
|
|
together,” which is exactly what seems to happen in your head as you
walk into a room and see a child with “syndromic features.” As a
pediatric orthopaedist, you may be the first to consider the diagnosis.
Although any particular syndrome can be rare, when taken together,
syndromes are a very important part of pediatric orthopaedics. The
incidence of orthopaedic genetic abnormalities is about 5/1000.1
Multiply this 0.5% by the population of your region and it is likely
that you will get a total number in the thousands. The point is: you
are going to have to deal with syndromes on a regular basis in
pediatric orthopaedics.
congenital hand abnormality, clubfoot, hemivertebrae), ask yourself “Am
I looking at a child with a syndrome?” Some syndromes are common enough
to have their own multidisciplinary program (neurofibromatosis, for
example). In such cases, these syndromes are easier to manage because
there are a lot of interested experts who can lend a hand. Other
syndromes are once-in-a-career rare. You may find that it is just you,
a textbook, and perhaps an interested geneticist trying to do the job.

limitations; remembering the name, orthopaedic manifestations, and
associated systemic problems of every known syndrome that one might
encounter will test those limitations. Therefore, to stay out of
trouble, back up your pattern recognition skills with a good syndrome
textbook,2 as well as a computer in
your clinic that has Internet access. With computer access, you can
search for a syndrome by name, or type OMIM into your favorite search
engine to get the On-Line Mendelian Inheritance in Man website.
with more gentle interrogation on the family history than an
orthopaedic surgeon would normally do.
consultant’s reports, all test results. Counsel family and construct
orthopaedic treatment plan.

genetic cause is a loaded issue. It carries the possibility of blame
assignment to one of the parents or to in-laws, with important
implications for future offspring. It is very important to get a
geneticist involved as early as possible in order to stay out of
trouble with syndromes.
a name and lose sight of the true goal: providing timely treatment for
the child’s problems. Associated problems of other affected tissues and
organ systems can be potential sources of trouble with syndromes. These
other manifestations may suddenly become pertinent when the child is
undergoing anesthesia for an orthopaedic procedure (Table 13-1).
Guide parents to support groups so they are not alone. Be alert to the
fact that a few hysterical parents on a website or chat room can spread
much
misinformation,
or libel a doctor or institution that they dislike. If you treat
several children with a given syndrome, you, too, should check out the
websites.
|
TABLE 13-1 Trouble When Operating on a Child with a Syndrome
|
||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
congenital malformation (1/660 live births), so there is a high
likelihood that most orthopaedists will care for several patients with
this condition. Staying out of trouble with Down syndrome means
focusing on the child and not abnormal radiographs. Down syndrome
patients can have all kinds of unusual physical exam and radiographic
findings, and yet function at a very high level. Focus on the problems
that will hamper function or cause pain.
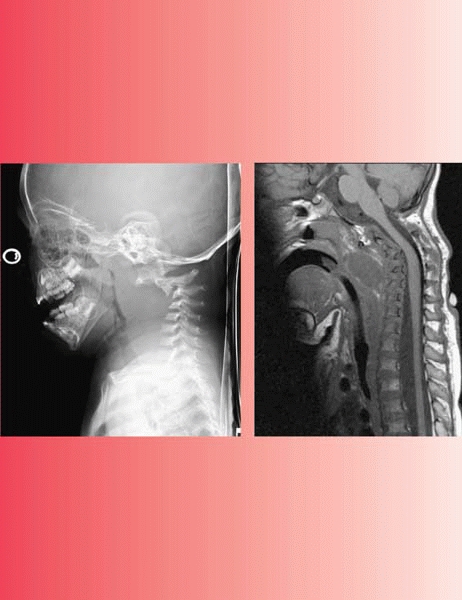
cervical instability, scoliosis, and spondylolisthesis. When evaluating
cervical spine instability, don’t just look at C1-C2, look at occiput
C1 as well (Fig. 13-1). A simplified and reasonable approach to cervical spine instability in Down syndrome is as follows:
-
Asymptomatic instability with ADI <10 mm: No treatment
-
Instability (ADI >10 mm) but no myelopathy: Check an MRI. If MRI shows cord impingement in flexion, then fuse.
-
Instability with myelopathy: Fuse.
 |
|
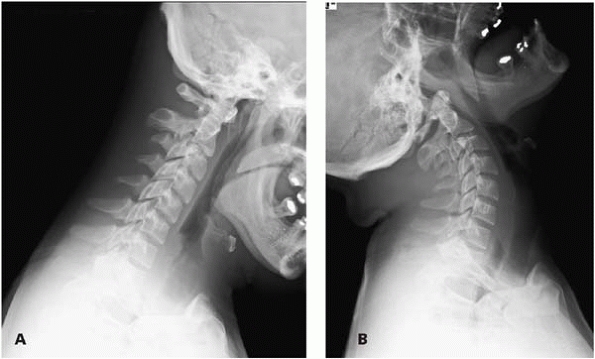
▪ FIGURE 13-1
Be alert to the fact that the instability in the cervical spine of a child with Down syndrome can be at the occcipitocervical junction, not just C1-C2. Note the relatively normal flexion film (A), but the movement of the occiput on C1 with extension (B). |
atlanto-dens interval is greater than 5 mm. Consider the risk and
benefits carefully, because attempts at fusion are known to be
associated with a very high complication rate. The use of wire
fixation, fusion to the occiput, halo immobilization, and carefully
constructed structural iliac crest bone graft has been reported to give
the best results.4
The hip can look good initially, but go on to complete dislocation
between the ages of 2 and 10 years. The acetabulum of the Down syndrome
hip can have a normal radiographic appearance, which belies the real
problem: extraordinary patholaxity of the soft tissues. Additionally,
behavior is a major component of problems in Down syndrome hips.
Children can be habitual hip dislocators, creating a challenge similar
to managing multidirectional shoulder instability in an adolescent with
generalized ligamentous laxity. In general, the surgical management of
hip instability in Down syndrome is extremely difficult. If you plan
surgery, do osteotomies, because you cannot rely on the soft tissues
for stability. Long-term postoperative bracing may help to avoid
redislocation. The complication rate of Down syndrome hip surgery can
approach 50%.5

 |
|
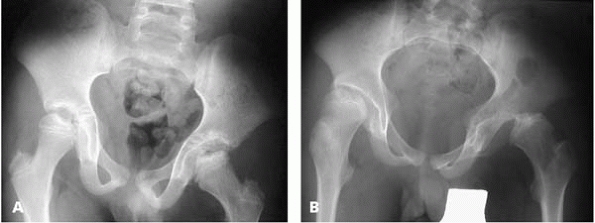
▪ FIGURE 13-2 The hips in Down syndrome make their own rules. A:
This boy presented at age 12 years with a few acute episodes of hip pain over the previous year, followed by refusal to bear weight for a few days, then a return to normal. The radiograph shows reduced, irregularly shaped femoral heads, and irregularly shaped acetabuli that seemed to provide good “coverage.” B: He returned at age 15 years with a painful, fixed dislocation of the left hip, subluxation of the right hip and shallow, dysplastic acetabuli. Could this trouble have been avoided? Can it now be successfully treated? |
 |
|
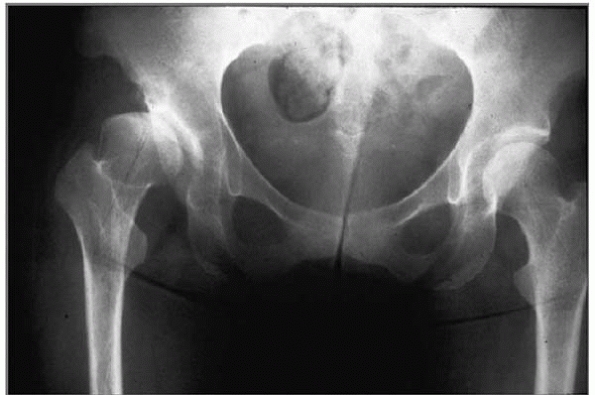
▪ FIGURE 13-3 Precocious osteoarthritis of the right hip in Down syndrome.
|

a Down syndrome patient who develops an abnormal gait or complains of
knee or thigh pain. Some authors have suggested that hypothyroidism may
play a role.6 It is well known that
children with Down syndrome are more likely to get avascular necrosis
(AVN) after an SCFE than otherwise normal children. A recent report
showed AVN in 5 of 8 patients.7
NF2. Because the orthopaedic problems are confined to NF1, you should
learn its diagnostic criteria. Neurofibromatosis is relatively common
(about 1/3000 infants), and many pediatric institutions have NF
programs, so you are certain to see affected children. Be alert that NF
can cause bone changes that are tumor-like in appearance (e.g., cysts
or scalloping) and that neurofibromas can undergo malignant
degeneration.

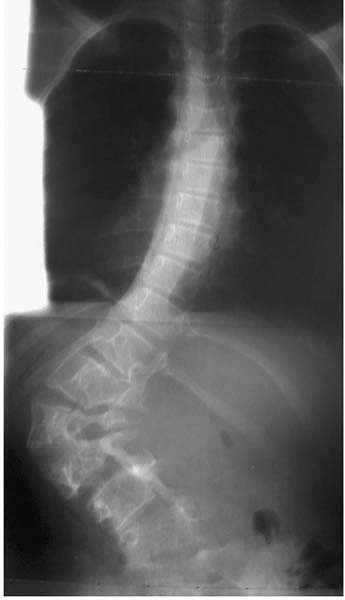
The curves can be short and sharp, and there can be rib penciling and
other findings that have been well described by Crawford and others (Fig. 13-4). Scoliosis and neurofibromatosis
can start early and progress relentlessly. Bracing is usually
ineffective. The enlarging neurofibroma can erode away at the pedicles
(screw fixation can be difficult). Fusion rates are lower in children
with NF, and families should be counseled that more than one operation
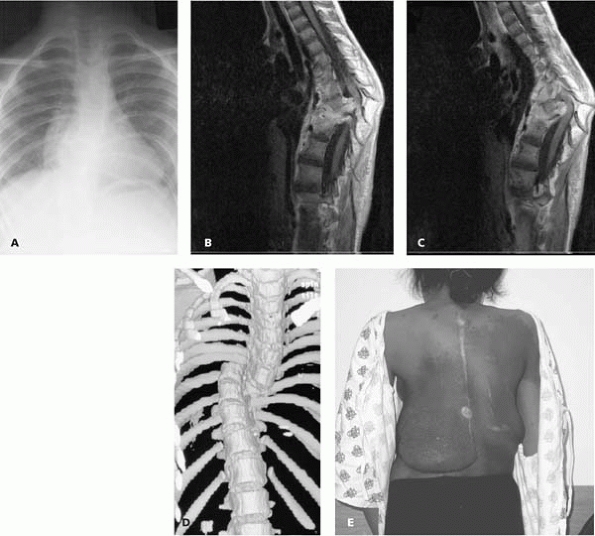
may be necessary. Dystrophic kyphosis is particularly dangerous (Fig. 13-5).
This spinal deformity can lead to paralysis fairly rapidly. Best
results have been attained by both anterior and posterior fusion, using
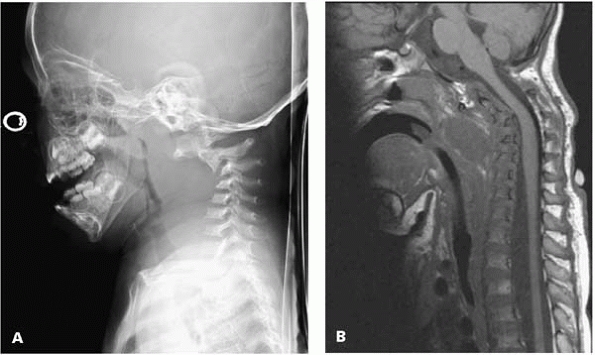
an anterior structural strut graft.8, 9, 10 Erosion of the pedicles in the cervical spine can lead to cervical instability over time (Fig. 13-6). Be sure to monitor involved children closely.
 |
|
▪ FIGURE 13-4 Scoliosis in a child with neurofibromatosis. Note the short, sharp curve that are classic in this NF spine trouble.
|
 |
|
▪ FIGURE 13-5
This young girl with neurofibromatosis presented with progressive neurologic deterioration. The initial thoracic spine radiograph (A) is difficult to interpret. Imaging is crucial in these cases: MRI (B,C) shows sharp thoracic kyphosis with cord impingement, and adjacent NF tissue, and the three-dimensional CT (D) shows what might be described as a dislocation of the spine in the mid-thorax. She underwent anterior decompression and posterior instrumented fusion, with a complete neurologic recovery. She returned several years later needing an extension to her fusion due to adding on below the curve. This clinical picture (E) demonstrates the cutaneous neurofibromas that can complicate the management of these cases. (Case courtesy of D. Drummond, MD.) |
 |
|
▪ FIGURE 13-6 This 2-year-old boy with NF developed severe cervical kyphosis (A). An MRI (B)
showed a large neurofibroma anteriorly, with impingement on the cord in flexion. He was managed with anterior and posterior fusion with halo vest immobilization. (Case courtesy of J. Dormans, MD.) |
the trouble that it causes. Perhaps the best testament to the trouble
with congenital pseudoarthrosis is the fact that amputation was a
common treatment (and still is for some cases).11 It is important to remember that congenital pseudoarthrosis affects not just the tibia (Fig. 13-7)
but can also affect the femur, clavicle, and either bone of the
forearm. To stay out of trouble, don’t do an osteotomy through bone
affected by neurofibromatosis. Clamshell bracing may be the best
protection in the very young. A comprehensive discussion of the risks
and benefits of different treatment methods is beyond the scope of this
book. Variable success has been reported with the Ilizarov device,12 microvascular bone transfer,13 and bypass grafting.14
To stay out of trouble treating congenital pseudoarthrosis of the
tibia, prepare yourself and the family for multiple procedures and
followup to maturity before success is declared. These cases may be
best handled by the small group of surgeons who have done several.
physical finding of rigid joint contractures. This category of
syndromes includes dozens of distinctly different diseases including
arthrogryposis multiplex congenita (AMC), Larsen syndrome,
Freeman-Sheldon syndrome and the pterygium syndromes. Children with AMC
often present difficult multiple orthopaedic problems. To stay out of
trouble, operate on joints early before adaptive changes secondary to
the contractures make it difficult to get satisfactory results. Do
osteotomies closer to the end of growth to avoid recurrence of
deformity.
don’t prescribe neonatal range of motion or other infant physical
therapy until birth fractures have been ruled out. In general, warn the
parents and the physical therapist against aggressive and forceful
manipulation that may cause fractures.
AMC is rarely successful. Recent reports of open reduction, followed by
only a brief period of postoperative spica casting, seem to be giving
better results than in the past (when orthopaedic surgeons often would
wait to see which children will walk and which would not).15,16
approximately 30 degrees, soft-tissue lengthening or release is
recommended in the first year of life. Knee extension contractures can
be treated at the same time as reduction of a dislocated hip,
but
knee flexion contractures should not: it is very hard to hold a hip
reduced when you are casting and splinting the knees in extension.
 |
|
▪ FIGURE 13-7 This 3-year-old with NF has a congenital pseudoarthrosis of the tibia that will be trouble for years to come. AP (A) and lateral (B) radiographs show a tibial pseudoarthrosis with an intact fibula. Prolonged casting (C,D) had no effect. One year later surgical management (E,F)
was begun. There is still no sign of healing, and the deformity has worsened. Many more procedures surely lie ahead. (Case courtesy of R. Davidson, MD.) |

In the upper extremities, it may be valuable to try to get one of two
rigidly extended elbows to flex to 90 degrees. Surgery on both elbows
is not recommended.
knees, be alert to the fact that you may have encountered Larsen
syndrome. These patients can have so many joint and spine problems that
they can spend much of their childhood preparing for and recovering
from orthopaedic surgery.

may be patellar malalignment as well as dislocations of the radial
head. If done early, manipulative treatment to reduce the knees may
result in distal femoral fractures.
spine segmentation. They develop kyphosis and myelopathy. Get a c-spine
series before the first birthday.
manage. To stay out of trouble, be alert that the popliteal artery will
be deep, but the sciatic nerve can be very superficial19
in the popliteal web, and thus at risk for accidental surgical
lengthening (not recommended). If full knee extension cannot be
achieved by soft-tissue surgery alone, consider shortening the femur.
of conditions. In this category of syndromes, the names change
frequently and new ones are added from time to time. Although each
mucopolysaccharidosis is rare, the entire group as a whole is not
unusual. It can be difficult to establish the diagnosis on physical
examination and radiographs in an infant. Be alert that children with
Hurler syndrome can have odontoid hypoplasia and a spinal canal
soft-tissue mass. Children with Morquio syndrome are infamous for the
trouble they cause the orthopaedist. Use braces after realignment
osteotomies to protect their unstable knees. The cervical spine in
Morquio is a major source of trouble.20
C1-C2 instability can lead to death even in the first year of life. Be
certain to get flexion/extension cervical spine films before any
general anesthetic is contemplated for a child with Morquio syndrome.21

upper extremity surgery, you will certainly be consulted on children
with VACTERL/VATER (acronyms to refer to congenital anomalies: vertebral, anal, cardiac, tracheal, esophageal, renal, limb/vertebral defects, imperforate anus, tracheoesophageal fistula, radial and renal
dysphasia). These consults can come from the neonatal intensivist or
general surgeon right after birth or the primary care doctor during the
first year of life. As you manage the spine or hand problem, stay out
of trouble by keeping in mind the high rate of congenital heart and
renal abnormalities.
autosomal dominant disorder. It can be difficult to make the diagnosis
because the clinical manifestations can vary greatly (Fig. 13-8).
If you are seeing a patient with long flat feet, scoliosis and
ligamentous laxity, it is wise to get the geneticist, cardiologist, and
perhaps the ophthalmologist involved if Marfan is suspected.
with Marfan syndrome is also a source of trouble. The spinal deformity
can follow the management guidelines of idiopathic scoliosis, but
bracing is less successful.22 The complications rate
of spinal fusion is higher in children with Marfan syndrome.23,24
Particular risks include pseudoarthrosis, progression outside the
fusion levels, and instrumentation levels and progressive kyphosis at
top or bottom of the instrumentation.25 Splenic rupture has recently been reported after spinal fusion in Marfan.26
Stay out of trouble by getting a cardiology consult before any surgery
is contemplated on children with Marfan syndrome. Some children may
need cardiac surgery first.
 |
|
▪ FIGURE 13-8 Steinberg’s thumb sign is useful in the diagnosis of Marfan syndrome.
|

Howard Steel recommended closing the triradiate cartilage to halt
protrusion before age 10 years.27
dysautonomia, also known as Riley-Day syndrome, if your practice
includes spinal deformity, particularly if there is a large Eastern
European Jewish population in your area. In this ethnic group, the rate
of Riley-Day syndrome is 1/3700. Stay out of trouble by understanding
some special features of spinal deformity in this group. In a large
percentage of cases, scoliosis starts early and progresses rapidly.
Bracing is less effective in children with familial dysautonomia than
in adolescent idiopathic scoliosis. Also, kyphosis is an important part
of the deformity in many.28 In
correcting their spinal deformities, try to manage the problem without
an anterior thoracic approach. Posterior osteotomies and pedicle screw
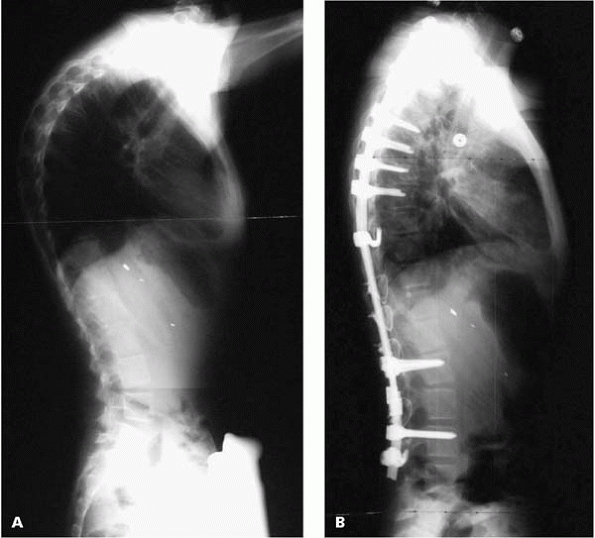
fixation may allow you to stay out of the chest (Fig. 13-9).
Be aware that the mortality after spine surgery is much higher in this
group than in patients with idiopathic scoliosis. Some children die in
childhood of pulmonary problems even without any thoracic surgery.

to pain in this syndrome can result in unrecognized or untreated
fractures. If you see a child with Riley-Day syndrome and
osteochondritis dissecans, the disease can behave more like a Charcot
joint. The lesion can be refractory to typical osteochondritis
dissecans management.
manifesting as limb hypertrophy, macrodactyly, or other size
incongruity. Although the bony overgrowth can be halted in
macrodactyly, there is not a good solution for the overgrown soft
tissue.
Involved feet often require amputation or ray resection. To stay out of
trouble with Proteus syndrome, exhaust nonoperative methods and counsel
parents extensively before attempting any surgical management.
Recurrences and complications are very common. In general, it is
considered better to amputate rather than debulk in cases of
macrodactyly.29
Simple epiphyseodesis of the overgrown leg is usually better than limb
lengthening, which can be fraught with complications. Angular
deformities can recur rapidly after surgical correction.
 |
|
▪ FIGURE 13-9 Kyphosis is often an important component of the spinal deformity in Riley-Day syndrome (A). This case was managed with a posterior instrument fusion without anterior release (B).
|

hypertrophy with cutaneous nevi and varicosities. The current theory is
that it is due to an abnormality of the walls of the veins of the
extremities.
likely to result in recurrence. If you plan to operate, it may be wise
to evaluate the full extent of the lesion with an MRI.
excessive bleeding during surgery, so it is essential to have blood and
blood products available when contemplating surgery.

To stay out of trouble, keep in mind that there is a 10% incidence of
abdominal tumors, especially Wilm tumor, associated with the
hemihypertrophy. Involve your referring pediatrician or your
institution’s oncology team in surveillance for these abdominal tumors.
syndrome can be a source of trouble. In this syndrome, they can have
very abnormal growth patterns, which can vary greatly between
individuals.30

hormones can lead to rapid worsening of scoliosis. Stay out of trouble
by working with your endocrinologist and following these children
closely while they are on growth hormones.
to you in your cerebral palsy clinic as a girl who seemed to be normal
in her first year or two, but then progressively develops dementia,
autism, ataxia, and repetitive hand motions. To stay out of trouble,
don’t ignore the scoliosis because you believe that Rett syndrome is
lethal. In some cases of Rett syndrome, life expectancy is normal.
Therefore, the spinal deformity should be managed with a full
understanding of life expectancy and careful counseling of the family.31
The spinal deformity is managed similarly to neuromuscular curves in
children with cerebral palsy, in whom segmental fixation and fusion to
the pelvis gives the best results. The onset of scoliosis is usually
before age 8 years, and rapid curve progression is usually detected
early in the second decade. In Rett syndrome, sagittal deformity with
excessive kyphosis can progress and necessitates close observation.
Orthotic treatment does not alter the natural history of scoliosis or
kyphosis. Indications for surgery are curve progression exceeding a 40-
or 45-degree Cobb angle or curves that cause pain or loss of function.32
conditions of greatly varying severity in which the fundamental problem
is a defect of Type 1 collagen. As such, it is easy to remember that
these children have fragile bones, but don’t forget about the
ligamentous laxity, spinal deformities, hearing loss, easy bruisability
and other manifestations. One potential source of major trouble may
occur at your first encounter. Fractures of infancy, and the
mishandling of the diagnostic dilemma of child abuse vs. OI, causes
lots of trouble for families, doctors and institutions33, 34, 35 (Fig. 13-10).
Diagnosis is easy when there is a family history of OI or other
features of the syndrome (e.g., blue sclera, etc.) or when the history
or injury constellation suggests abuse. Unfortunately, such history and
examination features are absent. In very ambiguous cases, work up both
child abuse and OI simultaneously, and enlist all the help you can from
Genetics and the appropriate authorities. To stay out of trouble,
expect that anything you write or say regarding an infant’s OI fracture
may end up in a court of law.
have some unexpected consequences. Be vigilant for growth arrests that
are caused by multiple injuries in the physeal region. Hypertropic
callus can be mistaken for osteogenic sarcoma,
leading to unnecessary amputation. Yet, osteogenic sarcoma can certainly occur in OI as it can in any child or adolescent.
 |
|
▪ FIGURE 13-10 Child abuse vs. OI.
|
fracture immobilization protocols further decrease bone density and
disuse atrophy. To avoid osteopenia, immobilize fractures only until
symptoms resolve. In children with very high fracture rates, a
“fracture kit,” containing a series of splints for each upper and lower
extremity limb segment, saves hospital visits and prolonged
immobilization in full casts.
treatment for the deformities associated with OI. Even in the best of
hands, the complication rate can be high, particularly when nailing is
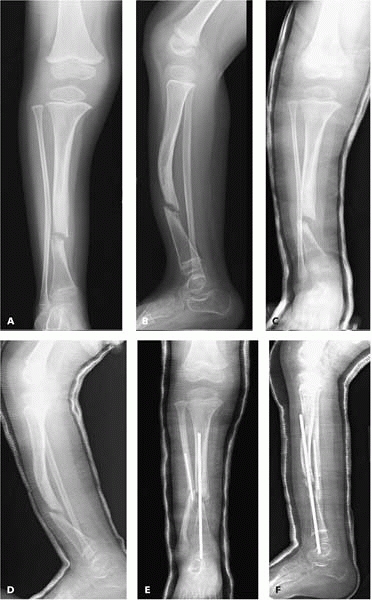
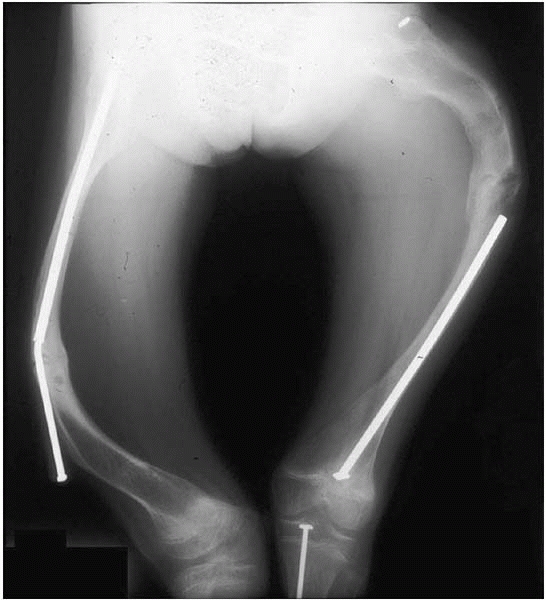
performed in young patients36,37 (Fig. 13-11).
Migration of the nail has been a particularly common source of trouble.38
When planning Sofield osteotomies, have every size of intramedullary
device available and be prepared to modify devices as needed. The
minimum internal diameter of the bone may be very hard to evaluate
preoperatively.39 Staging of
multiple osteotomies in four different limbs may be necessary to avoid
very high blood loss. Increasingly, Sofield osteotomies are being done
through small percutaneous incisions.40 Regarding timing after fracture, a Sofield osteotomy should either be done within a few days after fracture41
or after complete fracture healing. If the osteotomy is done 1 to 3
weeks after fracture, the orthopaedic surgeon may encounter excessive
bleeding from hyperemia of bone healing. Be prepared that there may be
no true medullary cavity. Reaming is sometimes necessary. The child
should be allowed to weight bear as soon as tolerated in casts or
braces to maintain maximum bone strength.
 |
|
▪ FIGURE 13-11
This single image illustrates the spectrum of trouble the orthopaedic surgeon may encounter when treating deformities and fractures in osteogenesis imperfecta. There has been separation of the Bailey Debow rod and subsequent removal in the left femur, causing refracture; in the right femur, there was bending of the Bailey-Debow rod, with progressive deformities on both sides. Lack of followup made management even more difficult. |
Corrective bracing can be difficult and detrimental. The children often
cannot tolerate the brace because of the excessive sweating related to
their condition. The corrective scoliosis brace can also deform the
soft ribs of a child with OI. For surgical correction, it is important
to plan an instrumentation strategy that involves multiple points of
fixation. Preoperative halo gravity traction has been used with some
success.44 Iliac crest autograft is
often insufficient, so the family should be consented for allograft.
Postoperative immobilization is tolerated poorly.
frequently than it was generations ago, you may occasionally be asked
to correct deformities or manage problems of children with rickets,
especially the forms related to an inherited metabolic disorder. To
stay out of trouble with the correction of angular deformities,
optimize the medical management first.45,46 Work with the patient’s pediatrician or
with the endocrinologist to be sure that the child is getting the
optimum doses of calcium and vitamin D (depending on the underlying
metabolic problem). In most cases, slight overcorrection of the angular
deformity is recommended to reduce the risk of recurrence. Finally,
think of SCFE in children with renal osteodystrophy who present with
hip, thigh or knee pain. Consider prophylactic pinning of the unslipped
opposite side because bilateral SCFE is very common in this population.
 |
|
▪ FIGURE 13-12
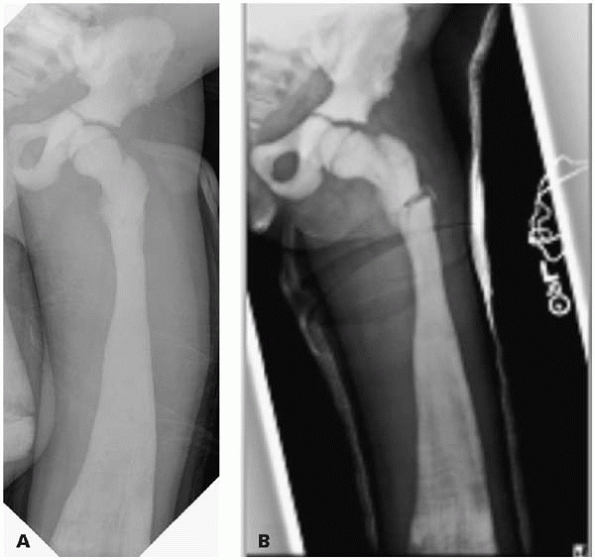
Treating fractures in osteopetrosis is trouble, particularly hip and proximal femur fractures. This nondisplaced subtrochanteric fracture (A) was treated with cast immobilization alone. Four months later (B), after prolonged casting and wedging, the fracture still shows little sign of healing. |
compromises the ability of these cells to reabsorb bone, leading to
very dense bone, fractures, and deformities. If you encounter a
fracture in a child with osteopetrosis, be alert that these fractures
can be slower to heal than you would normally expect. Corrective
osteotomies for coxa vara and other deformities can be difficult to
perform. Intramedullary fixation is difficult, as drills, saw blades,
and other tools become very dull and can break during work on this
extremely hard bone.47 Be alert to the fact that children with osteopetrosis are also at increased risk of osteomyelitis (Fig. 13-12).
-
Syndromes are unavoidable. Discover the syndrome before it finds you (and gets you in trouble).
-
Back up your pattern
recognition skills with a good syndrome textbook, as well as a computer
in your clinic that has Internet access. -
Get a geneticist involved as early as possible.
-
Don’t forget other organ systems: cardiac, renal, etc.
-
Down syndrome: focus
on the problems that hamper function or cause pain; look at the entire
cervical spine, and don’t do a fusion just because the ADI is increased. -
NF spinal deformity can lead to paralysis fairly rapidly.
-
Arthrogryposis: rule out birth fractures before starting PT; do osteotomies closer to the end of growth to avoid recurrence.
-
Larsen syndrome: cervical kyphosis and myelopathy can develop later in childhood.
-
Morquio syndrome: C1-C2 instability can lead to death, even before the 1st birthday.
-
Marfan syndrome: get a cardiology consult before any surgery is contemplated.
-
Klippel-Trenaunay-Weber: prone to excessive bleeding during surgery.
-
Child abuse vs.
osteogenesis imperfecta: in very ambiguous cases, work up both child
abuse and OI simultaneously, and enlist all the help you can from
Genetics and the appropriate authorities.
Regemorter N, Dodion J, Druart C, et al. Congenital malformations in
10,000 consecutive births in a university hospital: need for genetic
counseling and prenatal diagnosis. J Pediatr. 1984;104-3:386-390.
P, Di Silvestre M, Greggi T, et al. Surgical correction of dystrophic
spinal curves in neurofibromatosis: a review of 56 patients. Spine. 1999;24-21:2247-2253.
V, Doman I, de Jonge T, et al. Surgical treatment of spinal deformities
associated with neurofibromatosis type 1: report of 12 cases. J Neurosurg Spine. 2002;97-3:310-316.
BG, Pill SG, Drummond DS. Irreducible thoracic spondyloptosis in a
child with neurofibromatosis: a rationale for treatment. Spine. 2002;27-14:E342-347.
S, Catagni M, Donzelli O, et al. Congenital pseudarthrosis of the tibia
associated with neurofibromatosis-1: treatment with Ilizarov’s device. J Pediatr Orthop. 1997;17-5:675-684.
A, Brockman R. Congenital pseudarthrosis of the tibia. Long-term
followup of 29 cases treated by microvascular bone transfer. Clin Orthop 1995;314:37-44.
JM, Kendall BE, Crockard HA, et al. The odontoid process in
Morquio-Brailsford’s disease: the effects of occipitocervical fusion. J Bone Joint Surg Br. 1991;73-5:851-858.
HH. Protrusio acetabuli: its occurrence in the completely expressed
Marfan syndrome and its musculoskeletal component and a procedure to
arrest the course of protrusion in the growing pelvis. J Pediatr Orthop. 1996;16-6:704-718.
GJ, Vanpaemel LA, Engelbert RH, et al. Complications of the
Bailey-Dubow elongating nail in osteogenesis imperfecta: 34 children
with 110 nails. J Pediatr Orthop B. 1999;8-3:203-207.
C, Jadhav A, Bernstein RM, et al. Rod diameter prediction in patients
with osteogenesis imperfecta undergoing primary osteotomy. J Pediatr Orthop. 2001;21-4:515-518.
M, Garapati R, Zelle B, et al. Combination of femoral fracture
treatment and corrective osteotomy in a child with osteogenesis
imperfecta. Arch Orthop Trauma Surg. 2004;124-5:341-345.
RF, Bitan FD, Laplaza FJ, et al. Spinal deformity, pulmonary
compromise, and quality of life in osteogenesis imperfecta. Spine. 1999;24-16:1673-1678.
GJ, Finidori G, Engelbert RH, et al. Operative treatment of severe
scoliosis in osteogenesis imperfecta: results of 20 patients after halo
traction and posterior spondylodesis with instrumentation. Eur Spine J. 2000;9-6:486-491.
IH, Kim JK, Chung CY, et al. Deformity correction of knee and leg
lengthening by Ilizarov method in hypophosphatemic rickets: outcomes
and significance of serum phosphate level. J Pediatr Orthop. 2002;22-5:626-631.
M, Said SE, Glorieux FH, et al. Principles and results of corrective
lower limb osteotomies for patients with vitamin D-resistant
hypophosphatemic rickets. Clin Orthop. 1988;237:264-270.
DG, Newfield JT, Gillespie R. Orthopedic management of osteopetrosis:
results of a survey and review of the literature. J Pediatr Orthop. 1999;19-1:122-132.
ED, Beals RK. The hip joint in Down’s syndrome: a study of its
structure and associated disease. Clin Orthop. 1992 May;(278):101-107.