
clinical deformity, functional significance, and natural history.
Congenital spine deformities appear to result from disordered vertebral
formation occurring early in embryogenesis, presumably during
somitogenesis between the 5th and 8th week of gestation (1,2). The coexistence of vertebral
malformations and nonspinal visceral and musculoskeletal malformations
is common in congenital scoliosis and other congenital spine
deformities (3).
The broad range of congenital scoliosis originates from varied
combinations of vertebral anomalies caused by failures of formation,
failures of segmentation, and failures of midline fusion. Congenital
spinal deformities are of significance not just for the spinal
deformities themselves and the potential risk they pose to neurologic
structures, but also because they are associated with thoracic
deformity, loss of spinal mobility, and altered body shape; these may
occur either without treatment or in spite of it. Each congenital spine
deformity condition is associated with a specific deformity and natural
history. Although some are discovered by fetal ultrasound (4,5,6)
or are visible at birth, many instances of congenital scoliosis go
unrecognized until a later age. Congenital vertebral deformity may be
troublesome at birth or within the first 2 years of rapid growth, or it
may remain quiescent until the preadolescent growth acceleration (7).
Although generalizations and caveats are possible, the history of each
congenital spinal deformity remains unique. For many congenital
deformities early treatment is critical, because later on deformity
correction is either not possible or entails greater risk. Generally,
the goal of observation and treatment in congenital spinal deformity
should be prevention of severe deformity rather than restoration of
normal spinal contours. Nonsurgical treatment is of extremely limited
value and early surgical treatment remains the most conservative
treatment for progressive deformity (8,9).
producing a deformity in the coronal plane. The distinction between
congenital scoliosis and congenital kyphosis or lordosis may be
blurred, as many deformities are mixed, with aberrations both in the
coronal and sagittal planes. A list of other spinal anomalies and
syndromes in which congenital scoliosis is commonly present includes
occipitocervical anomalies such as occipitalization of C1 with atlantal
hypoplasia, Klippel-Feil deformity, Jarcho-Levin syndrome,
spondylocostal or spondylothoracic dysplasia, spinal dysgenesis, and
congenital spinal dislocation. Related anomalies that are generally
considered distinct from congenital scoliosis include spinal
dysraphisms and neural tube defects including spina bifida,
myelodysplasia, lipomyelomeningocele, caudal regression syndromes, and
other major disruptions of normal vertebral formation in which the
natural history of the underlying neurologic deficit usually
overshadows any associated congenital scoliosis.
distinct from scoliosis or kyphosis that occur in association with
congenital bone dysplasias; it is also distinct from other multiple
vertebral abnormalities associated with generalized anomalies in bone
development. For example, although the vertebral deformities associated
with spondyloepiphyseal or multiple-epiphyseal dysplasia or with
mucopolysaccharidoses originate early in gestation, they are caused by
a generalized failure of normal bone growth and development, and the
failure of normal vertebral shape and spinal growth are secondary. They
are not caused by early failures of vertebral segmentation or formation
as in congenital scoliosis. A clear distinction is also made between
congenital scoliosis or kyphosis and infantile idiopathic or
early-onset idiopathic scoliosis. Infantile idiopathic scoliosis may be
present in utero, but it originates from
normally formed vertebral segments and has a natural history quite
distinct from that of congenital scoliosis. The term congenital scoliosis
is often mistakenly applied to infantile idiopathic scoliosis, but the
two entities must be clearly separated for purposes of diagnosis and
treatment.
All congenital vertebral anomalies share a common etiologic mechanism
of failure of segmentation and/or formation, but may also exhibit a
mutually shared association with exposure to teratogens, maternal
diabetes (2,10,11,12,13,14)
and other syndromes that involve vertebral anomalies. Any vertebral
anomaly may be associated with malformations of other organ systems
including auditory anomalies, renal and renal-collecting-system
anomalies, congenital heart anomalies, visceral, uterine, and vaginal
anomalies, and upper extremity anomalies such as radial hypoplasia (3,15,16,17,18,19,20,21,22,23,24).
Likewise, any of the congenital vertebral anomalies may be identified
with a syndrome such as Goldenhar; Poland; Noonan; Crouzon, basal cell
nevus; vertebrae, anus, cardiovascular, trachea, esophagus, renal
system, and limb buds (VACTERL); coloboma, heart, atresia, renal,
genital, ear (CHARGE); and others (2,25,26,27,28,29,30,31,32,33,34,35).
not distinguish congenital scoliosis from idiopathic scoliosis, and the
true prevalence of congenital scoliosis is therefore unknown, partly
because so many vertebral malformations go unrecognized. Assessment of
a number of congenital scoliosis operations done at a large center
suggests that as many as 20% of cases serious enough to require surgery
among adolescents and children are congenital in etiology (36).
have permitted earlier and more comprehensive detection of fetal spinal
anomalies. Families may wish to understand the outlook for the child’s
spinal deformity even at the prenatal stage.
Fetal
screening is still an evolving field, and predictions of postnatal
anomalies by prenatal screening continue to be imperfect. Multiple
vertebral anomalies noted on prenatal screening may prove to have
minimal significance postnatally, whereas apparently localized
vertebral anomalies seen prenatally may result in more generalized
defects and syndromic associations with profound postnatal effects. It
is also difficult to accurately predict the postnatal course on the
basis of prenatal neural tube defects. Caution is urged in
prognosticating on the basis of prenatal ultrasound.
 |
|
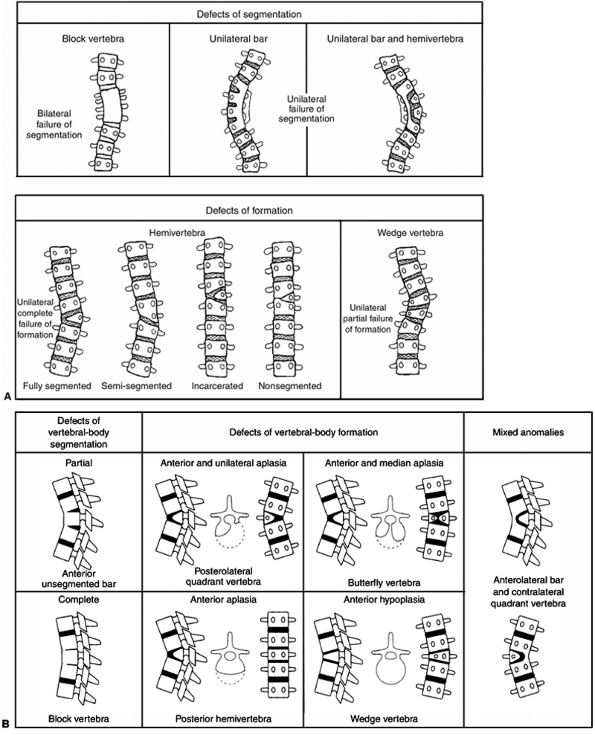
Figure 19.1 A:
Classification of congenital scoliosis. Failures of segmentation or formation, singly or in combination, describe nearly all congenital scoliosis. Hemivertebrae can be classified as fully segmented, semisegmented, or nonsegmented. When deficiencies of vertebrae above and below compensate for the hemivertebra, it is described as incarcerated. The growth potential of hemivertebrae can be estimated by the presence and thickness of superior and inferior endplates. A unilateral bar opposite a fully segmented hemivertebra is very likely to progress with growth. B: Classification of congenital kyphosis. Many instances of congenital scoliosis contain elements of congenital kyphosis. Nearly all congenital kyphotic deformities are progressive and are best treated by early in situ posterior fusion if the patient is less than 5 years and the curve is less than 50 degrees. If untreated, progressive kyphotic deformities with failure of formation may produce paraplegia. (A: From McMaster MJ, Ohtsuka K. The natural history of congenital scoliosis. A study of two hundred and fifty-one patients. J Bone Joint Surg Am 1982;64(8):1128–1147, with permission. B: From McMaster MJ, Singh H. Natural history of congenital kyphosis and kyphoscoliosis. A study of one hundred and twelve patients. J Bone Joint Surg Am 1999;81(10):1367–1383, with permission.) |
It would be potentially valuable to provide genetic counseling to
apprise the family of the exact risk of having a subsequent child with
a similar anomaly, but it is difficult to achieve (42). Wynne-Davies (43)
noted a risk of recurrence of 2% to 3% in siblings when multiple
vertebral defects were present, and multiple occurrences have been
documented in the same family (41). Some anomalies such as Klippel-Feil deformity have a clear risk of recurrence in other siblings (2,44). Purkiss et al. (45)
noted a 20% overall incidence of spinal deformity and 17% incidence of
idiopathic scoliosis in families of patients with congenital scoliosis.
For many families there is a sense of guilt associated with the birth
of a child with a congenital anomaly, because they are under the
mistaken
impression that environmental influences or parental factors have
contributed to the congenital anomaly. Although this view may be
correct for some teratogens (46) and for maternal diabetes (14),
most congenital vertebral defects represent a spontaneous occurrence.
Recent research has identified genetic associations for congenital
vertebral anomalies in laboratory animals. In the mouse model,
connections between the genes regulating somite formation and
segmentation have been associated with genes in the “notch” family (2,47,48,49).
Notch genes have been shown to regulate development of the somite and
vertebral precursors in the mouse, and in humans they are associated
with spondylocostal dysostosis and Alagille syndrome (48,49,50).
(including genetic trisomies) and an increased incidence of congenital
vertebral malformations are also well documented (2,19,25,44,45,51,52,53,54,55,56,57,58,59,60).
A few recognized syndromes are associated with congenital scoliosis,
and a knowledge of the genetic basis of these syndromes may help with
planning and screening for future siblings. The genetic markers
associated with some syndromes, such as spondylocostal dysostosis, have
been identified and may be of significance to families (1,47,48,49).
Continued elucidation of the control mechanisms of somitogenesis will
likely lead to identification of other genetic abnormalities among
patients with congenital vertebral anomalies.
normal vertebral development in early embryogenesis. Early disruption
of normal somitogenesis is presently agreed to be the source of the
failures of segmentation and formation seen in congenital scoliosis (1). Failure to form normal pairs of somites is reflected in hemivertebrae and wedged vertebrae and in hemimetameric shifts (61),
whereas failure in the temporal sequence of somitogenesis from cranial
to caudal manifests as segmentation failures such as block vertebrae
and multiple contiguous vertebral anomalies. Butterfly vertebrae are
presumed to originate from failure of anterior midline fusion between
somite pairs. Use of animal models and inspection of human embryos have
determined that somitogenesis occurs between the 5th and 7th week of
gestation. This is also the time of organogenesis for many organ
systems, and therefore it is not surprising that there is a
considerable association between malformations in the vertebral column
and malformations in the auditory, renal, cardiac, and visceral
systems. Signaling and formation of somite precursors for rib and limb
development occur during a similar interval,
hence rib and limb anomalies are commonly found in association with congenital vertebral anomalies (Fig. 19.2 A,B,C).
 |
|
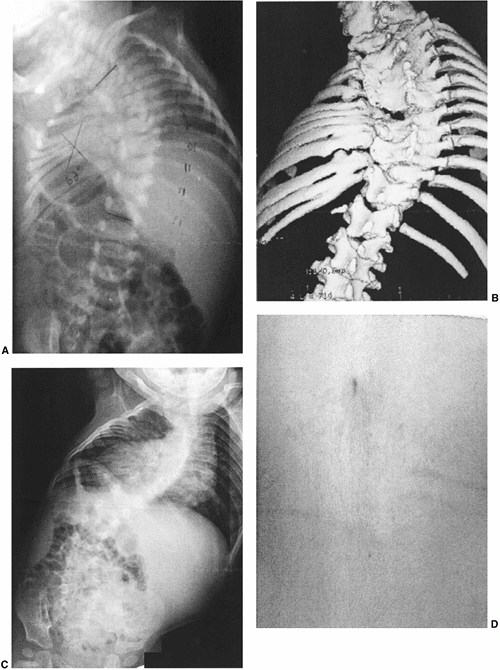
Figure 19.2 Anomalies associated with congenital scoliosis. A:
One-week-old infant with congenital scoliosis. The multiple vertebral anomalies seen here include vertebral bony bars, hemivertebrae, and wedge vertebrae. Other anomalies included ear anomalies and deafness, solitary kidney, imperforate anus, tethered spinal cord, atrial septal defect, hypoplastic lung, and radial hypoplasia. B: Three-dimensional computed tomography (CT) scan of the same patient at 10 months. Fused ribs on the concavity of the thoracic scoliosis will act as a tether, producing more deformity with growth. C: Upright radiograph at 10 months. A new curvature in the more normal thoracolumbar spine is compensatory to the upright position, but should suggest the possibility of spinal cord tethering. D: Cutaneous abnormalities such as this skin dimple over the thoracolumbar spine may indicate an underlying intraspinal anomaly. |
 |
|
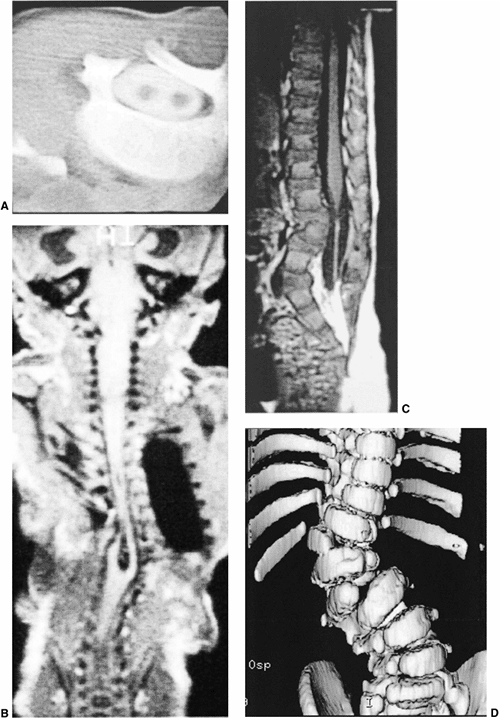
Figure 19.3 A:
Intraspinal anomalies accompanying vertebral anomalies are common. Indications for magnetic resonance imaging (MRI) include planned surgical intervention, abnormalities found on neurologic examination, and progressive curvature in the unaffected section of the spine. Diplomyelia is visible in this computed tomography (CT) myelogram. B: Diastematomyelia, diplomyelia, tethered spinal cord, and other anomalies are present in this infant with multiple vertebral anomalies. Surprisingly, the lower extremity neurologic exam is normal (MRI scan image). C: Tethered spinal cord with thickened filum terminale (MRI scan image). D: A CT scan with sagittal, coronal, or three-dimensional reformatting is helpful for understanding details of congenital vertebral anomalies. Two lumbar hemivertebrae are readily visible here. |
although the mechanism of action of these teratogens has not yet been
determined. Likewise, the association between maternal diabetes
(particularly type I diabetes) and fetal vertebral anomalies has been
well documented (63).
in these experiments vertebral anomalies and associated skeletal
defects have been similar to those seen in more extensive instances of
congenital scoliosis associated with anomalies of the ribs and
extremities. These same animal experiments have demonstrated that the
location and extent of skeletal malformations are partly dependent upon
the timing of the toxic insult, presumably because of the temporal
sequence of creation of somites from cranial to caudal. Oxygen
deficiency and carbon monoxide exposure, studied in mice, produce
considerable skeletal defects including vertebral malformations.
Although there is no direct evidence linking hypoxia or carbon monoxide
exposure in humans to congenital vertebral defects, there is an
increased incidence of Klippel-Feil syndrome noted among patients with
fetal alcohol syndrome (30,32,67).
Hyperthermia may also be a cause of vertebral malformation and has been
shown to disrupt normal vertebral development in animal models (12,68).
greatly. Many instances are unrecognized until an incidental x-ray film
shows an underlying vertebral anomaly. If balancing deformities are
present (Fig. 19.4) (hemimetameric shift), the
congenital scoliosis may be completely unapparent clinically except for
diminished spinal motion. Some congenital scoliosis deformities are
immediately apparent at birth, with major malformation of the spine and
an associated deformity or shortening (Fig. 19.2 A,B,C). Clinical features that may suggest congenital scoliosis or other vertebral
anomalies include signs of spinal dysraphism such as skin dimples (Fig. 19.2D) or hairy patches near the spine, limb paralysis (69)
or atrophy, and signs of chronic neurologic abnormality including foot
deformity, particularly if asymmetric. Spinal dysgenesis or congenital
spinal dislocation may be manifest as early lower-extremity paralysis
or dysfunction (70,71).
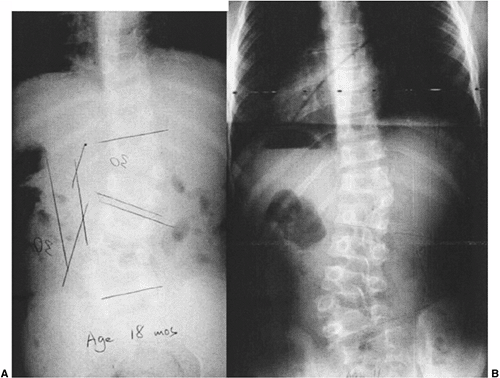 |
|
Figure 19.4 Nonprogressive, balanced, double hemivertebrae (“hemimetameric shift”). A: In a child 18 months of age, two hemivertebrae balance each other well. B: At 11 years of age, there has been extensive growth but no worsening of the untreated, balanced deformity.
|
and this shortness may be manifest as a relative limb–trunk
disproportion. Most anomalies involving multiple vertebral levels are
associated with loss of motion of the affected spinal segment. In
Klippel-Feil deformity this may manifest as a torticollis with
diminished motion of the cervico-thoracic junction or cervical spine.
Likewise, vertebral anomalies involving the entire thoracic and lumbar
spine may manifest not as deformity but as diminished mobility. Rarely,
vertebral anomalies may present as a mass or bump near the spine rather
than as a spinal deformity. Congenital lordosis caused by posterior
failures of fusion may appear as an abnormally concave area of spine
without scoliosis. Congenital anomalies at the cervicothoracic junction
may present as a supraclavicular mass or may mimic Sprengel deformity,
with a high-riding scapula. Congenital scoliosis centered at the
cervicothoracic level may produce a profound cosmetic deformity with
head tilt, shoulder elevation, and secondary spinal imbalance.
Hemivertebrae at the thoracolumbar junction may manifest as trunk
imbalance, whereas those at the lumbosacral junction may appear as
pelvic tilt or apparent leg-length inequality.
predictably in thoracic idiopathic, neurogenic, and paralytic
scoliosis, but is variable in congenital scoliosis. Some congenital
thoracic scoliosis is associated with little if any rotational thoracic
asymmetry, whereas other cases, particularly when many spinal segments
are involved or the deformity is progressive, manifest as severe
thoracic deformity (73). Rarely, well-balanced
congenital scoliosis with rib fusions may first present with
restrictive respiratory difficulty caused by a short, stiff thorax.
Congenital scoliosis, when progressive, worsens during periods of rapid
growth and may therefore first be noted during either early childhood
or adolescence.
can generally be made from plain radiographs. Careful assessment of
well-penetrated anteroposterior and lateral radiographs will usually
reveal nearly all the salient details of a congenital vertebral
anomaly. Absence of spinous processes, rib fusions, joined pedicles, or
absent or narrowed disc spaces may suggest an underlying congenital
anomaly. More extensive radiographic evaluation is needed
preoperatively for surgical planning, but not usually for
classification or observation. MRI is helpful for assessment of spinal
cord anatomy and will reveal vertebral body segmentation and formation
anomalies with accuracy, but it can be confusing in the assessment of
posterior elements.
Sagittal plane anomalies are often important concomitant deformities
that should be included in any description. If the failure of formation
or of segmentation occurs only on the right or left side, a scoliosis
results. If the failure occurs only anteriorly or posteriorly, then a
congenital kyphosis or congenital lordosis will occur. Most cases are
subtle combinations of both coronal and sagittal plane deformities, and
a more three-dimensional classification system is needed. Recognizable
patterns of failures of formation include wedge vertebrae and
hemivertebrae in congenital scoliosis, and type I (Winter) failures of
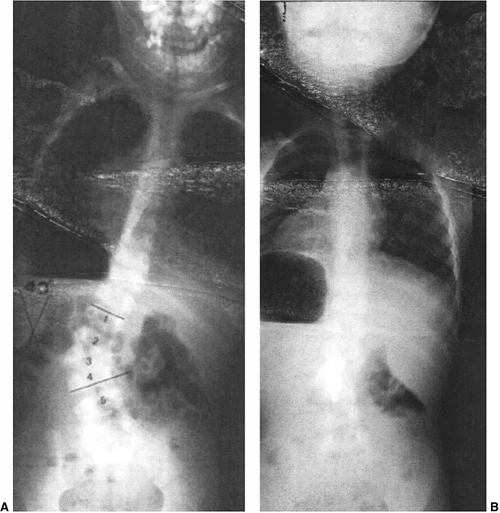
formation in congenital kyphosis (80,81) (Fig. 19.5). Wedged vertebrae result from a unilateral partial failure of
formation, and usually a pedicle is present bilaterally. Unilateral
complete failure of formation manifests as a hemivertebra, recognizable
by the absence of a contralateral paired pedicle. Hemivertebrae can be
subclassified as fully segmented, semisegmented, or nonsegmented on the
basis of the presence or absence of a full growth plate and disc
remnants at the cephalad and caudad sides of the hemivertebra (Fig. 19.1). Hemivertebrae can be either paired or unilateral and can be offset with a “hemimetameric shift” (Fig. 19.4). Failures of segmentation can result in block vertebrae (Fig. 19.6),
anterior or posterior unilateral bars, congenital lordosis, or type II
congenital kyphosis. Complete bilateral failures of segmentation
anteriorly result in a block vertebra. Unilateral anterior failure of
segmentation produces an anterior unilateral bar. Partial failure of
segmentation of the anterior portion of the disc and growth plate
results in a type II congenital kyphosis (Fig. 19.7).
Posterior failure of segmentation can be bilateral and symmetric,
producing congenital lordosis, or unilateral, creating a posterior
cartilaginous or bony bar. Complete anterior and posterior failure of
segmentation creates a block vertebra, whereas unilateral anterior and
posterior failure of segmentation causes a unilateral bar.
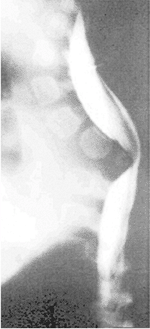 |
|
Figure 19.5
Congenital kyphosis (type I by Winter classification) with posterior hemivertebra. Neurologic function has regressed from normal to paraplegic by 9 months of age, the time of this computed tomography (CT) myelogram image. Anterior decompression and stabilization were needed. |
segmentation, failure of formation, and mixed failures of segmentation
and formation assumes that anterior spinal anatomy is similar to
posterior spinal anatomy. Clinical observation and CT scans (82,83)
show that it is not rare for anomalies in the posterior elements to
show no correspondence with the anomalous anterior elements. The
anterior elements, best seen on plain x-ray film or MRI, guide the
overall classification. Posterior elements are better seen by CT scan
and CT reconstructions.
posterior anomalies may explain some of the difficulty in accurately
predicting which deformities will be progressive. If, for example, an
anterior fully segmented hemivertebra is associated with a bilateral
posterior failure of segmentation over the same levels, there may be
slow progression rather than rapid progression, or the progression may
occur more into lordosis than into scoliosis, the posterior failure of
segmentation preventing or slowing the tendency of the fully segmented
hemivertebra to progress. The availability of the three-dimensional
reconstruction of CT scans makes it easier to understand and describe
these anomalies. Radiographic findings may also include associated
anomalies (Fig. 19.2). The presence of rib
fusions or of distortion of the thoracic cavity should be noted. The
presence of multiple rib fusions may act as a paraspinal tether
creating thoracogenic scoliosis, or may enhance progressive deformity
because of the vertebral bar that is commonly juxtaposed to rib
fusions. Examination of the underlying vertebral anatomy and an attempt
at classification is helpful for prognosis (Fig. 19.8).
If certain patterns are identified, such as a fully segmented
hemivertebra, the likelihood of rapid progression may be much higher.
If a unilateral bar
is
noticed, there is a great likelihood of continued progression of the
deformity. The combined presence of one or more fully segmented
hemivertebra as well as a cartilaginous bar and/or rib fusions is
highly likely to lead to progressive deformity with growth (7,8,9,74,75,76,79,84,85).
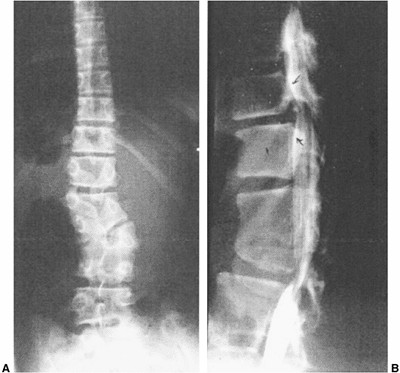 |
|
Figure 19.6
Adjacent segment hypermobility, sometimes with instability or stenosis, may occur adjacent to naturally occurring congenital or surgically created rigid segments of spine. A: A well-balanced untreated congenital scoliosis with increasing spasticity of the lower extremities. B: Myelogram reveals hypertrophic discs and spinal stenosis. Treatment was by decompression and fusion of the hypermobile segments. |
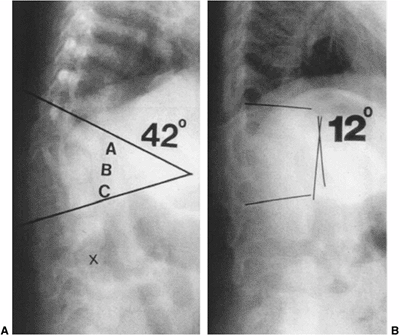 |
|
Figure 19.7 Early in situ fusion is the standard treatment for progressive congenital curves. Early posterior in situ
fusion with cast and/or instrumentation immobilization can produce progressive correction in patients younger than 5 years of age with moderate congenital kyphosis (less than 50 degrees) without neurologic involvement. A: Progressive type II congenital kyphosis at 13 months of age. At 14 months of age, the patient was treated with in situ posterior fusion and postoperative cast. B: At age 4 years, there is progressive correction of the kyphosis due to the posterior spinal fusion acting as a tether, while anterior growth continues. |
significance than overall deformity and the change in that deformity
over time with growth. Documentation of radiographic change with growth
is the mainstay of observation and surgical decision making. Comparison
of current with early x-ray films is needed in order to ascertain
whether there has been deformity progression. Radiographic change may
be very gradual, and archiving of original x-ray films is useful for
later comparison and remeasurement of Cobb angles.
assessment of deformity progression. Unlike idiopathic scoliosis, in
which a standard measure, the Cobb angle, is fairly reproducible within
and between observers, meaningful documentation of progression in
congenital scoliosis
requires
measurement of reference points that are consistent over time. It is
mandatory that congenital scoliosis be measured in a reproducible way
on the basis of reference to prior radiographs. Reference to only the
immediately prior radiograph may miss slow change and produce
cumulative error in measurement. Because change may occur slowly over
many years, families should be urged to archive original and
intermediate films for later comparison. Of necessity, initial
radiographs of an infant’s spine will be taken in the supine position,
but as the child grows older radiographs should be taken with the child
in the upright position. Caution is needed in interpreting a change in
deformity over this period of time if the comparison is between a
supine-position film and an upright-position film (Fig. 19.2 A,C).
Generally, both posteroanterior and lateral films are necessary. Subtle
worsening of congenital kyphosis or congenital lordosis may go
unrecognized if only posteroanterior films are taken.
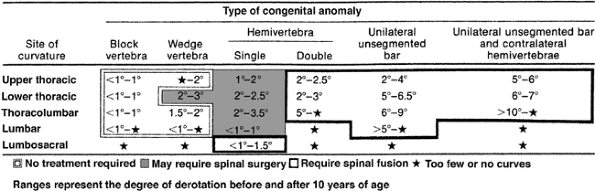 |
|
Figure 19.8
McMaster has compiled data about the likelihood of progression of congenital scoliosis associated with different vertebral anomalies, based on annual rate of progression. Double hemivertebrae, unsegmented bars, and unsegmented bars opposite hemivertebrae were noted to be rapidly progressive and to require spinal fusion. (From McMaster MJ, Ohtsuka K. The natural history of congenital scoliosis. A study of two hundred and fifty-one patients. J Bone Joint Surg Am 1982;64(8):1128–1147, with permission.) |
coronal and sagittal planes requires the consistent identification of
end points (86,87).
Identification of end points can be done easily in patients with
idiopathic scoliosis, but requires innovation and consistency in those
with congenital scoliosis. Cobb angle measurement of a pure coronal
deformity may be made by using the vertebral body endplates or,
commonly, the pedicles as endpoints. Measurement of a low lumbar spinal
deformity may be more easily achieved by using the tops of the iliac
crest as the lower end point, in essence measuring pelvic obliquity.
The choice of measurement end points must be consistent between films,
and usually it will be necessary to go back and remeasure films,
starting from infancy, with the same end points in order to determine
whether progression has occurred. It is appropriate to measure not only
the anomalous congenital segment but also the entire curve associated
with it. Attention to secondary curves is equally important, and all
curves should be measured at all intervals. A change in a secondary
curve may reflect an unrecognized change in the associated congenital
“primary” curve. A change in the normally segmented part of the spine
without any change in the congenital curve raises the possibility of
the change being caused by spinal cord tethering, and should prompt an
MRI examination of the intraspinal contents for anomalies (Fig. 19.3 A,B) and tethered spinal cord (Fig. 19.3C) if not already done (3,21,88,89,90).
Other indications of spinal deformity, including trunk imbalance,
apical vertebral deviation, occipital or cervical spine imbalance
relative to the pelvis, and appearance of sagittal plane posture and
sagittal plane balance as seen on lateral radiographs, may be of even
greater significance than the Cobb angle. Loder et al. (87)
found that intra- and interobserver error in Cobb angle measurement in
congenital scoliosis was as much as 13 degrees, thereby suggesting that
Cobb angle alone may be a poor marker for documentation of progression
in congenital scoliosis. Lonstein et al. (86),
however, showed that if sequential radiographs were measured and care
was taken to use the same end points, inter- and intraobserver error in
Cobb measurement of congenital scoliosis was similar to or better than
studies of measurement error in idiopathic scoliosis. Their observation
that sequential radiographs and reproducibility of measurement end
points over time enhance accuracy is of the utmost importance, and the
clinician should follow their advice to re-examine multiple prior
radiographs each time that the patient’s new radiographs are measured.
including screening for underlying renal anomalies, is mandatory. If
the child is less than 6 to 8 weeks of age, spinal ultrasound (91)
may be effective for the screening of intraspinal contents for such
major anomalies as low-lying conus, diplomyelia, dermoid,
diastematomyelia, syringomyelia, or tethered spinal cord. If there is a
high suspicion of intraspinal anomaly, however, MRI is needed and in
infants will usually require anesthesia or sedation.
reporting a rate of 37%. An MRI is clearly indicated prior to any
surgical intervention. Even if the surgical intervention does not
involve correction, a search for underlying intraspinal anomalies
should be made before altering the spinal anatomy by fusion or
instrumentation. Anomalies include associated syringomyelia with or
without Chiari I malformation, fatty filum terminale, intradural
lipoma, and occult split cord or diastematomyelia malformations. Major
intraspinal malformations increase the likelihood of progressive
deformity and progressive or acute neurologic loss if acute surgical
correction of the deformity is attempted (92). Any tethering lesions should be considered for surgical release (17,21,93,94,95).
Urodynamic testing may help establish the significance of a tethering
lesion in a patient who otherwise does not have a progressive deformity
or neurologic abnormalities of the extremities (96).
intervention or if progressive scoliosis is noted in a structurally
normally segmented area of the spine. Whether MRI should be performed
as a screening test in all individuals with congenital spinal anomalies
is, however, controversial. Some series recommend a screening MRI in
all individuals with vertebral malformations (3,88).
In young children, an MRI requires sedation or general anesthesia, and
thus other sources suggest that MRI screening needs to be done only
preoperatively or if neurologic abnormalities are present (90).
Subtle signs of root tension or root irritation such as limited neck
flexion or tight hamstrings, an extended trunk and extremity posture,
or asymmetric reflexes mandate a
screening
MRI. More obvious neurologic findings, including asymmetry in the girth
or strength of the extremities, weakness in the extremities, or
otherwise unexplained incontinence, should prompt an MRI of the
intraspinal contents. MRI of the spinal cord done in infancy may not
reveal anomalies that become visible later in childhood, and motion
artifact may obscure underlying anomalies. Some anomalies such as
intradural lipomas or syringomyelia may change over time. If a child
has had a normal MRI study in infancy but there are clinical signs of
intraspinal abnormalities at a later time, a subsequent MRI in later
childhood is potentially useful. If progressive deformity is noted in
the normal part of the spine, MRI is mandatory, as spinal cord
tethering or some intraspinal anomaly may be responsible for the change
in deformity.
spine, consideration should be given to dynamic flexion/extension views
periodically to assess instability.
associated with congenital scoliosis and other congenital spinal
anomalies at rates as high as 61% (3,15),
particularly in more extensive and mixed vertebral anomalies. Screening
of the kidneys and renal collecting system should be done in every
individual with congenital scoliosis. Asymptomatic obstructive uropathy
can occur; in one series (3), one third of the
patients noted to have renal anomalies required treatment. The
orthopaedic surgeon may be the only medical provider to recognize the
potential occurrence of renal anomalies in association with congenital
vertebral anomalies. Screening renal ultrasounds can be performed on
small children without sedation. If the kidneys and collecting system
are visualized incidentally on an MRI, a screening ultrasound is not
needed.
particularly with cervical anomalies and Klippel-Feil syndrome, in
which as many as one third of the patients may have hearing loss (29).
Major hearing deficits can go unrecognized in infants, particularly if
the child has other major health problems which distract from the
diagnosis. Infant auditory screening is probably sufficient to rule out
a major hearing deficit, but if it has not been performed, auditory
testing should be considered.
congenital scoliosis, but the spine in most scoliosis that is
associated with severe congenital heart disease is normally segmented (98,99).
A clinical examination will pick up manifestations of congenital heart
disease such as abnormal heart sounds, abnormal rhythm, and abnormal
oxygenation, but may not reveal significant underlying cardiac
anomalies. An echocardiographic examination of the heart is probably
warranted in individuals with congenital spinal deformities who will be
undergoing major surgical intervention, because the incidence of
cardiac anomalies is reportedly as high as 26% with congenital
scoliosis (3).
and congenital kyphosis is highly variable. Four mechanisms are
probably responsible for the evolution of deformity in congenital
vertebral anomalies.
failure of segmentation. Early fetal ultrasound of vertebral defects
sometimes reveals a major deformity early in embryogenesis. Asymmetric
growth of malformed vertebral elements is the presumed mechanism for
worsening of the deformity during the remainder of embryogenesis after
initial vertebral column formation. Asymmetric growth originates from
the presence of asymmetric tissue with growth potential, including both
vertebral cartilaginous endplates as well as vertebral cartilage
destined for endochondral growth. Examination of coronal or sagittal
cross sections of congenital vertebral pathologic specimens, or CT or
MRI scans, readily reveal an asymmetry in growth-related tissue. The
growth potential of any given area of tissue can be estimated from the
thickness of the growth plate seen on imaging, which is also juxtaposed
to disc material. If a thick, asymmetric disc space is seen on imaging,
it may be presumed that the growth potential of that area is also
asymmetric. Estimation of the growth potential of posterior elements is
less well understood.
asymmetric tethering of spinal growth by a cartilaginous or bony bar,
rib fusions, or even iatrogenic fusions. Tethering probably produces
progressive deformity only if there is substantial growth on the side
of the spine opposite the tethering structure. If an area with
substantial growth is juxtaposed to an area of complete absence of
growth or tethering, then progression will likely occur and may evolve
quickly (Fig. 19.9). Associated rib fusions may produce a tether adjacent to the spine with or without vertebral anomalies (Fig. 19.10A),
much like a thoracogenic scoliosis associated with burn scars.
Relatively normal growth may occur in the spine but if an adjacent
tethering structure is strong, the spine will be drawn into a
progressively deformed position with the ribs acting as a concave
tether. Campbell demonstrated growth in bony vertebral bars (100),
but bony or cartilaginous bars will reliably act as a tether if there
is considerable growth on the opposite side of the spine (79,85).
long-term action of gravity and posture. Just as idiopathic deformities
can progress in the presence of what was initially a normally formed
vertebral column, so also congenital scoliosis can deform over time
through asymmetric
growth
of previously symmetric structures. This progression is presumably
caused by the prolonged effect of gravity on an asymmetric or
out-of-balance spine, which best explains the rapid worsening of
previously stable congenital deformities seen during the preadolescent
growth acceleration (Fig. 19.11), the same period when progression of idiopathic spinal deformity is commonly noted.
 |
|
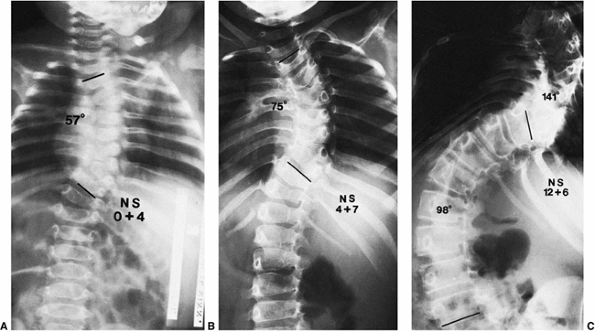
Figure 19.9 Relentless progression of untreated thoracic congenital scoliosis from 4 months of age (A) to 4 years of age (B) to 12 years of age (C).
Progression has occurred in the area of an unsegmented bar (segmentation defect) opposite hemivertebrae, with fused ribs in the concavity of the curve. |
 |
|
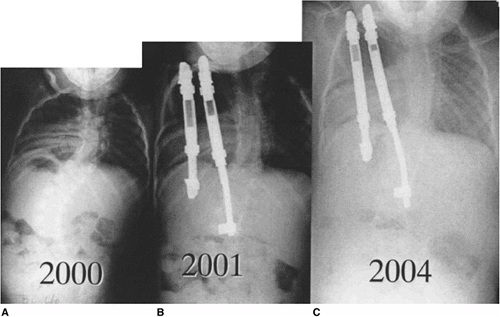
Figure 19.10
This two-and-a-half-year-old patient with vertebrae-anus-cardiovascular-trachea-esophagus-renal-limb-buds (VACTERL) syndrome underwent in situ fusion of progressive congenital thoracic scoliosis associated with concave fused ribs at 14 months of age. A: At 2½ thoracic curve progression has continued postoperatively. B: One year postoperatively, after expansion thoracostomies at two levels, insertion of rib-to-rib and rib-to-spine devices, and device lengthening. C: Three years postoperatively after vertical expandable prosthetic titanium rib (VEPTR) devices were outgrown and replaced with longer devices. |
 |
|
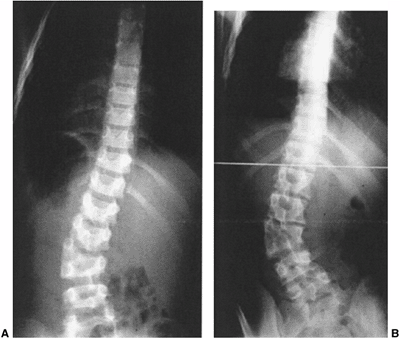
Figure 19.11 Congenital scoliosis may be stable during childhood but then progress with the preadolescent growth acceleration. A: A 12-year-old boy with a semisegmented hemivertebra noted incidentally on screening exam. B:
At 14 years of age, after 18 months of rapid growth, there has been a drastic increase in curvature. MRI was negative for tethering lesions. |
with intraspinal anomaly or tethered spinal cord. The mechanism of
deformity production is not clear in this instance, but the presence of
abnormal intraspinal elements and growth can combine to produce
progressive deformity either in a normally segmented spine or in
congenital scoliosis. MRI investigation is mandated when a progression
of deformity is noted in a part of the spine that was free from
congenital deformity, or when a previously stable congenital deformity
worsens rapidly.
is hampered by the rarity of the condition and the extreme variability
in severity of congenital spine deformities. The natural history of
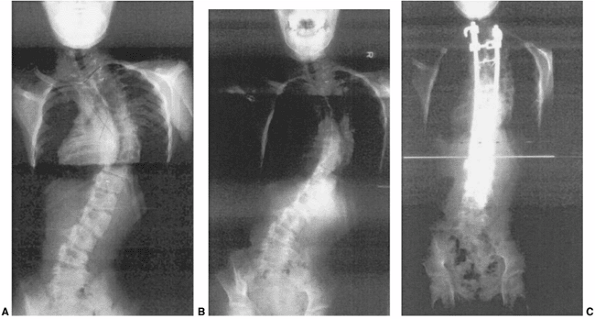
untreated congenital scoliosis ranges from relentless progression of
deformity (Fig. 19.9) with severe consequences for pulmonary and neurologic function, to no progression and no functional deficit (Fig. 19.12).
The natural history of congenital scoliosis is best seen as separate
but interrelated problems of short- and long-term concern: progression
of spinal deformity, spinal instability and function, pulmonary
function, and height.
during growth is well understood and documented. The natural history in
the short term ranges from anomalies with dire short term neurologic
consequences, such as spinal dysgenesis (55,70,71,101) or type I kyphosis (Fig. 19.5), to those with rapid deformity change during growth, such as fully segmented hemivertebrae opposite a bar (79,85) (Fig. 19.9),
to those which are completely stable through growth and adulthood. If
the congenital spine deformity is progressive, it generally mirrors
growth velocity, changing rapidly during the first 2 years, slowly
during childhood, and then rapidly again during the preadolescent rapid
growth phase (7) (Fig. 19.11).
Changes in deformity during childhood growth reflect anomalous,
asymmetric, or tethered growth, whereas those seen during
preadolescence may reflect these effects and the effects of gravity as
well. Worsening deformity in congenital scoliosis, congenital lordosis,
or congenital kyphosis may occur most rapidly during the first 2 years
of growth. Progression of the deformity depends upon the balance of
cross-sectional growth in the spine and the secondary effects of any
tethering structures. The presence of intact growth plates unbalanced
by similar growth forces on the opposite side of the spine,
particularly if a tether is present, leads to progressive deformity.
 |
|
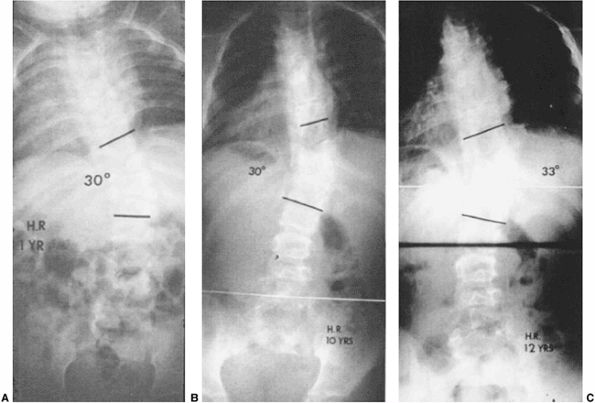
Figure 19.12 Some extensive congenital anomalies are either not progressive or only minimally progressive. A: Fully segmented hemivertebrae with concave rib fusions and other anomalies are noted at 1 year of age. B:
The patient was put under observation only, and no treatment was given. No significant progression has occurred by 10 years of age. C: At 12 years of age after preadolescent growth acceleration there has been slight progression. No treatment was needed. |
noting that progression depends on the type of deformity, its location
in the thoracolumbar spine, the length of spine involved, and the
amount of growth remaining (Fig. 19.8). A
unilateral bar opposite a contralateral hemivertebra is most likely to
progress by as much as 10 degrees per year during rapid growth (Fig. 19.9).
Unilateral bars or fully segmented hemivertebrae alone may also
progress rapidly. Wedge and block vertebrae are less likely to show
progression (Figs. 19.4 and 19.6).
Early prophylactic fusion without waiting for progression may be
justified for some deformities with a high likelihood of worsening such
as congenital kyphosis (Fig. 19.7) or a fully segmented hemivertebrae opposite a bony or cartilaginous bar (79,85,103).
the spine. Lumbosacral hemivertebrae and thoracolumbar hemivertebrae
may show more rapid progression of deformity than congenital
deformities contained within the thoracic spine and constrained by the
secondary stability of the chest. Rib fusions may act as a tether,
causing thoracogenic scoliosis that may either enhance or counteract
the tendency of vertebral deformity to progress (104,105) (Fig. 19.10A). During adulthood larger deformities may continue to progress (106,107), presumably due to the forces of gravity and imbalance acting through vertebral remodeling.
deformity during infancy rarely includes instability and neurologic
change. Neurologic loss caused by spinal canal encroachment may occur
in segmental spinal dysgenesis (11,55,70,71) or type I congenital kyphosis (102) (Fig. 19.5),
but is uncommon otherwise in congenital scoliosis unless there is an
associated intraspinal anomaly, stenosis, instability, or a major
kyphotic component in the deformity (92).
Spinal mobility is restricted in areas of marked congenital scoliosis,
and the concentration of motion stress at the junction between stiff,
abnormal congenital spine and mobile, normal spinal segments may
produce junctional degenerative changes, instability, or stenosis (Fig. 19.6).
Adjacent segment degeneration, instability, and late spinal stenosis
are best documented in association with Klippel-Feil syndrome (2), but can also be seen with congenital scoliosis (106).
adverse effects in overall growth and health that are independent of
any associated syndrome such as VACTERL or Klippel-Feil. Concomitant
with abnormalities of vertebral shape, there are consistently observed
deficiencies in spine size and height. Goldberg et al. (72)
have brought attention to the short stature associated with congenital
scoliosis, with and without treatment. One of the later consequences of
disordered growth of the thoracic spine is the effect on pulmonary
function (104,108). Campbell, Day, and others (104,108,109,110,111)
have documented the associations of both congenital and early-onset
scoliosis with pulmonary insufficiency. Pehrsson et al. have linked the
diagnosis of early-onset scoliosis with decreased adult survival
because of pulmonary insufficiency (111). It
seems clear that if congenital scoliosis involves the thoracic spine,
and if the thoracic spine is very short or the chest very distorted,
then the likelihood of major pulmonary insufficiency during adulthood
is increased (104). Campbell et al. have
documented the combination of interrelated thoracic spinal deformity
and chest cage distortion leading to compromise in respiratory function
in progressive congenital and other early-onset spinal deformities (104), and has introduced the useful concept of thoracic insufficiency syndrome to describe the interdependence of chest wall, spinal growth, deformity, and lung function (Figs. 19.2 A,B,C, 19.3, 19.10, 19.13, and 19.14).
By definition, thoracic insufficiency syndrome is the inability of the
spine and chest wall to support normal lung growth and function. In the
series studied by Campbell et al. (104),
thoracic insufficiency occurred in patients with fused ribs and
congenital scoliosis as well as in those with extensive spinal and rib
anomalies such as Jarcho-Levin, spondylocostal, and spondylothoracic
dysplasias (104,112).
Their description of thoracic insufficiency syndrome has led to a
heightened awareness of the potential short- and long-term effects of
early spinal deformity and associated chest wall deformity on pulmonary
function. The inherent shortness of the thoracic spine, associated
congenital rib fusions, distortion of the chest shape with progressive
spinal deformity, and “penetration” of the chest by lordotic thoracic
deformities all contribute to thoracic insufficiency by diminishing
thoracic volume and compliance (Fig. 19.14). Deformity of the lumbar spine, or thoracolumbar or lumbar kyphosis, may also create “secondary” thoracic insufficiency (104) by diminishing the space between thorax and pelvis, thereby in terfering with normal diaphragmatic excursion and respiration (16).
the natural history of the congenital scoliosis and the continuum of
treatment options available. Treatment may be early and prophylactic
for predicted progression of deformity or later and corrective for
existing deformity. In situ fusion is
commonly employed, and a choice must usually be made between simply
arresting the deformity with a relatively safe predictable procedure or
correcting the deformity with a more elaborate procedure. Overall a
reasonable goal of treatment of congenital scoliosis is to prevent the
development of a severe deformity. For many congenital deformities no
treatment is needed (Fig. 19.12), and the best
choice may be to perform as little surgery as possible. Generally,
waiting for more growth leads to worse deformity (Figs. 19.14A and 19.15A,B) and exposes the patient to the risk of a more dangerous operation.
accurate observation in congenital scoliosis. Difficulty with
radiographic measurement in congenital scoliosis has been well
documented, with a high inter-observer variability noted by Loder et
al. (87). In this frequently quoted study,
intraobserver error for progression in congenital scoliosis was as high
as 13 degrees. Facanha-Filho et al. (86)
documented a much smaller, acceptable inter- and intraobserver error
when care was taken to use the same measurement endpoints and expert
measurers were employed. Observation of change is best done by
reexamination and remeasurement of a range of radiographs begun at
infancy and followed up through the most recent radiograph. Choosing
endpoints for Cobb measurement is arbitrary, so the same endpoints,
whether they be pedicles or vertebral endplates, should be used for
each measurement. Archiving of films is crucial, and it may prove
useful to ask families to retain copies of all the films for the
purposes of repeat measurement. Change in congenital scoliosis may be
rapid in infancy and preadolescence, whereas during middle childhood
years the curve change can be very slow or subtle. Frequency of
radiographic observation should reflect prior behavior of the curve and
growth rate. Observation should include the Cobb angle for all curves,
but also measures of trunk balance. Cobb angle itself may be very
differently interpreted for the same curve. If the most congenitally
distorted section of the curve is measured, it may yield a much higher
Cobb angle than would the overall curve including the congenital
deformity. Other elements of trunk balance, such as deviation of C7
with reference to the sacrum, trunk shift, apical vertebral
displacement, and sagittal balance, should also be measured, and the
results may be more important than the Cobb angle to treatment
decisions. Secondary curves involving noncongenitally involved
vertebrae should also be measured and may be the most accurate
reflection of progression. The secondary curves may act as a marker or
indicator of otherwise unrecognized progression in the congenitally
deformed section of the spine. Films in infancy of necessity need to be
in the supine posture. Upright view x-ray films should be taken
when
practical. The interval of observation with x-rays depends upon growth
rate, curve severity, and prior curve behavior. Growth is most rapid
during the first 2 or 3 years of life, and during this time radiography
as frequent as every 3 or 4 months may be appropriate. If a curve has
remained stable for an interval of observation, it is reasonable to
extend the interval between subsequent observations. The need for early
detection of curve progression must be balanced against the theoretical
risks of repetitive diagnostic radiographs in patients with early-onset
scoliosis (113).
 |
|
Figure 19.13
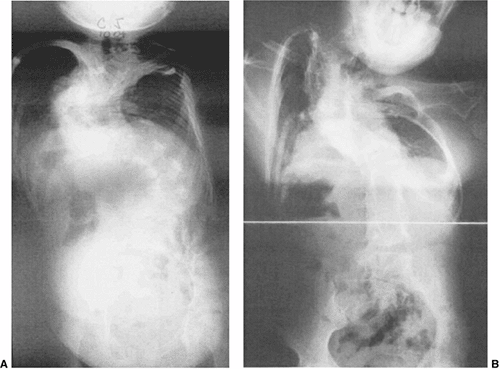
This 2-year-old boy has progressive congenital scoliosis, rib fusions, progressive thoracic deformity, and thoracic insufficiency syndrome. Tracheotomy was needed after birth. Ventilator dependence persists. A: Preoperative posteroanterior view. Note the disparity in space available for the left lung as compared to the right lung. B: Preoperative computed tomography (CT) scan with three-dimensional reformatting shows the block vertebrae and fused ribs. C: Postoperative posteroanterior view after two expansion thoracostomies through fused ribs, and insertion of one rib-to-rib and one rib-to-spine vertical expandable prosthetic titanium rib (VEPTR) device (Titanium Ribs). Space available for the previously constricted left lung is now increased, and shortly after the surgery ventilator requirements diminished. D: Postoperative lateral view after expansion thoracostomy and placement of two VEPTR devices. E: Two years after the initial procedure. VEPTR devices were lengthened every 6 to 9 months. Ventilator dependence diminished postoperatively, and 2 years postoperatively he is ventilator free. Space available for the left lung continues to be nearly that of the right. The thoracic spine continues to lengthen. The central portion of the devices will need to be substituted with longer ones after several more lengthenings. Fusion will be necessary near maturity. |
 |
|
Figure 19.14 The natural history of congenital scoliosis can include severe respiratory insufficiency, either with or without treatment. A:
A patient 10 years of age with severe congenital scoliosis. Misguided, ineffective orthotic treatment of congenital scoliosis while waiting for “maturity” for surgical intervention led to this severe deformity. The vital capacity of the lung is only 20% of normal and was not helped by subsequent surgical intervention. Earlier surgical intervention would have been preferable. B: Another patient at maturity after several spinal fusions for severe congenital scoliosis. Although there are reasonable overall spinal contours, there is also severe distortion of the thorax, which occurred early in the progression of deformity. Lung fields are small, and there is severe three-dimensional rotational deformity of the thorax, which inhibits thoracic movement on respiration. This patient’s restrictive lung disease necessitates full-time oxygen supplementation. |
treatment of congenital spine deformities, there being little evidence
of its efficacy. At times treatment of the noncongenital part of the
spinal deformity with spinal orthoses can be effective. If there is
progression of a deformity within the noncongenital part of the spine,
however, the possibility of spinal cord tethering should be
investigated with MRI; likewise, conditions such as thickened filum
terminale, syringomyelia, or intradural lipoma may be responsible for
curve progression, rather than the actual congenital deformity itself.
There is minimal documentation, and that only in limited circumstances,
about the efficacy of bracing in congenital spinal deformity. There is
no evidence for the efficacy of exercises or chiropractic in slowing
the progression of congenital spine deformities. James noted that the
curve progressed in more than 50% of cases of infantile scoliosis
treated with the advocated casts and Milwaukee braces (114).
Winter et al., however, reported that the Milwaukee brace was useful
for compensatory curves above and below a congenital curve, for long
flexible curves containing congenital elements, and for maintaining
trunk and spine balance following localized fusion during growth (115).
The potential negative long-term effect of casting and thoracic-lumbar
sacral orthosis (TLSO) treatment on the shape of the chest has caused
concern but is not well documented. If long-term bracing is utilized in
the child with
congenital
scoliosis, it should be nonrestrictive to the chest, as is the
Milwaukee brace technique, which carefully applies pressure in
localized areas rather than globally over the chest wall. One should be
sure that the nonsurgical treatment is providing some meaningful effect
rather than just taking up time and permitting development of a more
severe deformity (Fig. 19.14A).
congenital spinal deformity. Because the deformity associated with
congenital scoliosis is difficult to correct, early treatment is
preferable to late treatment of a deformity that has become severe.
Allowing the progression of a deformity will inevitably result in
greater risks and a more difficult final surgical procedure (Fig. 19.15).
assessment of intraspinal anomalies. The MRI should be used not just to
seek the presence or absence of congenital anomalies but to determine
whether a significant spinal stenosis or disc protrusion exists.
Because segment instability and degeneration with secondary stenosis
with hypertrophy of discs and ligamentum flavum may be present adjacent
to congenital cervical, thoracolumbar, or lumbar scoliosis, there may
be a relative stenosis at either end of a block of congenital
deformity. Dynamic flexion/extension radiographs may be helpful.
Awareness of a relative spinal stenosis preoperatively may be crucial
for planning operative fixation devices or osteotomies. Surgical
planning may include CT assessment using two- or three-dimensional
image reconstruction where possible (Figs. 19.2B, 19.3D, 19.13B, and 19.15D). Newton et al. and Hedequist and Emans (82,83)
have demonstrated the efficacy of three-dimensional CT scan in surgical
planning. Although the congenital anomaly may be readily visible on
preoperative plain x-ray film, the posterior anatomical anomalies may
not correspond to the anterior anomalies seen on plain radiographs. If
surgical correction is planned, and particularly if instrumentation is
planned, it is helpful to have adequate three-dimensional imaging.
Initial preoperative screening tests should also include an assessment
of renal anatomy if one has not already been done. If not already
investigated, a cardiac evaluation including cardiac echocardiography
may be helpful in ruling out substantial underlying congenital heart
disease
consideration should be given to correcting the anomaly prior to
surgical correction of the deformity. Simple deformities such as tight
filum terminale (Fig. 19.3C) or a small
intradural lipoma may be effectively dealt with at the time of the
operation, prior to actual deformity correction. An argument can be
made that more major deformities such as diastematomyelia or major
intradural anomalies should be corrected well in advance of a surgical
correction of the deformity itself (17,21,88,93,94)
in order to prevent the possibly additive adverse effects of deformity
surgery and intradural deformity. The group led by Transfeldt reported
that 3 of 38 patients (8%) experienced worsened neurologic symptoms
when scoliosis fusion surgery was done without previous syrinx
decompression (94). Detethering well in advance
allows one to distinguish neurologic deficits caused by the detethering
from those caused by the surgical procedure itself. Disadvantages of
detethering as a separate procedure include the need for a repeated
surgery on the same area and resultant increased difficulty of the
second procedure because of operative scarring.
deformity is complex. In trying to determine guidelines for how to deal
surgically with a progressive congenital spinal deformity, several
factors may help define an appropriate treatment. It is helpful to
assess how severe the deformity is. If the deformity itself is minor
but progressive, a simple in situ fusion
may suffice. If the deformity is severe and causes secondary distortion
of the remainder of the spine, then deformity correction, with its
increased risk, may be justified. Whether the deformity, and hence the
corrective procedure, involves a long or short section of spine can
influence the choice of treatment. Is there impingement by the
deformity on the intraspinal contents? Has the deformity primarily or
secondarily affected respiratory function (thoracic insufficiency
syndrome), and will the proposed treatment create or worsen thoracic
insufficiency? The decision to simply arrest rather than correct
progressive deformity assumes that the existing deformity is
acceptable. The acceptability of an existing deformity is relative. If
correction is very dangerous, then a more severe deformity can be
accepted. If correction is relatively safe, correction with the goal of
improving the deformity may be justified. Therefore, knowledge of all
techniques and an understanding of how much risk is involved are
necessary. Another factor to consider is the effect of the operation on
overall spinal growth (116). If a large segment of the spine is fused in a small child, the overall height of the child may be adversely affected (116)
and thoracic insufficiency syndrome rendered more likely. The
consequences of any fusion on the stability of adjacent segments must
also be considered. Arthrodesis for progressive deformity will make
more of the spine stiff, possibly creating or worsening functional
stiffness and degeneration or instability of adjacent segments. This
dichotomy between extending fusion and creating more junctional stress
is frequently encountered in Klippel-Feil deformity and
cervico-thoracic
deformity.
Extending the fusion to include more vertebrae may help arrest
progressive deformity or instability, but it also concentrates stresses
on a more localized section of spine. Finally, the dangers inherent in
the operation must be examined. The risks of neurologic loss and
intraoperative bleeding are increased by the need for a large degree of
correction or lengthening of the spinal canal, the presence of spinal
stenosis or neurologic deficits, older age of the patient, the need for
circumferential procedures, and other factors (92).
 |
|
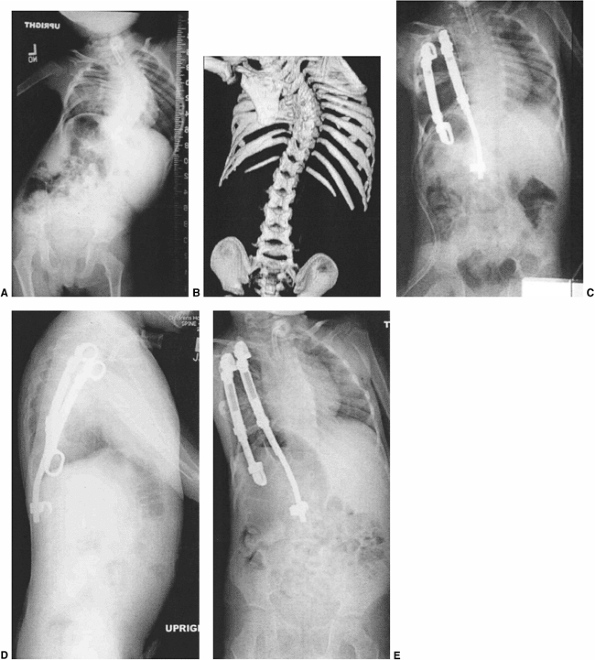
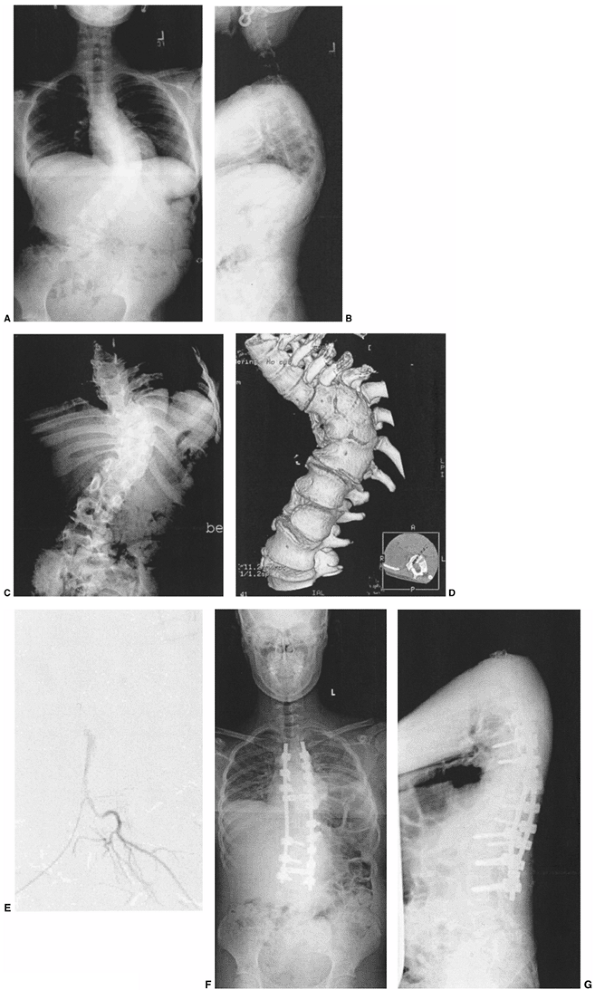
Figure 19.15
A 12-year-old patient with progressive congenital scoliosis treated with anterior and posterior osteotomy and fusion. Drastic progression of the curve occurred because of a mistaken decision to place the patient under observation and delay surgery until “maturity.” A: Preoperative posteroanterior upright view shows a rigid mid-thoracic congenital scoliosis. B: Preoperative lateral view shows substantial congenital kyphosis. Congenital kyphosis is thought to be strongly associated with postoperative neurologic deficit. C: Preoperative bending view shows rigid nature of curve. D: Three-dimensional computed tomography (CT) scan helps evaluate rigid thoracolumbar anatomy. E: Preoperative angiography of the spinal cord was performed because of the need for extensive anterior vertebrectomies. Angiography revealed major segmental feeding vessels at T8 and L3 (L3 vessel shown). Wedge resections and vertebrectomy were performed well away from L3 and T8 segmental vessels. Anterior and posterior procedures were staged. F: Postoperative posteroanterior radiograph with restoration of balance, and preservation of some low lumbar motion segments. G: Postoperative lateral radiograph with reduction in kyphosis. Resected wedges were larger posteriorly in order to improve kyphosis. The instrumentation exaggerated the shortening. An easier, less risky procedure could have been performed years earlier at the first presentation of progression. |
established technique and has been documented in groups led by Winter,
Lonstein, Lindseth, Dubousset, and Thompson (117,118,119) who report a variable epiphyseodesis effect (120,121,122).
Hemiepiphyseodesis can be done posteriorly, or both anteriorly and
posteriorly. When kyphosis is present hemiepiphyseodesis should be done
posteriorly, and when lordosis is present, anteriorly and posteriorly.
Hemiepiphyseodesis can be done as an “eggshell” transpedicular
procedure (119,123).
The anterior portion of an anterior and posterior hemiepiphyseodesis
can also be done thoracoscopically. Immobilization and passive
correction associated with the epiphyseodesis is a critical part of the
procedure, and is best done with a cast. All surgeons document that a
substantial portion of the correction is achieved at the time of the
initial casting and correction (117,118,119,123,124).
The optimal patient for hemiepiphyseodesis is 5 years or less in age,
with a curve that is moderate (50 degrees or less). Compared with a
localized wedge resection for hemivertebra, hemiepiphyseodesis involves
more of the spine, and to some extent builds in an existing deformity.
Further correction of the curves is slow after the initial cast
correction, and some surgeons report the unpredictability of results
following hemiepiphyseodesis (125). The major advantage of epiphyseodesis is that it is very safe and is unlikely to cause new neurologic problems. Posterior in situ fusion (in effect, posterior hemiepiphyseodesis) works well for early moderate kyphosis (Fig. 19.7).
fusion can be performed anteriorly, posteriorly, or both as dictated by
the direction of the deformity and extent of growth remaining. In a
lordotic deformity an anterior fusion is mandatory. In a
kyphotic
deformity posterior fusion alone may help correct the kyphosis by
acting as a tether while continued anterior growth produces some
correction of the kyphosis. In situ
fusion, as classically described, is performed without instrumentation;
however, the availability of modern down-sized instrumentation makes
their use feasible (128), probably diminishing the incidence of pseudarthrosis and length of time the patient must spend in a cast (129).
Correction of congenital vertebral deformity can be much more difficult
than correction of idiopathic deformity. Any correction of a congenital
vertebral anomaly carries with it some serious risk of neurologic
injury (92,128). Where
possible, early arrest of progressive deformity is the preferable
option. The underlying congenital vertebral deformities are unyielding
and, if correction is desired, much more than simple instrumentation
may be required. Bending, traction, or supine view x-ray films (that
remove the effect of gravity) may indicate the stiffness of the
congenital vertebral anomaly. Correction beyond that suggested by
bending position radiographs can be achieved by several means. Releases
through discs and facet joints, as with any severe idiopathic
deformity, will permit more correction only if the congenital deformity
is relatively normally segmented. Osteotomy of anterior vertebral bars
or posterior vertebral fusions may be necessary over areas of block
vertebrae or prior fusions (Figs. 19.15 and 19.17).
A decision as to whether deformity correction is worth the additional
risk must take into account the effects of correction on the remaining
mobile parts of the spine. Other risks associated with osteotomy or
releases include excessive bleeding and direct cord injury. Corpectomy,
vertebrectomy, or vertebral column resection may be necessary for
achieving any improvement of the most deformed sections of the spine (130,131).
Traction alone after release or osteotomy remains an option for slow
correction of deformity. Caution should be exercised in using traction
after vertebrectomy, osteotomy, or extensive release, as the segmental
instability of the spine created by the vertebrectomy or osteotomy may
be severe enough to make traction dangerous. In the case of a
preexisting neurologic deficit, traction (halo-gravity, halo-pelvic, or
halo-femoral) (132,133,134) may be either helpful or disastrous, depending upon the spinal anatomy and cause of the paralysis.
Particular care should be exercised if the deformity involves kyphosis,
because elongation of the spine with traction may draw the compromised
spinal cord more tightly against the kyphotic deformity before the
deformity itself improves (81).
 |
|
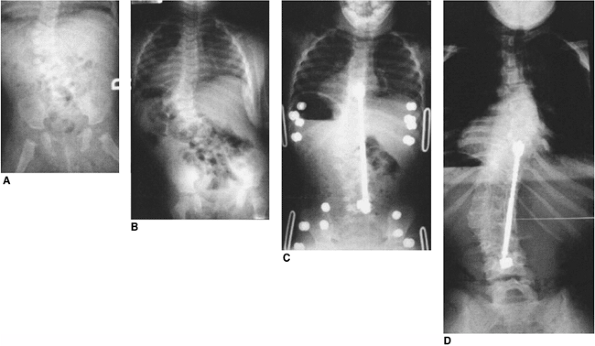
Figure 19.16 Successful early posterior fusion at 21 months of age in a patient with a slowly progressing curve since infancy. A: Infancy. B: Twenty-one months of age, showing progression. Myelogram was negative for tethering lesions. C: Thirty months of age after fusion and Harrington rod. D:
Twelve years of age. At maturity the patient is active, asymptomatic, and has normal pulmonary function. Early fusion for congenital scoliosis can be well tolerated. Excision of two hemivertebra might also have been successful. |
 |
|
Figure 19.17 This girl with progressive thoracic congenital scoliosis underwent thoracic in situ,
posterior-only fusion at 3 years of age with cast immobilization. Steady continued progression and increasing thoracic lordosis occurred postoperatively. Anterior and posterior fusion might have prevented the progression. A: At 11 years of age, after 8 years of progression, she declined further surgery. B: At 14 years of age before anterior and posterior osteotomies, further trunk imbalance was caused by fixed thoracic deformity. C: After anterior and posterior osteotomies at 15 years of age, balance has been restored. If the initial procedure had included an anterior fusion and more correction, the later procedure and osteotomies might not have been needed. |
The ideal indication for hemivertebra excision is an isolated, fully
segmented hemivertebra, at the thoracolumbar junction or below, that
has produced a single deformity (Fig. 19.18).
The rationale for early hemivertebra excision includes correction of
the deformity and presumed prevention of subsequent secondary deformity
in the remaining relatively normal spine. Excision of anterior and
posterior hemivertebra elements or “wedge resection” can be performed
through staged or simultaneous anterior and posterior approaches or
through a posterior approach alone. The choice of approach should
depend upon the experience of the surgeon and the location and
direction of the deformity. Anterior hemivertebrectomy followed by a
separate posterior hemivertebrectomy and posterior instrumentation is
the standard practice. The anterior and posterior incisions can be
performed at the same time if desired, although this makes the
posterior instrumentation more awkward to fit. Positioning of the
patient in a manner to permit this procedure has been described (143).
approaches includes the ability to readjust the amount of resection
either anteriorly or posteriorly in order to ensure satisfactory
correction. It is also feasible to perform a single posterior or
posterolateral approach for hemivertebra excision. A posterior-only
approach is easier in kyphotic than in lordotic deformities. When
performed on the thoracic spine, the posterior approach may be combined
with costo-transversectomy for added exposure. Difficulty in
controlling vertebral body bleeding may be encountered in any
hemivertebra excision but is probably greatest in a purely posterior
approach.
planned so as to include enough of the convex hemivertebra and adjacent
cartilage and disc and to extend far enough toward the concavity so
that complete correction can be achieved. Excising the hemivertebra
just to the midline where the bony hemivertebra may end will usually be
insufficient for substantial curve correction. A wedge resection
including the hemivertebra, hinged on the concave edge of the curve, is
generally necessary. Without extensive correction of the deformity
associated with the hemivertebra, the
major
advantage of early hemivertebra excision is not achieved.
Contraindications to hemivertebrectomy probably include the inability
to maintain the correction with a cast or instrumentation; the presence
of rigid curves above or below the excision, which will cause spine
imbalance; a major intraspinal anomaly in the area; and, probably,
location of the hemivertebra in the cervical spine, where the vertebral
artery complicates excision. Early hemivertebra excision is advocated
because, if allowed to progress, a deformity associated with the
hemivertebra may produce more structural deformity over a longer
section of the spine, eventually requiring longer fusion and more
growth arrest. By doing a hemivertebra excision early, one may restrict
the growth arrest to a short section of spine. The timing of
hemivertebra excision depends upon several factors. The longer the
child remains in an upright position with a severe deformity from a
hemivertebra, the more likely the secondary curves are to become
structural. On the other hand, hemivertebra excision in the very small
child, younger than 1 year, is made more difficult by the need for very
small instrumentation.
 |
|
Figure 19.18
Hemivertebra excision or wedge resection can correct substantial localized deformity, while not affecting the growth of the remainder of the spine. A: A 3-year-old with an isolated lumbar hemivertebra and a slowly progressing deformity. MRI is negative for tethering lesions. B: After simultaneous anterior and posterior excision and wedge resection of the hemivertebra and fusion of adjacent vertebrae, the patient was placed in cast immobilization for 2 months. The global spinal deformity remains corrected, although the operation was confined to a localized region. |
but it is probably safe to perform fusion or instrumentation before
that age without fear of creating spinal stenosis, as the canal
diameter at birth is already approximately two-thirds of the adult
dimension (148). Kim et al. (129)
found no instances of permanent neurologic deficit in wedge resections
done before the age of 5 years; in general, the earlier the procedure
was performed, the less likely neurologic injury was (92,129). Other series of hemivertebra excisions have also indicated that early excision makes neurologic injury less likely (141,144).
Injury may come from direct contusion of cord or roots or may come from
stretching of the concave roots as a new position is achieved. Some
hemivertebrae
are nonprogressive (Figs. 19.4 and 19.12)
and do not produce a deformity. Indications for hemivertebra excision
must therefore include either progressive deformity or an existing
deformity that has a considerable effect on the overall configuration
of the spine above and below. In the absence of specific
contraindications, lumbosacral, lumbar, or thoracolumbar locations are
optimal for hemivertebra excision, whereas deformity correction with
hemivertebra excision in the mid- and upper thoracic spine is limited
by the inflexible thorax, and probably entails greater neurologic risk
because of the presence of thoracic spinal cord at the level of the
operation. For an isolated hemivertebra in the midthoracic spine, in situ fusion or hemiepiphyseodesis may be a better choice than excision.
exists and contributes to congenital spinal deformity, osteotomy may be
required for achieving correction of deformity and balance (Figs. 19.15 and 19.17).
Generally both anterior and posterior osteotomies and/or release will
be needed, and can be staged or done sequentially. Vertebral column
resection (131) may be done in a similar
fashion. Anterior procedures can be osteotomies at each vertebral level
in the area of deformity or can be restricted to a limited number of
osteotomies if localized correction is planned. Posterior osteotomies
also can be done at multiple levels or locally. Pedicle subtraction
osteotomy has gained currency for treatment of adult spinal deformity (149) and has similar application in children’s congenital deformities. Transpedicular eggshell procedures may be carried out (150)
for previously fused pediatric congenital spine deformities. With any
vertebral osteotomy, the risk of neurologic injury is considerable, and
substantial bleeding can be anticipated. Osteotomy technique is well
described in the literature, but each procedure should include careful
localization and preoperative planning. Three-dimensional CT scans (Fig. 19.15D)
will help rationalize the decisions regarding an osteotomy and help the
surgeon localize the site of osteotomy. Inner edges of osteotomies
should be tapered and undercut so that there is no impingement on the
spinal canal at the time of closure. Correction after osteotomy should
emphasize avoidance of spinal column elongation (Fig. 19.15 F,G).
of a deformity. Traditional immobilization has included cast fixation
either with or without instrumentation. Cast immobilization has obvious
disadvantages but is entirely feasible in children. Maintenance of
correction with cast immobilization alone requires attention to details
relating to the cast. Many of the instrumentations used in small
children are not strong enough to permit immediate mobilization, and
therefore backup immobilization of instrumentation may be needed for
several months after the operation. Instrumentations used in fusion
with correction can be of various kinds. Preoperative evaluation of the
congenital vertebral anomaly, usually by CT scan, should include an
assessment of available anatomic anchor points for fixation (82,83).
Pedicles may be very distorted or absent and laminae may be distorted,
oblique, absent, bifid, or very thin. Canal diameters may be
restricted. Individualization of fixation, balancing risk against
stability, is needed. The increasing availability of downsized
instrumentation, either intended specifically for smaller children or
adapted from adult cervical applications, has made instrumentation of
deformity in smaller children more feasible and permitted a reduction
in the use of cast and brace immobilization postoperatively (128).
However, the posterior and especially anterior osseous elements in
small children are much less dense than the equivalent structures in
adults and do not permit the application of corrective forces via
anchor points as they would in adults. Where possible, correction and
instrumentation of congenital scoliosis should not involve elongation
of the spinal column, because the spinal cord in congenital deformity
is likely to tolerate elongation poorly.
Expansion Thoracostomy and Vertical Expandable Prosthetic Titanium Rib
Procedure
deformity have included early fusion. The rationale behind this
approach has been to avoid the development of more severe deformities
that would necessitate more extensive and dangerous procedures later in
childhood or adulthood. The use of early fusion to prevent further
progression of deformity assumes that the loss of growth associated
with early fusion is well tolerated. In general, this is true, and the
principle of early arrest of progressive deformity remains the mainstay
of surgical treatment. Some patients, however, particularly those with
an already shortened spine whose deformity would necessitate fusion of
a long portion of spine at an early age, and especially those with
associated chest wall anomalies, may be better treated by techniques
that permit some continued growth of the abnormal spinal elements while
still controlling the deformity. Expandable posterior rods and
instrumentation without fusion have been employed in children with
infantile and juvenile idiopathic deformities, but also in small series
of those with congenital deformity (132,151,152) (Fig. 19.19).
A major disadvantage of this treatment is the need for repetitive
lengthening procedures, but it may be an appropriate temporary choice
in the very young child with a deformity involving a large portion of
the spine.
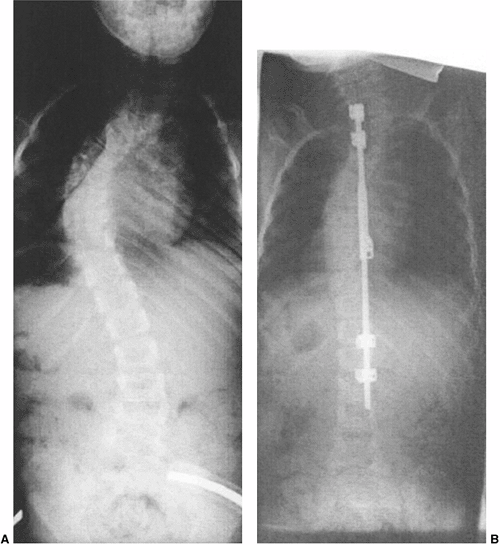 |
|
Figure 19.19
A 6-year-old patient with progressive congenital cervicothoracic scoliosis and noncongenital structural thoracic scoliosis. Both conditions are progressive. MRI is negative for tethering lesion. A: Preoperative posteroanterior view. B: Immediate postoperative posteroanterior view. Treatment consisted of in situ fusion of the cervicothoracic congenital scoliosis and instrumentation without fusion of the thoracic curve, and later included periodic rod lengthening. Fusion is planned later in growth. Dual extensible rods might be preferred now. [Currently, extensible rods are not approved by the U.S. Food and Drug Administration (FDA) for use without complete fusion.] |
Combined spine and thoracic deformity caused by congenital vertebral
deformities associated with multiple rib fusions is particularly
amenable to the use of this technique during growth, and is the
indication best documented by Campbell et al. (112).
By performing one or more expansion thoracostomies through the
constricted chest wall, either between ribs or through the rib fusion
mass, expansion of the chest and secondary control of the spinal
deformity without fusion is possible. The expandable devices attach rib
to rib, rib to spine, or rib to pelvis and are periodically expanded to
permit growth. Eventual fusion near the end of growth is anticipated,
but by expanding and controlling the shape of the chest while
permitting longitudinal growth of the spine, the chance of thoracic
insufficiency syndrome is minimized. Growth of the spine and growth of
concave bony bars has been demonstrated in association with this
technique (100). It was Campbell who brought
attention to thoracic insufficiency syndrome, pointing out that growth
of the spine and chest are intricately intertwined. Primary chest wall
deformity, such as massive areas of fused ribs, associated with
congenital spinal deformity acts as a powerful deforming force on the
spine. Treatment of the spine alone will often be defeated by the
tethering effect of rib fusions or rib abnormalities. In young
children, a primary
chest
wall deformity usually creates a secondary spine deformity, and
conversely, severe progressive spinal deformity usually creates a chest
deformity. While considering treatment of any early-onset spinal
deformity, one should keep in mind the need to prevent irreversible
chest wall deformity and thoracic insufficiency syndrome (104).
observation and surgical treatment of congenital scoliosis requires
that orthopedists acknowledge the impossibility of accurately
predicting progressive deformity in many instances of congenital
scoliosis, while at the same time considering potential long-term
problems, including the creation of thoracic insufficiency syndrome.
Traditional attitudes toward congenital scoliosis are outlined in this
chapter. Early fusion or early epiphyseodesis of progressive deformity
is strongly advocated. Prophylactic fusion for deformities that can be
accurately predicted to be progressive are similarly advocated. In my
experience, however, those deformities that are predicted to be
progressive do not necessarily prove to be so if observation is
continued, and in some instances early prophylactic fusion or
hemiepiphyseodesis are not successful in halting the progression of
deformity (Fig. 19.10A) and may in fact
contribute to long-term difficulties such as short stature, stiffness,
and thoracic insufficiency. This attitude is probably colored by a
referral practice, which includes many failures. Although it has been
the author’s practice to prophylactically fuse congenital kyphosis, a
more contemporary attitude toward congenital scoliosis has been to
conduct close observation and employ prophylactic procedures only for
congenital kyphosis or for hemivertebrae, which cause considerable
trunk imbalance. If multiple areas of the spine are involved in
progressive deformity, or if the procedure necessary to halt the
deformity would require a long segment of fusion in a small child, I
have chosen procedures that offer some preservation of spine length,
such as growing rods or expansion thoracostomy with titanium rib
insertion, instead of fusion.
practice. In my experience, many congenital anomalies predicted to be
progressive are not progressive or are not progressive until the
preadolescent growth spurt. Although most congenital kyphosis is indeed
progressive, and early posterior in situ
fusion is usually the best choice, some instances of nonprogressive
congenital kyphosis have been observed. Three-dimensional CT scan
visualization of posterior elements may explain why some anomalies are
not progressive; fully segmented hemivertebrae seen on plain x-ray film
may include nonsegmented posterior elements on the convex side of the
curve. Therefore the underlying posterior congenital anomaly acts as
its own “tether” on the convex side, preventing progression. This
phenomenon may also be the reason why occasionally congenital kyphosis
does not progress, with a spontaneous fusion of posterior elements
acting as a tether opposite the congenital kyphosis.
effect, at least in my hands, is much less effective than is suggested
by the literature, and that the hemiepiphyseodesis effect of further
correction after cast correction is infrequent in spite of careful cast
correction and immobilization. Hemiepiphyseodesis was developed as a
technique in an era when instrumentation was not available for the
congenital spine deformities in very young children and when
intraspinal imaging of anomalies was less advanced. With the
availability of small pediatric instrumentation (128), it now seems more effective to achieve correction over an area of deformity by instrumentation rather than by casting alone.
transpedi-cular hemiepiphyseodesis have, in my experience, been more
difficult than anticipated and less satisfactory. In very small
children, nearly complete obliteration of the pedicle is necessary in
order to achieve enough access to the endplates to assure growth
arrest. Destruction of the pedicles and further disruption of posterior
elements in some instances seems counterproductive to the goal of
achieving a solid fusion.
for surgical correction or the execution of epiphyseodesis or fusion.
If major rib fusions exist in the concavity of a curvature, it has been
the author’s observation that the rib fusions will usually continue to
deform the affected spine in spite of in situ
fusion, instrumented fusion, or osteotomy. With the present
availability of expansion thoracostomy, rib fusions should be taken
into consideration in surgical planning.
in growth, and severe instances of congenital scoliosis are rapidly
progressive during infancy and early childhood. As a result, there is a
focus on early progression of congenital scoliosis, and a false sense
of security often results if the deformity has not progressed by
mid-childhood. Families and caregivers alike are falsely assured that
if the deformity is nonprogressive during early childhood, it will
remain nonprogressive through the remainder of growth. Late,
considerable progression during the preadolescent growth spurt is often
unanticipated and can be devastating. Continued observational vigilance
is needed all through growth.
component has been highly successful (Fig. 19.7). Supplementation of posterior in situ
fusion with instrumentation lessens the need for cast immobilization.
The principle of early creation of a posterior tether opposite an
anterior congenital kyphosis with anticipated continued anterior growth
and later kyphosis correction is valid and effective. Because the
incidence of perioperative paralysis associated with congenital
kyphosis is high in late reconstructive procedures, particularly
circumferential procedures, early in situ posterior fusion seems warranted and is easily accomplished.
resection for localized deformity has been highly successful. If the
deformity involves a short section of spine, and particularly if this
short deformed section causes a compensatory deformity above or below
in a relatively normally structured region of spine, hemivertebra
excision or wedge resection should be strongly considered. Usually
fusion can be restricted to the segment just above and below the
hemivertebra, not involving any normal spine above or below with fusion
or instrumentation. Preoperative planning requires the demonstration by
CT scan of posterior elements suitable for fixation and corresponding
to what is seen in the anterior elements on plain x-ray film. Complete
correction of the deformity is necessary in this procedure, and enough
of the hemivertebra and associated disc must be excised to provide
correction. The excision must traverse to the concave corner of the
wedge in order to provide mobility for correction. Simply excising the
bony hemivertebra results in undercorrection and defeats the goal of
complete correction involving a short segment of spine. The use of
growing rod constructs in congenital scoliosis is poorly documented.
instrumented fusion of a short congenital segment of spine and growing
rod construct treatment of the longer, normally segmented section of
spine has worked well for large deformities involving normally
segmented spinal curves adjacent to congenital curves (Fig. 19.19).
Likewise, very long, relatively mobile sections of congenital spinal
anomaly in young children seem to be amenable to growing rod treatment,
provided the curve is relatively flexible and sufficient anchor points
are present. If a chest wall anomaly is present or there is severe
chest distortion in a young child, and thoracic insufficiency syndrome
is a consideration, then expansion thoracostomy with VEPTR treatment is
my preferred choice (Figs. 19.10 and 19.13).
If there is no considerable thoracic deformity and if the problem is
restricted to the spine, it is my preference to use a growing rod
construct. However, if the chest deformity is advanced, then expansion
thoracostomy, as described by Campbell et al., may be the preferable
way to control spinal deformity while still permitting growth and
improving chest volume and shape. The clearest indication for expansion
thoracostomy and VEPTR, in my opinion, is the combination of congenital
vertebral anomalies over a substantial section of spine and concave
fused ribs. If a spine operation alone is performed, the fused ribs
will continue to tether the spine, causing the deformity to worsen (Fig. 19.10A).
Combined treatment of concave fused ribs and extensive vertebral
anomalies by expansion thoracostomy and an expandable rib-to-spine and
rib-to-rib prosthesis has been highly effective in the experience of
Campbell et al. (112) and also in my practice.
Presumably most patients treated with expansion thoracostomy will need
a definitive spinal fusion at the end of growth.
such as growing rods and expansion thoracostomy makes the choice
between traditional fusion-based treatment and growth-sparing
procedures more complex. Several factors can be considered in choosing
a treatment: the age of the patient, and therefore the amount of growth
remaining; the length of the spine involved; and the availability of a
satisfactory, traditional, early fusion, hemiepiphyseodesis, or
hemivertebra excision procedure for that area of spine. Essentially, if
the deformity is easily treatable with a localized procedure such as
hemivertebra excision or wedge excision involving a short section of
the spine, then this sort of one-time, traditional, fusion-based
procedure is preferable to repetitive surgeries associated with growing
rod techniques or expansion thoracostomy. However, if the child is very
young and a long section of the spine is involved, early fusion-based
techniques will result in growth arrest over a longer, more significant
section of spine. In such an instance, where a lot of growth remains,
the choice of growing rods or expansion thoracostomy in the VEPTR
procedure may be preferable even though it involves multiple procedures
and multiple hospitalizations. Finally, if major rib fusions are
present, the child is very young, and a long section of thoracic spine
is involved in the curvature, then expansion thoracostomy with VEPTR
procedure is preferable.
congenital scoliosis it is of paramount importance to have specific
goals in mind. Treatment should maximize the length of the spine at the
end of growth and should minimize thoracic deformity; it should leave
as many segments of the spine mobile as is possible, and should place
the patient at as little risk as possible. Hospitalizations and
repetitive procedures should be kept to a minimum. For example, a
procedure such as hemivertebra excision for an isolated thoracolumbar
hemivertebra can be expected to diminish total spine height very
little, and calls for a single, brief hospitalization. A more complex
procedure such as expansion thoracostomy and VEPTR insertion will
require multiple procedures and likely be associated with some surgical
problems or complications, but will maximize the available spine length
at the end of growth. Early in situ fusion
for congenital kyphosis, although it may possibly result in less trunk
height than an anterior release and strut grafting, is a less complex
procedure, exposes the patient to a minimum risk of paralysis, and is
therefore preferable.
deformity both with and without treatment. It may be difficult to
separate the long-term complications of treatment from the natural
history of the underlying congenital spinal deformity itself. Failure
to recognize a progressive deformity or the potential significance of
congenital spinal deformity and to act in a timely fashion may be as
common a complication as those related to surgical treatment. Allowing
extensive progression of congenital spine deformity (Figs. 19.14 and 19.15A) may necessitate more complex, complication-prone treatment. As an example, early in situ posterior fusion for moderate congenital kyphosis (103)
is safe and reliably produces subsequent spontaneous correction with
further growth, whereas permitting extensive deformity progression may
necessitate later complex surgical procedures with a higher risk of
neurologic complications. Recognized complications associated with
treatment of congenital spine deformity include recurrent or
progressive deformity, neurologic injury, further loss of spinal
height, thoracic insufficiency syndrome, and deterioration of adjacent
segments.
or by “adding on” to the curvature above or below the prior surgical
treatment. Recurrent deformity can be addressed by extension of fusion
and instrumentation and osteotomy of prior fusions.
treatment of congenital spine deformity, notably congenital kyphotic
deformity (9,92,159,160,161). Early morbidity reports from the Scoliosis Research Society (160) and other reports (9,92)
note the considerable risk of neurologic complications associated with
surgery of congenital spine deformity, particularly congenital
kyphosis. Neurologic problems are associated with progression of the
deformity (159) as well as with surgical
treatment. Although type I congenital kyphosis may be associated with
paralysis if untreated, it is uncommon for untreated progressive
congenital scoliosis to cause neurologic loss unless there is a very
severe deformity, associated kyphotic deformity, lateral translocation
or dislocation (162), a spinal cord anomaly, or spinal cord tethering.
congenital spinal deformity explain the relatively high incidence of
neurologic injury associated with treatment. The final common pathway
for most perioperative neurologic complications is probably vascular
insufficiency of the spinal cord. The combination of an inherently
anomalous or insufficient vascular supply to the spinal cord (Fig. 19.15E)
coupled with corrective forces and change in position or shape of the
spinal canal may result in neurologic compromise more commonly in
surgeries for congenital deformities than in surgical correction of
idiopathic deformities. Apel documented changes in somatosensory evoked
potential (SSEP) monitoring with anterior segmental vessel ligation in
congenital scoliosis (163). This has also been
the author’s experience in several instances. The vascular supply of
the spinal cord associated with congenital scoliosis can be anomalous,
and wherever possible surgical procedures should consider the possible
effect of interruption of spinal cord blood supply during anterior
surgical procedures. Distraction of the spinal cord in an animal model
has been shown to be associated with diminished vascular flow, which
precedes the appearance of neurologic dysfunction (164,165).
injuries associated with correction of congenital spine deformity.
Wherever possible, acute surgical correction should shorten the spinal
column rather than distract the spinal cord. There may be value in
staging corrections or producing the correction slowly in traction in
order to allow monitoring of neurologic function when the patient is
conscious (134). Neurologic injury may also
occur from direct mechanical impingement on the spinal canal.
Translation or angulation of the canal associated with spinal column
correction may result in a new spinal stenosis or angular impingement
on the spinal cord.
preoperative imaging assessment of canal anatomy and dimension, and
adequate control, observation, and decompression of the spinal canal
during corrective maneuvers. Undercutting of lamina and procedures that
shorten the area of deformity may make impingement less likely.
Neurologic injuries associated with surgical treatment of congenital
spinal deformity may occur without obvious cause (preoperative stenosis
or spinal cord anomaly or tethering), and may be delayed (81).
be cause for thorough preoperative evaluation, and probably signifies
an increased risk of postoperative neurologic deficit. Early treatment
is preferred where possible, while the deformity is less severe. Also,
during early childhood the spinal cord appears more resistant to
injury, and there is a lower incidence of neurologic injury (129).
However, surgeons should realize that even with the most cautious
approach, there is still a risk associated with manipulation of the
congenitally anomalous vertebral column and spinal cord, and that the
incidence of neurologic injury is still much higher than with
idiopathic deformity. Standard treatment measures for neurologic loss
following surgery for congenital scoliosis
include
maximizing spinal cord perfusion and oxygenation, considering
diminishing correction or removing instrumentation, and evaluation for
direct mechanical compression of the cord by appropriate imaging such
as CT myelogram.
worsening of deficiencies in existing trunk height. Generally the loss
of trunk height is well tolerated (103,166,167)
and is not considerable compared with the deformity that would have
resulted if the condition had been allowed to progress. Goldberg et al.
have brought attention to the shortened spine height associated with
congenital spinal deformity and diminished trunk and spinal dimensions
after spinal arthrodesis (72,107). Campbell et al. have described thoracic insufficiency following spinal arthrodesis for congenital scoliosis (104)
associated with fused ribs. Patients with a preexisting short trunk,
chest wall anomalies, or other causes of restrictive lung disease may
not tolerate the further shortening associated with an extensive spinal
arthrodesis. In this select group, procedures such as instrumentation
without fusion or expansion thoracostomy with VEPTR insertion may be
preferable to an early extensive arthrodesis, acknowledging that
eventual arthrodesis will be needed at a later stage of growth.
Diminished height of the spine, thoracic insufficiency syndrome, and
the effects of early fusion (168) may not become apparent until the adolescent period, when the normal thoracic volume increases greatly.
the transition zone between spinal fusion and the mobile unfused spine
are acknowledged and documented phenomena associated with spinal
fusions in all ages (169,170,171).
A similar phenomenon of increased instability and degeneration of
adjacent segments occurs adjacent to congenital fusions that extend
into the cervical spine in the Klippel-Feil association (27,29,106,172).
Adjacent segment degeneration or transition zone instability can be
seen after fusion for congenital scoliosis; they may manifest as pain
or neurologic deficit associated with spinal cord compression at the
degenerated level caused by hypertrophy of the hypermobile disc,
ligamentum flavum, or associated osteophytes. A consideration of the
possible occurrence of adjacent segment degeneration should be factored
into the choice of surgical options in congenital scoliosis.
B, Tracy MR, Dormans JP, et al. Congenital scoliosis and vertebral
malformations: characterization of segmental defects for genetic
analysis. J Pediatr Orthop 2004;24(6):674–682.
RB, Moe JH, Lonstein JE. Posterior spinal arthrodesis for congenital
scoliosis. An analysis of the cases of two hundred and ninety patients,
five to nineteen years old. J Bone Joint Surg Am 1984;66(8):1188–1197.
A, Cordero D, Mejides A, et al. Caudal regression syndrome:
etiopathogenesis, prenatal diagnosis, and perinatal management. Obstet Gynecol Surv 1994;49(7):508–516.
A, Westbom L, Kallen B. Congenital malformations among infants whose
mothers had gestational diabetes or preexisting diabetes. Early Human Development 2001;61(2): 85–95.
PJ Jr, Kuklo TR, Taylor KF, et al. Intraspinal anomalies associated
with isolated congenital hemivertebra: the role of routine magnetic
resonance imaging. J Bone Joint Surg Am 2004;86-A(8):1704–1710.
SD, Westgate J. Solitary crossed renal ectopia associated with
unicornuate uterus, imperforate anus and congenital scoliosis. J Urol 1996;156(1):221.
J, Larson O, Ph DD, et al. Associated malformations in infants with
cleft lip and palate: a prospective, population-based study. Pediatrics 1997;100(2 Pt 1):180–186.
AS, Taylor TK, Smith GH, et al. Congenital abnormalities of the
urogenital tract in association with congenital vertebral
malformations. J Bone Joint Surg Br 2002;84(6):891–895.
ML, Coutts J, al-Roomi L, et al. Uniparental isodisomy for chromosome
16 in a growth-retarded infant with congenital heart disease. Prenat Diagn 1995;15(6):579–584.
de Gauzy J, Accadbled F, Sarramon MF, et al. Prenatal sonographic
diagnosis of the congenital dislocated spine: a case report. Spine 2003;28(2):E41–E44.
RB, Jabs EW, Graham GE, et al. Syndrome of coronal craniosynostosis,
Klippel-Feil anomaly, and Sprengel shoulder with and without Pro250Arg
mutation in the FGFR3 gene. Am J Med Genet 2001;104(2):112–119.
JC, Hailey H, Moore SJ, et al. Long term health and neurodevelopment in
children exposed to antiepileptic drugs before birth. J Med Genet 2002;39(4):251–259.
PD, Whittock N, Duncan J, et al. Novel mutations in DLL3, a
somitogenesis gene encoding a ligand for the Notch signalling pathway,
cause a consistent pattern of abnormal vertebral segmentation in
spondylocostal dysostosis. J Med Genet 2003;40(5):333–339.
NV, Ellard S, Duncan J, et al. Pseudodominant inheritance of
spondylocostal dysostosis type 1 caused by two familial delta-like 3
mutations. Clin Genet 2004;66(1):67–72.
AS, Ramirez N, Arroyo S, et al. Phenotype characterization and natural
history of spondylothoracic dysplasia syndrome: a series of 27 new
cases. Am J Med Genet 2004;128A(2): 120–126.
CP, Lee CC, Pan CW, et al. Partial trisomy 8q and partial monosomy 15q
associated with congenital hydrocephalus, diaphragmatic hernia, urinary
tract anomalies, congenital heart defect and kyphoscoliosis. Prenat Diagn 1998;18(12):1289–1293.
M, Iagaru N, Popescu-Miclosanu S, et al. Congenital hemihypertrophy.
Tendency to association with other abnormalities and/or tumors. Morphol Embryol (Bucur) 1983;29(1):39–45.
R, Littlejohn A, Gormley J. Aetiology and interrelationship of some
common skeletal deformities. (Talipes equinovarus and calcaneovalgus,
metatarsus varus, congenital dislocation of the hip, and infantile
idiopathic scoliosis.) J Med Genet 1982;19(5):321–328.
K, Winbladh B, Kallen B. Major malformations in infants exposed to
antiepileptic drugs in utero, with emphasis on carbamazepine and
valproic acid: a nation-wide, population-based register study. Acta Paediatr 2004;93(2):174–176.
RT, Hernandez MJ, Lerner AL, et al. The induction of congenital spinal
deformities in mice by maternal carbon monoxide exposure. J Pediatr Orthop 2000;20(5):662–666.
DF, Sandor GG, MacLeod PM, et al. Intrinsic defects in the fetal
alcohol syndrome: studies on 76 cases from British Columbia and the
Yukon Territory. Neurobehav Toxicol Teratol 1981;3(2):145–152.
RB, House JH. Congenital cervical scoliosis with unilateral congenital
nerve deficit in the upper extremity. Report of two cases. Spine 1981;6(4):341–346.
K, Weber M, Lorani A, et al. Prognostic significance of the Nasca
classification for the long-term course of congenital scoliosis. Arch Orthop Trauma Surg 2002;122(7):383–389.
RB, Moe JH, Lonstein JE. The surgical treatment of congenital kyphosis.
A review of 94 patients age 5 years or older, with 2 years or more
follow-up in 77 patients. Spine 1985;10(3):224–231.
DJ, Emans JB. The correlation of preoperative three-dimensional
computed tomography reconstructions with operative findings in
congenital scoliosis. Spine 2003;28(22): 2531–2534.
PO, Hahn GW, Fricka KB, et al. Utility of three-dimensional and
multiplanar reformatted computed tomography for evaluation of pediatric
congenital spine abnormalities. Spine 2002;27(8):844–850.
A, Yazici M, Alanay A, et al. The course of sagittal plane abnormality
in the patients with congenital scoliosis managed with convex growth
arrest. Spine 2004;29(5):547–552.
SW, Sarwark JF, Vora A, et al. Evaluating congenital spine deformities
for intraspinal anomalies with magnetic resonance imaging. J Pediatr Orthop 2001;21(4):525–531.
KM, Geley T, Freund MC, et al. US of the spinal cord in newborns:
spectrum of normal findings, variants, congenital anomalies, and
acquired diseases. Radiographics 2000;20(4): 923–938.
E, Alanay A, Akalan N, et al. Risk factors associated with corrective
surgery in congenital scoliosis with tethered cord. Turk J Pediatr 1997;39(3):373–378.
EG, Jaufmann BA, Kaste SC, et al. Successful staged surgical correction
of congenital segmental spinal dysgenesis and complete rotary
subluxation of the thoracolumbar spine in an infant. J Pediatr Surg 1996;31(7):960–964.
RB, Moe JH, Wang JF. Congenital kyphosis. Its natural history and
treatment as observed in a study of one hundred and thirty patients. J Bone Joint Surg Am 1973;55(2):223–256.
RM, Jr., Smith MD, Mayes TC, et al. The characteristics of thoracic
insufficiency syndrome associated with fused ribs and congenital
scoliosis. J Bone Joint Surg Am 2003;85-A(3):399–408.
JS, Davidson JA. Cor pulmonale presenting in a patient with congenital
kyphoscoliosis following intercontinental air travel. Anaesthesia 1999;54(4):361–363.
K, Larsson S, Oden A, et al. Long-term follow-up of patients with
untreated scoliosis. A study of mortality, causes of death, and
symptoms. Spine 1992;17(9):1091–1096.
RM, Jr., Smith MD, Mayes TC, et al. The effect of opening wedge
thoracostomy on thoracic insufficiency syndrome associated with fused
ribs and congenital scoliosis. J Bone Joint Surg Am 2004;86-A(8):1659–1674.
Doody M, Lonstein JE, Stovall M, et al. Breast cancer mortality after
diagnostic radiography: findings from the U.S. Scoliosis Cohort Study. Spine 2000;25(16):2052–2063.
RB. Convex anterior and posterior hemiarthrodesis and
hemiepiphyseodesis in young children with progressive congenital
scoliosis. J Pediatr Orthop 1981;1(4):361–366.
PM, Lindseth RE, DeRosa GP. Progressive congenital scoliosis treatment
using a transpedicular anterior and posterior convex hemiepiphyseodesis
and hemiarthrodesis. A preliminary report. Spine 1994;19(17):1933–1939.
AG, Marks DS, Sayampanathan SR, et al. Long-term results of combined
anterior and posterior convex epiphysiodesis for congenital scoliosis
due to hemivertebrae. Spine 1995;20(12):1380–1385.
J, Dubousset J. Combined anterior and posterior convex epiphysiodesis
for progressive congenital scoliosis in children aged < or = 5 year.
Eur Spine J 1994;3(2):120–125.
AG, MacEwen GD, Bose WJ. Transpedicular convex anterior
hemiepiphysiodesis and posterior arthrodesis for progressive congenital
scoliosis. Spine 1992;17(Suppl 8):S291–S294.
AA, Cil AA, Yazici MM, et al. The efficacy of convex hemiepiphysiodesis
in patients with iatrogenic posterior element deficiency resulting from
diastematomyelia excision. Spine 2003;28(8):799–805.
IT, Duman E, Akalin S, et al. [An evaluation of the types and the
results of surgical treatment for congenital scoliosis]. Acta Orthop Traumatol Turc 2003;37(4):284–298.
DJ, Hall JE, Emans JB, et al. The safety and efficacy of spinal
instrumentation in children with congenital spine deformities. Spine 2004;29(18):2081–2086.
V, Papin P, Marchesi D. Halo femoral traction and sliding rods in the
treatment of a neurologically compromised congenital scoliosis:
technique. Eur Spine J 1999;8(4): 329–331.
EL, Karol LA, Sanders J, et al. Efficacy of perioperative halo-gravity
traction in the treatment of severe scoliosis in children. J Pediatr Orthop 2001;21(4):519–524.
DC, Winter RB, Lonstein JE, et al. Excision of hemivertebrae and wedge
resection in the treatment of congenital scoliosis. J Bone Joint Surg Am 1995;77(2):159–171.
HH, Matsuda HH, Konishi SS, et al. Single-stage excision of
hemivertebrae via the posterior approach alone for congenital spine
deformity: follow-up period longer than ten years. Spine 2002;27(1):110–115.
M, Harms J. Posterior hemivertebra resection with transpedicular
instrumentation: early correction in children aged 1 to 6 years. Spine 2003;28(18):2132–2138.
GJ, Matricali B, van Persijn van Meerten EL. Postnatal development and
structure of the neurocentral junction. Its relevance for spinal
surgery. Spine 1996;21(6):661–666.
JE, Campbell DM, Golden MH, et al. Antenatal factors in the development
of the lumbar vertebral canal: a magnetic resonance imaging study. Spine 2003;28(13):1418–1423.
KH, Lewis SJ, Rinella A, et al. Pedicle subtraction osteotomy for the
treatment of fixed sagittal imbalance. Surgical technique. J Bone Joint Surg Am 2004;86-A (Suppl 1): 44–50.
MR, Graziano GP, Hensinger AR, et al. Transpedicular eggshell
osteotomies for congenital scoliosis using frameless stereotactic
guidance. Spine 2001;26:2289–2296.
A, Schulze Bertelsbeck D, Schmitt O. Five-year follow-up of
intermittent distracting rod correction in congenital scoli-osis. Eur J Pediatr Surg 2002;12(6):416–418.
RM Jr, Smith MD, Hell-Vocke AK. Expansion thoracoplasty: the surgical
technique of opening-wedge thoracostomy. Surgical technique. J Bone Joint Surg Am 2004;86-A (Suppl 1):51–64.
CL, Bridwell KH, Lenke LG, et al. Posterior arthrodesis in the
skeletally immature patient. Assessing the risk for crankshaft: is an
open triradiate cartilage the answer? Spine 1997; 22(12):1343–1351.
KL, Lonstein JE, Denis F, et al. The crankshaft phenomenon after
posterior spinal arthrodesis for congenital scoliosis: a review of 54
patients. Spine 2003;28(3):267–271.
JE, Winter RB, Moe JH, et al. Neurologic deficits secondary to spinal
deformity. A review of the literature and report of 43 cases. Spine 1980;5(4):331–355.
GD, Bunnell WP, Sriram K, et al. Acute neurological complications in
the treatment of scoliosis. A report of the Scoliosis Research Society.
J Bone Joint Surg Am 1975;57(3):404–408.
DM, Marrero G, King J, et al. Avoiding paraplegia during anterior
spinal surgery. The role of somatosensory evoked potential monitoring
with temporary occlusion of segmental spinal arteries. Spine 1991;16(Suppl 8):S365–S370.
Y, Owen JH, Lenke LG, et al. Use of sciatic neurogenic motor evoked
potentials versus spinal potentials to predict early-onset neurologic
deficits when intervention is still possible during overdistraction. Spine 1993;18(9):1134–1139.
M, Owen JH, Bridwell KH, et al. Effects of distraction on physiologic
integrity of the spinal cord, spinal cord blood flow, and clinical
status. Spine 1992;17(10): 1154–1158.
RB, Smith MD, Lonstein JE, et al. Congenital scoliosis due to
unilateral unsegmented bar: posterior spine fusion at age 12 months
with 44-year follow-up. Spine 2004;29(3): E52–E55.
RB, Lonstein JE. Congenital scoliosis with posterior spinal arthrodesis
T2-L3 at age 3 years with 41-year follow-up. A case report. Spine 1999;24(2):194–197.
J, Kassab F, Caubet JF, et al. Earlier and more extensive thoracic
fusion is associated with diminished pulmonary function. Outcome after
spinal fusion of 4 or more thoracic spinal segments before age 5. Paper presentation 101, Scoliosis Research Society Annual Meeting, Buenos Aires, Argentina, Sept. 6–9, 2004.
S, Schlenzka D, Poussa M, et al. Disc degeneration in young patients
with isthmic spondylolisthesis treated operatively or conservatively: a
long-term follow-up. Eur Spine J 1997;6(6):393–397.
JL, Elster AD, Ginsberg LE, et al. Klippel-Feil syndrome: CT and MR of
acquired and congenital abnormalities of cervical spine and cord. J Comput Assist Tomogr 1993;17(2): 215–224.