a serum uric acid
level of ≤ 5 mg/dL is achieved. Patients should be instructed to
immediately discontinue allopurinol if they develop a new rash. The
development of allopurinol hypersensitivity can rarely be associated
with fatal toxic epidermal necrolysis. The indications for using
allopurinol include three or more attacks in a year and one of the following: renal insufficiency, nephrolithiasis, or tophi.
increased frequency of gout attacks. Consequently, prophylaxis against
acute attacks is usually administered for a week before the initiation
of allopurinol and continued for 1 to 12 months in the form of oral
colchicine 0.6 mg once or twice daily. Small doses of NSAID (i.e.,
ibuprofen 400 mg once or twice daily) may be substituted if the risk
for NSAID associated toxicity is low.
a rational choice in patients with uric acid underexcretion. There are
some limitations to this approach: (1) normal renal function is
required for the uricosuric drugs to be effective; (2) increased
concentration of urine uric acid can result in acute stone
formation—uricosuric drugs should not be used in patients with a
history of stones; (3) serum uric acid reductions are likely to be
modest compared with reductions achievable with allopurinol; (4)
uricosuric drugs have a short half-life and must be given four times a
day. Probenecid and sulfinpyrazone are both uricosuric drugs used in
the long-term management of gout. Probenecid is usually begun at 500
mg/day and increased to 2000 to 3000 mg/day in four divided doses as
needed to
lower
the serum uric acid. Sulfinpyrazone can be initiated at 100 mg/day and
increased as needed to 800 mg/day in four divided doses. Increased
fluid intake is recommended to lessen the risk of acute urinary stone
formation. Just as with allopurinol, acute lowering of serum uric acid
with uricosuric drugs can precipitate acute gout attacks, prophylaxis
should be used.
thought to have gout were found to have crystals in synovial fluid that
were not MSU. Radiographs showed abnormal calcifications and later the
crystals in synovial fluid were identified as calcium pyrophosphate
dihydrate (CPPD).
intra-articular deposits are not well understood. There does not appear
to be an increased whole-body load of inorganic pyrophosphate (PPi).
Plasma, serum, and urinary levels of PPi are no different in most
patients with CPPD crystal deposition than normal. Synovial fluid PPi
concentrations are higher than plasma in many arthropathies but are
highest in patients with CPPD crystal deposition. Accumulated evidence
suggests that elevated intra-articular PPi concentrations are more
likely due to excess production of PPi by cartilage than to impaired
PPi clearance mechanisms. Impaired PPi hydrolysis may be an important
factor in some cases. Breakdown of PPi in vitro
by inorganic pyrophosphatases, which are widely distributed in human
tissue, is inhibited by high calcium and low magnesium. This may
partially explain the association of CPPD deposition with
hyperparathyroidism and hypomagnesemia.
uncommonly before the sixth decade. The incidence of chondrocalcinosis
on radiographs steadily rises and may exceed 30% of the asymptomatic
population over 85 years old. CPPD deposition also occurs more
frequently in certain metabolic diseases such as hypothyroidism,
hyperparathyroidism, hemochromatosis, hypomagnesemic states, and
hypophosphatasia.
attacks of inflammatory arthritis (monoarthritis, often termed
pseudogout, or oligoarticular arthritis) commonly occur first in the
knees. However, the wrist, elbows, ankles, and shoulders are often
involved. Involvement of the first MTP occurs, but is uncommon. Acute
attacks are usually self-limited, lasting several days to weeks. Acute
attacks may occur in a postoperative patient and, like in gout, may
uncommonly coexist with a septic joint.
arthritis, commonly involving the knees, wrists, and elbows.
Involvement of the small joints of the hands and feet can be seen but
is uncommon. Low-grade, chronic joint pain and swelling can be seen in
some with evidence of degenerative joint disease. Some patients will
present with what appears to be a disintegrating, neuropathic joint as
a result of CPPD crystal deposition. Finally, many have no clinical
symptoms but chondrocalcinosis can be demonstrated on radiographs.
calcium pyrophosphate crystals within the synovial fluid of an affected
joint. Compensated, polarized light microscopy will reveal positively
birefringent, rhomboid-shaped crystals found within PMNs. Other tests
on synovial fluid are of limited use. Synovial fluid leukocyte counts
are similar to those seen in gout.
calcification of the fibrocartilage and hyaline cartilage of involved
joints. The most commonly calcified fibrocartilage includes the knee,
the pubic symphysis, and the triangular fibrocartilage of the wrist.
Hyaline cartilage calcification tends to be more linear and parallels
the subchondral bone. Joint space narrowing, subchondral sclerosis, and
subchondral cysts can also be seen. Isolated patellofemoral joint space
narrowing with osteophytes suggests the diagnosis of CPPD.
that used for gout and includes NSAIDs, corticosteroids, colchicine,
and intra-articular corticosteroids (if infection is excluded).
Urate-lowering therapy is ineffective in CPPD.
calcium phosphate crystals) can deposit into articular tissue and cause
an acute, subacute, or chronic arthritis as well as a periarthritis
(bursitis, tendonitis). The periarthritis may be associated with
radiographic calcifications at the sites of inflammation. Commonly
affected areas include the shoulder, the greater trochanter, the
epicondyles, and tendinous attachments about the wrist and knee. The
“Milwaukee shoulder” is the term applied to a progressive, erosive
arthritis of the shoulder seen in older women. Calcium hydroxyapatite
crystals are difficult to identify on polarized microscopy due in part
to their small size. NSAIDs, colchicine, and oral and intra-articular
corticosteroids have been used successfully in therapy.
which literally means “hard skin,” is the key feature linking a
heterogeneous group of conditions, commonly referred to by the same
name. Scleroderma can be subclassified into disease of the skin only,
or localized scleroderma, versus disease with internal organ involvement, or systemic sclerosis. Systemic sclerosis (SSc) is further classified by the extent of skin disease into diffuse cutaneous systemic sclerosis (dcSSc), versus limited cutaneous systemic sclerosis (lcSSc) (Table 6-3).
By definition, limited skin involvement means the face, neck, feet, and
upper extremity involvement that is most often distal to the wrist and
virtually always distal to the elbow, whereas diffuse skin involvement
includes the trunk and proximal extremities. This classification system
is clinically useful because the pattern and extent of skin disease,
coupled with the specific type of autoantibodies detected, correlates
well with the pattern of organ involvement and overall disease
mortality and morbidity. In rare cases SSc can occur without
significant skin disease, or systemic sclerosis sine scleroderma.
It may also occur as part of an overlap syndrome with other autoimmune
diseases such as systemic lupus erythematosus, dermatomyositis,
rheumatoid arthritis, and Sjögren’s syndrome. The combination of
Raynaud’s phenomenon, with either abnormal nailfold capillaries or
SScspecific serologic markers is so predictive of underlying SSc, that
it is often referred to as “pre-scleroderma.”
the United States of about 240 per million, and an incidence of about
20 per million per year. It is more common in women than men by a ratio
of about 4:1. The cause is unknown, but it most likely involves an
interaction between genetic and environmental factors. The pathogenesis
can best be viewed as an interacting triad of pathological immune
system activation (as evidenced by autoantibodies to nuclear antigens),
endothelial cell dysfunction (with resulting small vessel vasculopathy
and tissue ischemia), and fibroblast dysfunction (with increased
synthesis and deposition of extracellular matrix), all culminating in
end organ damage, fibrosis, and atrophy.
skin, musculoskeletal, pulmonary, renal, cardiac, and gastrointestinal
systems. Organ-specific manifestations will be discussed first,
followed by a discussion of characteristics that distinguish lcSSc and
dcSSc.
especially Raynaud’s phenomenon (RP), which is often the first sign of
SSc. RP is an exaggerated vasoconstrictive response of terminal
arteries to stimuli such as cold or emotional stress, most noticeable
in the digits, and manifested by coldness, ischemic pain, and striking
color changes: pallor (white), followed by acrocyanosis (blue), then
hyperemia (red) on reperfusion. In SSc-associated RP, vessel wall
morphology is abnormal. Small and medium arteries show abnormal smooth
muscle cell activation, with markedly increased α2c adrenergic
reactivity. Over time progressive intimal hyperplasia and fibrosis
reduces the effective vessel lumen and limits dilatation, leading to
chronic tissue ischemia, ulceration, and infarction.
|
TABLE 6-3. Classification of Scleroderma and Subtype Associations
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||
examine capillary loops proximal to the fingernail cuticle (nailfold).
In SSc this exam will reveal abnormally dilated and tortuous capillary
loops, sometimes with areas of vessel loss. When RP is accompanied by
this finding, even in the absence of autoantibodies, it is strong
evidence for an underlying autoimmune disease. Telangectasias
are dilated venules and capillaries, which form small (2 to 7 mm)
polygonal macules on the hands, face, lips, oral, and intestinal mucosa.
system manifestations, including some of the pulmonary,
gastrointestinal, and renal manifestations discussed in the following
sections.
in early stages become diffusely edematous or “puffy” in appearance. As
the swelling subsides, the skin becomes thickened and tough, and
eventually “hidebound” to the deeper tissues. The normally plump finger
tuft atrophies, causing the distal phalanx to shrink behind the
fingernail. The face and neck may also be involved. Perioral skin may
take on a puckered appearance with radial folds, and the oral aperture,
measured as the inter-incisor distance, decreases. Skin will feel tight
and sometimes pruritic. Scleroderma of the fingers is called sclerodactyly. When the hands are
involved proximal to the fingers, but the trunk is spared, this is termed acrosclerosis.
The hands and face are involved in both major disease subtypes, but
rapid progression to involve the trunk and proximal extremities
indicates the diffuse cutaneous form of SSc.
calcium phosphate. Usually a late manifestation, it eventually occurs
in up to a third of patients. Lesions can persist for years, and can
occur anywhere, but are most often found in the distal finger tufts,
near the proximal interphalangeal joints, along extensor surfaces, or
near other bony prominences, such as the elbows and knees. Calcinoses
may be painful, especially when they occur in highly innervated
locations such as the fingertip. They will sometimes ulcerate and leak
a whitish material, and secondary infection is a common complication.
as many as 20% will have arthritis, mainly affecting the finger joints.
Synovial biopsy reveals fibrosis rather than synovitis. Radiographs
will sometimes show joint erosions, or the virtual disappearance of the
distal phalanx, termed acroosteolysis.
Flexion contractures of the finger joints result from tightening of the
periarticular skin, fascia, and tendons. Tendon friction rubs—a
palpable or audible crepitus with movement of the tendon —occur in over
50% of patients with dcSSc, and occasionally in those with lcSSc.
However, a true primary myopathy with proximal muscle weakness and mild
elevation of creatinine kinase has been reported. Muscle biopsies
reveal interstitial fibrosis and fiber atrophy, without much evidence
of inflammatory cell infiltration.
majority of patients, and at least half are symptomatic. Though any
level of the tract may be involved, the most common symptoms arise from
the esophagus. Esophageal hypomotility and incompetence of the distal
sphincter lead to recurrent acid reflux, which in turn can cause
erosive esophagitis, Barrett’s esophagus, and stricture formation.
Patients report dysphagia, acid reflux, or regurgitation. Gastric or
intestinal disease results in loss of normal peristalsis, bacterial
overgrowth, malabsorption, and resulting malnutrition. Patients report
prolonged satiety or loss of appetite, weight loss, bloating,
constipation, and diarrhea. Telangectasias in the gut will sometimes
bleed and cause anemia.
disease-related death in SSc, and over 70% patients have some form of
lung involvement. There are two main types: interstitial lung disease
(ILD) (also called fibrosing alveolitis or pulmonary fibrosis); and
vascular disease of the lung leading to pulmonary arterial hypertension
(PAH). Other pulmonary complications include aspiration pneumonia due
to severe esophageal reflux, pleural fibrosis, bronchiectasis, and
spontaneous pneumothorax.
occur in lcSSc, and usually presents as dyspnea on exertion and cough.
Examination will reveal bilateral basal lung crepitations. The 8-year
survival of patients with dcSSc and ILD is about 50%.
about 10% of these patients, and is associated with high mortality.
Life expectancy is less than 1 year once gas exchange drops to 25% of
the normal value. Patients report an insidious onset of dyspnea on
exertion. Lower extremity edema and hepatic congestion will occur with
advancing disease.
crisis,” is much more common in dcSSc than in lcSSc, and is actually
another manifestation of SSc vasculopathy, so it is not surprising that
it often begins during cold weather. As renal arterioles become
narrowed, afferent renal blood flow decreases, activating the
renin-angiotensin system, resulting in hypertension, which, if
untreated, can quickly progress to malignant hypertension and renal
failure. Patients may present with ophthalmic evidence of hypertensive
retinopathy (hemorrhages, cotton wool exudates, and papilledema).
fibrosis, clinically significant myocardial dysfunction is uncommon.
Radionuclide-exercise studies may reveal fixed thallium perfusion
abnormalities, even when angiography is normal, indicating that the
lesion is due to small vessel disease. Clinically significant
pericardial disease may occur in up to 10% of patients, but frank
tamponade or hemodynamically significant effusions are very rare.
and eye symptoms may occur with fibrosis of exocrine tear and salivary
glands. SSc spares the central nervous system, but peripheral nerve
entrapment syndromes, such as carpal tunnel syndrome or facial nerve
palsies, sometimes occur secondary to fibrosis and sclerosis of
surrounding tissue. Osteoporosis may occur, usually as a consequence of
malabsorption and malnutrition from GI disease.
classified as lcSSc, where skin disease spares the trunk and proximal
extremities, and is frequently limited to the hands or fingers. LcSSc
is sometimes referred to as the CREST syndrome, an acronym for the most
common disease manifestations: calcinosis cutis, Raynaud’s phenomenon (RP), esophageal dysmotility, sclerodactyly, and telangectasias.
LcSSc tends to have a longer and more indolent disease course than
dcSSc, with lower overall disease mortality and generally later onset
of significant internal organ disease. RP may be present for years
before other signs of disease appear. However, RP tends to be more
severe in lcSSc, with a higher incidence of digital ulcers and
necrosis. About 10% of patients will develop significant PAH, and this
group will have a very poor prognosis. ILD can occur, but is less
common than in dcSSc. Renal crisis is rare.
extensive skin involvement that defines dcSSc. Early clues that a
patient will develop this pattern include appearance of skin disease
before or within one year of onset of RP, dilated capillary loops with dropout
at the nailfold, tendon friction rubs, and early onset of ILD, renal
crisis, or diffuse GI disease. ILD occurs frequently, and mortality can
be high. However, in some patients the disease course is one of initial
worsening, followed by a plateau, and then some improvement or
stabilization thereafter. Diagnosis and organ-specific treatment early
in the disease may significantly improve the prognosis.
|
TABLE 6-4. Common Serum Autoantibody Associations in Systemic Sclerosis
|
|||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||||||||||
essential for diagnosing early SSc. A complete review of systems is
key, as patients seeking evaluation for puffy hands may not
spontaneously report mild dyspnea, or new nocturnal heartburn. Patients
with dcSSc, should be taught to monitor their own blood pressure
frequently, and report any consistent elevation. Assessment of skin
tightness, and use of a standard scoring system, can help to detect
rapidly progressive disease. The most commonly used system involves
measurement at 17 sites, scoring each site on a scale of 0 (normal) to
3 (hidebound skin). Skin biopsy is rarely needed to confirm scleroderma
and should generally be avoided as healing may be impaired. Radiographs
of the hands may detect acro-osteolysis, calcifications, or joint
erosions, but will have little impact on the overall disease management.
antinuclear antibodies (ANA), and the pattern detected on
immunofluorescence is very important (Table 6-4).
An anticentromere pattern (ACA) is detected in 70 to 80% of patients
with lcSSc, but is unusual in patients with dcSSc. ACA correlates
positively with PAH and negatively with
ILD or renal disease. A speckled ANA pattern may indicate antibodies to
topoisomerase-1 called anti-Scl-70, which can be measured directly in a
separate assay. Anti-Scl-70 antibodies are found in about 30% of
patients with dcSSC but fewer patients with lcSSc, and correlate positively with ILD and negatively
with PAH. Autoantibodies to several other nuclear antigens have been
linked with SSc. As many as 20% of patients with dcSSc will have an
antinucleolar or speckled ANA pattern due to antibodies to RNA
polymerase I and III, a finding that correlates with renal disease. The
false-positive rates for detection of ACA and anti-Scl-70 among
disease-free controls are both very low, giving these tests a high
degree of specificity for SSc. However, negative antibody tests,
including ANA, do not rule out SSc.
laboratory, functional, and imaging studies are useful to detect and
monitor disease in each affected organ system. Serum creatinine should
be checked periodically in patients with dcSSc to monitor renal
function. Oxygen saturation or arterial blood gas profile, pulmonary
function tests including DLCO, and chest radiographs, should be
obtained at diagnosis of SSc, and repeated with any new respiratory
symptoms to detect onset of ILD or PAH. Additional studies may include
high-resolution chest CT to confirm fibrosis, and/or echocardiography
with Doppler to estimate pulmonary artery pressure. Invasive studies
such as bronchoscopy for bronchoalveolar lavage, or right-heart
catheterization to directly measure pulmonary artery pressure, may be
needed to confirm a diagnosis before beginning treatment with high-risk
medications. Gastrointestinal disease can be detected and monitored
with radiographic studies such as barium swallow, gastric emptying
tests, or abdominal CT. Esophageal manometry, upper endoscopy, or
colonoscopy may also be needed.
for all SSc manifestations. Disease management is therefore primarily
organ based. Search is under way, however, for treatments that will
prevent tissue fibrosis, and thus reduce the risk of end organ failure.
SSc. Occupational therapists can help to educate patients on how to
keep warm, protect their fingers while performing activities of daily
living, and maintain flexibility. Because nicotine will worsen RP, and
smokers have a significantly higher risk for finger ulcers and digital
necrosis, patients must be advised that smoking cessation is absolutely
necessary. Medications that trigger vasospasm must be avoided.
Hypothyroidism will worsen Raynaud’s, so hypothyroid patients must be
on adequate replacement hormone. Some antihypertensives, such as
β-blockers, may worsen RP, or at least limit use of other
antihypertensives that will more effectively treat RP.
most frequently to treat RP, and efficacy is well established. Use may
be limited, however, by intolerable side effects such as postural
hypotension, dependent edema, and significant gastroesophageal reflux,
especially in those who already have an incompetent esophageal
sphincter. ACE inhibitors and angiotensin receptor blockers have been
used with mixed success. They are generally well tolerated, and do not
worsen esophageal function. Platelet serotonin may play a role in the
pathogenesis of RP. The selective serotonin-reuptake inhibitor,
fluoxetine, has been reported to be beneficial, and was superior to
nifedipine in one small trial. Most authors recommend daily aspirin (81
to 325 mg) to prevent formation of microthrombi, though actual benefit
is unproven. Anecdotal reports support use of pentoxifylline, which may
improve microcirculation. There are promising reports that the
phosphodiesterase inhibitor, sildenafil, is effective and well
tolerated for short-term management of severe Raynaud’s, but controlled
trials are needed to prove efficacy in SScassociated RP.
treat impending digital infarction. Topical nitroglycerine may improve
circulation to the affected digit, but intolerance may limit its use.
Several synthetic prostacyclins have been used for this indication, but
the need for constant IV infusion limits use to the hospital setting,
and there may be rebound worsening once infusion is stopped.
the lungs.” Unfortunately, it does not respond adequately to most of
the oral medications used to treat Raynaud’s. The endothelin receptor
blocker, bosentan, has been shown to help stabilize pulmonary function
in patients with significant PAH. Controlled trials showed slight
functional improvement (as measured by 6 minute walking distance) in
bosentan-treated SSc patients, while untreated control patients showed
significant continuing decline. Bosentan may also be effective for
severe
digit-threatening Raynaud’s, but cost will likely limit any off-label
indication. Intravenous prostaclycin analogues are effective in PAH but
costly. Administration by continuous subcutaneous infusion, or by
inhalation, is showing promise in clinical trials. Sildenafil and other
oral phosphodiesterase inhibitors may also have a role in management of
PAH, either alone or in combination with the synthetic prostacyclins.
SSc-related ILD. The goal is to treat the disease with aggressive
immunosuppression in the early inflammatory phase and thereby reduce
the risk of progressive fibrosis and decline in function that may
follow. Retrospective data supports this approach but prospective
studies are needed to confirm this strategy. Cyclophosphamide, either
by daily oral dose or pulse IV dosing, is the current drug of choice
for treatment of ILD. Some recommend adding daily low dose prednisone,
but there is limited evidence that this is helpful.
hypertension and SSc. Renal crisis was once the major cause of death in
SSc, but ACE inhibitors have proven to be very effective at controlling
hypertension and reducing the rate of subsequent renal failure. Studies
have shown long-term benefit, even if creatinine levels increase after
starting an ACE inhibitor.
gastroesophageal reflux, and to reduce the risk for esophagitis and
recurrent aspiration. Prokinetic agents such as metoclopromide may help
patients with delayed gastric emptying and poor appetite. Cyclic oral
antibiotics are used to treat bacterial overgrowth in advanced disease.
Dieticians may be needed to guide the use of oral supplements when
malabsorption leads to malnutrition. Some patients do eventually
require total parenteral nutrition.
degree of efficacy. The fact that sclerosed skin may spontaneously
start to soften many years after disease onset complicates
interpretation of long-term studies. Many agents reported to be
beneficial in small open trials have not shown benefit when studied in
large double-blind studies. D-Penicillamine has shown promise in some
trials but not in others. Studies on methotrexate have shown benefit or
at least a trend favoring treatment, but further studies are needed.
Minocycline inhibits cation-dependent matrix metaloproteases, which
have been implicated in the tissue damage that follows inflammation in
SSc. Minocycline 100 mg twice/day did seem to soften the skin of
several patients with advanced dcSSc in one small open-label trial, but
this needs to be repeated in a larger trial. Corticosteroids play no
role in managing the skin disease of SSc and topical steroids are
contraindicated.
and resulting poor healing of wounds, surgical procedures are
relatively contraindicated. Circumstances will arise, however, which
necessitate surgery, such as severe esophageal strictures, and small or
large bowel obstructions.
circumstances. Patients with severe carpal tunnel syndrome may benefit
from surgical release. When sclerodactyly leads to severe (> 90°)
contracture of the PIP joints, arthrodeses or joint replacements may be
of benefit. Debridement and amputation of ischemic digits will
occasionally be indicated to address secondary infection, but should be
done conservatively, to allow the maximum possibility of spontaneous
healing. In general, this should follow aggressive medical therapy with
a parenteral prostacyclin and possibly a trial of bosentan.
extremity RP, and can now be performed thorascopically, but they rarely
provide long-lasting benefit, probably due to accessory sympathetic
pathways and the upregulation of sympathetic receptors on vascular
smooth muscle following denervation. Lumbar sympathectomies, by
contrast, have proven very effective for Raynaud’s of the feet with
lasting benefits.
to relieve pain and ulceration of severe RP, including cases secondary
to SSc. Yee and colleagues (1998) report very
favorable outcomes in their series of 13 procedures in 9 patients. They
conclude that adventitial stripping of the arteriole both denervates
and decompresses the vessel, which is why their procedure was effective
even in patients who showed no discernable response to sympathetic
blockade prior to surgery. This procedure should be considered when
medical management fails or is contraindicated by other medical
conditions.
(present at rest but exacerbated by activity), fatigue, and sleep
disturbance. The American College of Rheumatology defined two criteria
for diagnosing fibromyalgia in 1990: (1) a history of widespread pain
and (2) pain in at least 11 of 18 defined tender point sites on digital
palpation with a force of 4 dynes/cm2 (Figure 6-10).
although pain may be concentrated in the neck and shoulder girdle area,
in the buttocks and thighs, or in an area previously injured. Typically
the patient cannot point to an exact location for the pain, nor can it
be localized anatomically to particular muscles, particular joints, or
to the area of distribution of particular nerves. Stiffness may be
present, but it is not relieved by heat or gentle activity, and thus
differs from the morning stiffness characteristic of inflammatory
disorders such as rheumatoid arthritis or polymyalgia rheumatica.
Usually there is a clear trend toward diminishing motor activity over
months or years, and patients may feel disabled by their pain.
off during the daytime. On prompting, they describe disordered sleep
hygiene of long duration: difficulty falling asleep, shallow sleep with
frequent awakening, and arising unrefreshed.
hands and/or feet—because the whole hand or limb is involved in
fibromyalgia, it can be distinguished from nerve entrapment syndromes
where the numbness conforms to the area served by a particular nerve;
(2) irritable bowel syndrome, characterized by alternating diarrhea and
constipation; (3) dysuria with normal urinalysis; and (4) a history of
having taken many different pain medications without relief. Periods of
more intense symptoms typically follow events that force reduced motor
activity (e.g., hospitalization for surgery), intensified sleep
disturbance (e.g., assuming care of an elderly parent), a painful
injury (e.g., a motor vehicle accident), or a period of increased
stress or muscle tension (e.g., combat duty lawsuit). This is how
susceptible patients with rheumatoid or osteoarthritis can develop
superimposed fibromyalgia symptoms.
Fingertip pressure on these sites, which would be judged mildly
unpleasant by a normal subject, produces an aversive reaction and is
perceived
as painful by fibromyalgia patients. While the ACR criterion of 11 of
18 tender points is a useful general guide for diagnosing fibromyalgia,
strict conformance with this criterion is not necessary in the clinic.
Some patients have palpable tender knots representing localized areas
of muscle spasm. Trigger points, where local pressure produces referred
pain over an entire region, is unusual in fibromyalgia. Joint and
neurologic exams are normal. So is muscle strength, although “breakaway
weakness” due to pain may be observed. In especially severe cases, the
tenderness may be generalized to the point that control points or even
gentle pinching of skin are also painful, a condition called allodynia.
 |
|
FIGURE 6-10.
Fibromyalgia tender points. The ACR published clinical criteria for fibromyalgia in 1990, emphasizing the characteristic nine pairs of tender points: insertion of cervical paraspinals on the occipital condyles, cleidomastoid muscle over lateral transverse processes of C5-7, belly of trapezius, supraspinatus, wrist extensor insertion on lateral epicondyle, pectoralis insertions on 2nd rib near sternum, upper outer buttock, gluteus insertion on greater trochanter, and insertion of vastus medialis on medial condyle of the femur. The diagram also shows a common tenth tender point over the rhomboids. Control points: middle of forehead; volar aspect of midforearm; thumbnail; muscles of anterior thigh. |
ages are susceptible, but the pain pattern often begins in the teens.
Community surveys, confirmed with tender point examinations, have
estimated that pain thresholds tend to be stable over time in a given
individual, and that females whose pain threshold falls in the lower 5%
tend to develop fibromyalgia symptoms when they encounter a prolonged
reduction in motor activity or a prolonged deprivation of deep sleep.
Family histories positive for fibromyalgia are common.
that peripheral stimulation of muscle pressure receptors produces
activation of thalamic regions typical of pain rather than touch.
Impulses interpreted centrally as pain are carried by peripheral
myelinated or unmyelinated nerve fibers in fibromyalgia, whereas in
normals they are confined to unmyelinated fibers. In fibromyalgia, as
in other chronic pain states, there is progressive failure of
inhibitory neurons in the spinal cord to focus afferent sensory
impulses appropriately, leading to “central sensitization,” which
widens the range of sensory impulses perceived as pain, and also
enlarges the area of pain, making it difficult for the patient to
localize pain. Research in progress focuses on the role in central
sensitization of endorphins (regenerated by exercise and deep sleep),
serotonin, and other neurotransmitters in the central nervous system.
biochemical changes in fibromyalgia muscles can be summarized as
showing many abnormalities typical of fatigued incompletely relaxed
muscle, but no primary physiologic abnormalities of muscle have been proven.
psychiatric conditions has recently been clarified. In general,
psychological profiles of fibromyalgia patient resemble those of other
chronic pain patients. The only psychiatric diagnoses more frequent in
fibromyalgia patients than in controls are mood and anxiety disorders.
Furthermore, in community-based surveys, this association only occurs
among the subset (about one-third) of persons with fibromyalgia who
seek medical intervention for their symptoms, and does not apply to the
others who learn from experience how to control their own symptoms. One
symptom characteristic of inadequately treated depression is an
inability to initiate and carry out a treatment program, which may make
it especially difficult for such a patient to control their
fibromyalgia symptoms. Patients with anxiety disorders have difficulty
tolerating the short-term muscle pain experienced during exercise,
fearing that they will damage themselves. Thus, the psychiatric
associations with fibromyalgia have less to do with primary pathology
than with the probability of treatment success.
of fibromyalgia other than normal values. The initial workup should
focus on ruling out fibromyalgia mimics or exacerbating conditions such
as hypothyroidism (TSH), diabetes (Hemoglobin A1c) or inflammatory
conditions (ESR and CRP). If muscle spasm is prominent, creatinine,
electrolytes, calcium, and magnesium may be useful. Anemia should be
ruled out as a cause of fatigue. Rheumatoid factor and ANA are
meaningless in the absence of specific clinical features of those
diseases. Males should be tested for hypogonadism (free and bound
testosterone).
visit the physician, get treated with a medicine, and recover. This
paradigm does not apply in fibromyalgia, where the success or failure
of treatment depends on the patient, who must faithfully carry out his
or her own treatment program on a
daily
basis over many weeks to alleviate his or her pain. The physician’s
role is to rule out alternative diagnoses, educate the patient, and
arrange the first meeting with the physical therapist who will act as
“coach.”
(1) patient education; (2) therapeutic exercise; and (3) achievement of
stage 3 sleep. Analgesic drugs have little impact on fibromyalgia
symptoms, and thus play a minor role, especially when the patient is
severely symptomatic.
fibromyalgia is psychosomatic, and many are reassured to learn that it
is a recognized medical disorder with a physiologic basis. The two key
points are the relationship of symptoms to deprivation of sleep and
reduced physical activity. The essential message is that by following
the treatment plan the patient will learn how to control his or her
symptoms, but that this control is won slowly over weeks or months.
Patients should be taught to identify and avoid the exacerbating
circumstances in their own lives. Examples include stress causing
muscle spasm or sleep interruption or occupations entailing tonic
muscle tension (orchestra conductors, keyboard operators, bus drivers).
They must learn that chronic pain does not have the protective warning
function we all associate with acute pain, and that it is not harmful
to engage in painful exercises today in order to decrease the pain you
will experience next month.
not only to apply the standard treatments, but also to direct
appropriate patients to second line treatments such as ultrasound,
biofeedback (for muscle relaxation), and acupuncture. Such a therapist
should also be able to individualize the exercise program for each
patient and make adjustments when other health events occur (e.g.,
abdominal surgery, because of the prolonged inactivity, usually
requires returning to an earlier stage of the treatment protocol).
treatment protocol: (1) gentle stretching, (2) graduated conditioning,
and (3) transcutaneous nerve stimulation.
muscles that are incompletely relaxed, which will hurt if contracted
too vigorously. Tai Chi-style slowmotion stretching toward the limit of
each motion promotes relaxation and produces short-term pain relief,
which can be applied several times per day. Contrasting activities to
be avoided in the early stages of treatment are quick forceful
contractions. An athlete needs to warm up before running a dash to
avoid painful muscle spasm, whereas the fibromyalgia patient needs to
warm up in order to perform normal daily activity.
begins with a low level of gentle exercise, designed to accelerate
breathing and heart rate, and cause less than an hour of delayed pain.
Swimming or moving limbs through a full range of motion in a swimming
pool three times per week is ideal, but walking and other gentle
land-based exercises are an effective second choice. A gradual increase
in the intensity and duration of exercise occurs until pain is
controlled. Exercises designed to build strength are especially painful
and may be counterproductive in the early stages of treatment. Normal
daily activities are encouraged, but do not substitute for therapeutic
exercise.
stimulators (TENS) help to relax thin muscles lying close to the skin,
so they work much better in the neck and shoulder region than in the
low back and buttocks. A brief trial with the therapist will usually
show whether TENS will work in a particular patient, who can then rent
a unit on prescription and use it at home or at work.
well-designed program on his or her own; prolonged “hands on” treatment
by a therapist is generally unnecessary, whereas periodic reassessments
for advancing the program are helpful. The most common reason for
failure of a treatment program is that the patient has not been doing
the exercise. Reassessment for undertreated depression should be
considered in such a patient. Fibromyalgia patients should be strongly
encouraged to seek accommodations in their work environment (such as
short breaks for stretching) so they can remain productive. Social
security disability from fibromyalgia is unusual.
fibromyalgia are the tricyclics, such as amitriptyline, trazodone,
nortriptyline, desipramine, or cyclobenzaprine. A dose given 2 hours
before bedtime should increase the proportion of stage 3 sleep.
Intolerable morning grogginess may necessitate cutting the dose in half
or taking it earlier. Lack of effect over several nights means a higher
dose is needed. Lack of effect plus morning grogginess means a
different tricyclic should be selected. Usually the beneficial effects
of a regimen become evident between 2 weeks and 3 months. Sleep apnea
may need to be ruled out by polysomnography, and treated if present for
progress to be made.
other analgesics is so typical of severe fibromyalgia that it is
considered to have diagnostic importance. As the patient’s pain
responds to exercise and treatment, the response to conventional
analgesics (such as ibuprofen, Tylenol, or tramadol) reemerges. Opioids
may be partially effective in some patients. However, breakthrough pain
increases with time, leading to dose escalation. Also narcotics can
impair deep sleep. Narcotic rebound pain can be difficult to
distinguish from fibromyalgia. For these reasons, opioids are not
recommended in fibromyalgia.
College of Rheumatology Subcommittee on Osteoarthritis Guidelines.
Recommendations for the Medical Management of Osteoarthritis of the Hip
and Knee. Arthritis Rheum 2000 update;43:1905-1915. Also available at: http://www.rheumatology.org/publications/guidelines/oa-mgmt/oa-mgmt.asp Following
the principles of evidence-based medicine, the ACR subcommittee
provides a detailed review of current knowledge regarding many modes of
treatment for OA of the hip and knee, both pharmacologic and
nonpharmacologic, and provides a consensus recommendation for the
nonsurgical management of OA in the hip and knee. The most current
version of these guidelines can be found at the above web address.
DT, Zhang Y, Hannan MT et al. Risk factors for incident radiographic
knee osteoarthritis in the elderly. The Framingham Study. Arthritis
Rheum 1997;40:728-733. An excellent study demonstrating the correlation between weight and incident radiographic knee OA in an elderly population.
article provides an authoritative review of normal joint tissue
physiology, and the epidemiology, genetics, and pathology of
osteoarthritis. Kraus also reviews the clinical features of OA, and
discusses treatment in terms of both primary and secondary
intervention, including the concept of chondroprotection.
makes a strong argument that exercise is integral to the successful
management of OA. This article includes exercise recommendations and
safety considerations.
article reviews the latest science on the role of cytokines in the
pathogenesis of RA and describes several potential therapeutic targets
with the use of anticytokine therapy.
ED Jr., Schur PH. Risk factors for and possible causes of rheumatoid
arthritis. In: Rose, BD, ed. UpToDate, Wellesley, MA, 2003. This article reviews the epidemiology of rheumatoid arthritis as well as the pathogenesis.
ED Jr., Schur PH. Overview of the systemic and nonarticular
manifestations of rheumatoid arthritis. In: Rose, BD, ed. UpToDate,
Wellesley, MA, 2003. This article reviews the
complications of the disease as well as the drugs used to treat RA
(such as steroid induced osteoporosis, entrapment syndromes, etc.)
article reviews the clinical features of RA including extra-articular
features, as well as discussing factors leading to a poorer outcome.
PJW, Maini RW. Diagnosis and differential diagnosis of rheumatoid
arthritis. In: Rose, BD, ed. UpToDate, Wellesley, MA, 2003. This
article reviews the diagnostic features of RA, including laboratory
testing, the other forms of arthritis that may present similarly to RA
and discusses a rationale approach to making the diagnosis
review summarizes the common musculoskeletal manifestations of SLE from
an orthopaedic viewpoint, and gives an overview of surgical
interventions commonly used.
excellent review summarizes the epidemiology, diagnosis, and medical
and surgical management of osteonecrosis in SLE patients.
review describes the features of lupus arthritis of the hands and
wrists and the conservative and surgical approaches to its sequelae.
DS, Gilkeson G, St Clair, EW. Systemic lupus erythematosus: diagnosis
and treatment. Med Clin North Am 1997;81:113-128. This
review details the clinical and laboratory diagnostic criteria for SLE,
and summarizes the medical therapeutic approach for SLE by organ
involvement.
EM, Cohen AS, Fries JF et al. Revised criteria for the classification
of systemic lupus erythematosus. Arthritis Rheum 1982;25:1271-1277. This
paper describes the 11 clinical and laboratory features that are used
in the diagnosis of patients with SLE. These diagnostic criteria were
developed for the standardization of subjects for research studies, but
are now used frequently in substantiating the diagnosis of SLE.
M, van der Linden S, Juhlin R et al. The European Spondyloarthropathy
Study Group preliminary criteria for the classification of
spondyloarthropathy. Arthritis Rheum 1991;34:1218-1227. A very comprehensive helpful presentation of the wide spectrum of clinical features within the family of spondyloarthropathies.
PJ. Disease-modifying antirheumatic drug therapy for
spondyloarthropathies: advances in treatment. Curr Opin Rheumatol
2003;15:205-212. A review of recent progress in disease-modifying therapy for spondyloarthropathy, including the newer biologic agents.
RE, Southwood TR, Baum J et al. Revision of the proposed classification
criteria for juvenile idiopathic arthritis: Durban, 1997. J Rheumatol
1998; 25(10):1991-1994. This article describes revisions of the International League of Associations for Rheumatology (ILAR) criteria for JIA.
article presents an overview of JRA utilizing the new JIA
classifications. It offers a review of the genetics, prognosis, special
aspects of the disease, and treatment in JIA. Web sites with
current information about JIA: Arthritis Foundation:
http://www.arthritis.org American College of Rheumatology:
http://www.rheumatology.org
textbook is popular with medical students and residents interested in
learning more about rheumatic diseases. The succinct book provides
sufficient detail, without being overwhelming.
JD. The changing spectrum of rheumatic disease in human
immunodeficiency virus infection. Semin Arthritis Rheum
2000;30:147-166. This book reviews rheumatic disease related to HIV.
concise book is extremely helpful to find answers quickly to virtually
all questions on rounds, in the clinic, and on exams. It is a favorite
with residents and fellows.
Carrera, and McCarty review an additional 15 cases of Milwaukee
shoulder syndrome describing common clinical features and discuss
potential predisposing factors.
provides a thorough review of gout, calcium pyrophosphate deposition
(CPPD), and basic calcium phosphate crystalline disease.
briefly reviews the clinical presentation of gout, followed by a more
in-depth discussion of the therapy of gout with supporting references.
LK, Wigley FM. Management of Raynaud’s phenomenon and digital ischemic
lesions in scleroderma. Rheum Dis Clin North Am 2003;29:293-313. The
authors review the quantity and quality of evidence supporting both
common and less common treatments for Raynaud’s, and lay out a
practical approach for disease management.
AT, Clements PJ, Furst DE. Update on disease-modifying antirheumatic
drugs in the treatment of systemic sclerosis. Rheum Dis Clin North Am
2003;29:409-426. The author reviews existing data for and against a series of agents used for the long-term management of SSc.
TA. Natural history of systemic sclerosis and the assessment of disease
activity, severity, functional status, and psychologic well-being.
Rheum Dis Clin North Am 2003;29:255-273. Medsger
summarizes data from several large case series to define the frequency
of different disease manifestations in early versus late disease, and
in lcSSc versus dcSSC; and also discusses the tools used to measure
disease activity.
AM, Hotchkiss RN, Paget SA. Adventitial stripping: a digit saving
procedure in refractory Raynaud’s phenomenon. J Rheumatol
1998;25:269-276. The authors describe their
generally favorable experience with digital microsurgery for severe
Raynaud’s in patients with systemic disease and conclude that efficacy
is due to both denervation and decompression of the ischemic vessel.
RM. Emerging concepts in the neurobiology of chronic pain: evidence of
abnormal sensory processing in fibromyalgia. Mayo Clin Proc
1999;74:385-398. This is a clearly written summary of the neurophysiology of fibromyalgia and its relationship to other chronic pain syndromes.
S, Bell MJ, Reynolds WJ et al. Comparison of amitriptyline,
cyclobenzaprine, and placebo in the treatment of fibromyalgia.
Arthritis Rheum 1994; 37:32-40. This article describes randomized double-blind placebo-controlled trial of two tricyclics in fibromyalgia.
study of fibromyalgia patients showed 66% were “a little or a lot
better” and 55% said they “felt well” after 10 years, but most still
had some symptoms.
F, Smythe HA, Yunus MB et al. The American College of Rheumatology 1990
criteria for the classification of fibromyalgia: report of the
Multicenter Criteria Committee. Arthritis Rheum 1990;33:160-172. This
article reviews different criteria and proposes standard criteria to
diagnose fibromyalgia. It underscores the difficulties one encounters
in diagnosing these patients.
F, Ross K, Anderson J et al. The prevalence and characteristics of
fibromyalgia in the general population. Arthritis Rheum 1995; 38:19-28.
This article describes a population-based study of fibromyalgia prevalence.