
medicine. Lead by the Human Genome Project, genetic information and
concepts are changing the way disease is defined, diagnoses are made,
and treatment strategies are developed. The profound implications of
actually understanding the molecular abnormalities of many clinical
problems are affecting virtually all medical and surgical disciplines.
Importantly, genetic technologies will increasingly drive biomedical
research and the practice of medicine in the near future.
aware of the genetic cause of its inherited disorders in order to make
appropriate referrals for genetic counseling and to refine the
prognosis and natural history in each individual patient. Current
management revolves around treatment to prevent or minimize medical
complications, psychosocial support of patients and their families, and
modification of the environment where appropriate. Gene discoveries
will allow the development of tests to detect disease or to quantify
the risk of disease. Furthermore, applying this knowledge is the best
hope for developing strategies to modify the pathologic effect of the
gene (drug therapy), repair the gene (gene therapy), or for approaches
to restore lost or affected tissue (tissue engineering).
Instead
of an empiric trial-and-error approach to therapy, it may become
feasible to tailor treatment to the specific molecular malfunction.
abnormalities and the power and speed of current genetic and
developmental biology information, a few selected disorders will be
discussed in this chapter. The discussion will address general concepts
on the genetic bases of musculoskeletal disorders, current
classifications, and it will focus on the clinical characteristics of
some of the most common disorders, including natural history and
treatment options, and it will reflect the most recent developments in
the understanding of their pathogenesis.
organ system, composed of 206 bones with many different shapes and
sizes. Like every other organ system, the skeleton has specific
developmental and functional characteristics that define its identity
in biologic and pathologic terms. For normal skeletogenesis to take
place, the coordination of temporal and spatial gene expression
patterns is a crucial prerequisite. Any disturbances in these processes
will lead to abnormalities of the skeleton.
is an unparalleled example of integrated cell behavior. After
fecundation, the single cell divides many times to produce the
trillions of cells of the organism, which form structures as complex
and varied as the eyes, limbs, heart, or the brain. Development is
essentially the emergence of organized and specialized structures from
an initially very simple group of cells. Therefore, during development,
differences are generated in the embryo that leads to spatial
organization, changes in form, and the generation of different cell
types. Since each cell has the same genetic instructions, it must
interpret this information with regard to time and space.
condensing into tissue elements outlining the pattern of future bones
(the patterning phase). Shortly thereafter, cells within these
condensations differentiate along the chondrocytic pathway. Subsequent
growth generates cartilage models (anlagen) of the future bones. The
cartilage anlagen will be replaced by bone and bone marrow in a process
called endochondral ossification. Finally, a process of growth and
remodeling will result in a skeleton that is well adapted to its
function as an organ not only for movement and internal organ
protection, but also for blood cell production and regulation of
calcium homeostasis.
temporal arrangements of cell activities are organized within the
embryo so that a well-defined structure develops. Pattern formation is
critical for the proper development of every part of the organism. In
the developing limb, for example, pattern formation enables the cells
to know whether to make the upper arm or the fingers, and where the
muscles should form.
mechanism where the cells first acquire a positional identity, which
determines their future behavior. The ability of cells to sense their
relative positions within a limited population of cells and to
differentiate according to this position has been the subject of
intense research. Interestingly, pattern formation in many systems has
similar principles, and more striking, similar genes. It is essentially
pattern formation—the size, shape, number, and arrangement of the
bones—that makes human beings different from rabbits or chimpanzees.
become structurally and functionally different from each other, ending
up as distinct types as osteoblasts, chondrocytes, or muscle cells.
Because each cell of the organism has the same genetic material, the
achievement and persistence of the differentiation state depends on a
series of signals that ultimately control the transcription of specific
genes. In humans, the zygote gives rise to about 250 clearly
distinguishable cell types.
of a finite number of discrete kinds of cells, each with its peculiar
repertory of biochemical activities and possible morphological
configurations. When cells achieve a distinctive state of
differentiation,
they do not transform into cells of another type. Differentiation leads
to a stable, irreversible set of cellular activities.
called dysostoses: these disorders affect only specific skeletal
elements, leaving the rest of the skeleton largely unaffected. In
contrast, mutations in genes that are involved primarily in cell
differentiation cause disorders called osteochondrodysplasias, which
affect the development and growth of most skeletal elements in a
generalized fashion. Many genes have important functions in both of
these processes so that some inherited disorders can display features
of both dysostoses and osteochondrodysplasias. Genes used during
skeletal development may also be important in other organs so that when
mutated, the resulting skeletal defects are part of a syndrome.
abnormalities occur in 6% of all live births. Twenty percent of infant
deaths are due to congenital anomalies. About 3% of newborns have
significant structural abnormalities. At present, the cause of
approximately 50 to 60% of birth defects is unknown. Chromosomal
abnormalities account for 6 to 7% of the abnormalities. Specific gene
mutations cause 7 to 8%. Environmental teratogens are responsible for 7
to 10% of defects. Combined genetic predisposition with environmental
factors causes the remaining 20 to 25 % of congenital abnormalities.
of clinically distinct and genetically heterogeneous conditions
comprising more than 150 forms. Although individually rare, the
different forms produce a significant number of affected individuals,
with significant mortality and morbidity. Clinical manifestations range
from neonatal lethality to congenital malformations, spinal and limb
deformities to only mild growth retardation. Importantly, secondary
complications such as early degenerative joint disease and
extraskeletal organ involvement add to the burden of the disease.
difficult to diagnose, and many attempts have been made to delineate
single entities or groups of diseases to facilitate the diagnosis. As
mentioned earlier skeletal disorders have been subdivided traditionally
into dysostoses, defined as malformations of individual bones or groups
of bones; and osteochondrodysplasias, defined as developmental
disorders of cartilage and bone. The criteria used for their
distinction has been based on a combination of clinical, radiographic,
morphologic, and, in a few instances, biochemical characteristics. The
modes of genetic inheritance and extraskeletal abnormalities have also
been used.
classification has progressed to the present reconsideration and
regrouping of the disorders according to their molecular pathogenesis.
The International Working Group on the Classification of Constitutional
Disorders of Bone updated the classification in 2001. The major change
was the addition of genetically determined dysostoses to the skeletal
dysplasias. However, it is now becoming increasingly clear that several
distinctive classifications are needed that reflect, on the one hand
the molecular pathology, and on the other, the clinical signs and
symptoms. Several reviews of the rapidly changing molecular basis of
the skeletal dysplasias have been published, focusing either on a
molecular-pathogenetic classification; on more specific aspects such as
transcriptional deregulation; or on a combination of
molecular-pathology and developmental biology of the musculoskeletal
system. These new concepts directly link the clinical phenotype to key
cellular processes of skeletal biology and should assist in providing a
framework accessible to clinicians as well as basic scientists for
future understanding of these disorders. It is likely that future
insights will lead to reclassification.
of the Osteochondrodysplasias,” now called “Nosology,” has been
recently revised to reflect the molecular and pathogenetic
abnormalities underlying these disorders. This classification uses
similar criteria to those of the functional classification proposed in
the 8th edition of The Metabolic and Molecular Bases of Inherited Disease.
information and services relevant to the genetics of musculoskeletal
disorders. One database that contains a wealth of clinical and genetic
data is the Online Mendelian Inheritance in Man (OMIM). It provides
free text overviews of genetic disorders and gene loci, with the
correspondent mouse correlate. In general, congenital abnormalities of
non-Mendelian inheritance, chromosomal abnormalities, and single case
reports are not included. The total
number
of entries exceeds 11,000, but most importantly, it is linked to a
wealth of other genetic databases allowing the users to obtain
information on gene structure, map location, function, phenotype, or
literature references. It is found at
http://www3.ncbi.nlm.nih.gov/entrez/query.fcgi ?db/OMIM.
of skeletal dysplasias is to establish precisely what the products of
the affected genes do during skeletal development and how mutations
disturb these functions to produce the characteristic phenotype.
Despite the many hypotheses generated from the work in human genetics
and the knowledge that has been gained from animal models, there
remains a relatively poor understanding of how these genes interfere
with skeletal development. Unraveling this mystery and defining it in
molecular and cellular terms will be the challenge for the near future.
prenatal screening, more patients with skeletal dysplasias are being
diagnosed before birth. When there is suspicion of a skeletal dysplasia
on ultrasound, the femoral length is the best biometric parameter.
Further testing may be performed, if indicated, by chorionic villous
sampling and mutation analysis.
defined as height more than two standard deviations below the mean for
the population at a given age. The resultant growth disproportion is
commonly referred to as “short trunk” or “short limb.” The short-limb
types are further subdivided into categories based on the segment of
the limb that is affected. Rhizomelic refers to shortening of the
proximal part of the limb; mesomelic refers to the middle segment; and
acromelic to the distal segment.
bone development, several aspects of the medical history and the
physical exam should be investigated. A history of heart disease,
respiratory difficulty, immune deficiency, precocious puberty, and
malabsorption should be sought because they are associated with some of
these disorders. Birth length, head circumference, and weight should be
recorded, and pertinent family history of short stature or dimorphism
should be sought. The height and weight percentile should be determined
using standard charts. The physical examination should include careful
characterization of the patient facies, and presence of cleft palate,
abnormal teeth, position of the ears, and extremity malformations. A
thorough neurological evaluation is needed because of the frequent
incidence of spinal compromise in many of these syndromes.
radiographs are used to identify the area of bone involvement. The
so-called skeletal survey may vary from institution to institution, but
it should include the following views: skull (AP and lateral);
thoracolumbar spine (AP and lateral); chest; pelvis; one upper limb;
one lower limb; and left hand. Flexion-extension views of the cervical
spine should be ordered if instability is suspected. In some instances,
imaging of other family members suspected of having the same condition
may be helpful.
alkaline phosphatase, serum thyroxin, and protein to rule out metabolic
disorders. Urine should be checked for storage products if a
progressive disorder is found. Referral to a pediatric geneticist is
often very helpful in reaching a diagnosis in complex cases, in
providing genetic counseling to the family, and to manage the many
medical problems associated with these disorders.
from hypoplastic femur, congenital coxa vara, congenital short femur
with coxa vara, and proximal femoral focal deficiency (PFFD). This
later term represents a severe disturbance in the growth of the femur
with significant shortening, abnormality of the hip, and it is commonly
associated to fibular deficiency. The cause is unknown, but in some
cases the combination of femoral deficiency and abnormal facies is
believed to be an autosomal dominant malformation.
the radiographic evaluation of the femoral and hip involvement. In
class A, a femoral head is present with an adequate acetabulum, but the
femur is shortened and there is significant coxa vara. In class B, the
femoral deformities are similar, but there is no connection between the
femoral shaft and the femoral head. In class C, the deformities are
more extensive with no femoral head with results in
a
poorly developed acetabulum. Finally, class D shows no development of
the proximal femur or acetabulum, and the deformity is frequently
bilateral.
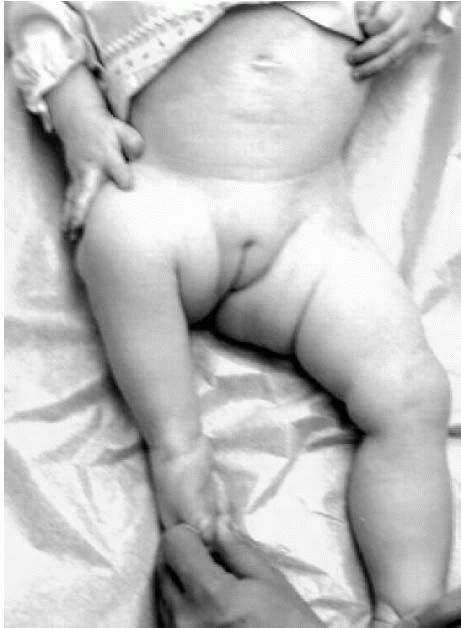
similar appearance and should be easily recognized. The affected site
is shortened with the foot and ankle frequently at the level of the
contralateral knee. The hip is flexed, abducted, and externally
rotated, with flexion contracture of the knee and anterior instability
secondary to absence of the cruciate ligaments (Figure 9-1).
Although the hip abductors and extensors are present, they are unable
to function properly because of the abnormal anatomy of the proximal
femur. Frequently there is an associated fibular deficiency with or
without foot deformity.
limitations the patient will experience, including shortening of the
limb, hip function and its relationship to the alignment of the
extremity, the deficiency in the muscles around the hip that will
result in a significant lurch, and the instability of the knee and
foot. Therefore, there are more options and variations in the treatment
of this complex deformity than in any other congenital limb deficiency.
 |
|
FIGURE 9-1.
Proximal femoral focal deficiency. Note the affected site is shortened with the foot and ankle frequently at the level of the contralateral knee. The hip is flexed, abducted, and externally rotated with flexion contracture of the knee. |
medical team after considering all surgical alternatives and functional
results. If the predicted leg length discrepancy is less than 20 cm at
maturity, the child may be a suitable candidate for a limb lengthening
procedure. If the discrepancy is more than 20 cm, the options are
prosthetic management with ankle disarticulation; ankle disarticulation
and knee fusion; ankle disarticulation and femoralpelvic fusion;
rotationplasty and knee fusion; and rotationplasty and femoral-pelvis
fusion.
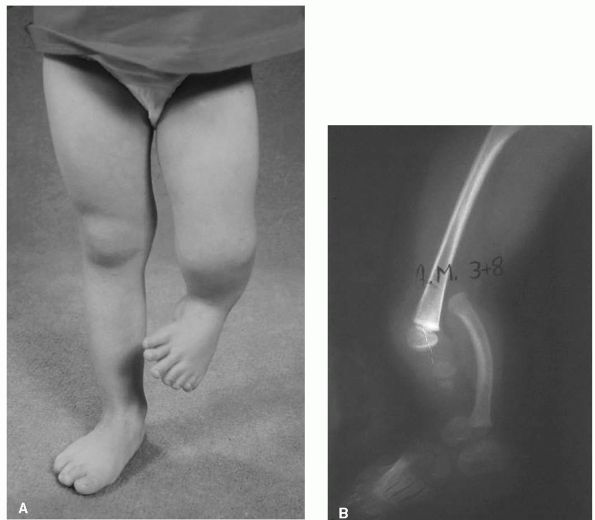
deficiency. It is characterized by a complete or partial absence of the
tibia with bowing, limb shortening, and an intact fibula. The tibial
deficiency may have a relatively normal knee and foot (intercalary
deficiency), or it may be associated with absence of the medial portion
of the foot (terminal deficiency). The knee may present a flexion
contracture and the foot is rigidly held in varus and supination. Other
findings include shortening of the femur, polydactyly and upper
extremity abnormalities, and other organ systems (Figure 9-2A). The cause is unknown, but familial occurrences have been reported.
radiographic findings. The simplest is that of Kalamchi and Dawe. Type
I is complete absence of the tibia (Figure 9-2B).
Type II is absence of the distal tibia but with a good proximal tibial
portion. Type II is distal tibiofibular diastasis. An unusual type is
fibular dimelia in which the tibia is absent and the fibula is
duplicated.
tibial deficiency, the magnitude of the limb shortening, the severity
of the foot deformity, and importantly, the presence of active knee
extension, which implies an adequate active quadriceps muscle and
insertion on the tibia. Patients with a complete absence of the tibia
can be treated by knee disarticulation and prosthetic fitting.
Alternatively, the fibula can be fused to the femur to warrant an
increase in the length of the stump and to provide a longer lever arm
for the prosthesis. In patients with absence of the tibia but with a
good quadriceps mechanism, the proximal fibula can be transferred to
the femoral
notch,
which will lead to a hypertrophy of the fibula. Those patients with a
portion of the tibia present, a proximal tibiofibular fusion and
amputation of the foot with prosthetic replacement will provide an
excellent result. In cases of distal tibiofibular diastasis, a fusion
of the calcaneus to the fibula is indicated.
 |
|
FIGURE 9-2. Tibial deficiency. (A)
The knee may present a flexion contracture and the foot is rigidly held in varus and supination. Other findings include shortening of the femur and polydactyly. (B) Type I tibial deficiency with complete absence of the tibia. |
imply a congenital absence of all or part of the fibula, and they
encompass a spectrum of abnormalities related to abnormal growth and
development of the fibula. The precise cause of fibular deficiency is
unknown and the deformity normally occurs sporadically. The resultant
limb deficiency is usually a terminal type with associated foot
abnormalities. In addition, up to 50% of the patients experience an
associated femoral shortening that is variable in severity.
the anatomic and radiographic appearance of the deformity, but most of
these classifications do not provide satisfactory guidelines for
management and they do not take in account the shortening of the limb,
one of the most significant factors when making management decisions.
Recently, Birch and collaborators have proposed a functional
classification based on the functionality of the foot and the limb
length discrepancy. However, given the large variation in the different
aspects of fibular deficiency, including parent’s expectations, any
classification only provides a general guide for management and a
method for comparison of treatment results.
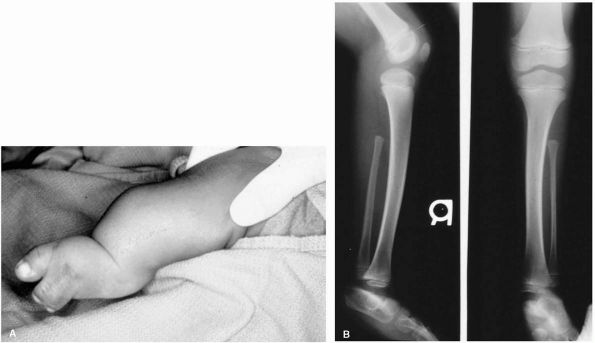
to severely affected. The typical characteristics include a valgus foot
that often has one or several rays missing. The ankle is in valgus
because of the absence of the lateral malleolus and angulation of the
distal tibia. A ball-and-socket ankle has been classically described.
In severe deficiencies, the foot is in rigid equinus (Figure 9-3A).
There is also shortening of the femur, variable anterior bowing and
shortening of the tibia, and variable valgus instability of the knee
with hypoplasia of the lateral femoral condoyle and patellar
abnormalities such as hypoplasia, patella alta, or subluxation (Figure 9-3B).
management of the foot deformity as well as of the limb length
discrepancy. If the foot has significant lateral deficiency and it is
rigid, then amputation (Boyd or Syme type) is generally considered.
Prosthetic fitting is then provided with excellent
results.
In patients with a plantigrade and functional foot, the limb length
discrepancy can be addressed by epiphysiodesis of the contralateral
side, femoral or tibial lengthening, or a combination of techniques.
 |
|
FIGURE 9-3. (A)
The typical characteristics of fibular deficiency including a valgus foot with several rays missing. The ankle is in valgus and rigid equinua because of the absence of the lateral malleolus and angulation of the distal tibia. (B) Partial fibular deficiency. |
skeletal dysplasia was SOX9 in camptomelic dysplasia. It is a severe
and rare form of dysplasia that is sometime fatal. This dominantly
inherited condition is characterized by congenital bowing and
angulation of long bones (camptomelia), primarily involving the tibias
and femurs, and disproportionate short-limb stature. There is relative
macrocephaly, distinctive face (flattened face with a high forehead),
low nasal bridge, and a specific pattern of defective mineralization
including areas of the spine with progressive scoliosis. There is also
defective cartilage in the tracheal rings and lower respiratory tract
that may cause respiratory failure. Extraskeletal features include XY
sex reversal and developmental defects of the heart and kidneys.
cartilage matrix proteins such as types II and XI collagen and
aggrecan. Hence, the skeletal manifestations are in part caused by
decreased expression of these molecules and explain some of the
phenotypic overlap with some of the severe type II collagenopathies.
The bowing of the bones appears to be due to an abnormality in the
formation of cartilage during fetal development (dischondrogenesis).
deformities. However, there is a high degree of complications such as
pseudoarthrosis and neurological complications.
affected, this is a true dysplasia because there are numerous
abnormalities in all parts of the skeleton, primarily those bones of
membranous origin. It is transmitted as an autosomal dominant
condition, and the defect is in the RUNX2 gene, which encodes for a
transcription factor required for osteoblasts differentiation.
Approximately two-thirds are familial and the rest are new mutations.
years of life. Classic features include a widening of the cranium and
dysplasia of the clavicles and pelvis. The patients have mildly to
moderate short stature. There is bossing in the frontal parietal and
occipital regions. There is maxillary micrognathism and common cleft
palate and dental abnormalities. The clavicles are partially or
completely absent (10% of the time). This defect causes the shoulders
to look droopy, the chest to be narrow, and the neck longer. The defect
may be palpable. When it is bilateral, the classic diagnostic feature
is that the child can touch the shoulders together, an ability that
helped one college wrestler to escape holds. Brachial plexus irritation
occurs in rare occasions. The pelvis shows bilateral involvement with
wide symphysis. The iliac wings appear small and coxa vara may occur,
causing limitation of abduction and Trendelenburg gait. Scoliosis and
syringomyelia have been described, and it is recommended to obtain a
MRI in patients with progressive scoliosis.
vara is treated by corrective femoral osteotomies. Scoliosis should be
treated as idiopathic scoliosis. If there is brachial plexus irritation
with pain and numbness, excision of the clavicular fragments can be
performed to decompress it.
genetic skeletal disorders. At least seven discrete types have been
described, ranging from mild disease with normal life expectancy to a
lethal form. Approximately 80 to 90% of patients have mutations in one
of the two genes encoding type I collagen (COL1A1 and COL1A2). However,
in spite of the large number of mutations already identified in OI, the
precise mechanisms by which different mutations cause different
phenotypes are not clearly understood.
an increased susceptibility for fractures to occur, short stature,
laxity of ligaments, hearing loss, and depending on the subtype, blue
sclera, dentinogenesis imperfecta, respiratory insufficiency, excessive
sweating, and early bruisability. The severity of involvement ranges
from the severe cases of a crushed stillborn fetus, to an infant with
multiple or unusual fractures, to an almost symptom-free adult.
Sillence classification. Type I is a milder form of OI with onset of
fractures after birth (most preschool age). Fractures heal without
deformity, and their incidence decreases after puberty. Type I patients
are further divided into two subgroups based on the presence of
dentinogenesis imperfecta. Type II demonstrates more severe involvement
(dark blue sclera, concertina femurs, beaded ribs) with most infants
dying in the perinatal period. Type III develops severe progressive
deformities in the extremities and spine, and these patients usually
have in utero fractures and many die in infancy and early childhood
from respiratory insufficiency. These patients have normal sclerae and
hearing. Type IV demonstrates severe osteoporosis, bowing of the bones,
and increased susceptibility to fractures, but they lack blue sclera.
Many of these patients have short stature. The most severely affected
surviving patients often have this type of disease. Type IV patients
are also subdivided based on the presence or lack of dentinogenesis
imperfecta. Type V patients have bone fragility, but not blue sclera or
dentinogenesis imperfecta. These patients are characterized by three
distinctive features: the presence of hypertrophic callus formation at
fracture sites, calcification of the interosseous membranes between the
bones of the forearm, and the presence of a radio-opaque metaphyseal
band immediately adjacent to the growth plates on radiograph. Type VI
patients have moderate deformity, not blue sclera or dentinogenesis
imperfecta. The distinctive features of this OI type VI are the fish
scale-like appearance of the bone lamellae and the presence of
excessive osteoid upon histological examination.
osteopenia though it varies in its severity according to the severity
of involvement. Type I patients have minimal alterations with thinning
of the cortices and decreased trabecular bone. In type II patients
there is bone shortening and widening, and they have “crumpled
concertina” appearance. Type III patients have narrow diaphyses with
increased flaring and enlargement of the metaphyses and epiphyses.
Typically the long bones are deformed due to multiple fractures. Pelvic
radiographs demonstrate protrusio acetabuli. Spine radiographs
demonstrate platyspondyly, biconcave vertebrae, and varying degrees of
scoliosis, kyphosis, or spondylolisthesis. Some patients will have
cranial osteoporosis with wormian bones and flattening of the occiput
(tam-o’-shanter skull).
mutant gene defect in OI. The treatment of OI has been focused on
maximizing patient’s function,
preventing
deformity and disability as a result of recurrent fractures, correcting
deformities that have developed, and monitoring potential complicating
conditions associated with OI.
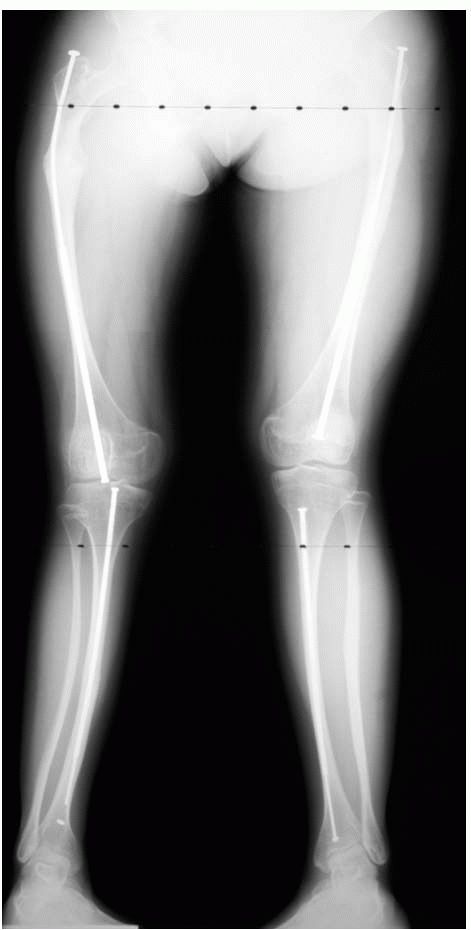
sometimes difficult. Fractures heal readily, often with exuberant
callus, but this is plastic and easily deformed. As a result, bone
deformities and shortening occur. Closed treatment is often used with
lightweight splints or braces. Devices such as the parapodium may help
a child to acquire an upright position. When conservative treatment
fails, intramedullary rodding is indicated. In addition, multiple
corrective osteotomies with intramedullary fixation have been accepted
for managing recurrent fractures (Figure 9-4). Scoliosis treatment in patients with OI still remains a challenge.
underlying biology of this disorder, the medical treatment of OI is
undergoing major improvements. Two new approaches have been studied;
one using bisphosphonates and the other using bone marrow
transplantation.
that inhibit bone resorption, therefore increasing bone mass overtime.
They have been extensively used in adults with osteoporosis, but they
have recently explored in children. Several cohort studies of patients
with OI have demonstrated improved bone mass after the administration
of intravenous pamindronate. This increase in bone mass is mainly due
to an increase in the thickness of the cortex, which resulted in a
significant reduction of fracture rates. Fracture healing was not
affected; neither was longitudinal bone growth. Patients undergoing
treatment also described less fatigue and less chronic pain, and they
had minimal side effects. Although very encouraging, many questions
still need to be resolve regarding when to start treatment, duration,
long-term efficacy, and the safety of the treatment.
transplantation. Bone marrow contains nonhematopoietic mesenchymal stem
cells (MSC) that can differentiate, among other cell types, into
osteoblasts. The results are very encouraging and demonstrated good
engraftment of the cells in vivo with improvement of the osteopenic
phenotype.
has an estimated incidence of approximately 1 in 10,000. Approximately
25% of cases occur in the absence of a family history representing new
mutations. The syndrome involves many systems (skeletal, ocular,
cardiovascular, pulmonary, skin and integument, and dura), but its more
prominent manifestations are skeletal, ocular, and cardiovascular.
Mutations in the gene encoding type 1 fibrillin (FBN1) have been
reported in Marfan syndrome patients. Fibrillin-1 represents the major
component of microfibrils, which are found in many types of connective
tissue, including bone. To date over 500 mutations have been identified
in the FBN1 gene.
Three
categories of mutations have been described: (1) missense mutations,
(2) small insertion or deletions, mutations causing premature
termination of translation, and (3) exon-skipping mutations. Presently
no definite genotype/phenotype correlations have been identified. To
facilitate the identification of different mutations, a “Marfan
database” has been developed that includes not only molecular but also
clinical data. It is only through a large collaborative international
effort that genotype and phenotype correlations will eventually be
identified.
 |
|
FIGURE 9-4. Osteogenesis imperfecta, type I. Treatment by intramedullary rodding.
|
it is of great importance to confirm or exclude the diagnosis in family
members at risk as early as possible because of the potential fatal
complications of the disease (aortic dissection and rupture). At
present, diagnosis for most cases is still based on thorough clinical
examination, including measurements of body proportions,
echocardiography of the aorta, slit-lamp ophthalmologic evaluation, and
radiographs. In the absence of a family history, patients are diagnosed
based on involvement of the skeletal system and two other systems with
at least two major manifestations (ectopia lentis, aortic
dilation/dissection, dural ectasia, or molecular data). In the presence
of a positive family history, an affected person should display one
major criterion in an organ system and involvement of a second system.
Within a given family, however, considerable heterogeneity may be
present. It is not uncommon for milder involved patients to have the
diagnosis go unrecognized until another family member is diagnosed. The
phenotype of the Marfan syndrome remains incompletely defined. Most
manifestations are age dependent and are difficult to quantify.
Molecular data are becoming increasingly important to better
characterize Marfan syndrome and to study its natural history.
patients with Marfan syndrome. Most characteristic is tall stature and
disproportionately long, thin limbs (dolichostenomelia). This can be
confirmed by demonstrating a smaller than normal upper segment (head to
pubic symphysis) to lower segment (pubic symphysis to plantar surface)
ratio or an arm span that exceeds the patient’s total height by at
least 7.5 cm. Patients frequently have arachnodactyly (abnormally long
and slender digits) and ligamentous laxity. This combination of joint
laxity and long digits results in several clinical signs that are
indicative of, but not pathognomonic, for Marfan syndrome. One such
sign is the thumb sign described by Steinberg. This is positive if the
opposed thumb projects past the ulnar border of the clenched fist. The
wrist sign is the ability of the patient to encircle the opposite wrist
with the thumb and small finger, with the thumb overlapping the distal
phalanx of the small finger. The crossover leg sign is the ability of
the patient to touch the floor with the foot of the crossing-over leg.
Joint laxity is another hallmark of the disease, and many patients have
recurrent instability of the patella, shoulder, hip, and thumb. Marked
flatfeet (pes planovalgus), genu valgum, and genu recurvatum are
typical.
(chest depression) or pectus carinatum (pigeon chest). The chest wall
deformities in Marfan patients are due to longitudinal overgrowth of
the ribs. A severe pectus deformity can become clinically significant
by reducing total lung capacity, forced vital capacity, and forced
expiratory volume. Additionally, the occurrence of a severe pectus
deformity in association with scoliosis may further compromise the lung
capacity of patients with Marfan syndrome. It is recommended that
patients with Marfan syndrome have repair of the pectus deformity done
after skeletal maturity to minimize the chance of recurrence with
longitudinal growth of the ribs.
with Marfan syndrome. Abnormalities include thoracic lordosis,
thoracolumbar kyphosis, flat-back deformity, and spondylolisthesis.
Scoliosis is one of the most common and important manifestations of
Marfan syndrome from an orthopaedic perspective with an incidence of
62%. The curve pattern frequently is either a single right thoracic
curve or a double major curve pattern. The thoracic curve is most
commonly lordoscoliotic. The thoracolumbar junction is prone to
kyphosis, probably related to the underlying ligamentous laxity. The
treatment regimen for scoliosis in Marfan syndrome and adolescent
idiopathic scoliosis is similar but the results of treatment are often
different. Specifically, scoliosis in Marfan syndrome has a higher
tendency toward progression and responds less well to bracing than
adolescent idiopathic scoliosis. Both family and physician should be
aware of these facts. In addition, back pain is frequently associated
with scoliosis in Marfan syndrome.
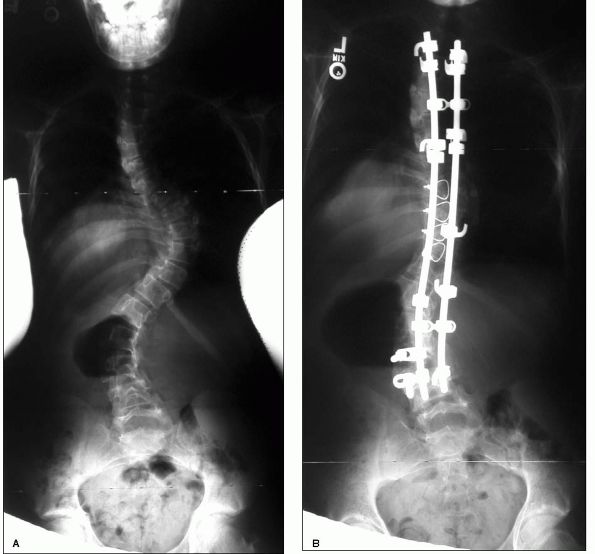
Spinal fusion with autogenous bone graft and rigid internal fixation
should be considered in patients with curves that have progressed
beyond 40° (Figure 9-5A,B). The surgeon should
be aware that there is significantly smaller pedicle widths and laminar
thickness in patients with Marfan syndrome as well as widened
interpedicular distances in the lumbar spine. These osseous vertebral
anomalies may be responsible for some reported postoperative
complications including fracture dislocation and pseudarthrosis.
Preoperative cardiac evaluation is critical and frequently determines
whether a patient is a suitable candidate for extensive spine surgery.
 |
|
FIGURE 9-5. (A) and (B). Scoliosis in a patient with Marfan syndrome. The deformity was treated by posterior spine fusion and instrumentation.
|
and is postulated to be due to increased ligamentous laxity resulting
from underlying connective tissue pathology. Acetabular protrusio can
be seen radiographically in as many as half of patients with Marfan
syndrome. Patients can be asymptomatic or have symptoms of hip pain and
stiffness. Because of the lack of a direct relation between protrusio
and hip symptomatology treatment recommendations are difficult to make.
reported incidence of 63% in Marfan syndrome and can result in back
pain and headaches. It usually occurs in the most caudal portion of the
spinal column
causing
bone erosion and anterior meningoceles. Dural ectasia is thought to
result from fibrillin deficiency resulting in weakness of the dural
sac. There has been a reported direct correlation between the size of
the dural ectasia and the presence of pain. Posterior laminectomy has
been used as a means of relieving back pain thought to be secondary to
dural ectasia.
(dislocated lens) due to lax suspensory ligaments. The dislocation is
superior and lateral and may not be recognized unless a slit-lamp
examination is performed. Ectopia lentis occurs in more than half of
involved patients. Other ocular manifestations include myopia, retina
detachments, strabismus, cataract, and glaucoma.
with Marfan syndrome and are the most common cause of death in this
patient group. For this reason a complete cardiology workup should be
done on all Marfan syndrome patients that includes an electrocardiogram
and echocardiogram. The most serious complication is dilation of the
ascending aorta and associated aortic regurgitation. This often leads
to a dissecting aortic aneurysm. Mitral valve prolapse and mitral valve
regurgitation are the most common significant cardiac problems in
children. Many patients are being treated with medications in an
attempt to delay the onset of serious cardiovascular abnormalities.
widely known and commonly occurring skeletal dysplasia. It is most
commonly inherited in an autosomal dominant fashion though autosomal
recessive forms have been described as well. Its prevalence is
estimated at 1 in 10,000 individuals. The predominant feature of
multiple epiphyseal dysplasia is the delayed and irregular ossification
of numerous epiphyses. In most cases there is pain and stiffness in the
joints, with hips and knees being most commonly affected. In general,
affected patients have mild short stature and early onset
osteoarthritis.
separate forms: Ribbing’s dysplasia, having mild involvement, and
Fairbanks dysplasia, a more severe type. However, with the current
genetic understanding, multiple epiphyseal dysplasia is now considered
to represent a continuous spectrum from mild to severe and these
eponyms have been abandoned.
heterogeneity. To date, mutations have been reported in 40 patients or
families with multiple epiphyseal dysplasia, and 15 of these are
allelic with pseudoachondroplasia and result from mutations in the gene
encoding cartilage oligomeric matrix protein (COMP). MED can also
result from mutations in the genes encoding the α1, α2, and α3 chains
of type IX collagen, COL9A1, COL9A2, COL9A3, respectively. Furthermore,
mutations in the gene encoding matrilin-3, a member of the matrilin
family of extracellular oligomeric proteins, can cause a distinctive
mild form of MED. Finally, it has been demonstrated that a form of
recessively inherited MED, with a distinctive clinical presentation
including clubfoot and bilateral double-layered patellae, can result
from mutations in the solute carrier family 26, member 2 gene
(SLC26A2). Therefore, MED is one of the more genetically heterogeneous
of the bone dysplasias.
of joint stiffness and contractures, lower extremity pain, angular
deformities of the knees, gait disturbance, or short stature. Depending
on the severity of the epiphyseal dysplasia, symptoms may develop as
early as 4 or 5 years. It is not uncommon, however, for patients with
milder forms to go unrecognized until young adult life. Most patients
have minimal short stature and are above the third percentile for
standing height; so true dwarfism is not present. The face and spine
are normal. There are no associated neurologic findings. Intelligence
is not affected. The epiphyses of the upper extremities can be involved
but patients rarely complain of any significant symptoms in this area.
Mild limitation of motion in the elbow, wrist, and shoulder is
occasionally found.
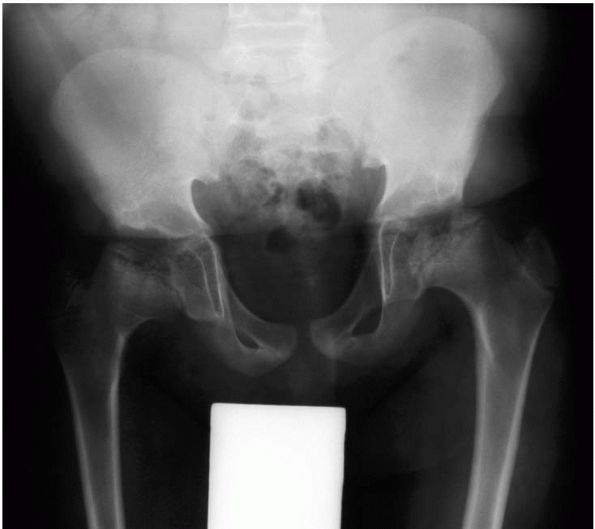
appearance of the ossification centers. When the epiphyses do appear,
they are fragmented, mottled, and flattened. The more fragmentation
there is in the capital femoral epiphysis, the earlier onset of
osteoarthritis (Figure 9-6).
appearance may be easily confused with those of bilateral
Legg-Calvé-Perthes disease. Several radiographic clues may be helpful
in differentiating the two. In Legg-Calvé-Perthes disease, usually one
hip is involved before the other, so that each hip is in a different
stage of the disease. This is not the case in MED. In addition,
acetabular changes are primary in MED and are more pronounced.
Metaphyseal cysts are seen in Legg-Calvé-Perthes disease, but not in
MED. Radiographs of the knees, ankles,
shoulders,
and wrists should be obtained in any child when the diagnosis of
Legg-Calvé-Perthes disease is being entertained to rule out MED.
 |
|
FIGURE 9-6.
Multiple epiphyseal dysplasia. Radiograph of the pelvis demonstrating a fragmented, mottled, and flattened femoral head, and corresponding changes in the acetabulum. |
knees often demonstrate flattening of the femoral condyles as well as a
genu valgum deformity. Osteochondritis dessecans may be superimposed.
Lateral radiographs of the knees demonstrate a double-layered patella
in some patients. When this is present, it is characteristic for MED.
The ankles in MED are also in valgus due to deformity in the talus
predominantly. Upper extremity involvement is less severe. The
metacarpals and phalanges usually are short with irregular epiphyses.
MED is distinguished from spondyloepiphyseal dysplasia by the absence
of severe vertebral changes. Mild endplate irregularities may be
present.
MED seek orthopaedic care in adolescence. Containment surgery can be
considered for those hips that show progressive subluxation. Although
the principle of coverage is the same as that used in Perthes disease,
there is often preexisting coxa vara in hips with MED, which
contraindicates use of a proximal femoral varus osteotomy. In those
cases, a shelf acetabular augmentation can improve coverage of the
misshapen femoral head. If hinge abduction is present on arthrography,
a valgus proximal femoral osteotomy may improve congruency and
therefore relieve pain. Osteotomies may be helpful in realigning
angular deformities at the knees. For optimal surgical correction, the
site of the deformity must be ascertained preoperatively as either the
distal femur, proximal tibia, or both. Degenerative joint disease is
the biggest problem, and it usually occurs in the second or third
decade. If the femoral head is well formed at maturity, the onset of
arthritis is delayed. The hip is the most common location of arthritis
in this patient group and often leads to total joint arthroplasty.
that has a prevalence of approximately four per million, making it one
of the more common skeletal dysplasias. It is characterized by
involvement of both the epiphyses and metaphyses with affected
individuals having significantly short stature and a predisposition to
premature osteoarthritis. The spine is also involved with this disorder.
autosomal dominant trait. The molecular genetics of
pseudoachondroplasia have been extensively studied and it now appears
that this disease results almost exclusively from mutations in the gene
encoding cartilage oligomeric matrix protein (COMP). The COMP gene
consists of 19 exons, and the majority of mutations to date (95%) are
clustered within exons 8 to 14, which encode the type III repeats. The
fact that most of these mutations are in the conformationally sensitive
type III repeats indicates that this region is critical for protein
function. The remaining 5% of mutations are in exons 16 and 18, which
encode specific segments of the C-terminal globule.
resulting from COMP mutations has been well documented. Abnormal COMP
is retained within the rough endoplasmic reticulum of cartilage,
tendon, and ligament cells. This results in the secondary retention of
type IX collagen, chondroitin sulfate proteoglycan 1 (aggrecan), and
link protein. This retention of proteins leads to a reduction in the
amount of these molecules available for interactions within the
extracellular matrix of cartilage resulting in cell death and the
phenotypic picture of pseudoachondroplasia. Additional studies are
needed to delineate the mechanism leading to excessive retention of
proteins in order to develop treatment modalities.
disease to provide molecular diagnosis for because it results almost
exclusively from mutations
along
a very compact region in the COMP gene. Molecular diagnosis for
pseudoachondroplasia is currently provided on a commercial basis
(www.genetests.com) and also as part of the service provision of the
European Skeletal Dysplasia Network (ESDN) for research and diagnosis
(www.esdn.org).
and are usually diagnosed at 2 years of age after the onset of a
waddling gait and at the time rhizomelic shortening becomes noticeable.
Adult height ranges from 106 to 130 cm. Growth curve charts specific to
pseudoachondroplasia are available. The clinical features are limited
to the skeleton. The skull and facies in pseudoachondroplasia are
normal, and this is helpful in differentiating it from achondroplasia,
in which frontal bossing and midface hypoplasia are present.
Abnormalities of the lower extremities are common and include genu
valgum and varum deformities. Some patients develop windswept deformity
of the knees, in which genu valgum is present on one side and genu
varum on the other. The joints are extremely lax, especially in
childhood and adolescence, and there is a predisposition to early
osteoarthropathy. The large weight-bearing joints (hips and knees) are
the ones most often affected, and approximately one-third of patients
need total hip replacement by their mid-30s. Scoliosis may occur in
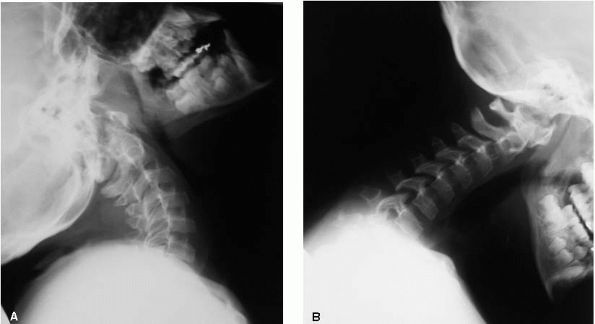
adolescence but is generally not severe. Cervical spine instability is
seen in 10 to 20% of individuals (Figure 9-7). Development milestones and intelligence are normal and premature mortality is not a reported problem.
 |
|
FIGURE 9-7. Patient with pseudochondroplasia and cervical spine instability.
|
epiphyses and metaphyseal changes. Hand radiographs reveal delayed
epiphyseal ossification resulting in delayed bone age. In the long
bones, these changes are seen as epiphyseal ossification delay. When
the epiphyses do ossify, they appear irregular and fragmented. The hip
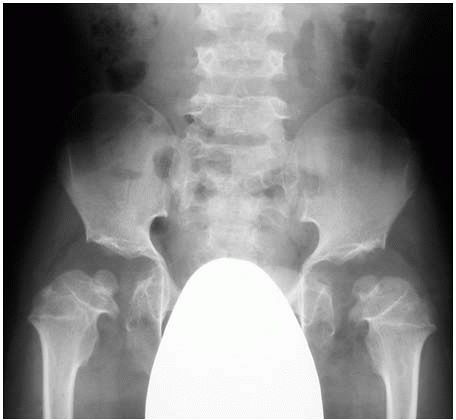
and knee are most severely affected (Figure 9-8).
In the pelvis, there is delay in ossification of the capital femoral
epiphysis, and when ossified they are small and flattened. The femoral
heads may resemble what is seen in other spondyloepiphyseal dysplasias
or bilateral Legg-Calve-Perthes disease. Sclerosis and irregularity of
the acetabular roof are commonly observed. Subluxation of the hips
often occurs and degenerative arthritis develops in response to the
incongruity. The vertebral changes in pseudoachondroplasia are
characteristic and consist of anterior beaking in childhood that
resolves in adolescence. The interpedicular distance in the lumbar
spine is normal in pseudoachondroplasia, unlike achondroplasia.
Odontoid hypoplasia may be present resulting in atlantoaxial
instability.
significant angular deformities of the lower extremities that require
corrective osteotomies. Careful preoperative assessment is necessary to
properly realign the mechanical axis through the hip, knee, and ankle.
For instance, in genu varum associated with
achondroplasia,
the deformity is present solely in the tibia; however, in
pseudoachondroplasia the deformity is often present in both the femur
and the tibia, requiring osteotomies in both the distal femur and the
proximal tibia. Care must also be taken in assessing the contribution
of ligamentous laxity to the bowing deformity. After corrective
osteotomy, recurrence of the deformity with growth is not uncommon.
 |
|
FIGURE 9-8. Patient with pseudoachondroplasia at age 8. Radiograph of the pelvis with severe proximal femoral and acetabular deformities.
|
is a frequent problem in pseudoachondroplasia. Patients with
symptomatic subluxation or incongruity may benefit from a realignment
osteotomy of the proximal femur. Varus osteotomy of the proximal femur
usually creates more incongruity. If hinge abduction is present,
demonstrated by the femoral head levering out of the joint with
abduction of the hip, a proximal femoral valgus osteotomy may improve
joint congruity and improve abductor function. Before performing a
proximal femoral valgus osteotomy, preoperative arthrography should be
performed to demonstrate improved congruity with 15° to 20° of flexion
and adduction of the femur. Abduction of the hip should demonstrate
hinge abduction of the femoral head. Reconstructive pelvic osteotomies,
such as the Salter osteotomy or the triple innominate osteotomy of
Steel, are contraindicated in pseudoachondroplasia because a concentric
reduction is not present preoperatively, which is a prerequisite for
these osteotomies. Salvage procedures such as the shelf augmentation or
the Chiari osteotomy can be done in select cases. As many as 50% of
adult patients have undergone total hip arthroplasty.
chondrodysplasia in humans, occurring in about 1 in 30,000 live births.
A single gene defect (a mutation in the FGFR3 gene) has been
established for this disorder. Initial reports, confirmed subsequently
in many other laboratories, demonstrated that more than 97% of patients
with achondroplasia carried the same mutation, a G to A change at
nucleotide 1138, and that the remaining patients had a G to C change at
the same nucleotide. Very recently it has been demonstrated that, as
previously expected, FGFR3 mutations in sporadic cases of
achondroplasia occur exclusively on the parentally derived allele.
recognized, similar observations regarding the conserved nature of
FGFR3 mutations and resulting phenotype have been made regarding
hypochondroplasia, the lethal thanatophoric dysplasia, SADDAN (severe
achondroplasia with developmental delay and acanthosis nigricans), and
recently two craniosynostosis disorders: Muenke coronal
craniosynostosis and Crouzon syndrome with acanthosis nigricans. More
importantly, the relationship between mutations in the FGFR3 gene and
other FGFR genes, and the phenotypes that result from these mutations
have improved our understanding of these disorders, and it has been
observed that there is a highly conserved relationship between
mutations at a particular amino acid and the resulting phenotype. The
skeletal manifestations of achondroplasia are related to a defect in
endochondral bone formation. The resulting growth disturbances are
variable, affecting proximal segments to a greater extent than the
distal segments of the limbs (rhizomelia), and with relatively minor
involvement of the growth of the spine.
appearance of a person with achondroplasia has numerous features that
are uniform and predictable. Intelligence is normal, and life
expectancy is not significantly diminished. The predicted adult height
is 132 cm for men and 122 cm for women. Obesity is more common than in
the general population. Developmental milestones are met later in
children with achondroplasia than the averagestature children.
bossing, midface hypoplasia, flattening of the nasal bridge, and
prominent mandible. The foramen magnum is frequently narrowed and it is
associated with neurological complications from compression of the
brain stem (quadriparesis, spasticity, sleep apnea and respiratory
insufficiency, and sudden death). The spine length is in the lower
range of normal whereas the extremities are much shorter than normal,
with the proximal segments—the humeri and femora—the most foreshortened
(rhizomelic). There is kyphosis of the thoracolumbar junction during
infancy, and it usually improves with increasing age. Scoliosis is
rare. Hyperlordosis of the lumbar spine increases with age, and there
is a high incidence of symptomatic spinal stenosis (narrowing of the
interpedicular distances with shortening of the pedicles). Clinically,
patients will present with low back and leg pain, paresthesias,
dysesthesias, weakness, or bowel and bladder incontinence.
patients may have asymptomatic radial head dislocations. Patients have
a classic “trident hand” characterized by a persistent space between
the long and ring fingers. The main functional limitations of the upper
extremities are related to shortening of the humeri, which lead to
difficulties in personal hygiene and dressing. Radiographically, the
pelvis is broad with a diminished vertical height. The iliac crest has
a square appearance and the superior acetabular roof is horizontal.
There is flaring of the distal femoral metaphysis. Genu varum is very
common, with ligamentous laxity and the fibula overgrow the tibia.
Internal tibial torsion is common with varus of the ankle (Figure 9-9).
in the first 2 years of life for signs of foramen magnum stenosis. If
the diagnosis is made and symptoms are persistent, decompression of the
brain stem is indicated. In some patients associated hydrocephalus will
require shunting. Problems of the ear, nose, and throat are frequent
secondary to the facial abnormalities. Recurrent otitis media may
result in hearing loss, thus early hearing screening should be
performed. Maxillary hypoplasia leads to dental crowding and
malocclusion, which may require orthodontic treatment. Sleep apnea
treatment, if necessary, begins with adenotonsillectomy and may
progress to include more complex procedures.
kyphosis, spinal stenosis, shortening of the extremities and angular
deformities of the knees. Kyphosis is noncongenital and it is centered
in the thoracolumbar junction. Treatment may be indicated to prevent
further development of the deformity and to assist in those that do not
correct with time (bracing); and in adulthood, to correct surgically
those cases in which kyphosis contribute to symptomatic spinal
stenosis. Spinal stenosis is the most serious problem and usually
develops in the third decade of life. Spinal decompression is indicated
as soon as the diagnosis is made. Limb lengthening remains
controversial but is gradually gaining greater acceptance. If the lower
extremities are lengthened, the humeri should be lengthened also to
facilitate personal care. Treatment of the genu varum usually requires
surgery because bracing is not effective. Fibular head epiphysiodesis,
fibular shortening, and tibial osteotomies can be performed to correct
the deformity usually not until age 4 at the earliest. Interestingly,
severe degenerative arthritis is not common in adults with
achondroplasia.
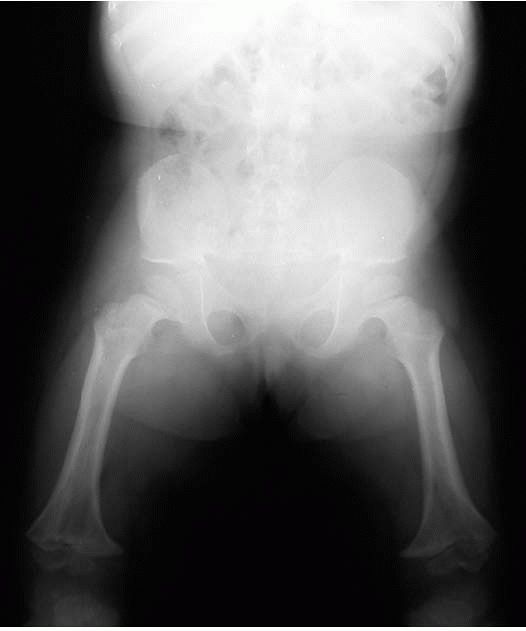 |
|
FIGURE 9-9. Characteristic radiographic appearance of achondroplasia.
|
disorders in which dietary intake of vitamin D is insufficient to
achieve normal mineralization of the growing bone. There are four types
of vitamin D-resistant rickets: phosphate diabetes (i.e., failure of
the reabsorptive mechanism for phosphate); failure of production of 1,
25-vitamin D (i.e., vitamin dependent rickets); end-organ insensitivity
to 1, 25 vitamin D, and renal tubular acidosis.
differentiated on the basis of their resistance to the ordinary
treatment doses of vitamin D and were found to have normal or near
normal levels of calcium, PTH, and vitamin D, but significantly
decreased serum phosphate and abnormal urinary excretion for phosphate,
water, amino acids, glucose, bicarbonate, ketone bodies, and glycine.
2 years of age, and the disease is suspected because of the family
history, laboratory determination of phosphorus concentrations can lead
to the diagnosis in infants as young as 3 months. The usual presenting
complaints are delayed walking, short stature, and angular deformities
of the lower extremities (genu varum, although genu valgum may happen
in some cases). Systemic manifestation such as irritability and apathy
are minimal. The “rachitic rosary” may also occur. Spinal stenosis and
kyphosis has been described. Radiographically there is marked increase
in axial height and widening of the growth plates, cupping, and thin
and indistinct cortices and fuzzy, poorly defined trabecular bone
(osteopenia). Coxa vara may be present as well as lateral bowing of the
femur and tibia. Looser lines can be seen in the rib cage of a child
with florid rickets, resulting from accumulation of osteoid in the bone
matrix.
|
TABLE 9-1. Mucopolysaccharidoses
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
nephrologist with expertise in metabolic bone disease. The usual
treatment consists of oral neutral phosphate replacement and the
administration of vitamin D, and the correction of the metabolic
abnormality present. The orthotic management to correct the lower
extremity deformities has proven ineffective. If patients experience
pain and increased deformities, surgical correction of the angular
deformities should be performed. Multilevel osteotomy is usually
required with intramedullary fixation or external fixation. Because of
a high risk of recurrences in the younger patient, surgery should not
be performed in early childhood.
this group of genetic disorders, and these disorders are among the
first skeletal dysplasias to be described and also among the first to
be understood at the biochemical level. There are at least 13 types (Table 9-1), and each type produces a particular
sugar in the urine because of a specific enzyme defect. The incidence is about 1 in 20,000 live births.
glycosaminoglycans (heparin sulfate, dermatan sulfate, keratan sulfate,
and chondroitin sulfate) by lysosomal enzymes is abnormal leading to
intracellular accumulation of these incompletely degraded compounds in
the lysosomes themselves. They are classified based on their enzyme
deficiency and the type of substance that accumulates. The most common
types are Morquio’s and Hurler’s syndromes.
tissues such as the brain, the viscera, and the joints. This
unremitting process leads to the clinical progression of the disorders.
The child is normal at birth, being biochemically detectable by 6 to 12
months of age, and clinically symptomatic by 2 years of age. All these
disorders lead to abnormally short stature. And in some cases, there is
severe mental retardation (Hurler’s, Hunter’s, and Sanfilippo’s). There
are also abnormalities of the skull (enlarged, with thick calvarium)
and facies (coarse, gargoyle), and deafness. In some cases there is
hepatosplenomegaly and cardiovascular abnormalities. Radiographically,
the clavicles are broad and the scapulae are short and stubby. The
vertebral bodies are ovoid and scoliosis and kyphosis are frequent (Figure 9-10). There is acetabular dysplasia, coxa valga, and the iliac wings are flared.
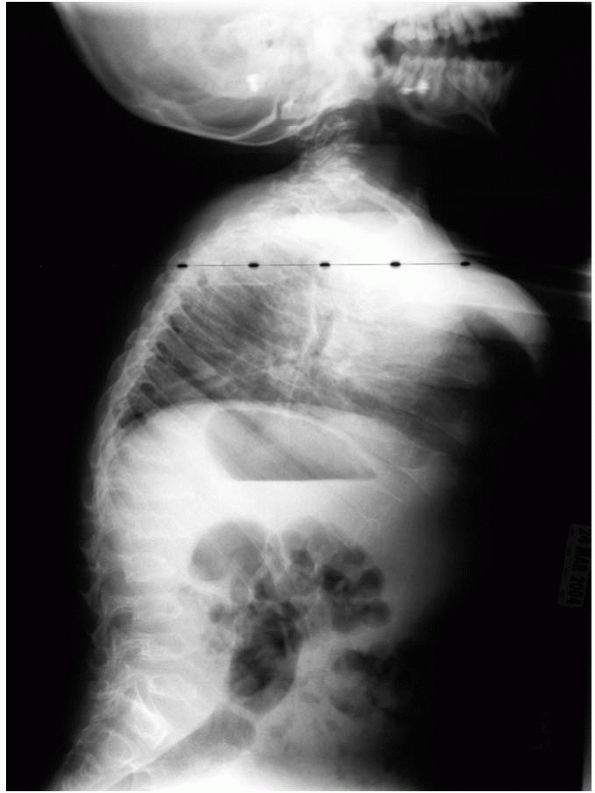 |
|
FIGURE 9-10. Spine of a patient with Morquio’s. Platyspondyly with anterior beaking and mild kyphosis of the thoracolumbar junction.
|
in the first two decades of life if untreated. Treatment is evolving
and some of these disorders have been treated successfully with bone
marrow transplantation. The preferred donor is an HLA-identical
sibling. Following successful transplantation, accumulation of the
mucopolysaccharide stops, and there is improvement in the coarse
facies, hepatosplenomegaly, and partially in the hearing. Research is
currently under way in the field of gene therapy for some of these
syndromes.
functionally impairing musculoskeletal deformities. Hip flexion
contractures and dysplasia often require surgical reconstruction,
including reduction, femoral and pelvis osteotomies. Cervical
instability may be present and C1-C2 fusion and halo immobilization may
be necessary. Kyphosis requires orthotic treatment or even surgical
spine fusion. Genu valgus may be treated with corrective osteotomies.
aclasia, is a highly penetrant, autosomal dominant trait characterized
by slightly stunted growth of long bones and multiple osteochondromas.
Osteochondromas are cartilage-capped excrescences of bone that develop
at the growth plate level during growth. These osteochondromas are
indistinguishable morphologically from the solitary cases. HME has an
incidence of about 1 in 50,000 live births. The median age at the time
of diagnosis in affected individuals is approximately 3 years. By the
second decade of life, nearly all affected individuals will have
exostoses as the penetrance of the disorder has been found to be 96 to
100%. Many patients with HME require resection of the lesions due to a
mass effect or neurovascular impingement symptoms. Importantly, up to
3% of patients with HME will eventually develop a malignant
chondrosarcoma.
genetics have permitted a better understanding into the molecular
players underlying these lesions. Linkage analysis has located three
etiological genes for HME, EXT1, EXT2, and EXT3. Interestingly,
mutations in any of these genes demonstrate very similar clinical
manifestations. These EXT loci have defined a new class of putative
tumor suppressor genes, to which have been recently added three related
genes, EXTL1, EXTL2, and EXTL3.
associated with loss of heterozygosity at one or more of the EXT loci,
a neoplastic model of pathogenesis has been suggested. The Knudson
“two-hit” theory of carcinogenesis derived from familial retinoblastoma
has been applied to HME. Both copies of the EXT1 gene have been
observed to be deleted and gene losses and mutations have been observed
in chondrosarcomas arising from osteochondromas. However, it is still
unclear how EXT1 and EXT2 can function as tumor suppressors.
the joints. Numerous sites can be involved, typically five or six
exostoses can be found in the upper and lower extremities. The most
common locations are distal femur (70%), proximal tibia (70%), humerus
(50%), and proximal fibula (30%). Over time, the extremities will
shorten in relation to the trunk, and legs will grow unequally. As the
lesions enlarge, they may cause discomfort secondary to mechanical
pressure to adjacent soft tissues and muscles. They rarely cause
neurological dysfunction. Often, patients complain of an undesirable
cosmetic appearance. Valgus deformity of the knee and ankle are not
uncommon, and osteochondromas of the proximal femur may lead to
dysplasia of the hip, which may require corrective osteotomies. In
adults, sarcomatous transformation will present as a painful and
enlarging mass in an area of previous deformity.
excision. However, not all the exostoses should be removed. Established
indications for surgery include growth disturbances leading to angular
deformities or hip dysplasia; functional limitation of joint range of
motion; spinal cord compression with neurological compromise; painful
mass and obvious cosmetic deformity, and rapid increase in the size of
the lesion. Deformities in the forearm should be treated early to
prevent further progression and to reduce disability. Knee osteotomies
are associated with a high incidence of peroneal nerve palsy.
in transcription factors result in an array of defects affecting
craniofacial, appendicular, and axial skeletal development.
authors present a classification based on a combination of molecular
pathology and embryology, taking into account the importance of
development for the understanding of bone disease.
A, Bonafe L, Rimoin DL. Molecular-pathogenetic classification of
genetic disorders of the skeleton. J Am Med Genet 2001;106:282. The
authors present a classification based on the molecular and
pathogenetic aspects of the disorders of the skeleton, with an attempt
to identify the metabolic pathways, signaling cascades, and regulatory
networks underlying these disorders.
GT. Proximal femoral focal deficiency: definition, classification, and
management. In: Proximal Femoral Focal Deficiency: A Congenital
Abnormality. Washington, DC: National Academy of Sciences, 1969:1. Classic
article on the clinical and radiographic characteristics of PFF, with a
discussion on its classification and treatment guidelines.
LA, Meyer LC, Warren FH. Proximal femoral focal deficiency: natural
history and treatment. Clin Ortho Rel Res 1982;162:135. The authors discussed the natural history of the deformity and options of treatment.
G, Yigiter K, Bayar K, Erbahceci F. Effectiveness of prosthetic
rehabilitation of children with limb deficiencies present at birth.
Prosthet Orthot Int 1999; 23:130. Discussion of the most important aspect of prosthetic rehabilitation in children with limb deficiencies.
FW. Construction of a knee joint in congenital total absence of the
tibia (paraxial hemimelia tibia). J Bone Joint Surg 1965;47A:695. Classic description of the fibular transfer for complete tibial deficiency.
D, Barnes J, Lloyd-Roberts GC. Congenital aplasia and dysplasia of the
tibia with intact fibula: classification and management. J Bone Joint
Surg 1978;60B:31. The authors propose a classification with recommendations for treatment based in a review of 20 patients.
classification of tibial deficiency based on its radiographic
appearance. Results of treatment of 21 patients are presented.
authors examine 87 cases from the literature using the minimal
requirements for a good result. They found that 53 of the 55 cases of
Jones type I had a poor result. It emphasizes the need for strong,
active knee extension.
authors reviewed the treatment results of 57 patients (71 limbs) with
tibial deficiency. An ablative procedure was performed on 61 of the
limbs. Brown’s procedure yielded less than satisfactory results.
JG, Lincoln TI, Mack PW. Functional classification of fibular
deficiency. In: Herring JA, Birch JG eds. The Child with a Limb
Deficiency. Rosemont, IL: American Academy of Orthopaedic Surgeons,
1998;161. The authors present a functional classification of fibular deficiency and recommend treatment option for each type.
JJ, Glancy GL, Chang FM et al. Fibular hemimelia: comparison of outcome
measurements after amputation and lengthening. J Bone Joint Surg
2000;82:1732. The purpose of our study was to
compare the outcome after amputation with that after tibial
lengthening, specifically with regard to activity restrictions, pain,
satisfaction, complications, number of procedures, and cost, in
children with fibular hemimelia. The study demonstrated that children
who undergo early amputation are more active, have less pain, are more
satisfied, have fewer complications, undergo fewer procedures, and
incur less cost than those who undergo lengthening.
of 33 patients treated with contemporary methods. The authors concluded
that surgical treatment and prosthetic rehabilitation yield excellent
results, both short and long term.
JA. Syme’s amputation for fibular hemimelia: a second look in the
Ilizarov era. Instructional Course Lectures 1992;41:435. Discussion of the results of Syme’s amputation in fibular deficiency.
D, Boyd NA, Fixsen JA et al. The natural history and management of
congenital short tibia with dysplasia or absence of the fibula. J Bone
Joint Surg 1977;59B:267. The authors note that the leg length discrepancy remains constant throughout childhood.
new classification system for fibular hemimelia is proposed based on
the authors’ experience with 32 patients (33 involved limbs)
representing a spectrum of involvement. The data demonstrate the broad
and unpredictable relationships among the fibula, ankle, and foot in
this disorder. Because of this variability and unpredictability of the
multiple relationships, limb salvage criteria should also include the
nature of the foot and ankle and not merely depend on the length
discrepancy or the presence or absence of the fibula.
study of Syme’s type amputation for fibular deficiency with severe
shortening of the limb and equinovalgus deformity of the foot and ankle.
JI, Grissom LE, Harcke HT. Radiographic characteristics of
lower-extremity bowing in children. Radiographics 2003;19:204. This
article reviews lower-extremity bowing conditions in infants and
children. Recognition of these pathologic conditions is important for
differentiating those that will resolve spontaneously from those that
require surgery or other treatment.
spinal abnormalities were found in eight patients (average age of 6
years, 5 months) with camptomelic dysplasia. This study clarifies that
patients with camptomelic dysplasia are surviving longer than
previously expected and therefore should have their spinal deformities
treated aggressively.
I, Baumert U, Hrala BP, Mussig D. Dentomaxillofacial variability of
cleidocranial dysplasia: clinicaoradiological presentation and
systematic review. Dentomaxillofac Radiol 2003;32:347. Review
of authors’ series (24 patients) and from the literature (259 cases)
with documentation of the most common caraniofacial abnormalities
observed in these patients.
A, Salvi S, Casali C et al. Six novel mutations of the RUNX2 gene in
Italian patients with cleidocranial dysplasia. Hum Mutat 2003;22:104. Report of clinical and molecular findings in 14 patients with this condition.
of clinical characteristics and a more complete delineation of clinical
complications associated with this condition. Management
recommendations based on the results of this study are included.
incidence and severity of spinal deformities in patients with this
condition varies with age (26% < 5 years and 80% for those older
than 12).
JR, Schwarze U, Wang PR et al. Gene targeting in stem cells form
individuals with osteogenesis imperfecta. Science 2004;303:1198. The
authors have used adeno-associated virus vectors to disrupt
dominant-negative mutant COL1A1 genes in MCS from individuals with this
condition, demonstrating successful gene targeting.
JG, Studwick WJ, Rinsky LA et al. Complications of intramedullary rods
in osteogenesis imperfecta: Bailey-Dubow rods versus nonelongating
rods. J Pediatr Orthop 1988;8:645. Evaluation of these two techniques with a complication rate of 69% for the Bailey-Dubow and 55% for the rigid rods.
of the molecular changes seen in osteogenesis imperfecta, the current
treatment options and the gene therapy approaches being investigated as
potential future treatments for this condition.
cross-sectional study comparing the prevalence and size of dural
ectasia in patients with Marfan syndrome with or without pain.
experience of the authors with 22 severely affected infants diagnosed
with Marfan syndrome. Morbidity and mortality may be high during
infancy and prompt recognition can facilitate management and counseling.
L, Levran O, Ramirez F et al. A molecular approach to the
stratification of cardiovascular risk in families with Marfan’s
syndrome. N Engl J Med 1994;331:148. The goal of
this study was to develop a widely applicable method of molecular
diagnosis. The results demonstrated that the various clinical
phenotypes may be due not to the single fibrillin mutations, but rather
to different genetic alterations.
C, Rosenthal A, and Nadas AS. Cardiac manifestations of Marfan syndrome
in infancy and childhood. Circulation 1973;47:587. Description of the clinical features associated with the cardiac abnormalities in this condition.
study analyzed the prevalence, inheritance, progression, and functional
implications of spinal deformity in Marfan syndrome.
MD, Chapman KL. Pseudoachondroplasia and multiple epiphyseal dysplasia:
mutation review, molecular interactions, and genotype to phenotype
correlations. Hum Mutat 2002;19:465. Excellent
review of the molecular abnormalities of these conditions, and
discussion of the correlation between genotype and phenotype.
RR, Ponseti IV, Maynard JA. Pseudoachondroplastic dwarfism: a
rough-surfaced endoplasmic reticulum storage disorder. J Bone Joint
Surg 1973;55A:475. Description of the ultrastructural abnormalities that differentiates this disorder from other skeletal dysplasias.
JF, Wynne-Davies R, Fulford GE. Bilateral failure of the capital
femoral epiphysis: bilateral Perthes disease, multiple epiphyseal
dysplasia, pseudoachondroplasia, and spondyloepiphyseal dysplasia
congenita and tarda. J Pediatr Orthop 1983;3:297. The authors conclude that Perthes disease can be differentiated radiographically from other skeletal dysplasias.
study delineates the natural history of this condition based on the
follow up of 79 patients. Premature osteoarthritis was the major health
problem for these individuals.
NJ, Jensen FO, Bankier A et al. Development of the hip in multiple
epiphyseal dysplasia. Natural history and susceptibility to premature
osteoarthritis. J Bone Joint Surg 1990; 72B:1061. Premature
osteoarthritis was a frequent outcome and was almost inevitable before
the age of 30 years in those with incongruent hips.
JA, Ippolito EG, Ponseti IV, Mickelson MR. Histochemistry and
ultrastructure of the growth plate in achondroplasia. J Bone Joint Surg
1981;63A:969. Detailed analysis of the histological and ultrastructural abnormalities of the growth plate in this condition.
R, Thompson LM, Zhu YZ et al. Mutations in the transmembrane domain of
FGFR3 cause the most common genetic form of dwarfism, achondroplasia.
Cell 1994;78:335. Description of the genetic abnormality underlying this condition.
IH, Kim JK, Chung CY et al. Deformity correction of knee and leg
lengthening by Ilizarov method in hypophosphatemic rickets: outcomes
and significance of serum phosphate level. J Pediatr Orthop
2002;22:626. The authors evaluated 14 patients
with this condition and found that the healing index correlated with
the biochemical parameters. They suggested a serum phosphate of 2.5
mg/dL as the cutoff point for surgical indications.
of the pathogenic mechanism, clinical and laboratory features of these
conditions, and discussion on the differential diagnosis.
J. The mucopolysaccharidoses: a heterogeneous group of disorders with
variable pediatric presentations. J Pediatr 2004;144:S27. Review of the most current understanding in these conditions with discussion of the clinical features and management strategies.
M, Grewal S, Peters C. Hematopoietic stem cell transplantation for
mucopolysaccharidoses and leukodystrophies. Clin Pediatr 2004;216:163. Recent update on the use of stem cell transplantation for these disorders.
JE, Clarke LA, Beck M et al. Enzyme replacement therapy for
mucopolysaccharidosis I: a randomized, double-blind,
placebo-controlled, multinational study of recombinant human
alpha-L-iduronidase (laronidase). J Pediatr 2004;144:581. This
study confirms that laronidase significantly improves respiratory
function and physical capacity, reduces glycosaminoglycans storage, and
has a favorable safety profile.
JA, Simpson Ah, Porter DE et al. Wrist and forearm dysfunction in
hereditary multiple exostosis. J Hand Surg 2003;28 (Suppl 1):26. Description and management strategies of wrist and forearm dysfunction in patients with this condition.
CR, Cole WG, Haynes R et al. Reevaluation of a genetic model for the
development of exostosis in hereditary multiple exostosis. Am J Med
Genet 2002;112:1. Review of the current understanding of the molecular abnormalities and their pathogenic implications for this condition.
KJ, Feinberg JR, Levenda A et al. Natural history of multiple
osteochondromatosis of the lower extremity and ankle. J Pediatr Orthop
2002;22:120. The authors evaluated 38 patients
with an average age of 42 years at follow up. They found measurable
decreases in ankle function and suggest that correction or prevention
of excessive tibiotalar tilt may be warranted to improve outcome.
BM, Crawford BE, Esko JD. Hereditary multiple exostoses and heparan
sulfate polymerization. Biochem Biophys Acta 2002;1573:346. An
overview of HME, the EXT family of proteins, and possible models for
the relationship of altered HS synthesis to the ectopic bone growth
characteristic of the disease.