
in providing structural support for the body, bone also plays a crucial
role in maintaining mineral homeostasis in serum. The serum levels of
calcium and phosphorus need to be maintained under tight control to
allow for the normal function of a variety of cells. The cancellous
bone has a tremendously large surface area that allows for the rapid
transfer of minerals stored in it, such as calcium, to the serum. This
process occurs at over a million sites in the human skeleton, mediated
by osteoblast and osteoclast cells. A variety of endocrine, metabolic,
and cellular factors are crucial for maintaining this tight homeostatic
balance. Not only do these various factors maintain serum minerals at
their proper levels, but they also act to regulate the amount of bone
present. Because of the interrelationship between these metabolic and
endocrine factors on the one hand, and the distribution of minerals
between the bone and serum on the other, metabolic and endocrine
disorders alter the quantity and quality of bone. This same
interrelationship occasionally works in reverse; that is, disorders
that alter bone structure dysregulate the serum mineral balance (1).
skeleton is very different in children and adults; this is because many
endocrine and metabolic factors affect the growth plate. Chondrocytes
in the growth plate go through a coordinated process of
differentiation, beginning with a proliferative phase at the epiphyseal
side of the growth plate and progressing to terminal differentiation
and apoptotic cell death at the metaphyseal side of the physis.
Terminal differentiation is associated with the expression of type X
collagen and the formation of scaffolding for bone formation. Blood
vessels located adjacent to the physis in the metaphyseal bone bring
pleuripotential mesenchymal cells to the region; these cells
differentiate into osteoblasts and produce new bone on the scaffolding
left behind by the growth-plate chondrocytes. This coordinated
differentiation process results in the longitudinal growth of long
bones. The process of growth-plate chondrocyte differentiation needs to
be tightly regulated because, if growth-plate chondrocytes on one side
of the body go through this process at a different rate from those on
the other side, a limb length inequality would result. The process of
growth-plate chondrocyte maturation is regulated by both local and
systemic factors (2). In those conditions in
which these systemic factors are dysregulated, as is the case in
several endocrinopathies, there is an associated growth-plate
abnormality (3). In addition, some endocrine
factors that regulate bone mineral homeostasis, such as thyroid
hormone, also regulate the growth-plate chondrocytes. Therefore,
whereas thyroid hormone dysregulation has implications for bone density
in adults, in growing children thyroid hormone dysregulation can also
cause an abnormality in the growth plate.
osteoclast cells that add to or break down bone. These cells are
regulated by local and systemic factors, some of which can be modulated
by the mechanical environment. All of these factors are interrelated in
a complex way that has still not been completely elucidated.
down new bone in the form of osteoid. These cells are derived from
pleuripotential stromal precursor cells (sometimes called mesenchymal stem cells)
and are the active cells that lay down new bone during skeletal growth
and remodeling. These cells produce alkaline phosphatase, an enzyme
that is often used to identify osteoblasts and osteoblastic activity.
As the bone matures, osteoblasts become encased in the new bone. Once
the osteoblasts become encased in osteoid, they become relatively
quiescent and are termed osteocytes. In
mature bone, osteocytes are located extremely far away from neighboring
cells and communicate with other cells through long cytoplasmic
processes. The osteocytes remain quiescent until stimulated by hormonal
or mechanical factors to begin to reabsorb or lay down bone. Although
osteoblasts and osteocytes are thought of as cells responsible for
building new bone, they are also able to reabsorb small quantities of
bone; they are able to do this rapidly in comparison to osteoclasts,
which require cellular differentiation and recruitment in order to
reabsorb bone. Therefore, osteoblasts and osteocytes are the first
cells that the body activates when bone reabsorption is required (4).
After differentiation and recruitment to the site on the bone where
they are required, osteoclasts are able to reabsorb bone in a very
robust manner. Osteoclasts form a ruffled border that attaches to the
osteoid and secretes proteins that degrade the bone matrix. As such,
osteoclasts form active reabsorption cavities called Howship lacunae.
There is an intimate relation between osteocyte and osteoclast
activities, and many of the signals that activate osteoclasts are
mediated by osteocytes. For instance, parathyroid hormone (PTH) does
not directly regulate osteoclast activity,
but
conveys information via osteocytes, which produce secondary factors
that regulate the differentiation of monocytes to osteoclasts. The
major signaling pathway that is used by osteocytes to regulate
osteoclasts involves a member of the tumor necrosis factor superfamily
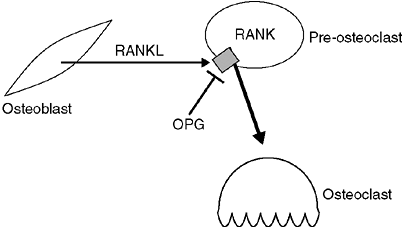
called RANKL,
its receptor, RANK, and a circulating inhibitor, osteoprotegerin (OPG).
The receptor, RANK, is present on osteoclast precursor cells; when
stimulated, it causes these precursors to differentiate into active
osteoclasts. RANKL is produced by osteocytes that are stimulated to
reabsorb bone. OPG also binds to RANKL, inhibiting its ability to
activate RANK, and thereby inactivating osteoclasts. The balance
between OPG and RANKL regulates the number of osteoclasts available (Fig. 7.1) (4,5).
Because OPG inhibits osteoclast production, its use represents a
promising approach to inhibiting osteoclastic activity and,
consequently, it has the potential to be developed into a useful
therapy for osteoporosis, neoplastic bone loss, and even the loosening
surrounding total joint implants (6,7).
understanding genes that regulate how bone cells develop. Much of this
information is covered in several review articles (1,2)
and is beyond the scope of this book. For the purpose of this chapter,
we consider three modulators of bone density: (a) calcium homeostasis,
(b) hormone factors, and (c) physical forces.
regulate bone mass. Calcium mobilization overrides the other functions
of the skeleton. Calcium deficiency resulting from renal disease,
malabsorption, or a diet poor in calcium invariably causes bone loss,
which cannot be overcome by modulating any of the other factors that
regulate bone mass. The effects of hormones such as estrogen seem to be
more potent than the effect of physical forces; this is suggested by
the facts that exercise has only limited ability to maintain or restore
bone mass in postmenopausal women, and amenorrhoeic marathon runners
lose bone. Of the three modulators of bone mass—calcium availability,
hormonal factors, and physical forces—the last has the least pronounced
effects, although this is the one that orthopaedic surgery concentrates
most of its efforts on (4).
 |
|
Figure 7.1
Expression of RANKL by osteoblasts and osteocytes activates RANK receptor on preosteoclasts to cause differentiation into active osteoclasts. Osteoprotegerin (OPG) is a circulating factor that can also bind to RANK, but inhibits its ability to cause differentiation to osteoclasts. |
conductivity, and contractility of smooth and skeletal muscle, and in
the irritability and conductivity of nerves. Small changes in
extracellular and intracellular calcium levels lead to dysfunction of
these cells. In the case of neurons, cellular activity is inversely
proportional to calcium ion concentration, whereas for cardiac myocytes
there is a direct proportionality. Therefore, decreases in ionic
calcium concentration can lead to tetany, convulsions, or diastolic
death. Conversely, increases in the concentration of calcium can lead
to muscle weakness, somnolence, and ventricular fibrillation. It is
obviously important for the body to guard the concentration of ionized
calcium, thereby providing a rationale for the overriding importance of
calcium homeostasis in modulating bone density (8,9).
and excreted primarily by the kidney. Therefore, diseases that affect
gut absorption or renal function have the potential to dysregulate
normal calcium homeostasis and bone mass. In addition, some conditions
that cause massive loss of bone mass, such as widespread metastatic
disease or prolonged bed rest, can also alter serum calcium levels.
Almost all of the body’s calcium is stored in the bones and is held in
the form of hydroxyapatite, a salt that is composed of calcium,
phosphorus, hydrogen, and oxygen (CaHPO) in very tiny crystals embedded
in the collagen fibers of the cortical and cancellous bone (10,11,12,13).
The small size of the crystals provides an enormous surface area, and
this factor, combined with the reactivity of the crystal surface and
the hydration shell that surrounds it, allows a rapid exchange process
with the extracellular fluid (ECF). This process converts the
mechanically solid structure of bone into a highly interactive
reservoir for calcium, phosphorus, and a number of other ions (12,14).
of body fluids, calcium and phosphate concentrations in serum exceed
the critical solubility product, and are predicted to precipitate into
a solid form. It is thought that various plasma proteins act to inhibit
the precipitation and
keep
these ions in solution. This metastable state is important for bone
structure, as it allows the deposition of hydroxyapatite with a minimal
expenditure of energy during bone formation. Unfortunately, it also
makes the occurrence of ectopic calcification and ossification easy,
because of increments in the levels of either or both of these ions.
membranes, and requires an active transport machinery to move it into
or out of cells (8,12,14,15,16).
Although the mechanism to control this transport is regulated, in large
part, by the action of the active form of vitamin D, PTH, and the
concentration of phosphate (14,17,18),
a variety of other cell-signaling pathways also play their roles in
transporting calcium across cell membranes. These other cell-signaling
pathways, however, seem to act in specialized cell types under specific
physiologic states and, as such, are likely to play only a small role
in regulating the total serum calcium level. Therefore, PTH, vitamin D,
and phosphate are the three factors that play the most crucial roles in
the calcium transport process and, consequently, in maintaining the
normal extracellular soluble calcium level.
the expression level of PTH is regulated by the serum level of ionized
calcium. When serum calcium levels are low, there is an increase in PTH
expression and protein production, leading to increased PTH levels in
the serum. There are four parathyroid glands, and any one gland has the
potential to produce enough PTH to maintain calcium homeostasis. This
fact is of importance in the surgical management of thyroid neoplasia,
in which it is preferable to maintain the viability of at least one
parathyroid gland. PTH binds to a family of cell membrane receptors
called parathyroid hormone receptors (PTHR) that activate a number of
cell-signaling pathways. The pathway that has been studied the most in
the control of calcium is the one that regulates adenyl cyclase
activity, resulting in an increased cellular level of cyclic adenosine
monophosphate (cAMP). cAMP renders the cell membrane more permeable to
ionic calcium, and it induces the mitochondria, which are intracellular
storehouses for calcium, to release their calcium. These actions
increase the intracellular concentration of calcium, but do not promote
transport to the extracellular space, a function that also requires
vitamin D. PTH acts with 1,25-dihydroxyvitamin D to facilitate cellular
calcium transport in the gut, the renal tubule, and in the lysis of
hydroxyapatite crystals (16,17,19).
PTH directly stimulates osteoblasts to begin to degrade the surrounding
calcium-rich osteoid. Osteoclasts do not contain receptors for PTH, but
are stimulated by PTH activation in osteoblasts through induction of
the expression of RANKL, which activates osteoclasts (19,20).
Another action of PTH is to diminish the tubular reabsorption of
phosphate, thereby causing the renal excretion of phosphate (19,21,22).
and are converted to calciferol and cholecalciferol by ultraviolet
light, a process that occurs in the skin. In the absence of ultraviolet
light, this conversion cannot occur; this explains why vitamin D
deficiency is associated with prolonged periods indoors away from
ultraviolet light sources, such as in chronically ill individuals, or
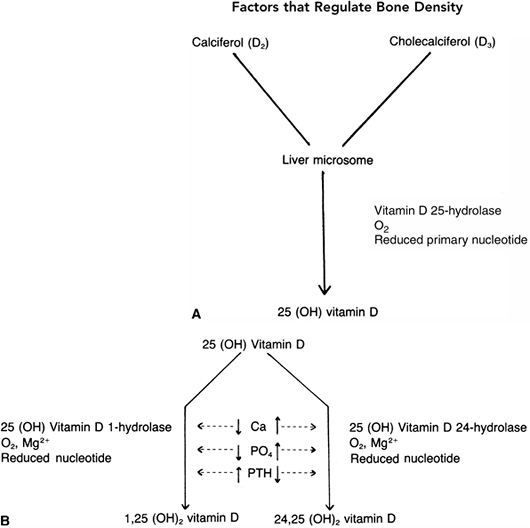
in individuals living in extremely cold climates (10,24). The compounds are then transported to the liver, where they are converted to 25-hydroxyvitamin D by a specific hydrolase (25,26,27,28,29).
Severe liver disease or the intake of drugs that block hydrolase
activity will inhibit the production of 25-hydroxyvitamin D, also
potentially leading to vitamin D deficiency. The final conversion
occurs in the kidney. In the presence of specific hydrolases and a
number of biochemical cofactors, 25-hydroxyvitamin D is converted to
either 24,25-dihydroxyvitamin D or 1,25-dihydroxyvitamin D. The latter
serves as the potent calcium transport promoter (30–32). A low serum
calcium level and a high PTH level cause conversion to the 1,25 analog,
whereas a high serum calcium level, a higher serum phosphate level, and
a low PTH level favor formation of 24,25-dihydroxyvitamin D, which is
less potent in activating calcium transport (Fig. 7.3) (30,33,34,35,36).
Serum phosphate also plays an important role here, because a high
concentration of phosphate shunts the 25-hydroxyvitamin D into the
24,25-dihydroxy form. Although the 24,25-dihydroxy form is less active
in regulating calcium levels, it has an important role in growth-plate
chondrocytes. This crucial role of the kidney in converting vitamin D,
as well as its important role in excreting excess calcium and
phosphorus, explains the particularly deleterious effect of renal
failure on bone homeostasis, causing vitamin D deficiency as well as
directly dysregulating normal calcium excretion. Because of the crucial
role of vitamin D in calcium metabolism, the National Academy of
Sciences and the American Academy of Pediatrics recommends 200 IU per
day of vitamin D (37). This dose will prevent
physical signs of vitamin D deficiency and maintain serum
25-hydroxyvitamin D at or above 27.5 nmol per L (11 ng per mL). The
generic name of 1,25-dihydroxyvitamin D is calcitriol.
Recent studies found that vitamin D also plays a variety of
extra-skeletal roles, including in modulation of the immune response,
and as a chemoprotective agent against certain cancers (38).
for calcium during periods of rapid bone growth. Recommendations from the American Academy of Pediatrics are summarized in Table 7.1 (8,24,39,40).
Adequate calcium in the diet during adolescent years is important for
the maintenance of bone mass over the long term, and orthopaedists
should counsel their patients about the importance of including
appropriate amounts of calcium, as well as vitamin D, in their diet.
Several dietary factors alter calcium absorption. Calcium salts are
more soluble in acid media; therefore the loss of the normal
contribution of acid from the stomach reduces the solubility of the
calcium salts and decreases the absorption of the ionized cation. A
diet rich in phosphate may decrease the absorption of calcium by
binding the cation to HPO2-4 and precipitating most of the ingested calcium as insoluble material (12,41).
Ionic calcium can be chelated by some organic materials with a high
affinity for the element, such as phytate, oxalate, and citrate.
Although these materials may remain soluble, they cannot be absorbed (12,41,42,43). Calcium, in the presence of a free fatty acid, forms an insoluble soap that cannot be absorbed (41,44).
Disorders of the biliary or enteric tracts, associated with
steatorrhea, are likely to reduce the absorption of calcium because it
forms an insoluble compound and because the fat-soluble vitamin D that
is ingested is less likely to be absorbed under these circumstances (45).
 |
|
Figure 7.2 The conversion of vitamin D from the skin or from dietary sources takes place in the liver and kidney. A: In the liver, the enzyme vitamin D 25-hydrolase acts to form 25-hydroxy vitamin D. B: The second conversion of vitamin D takes place in the kidney, where at least two pathways have been described. The maintenance pathway [when the need is minimal, as defined by a normal calcium and phosphorus and low parathyroid hormone (PTH)
level] occurs in the presence of a specific enzyme (25-hydroxyvitamin D 24-hydrolase) and results in the less active 24,25-dihydroxyvitamin D. If calcium transport is required, as signaled by the presence of low serum calcium and phosphorus levels and a high PTH level, the body converts the 25-hydroxyvitamin D to the much more active form, 1,25-dihydroxyvitamin D. |
|
TABLE 7.1 DIETARY CALCIUM REQUIREMENTS
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
gastrointestinal (GI) tract than is calcium, and is freely transported
across the gut cell to enter the extracellular space, in which it
represents a major buffer system. Transport into and out of the bone is
passive and is related to the kinetics of the formation and breakdown
of hydroxyapatite crystals. Tubular reabsorption of phosphate, however,
is highly variable, with reabsorption ranging from almost 100% to less
than 50%. The principal factor that decreases tubular reabsorption of
phosphate is PTH.
 |
|
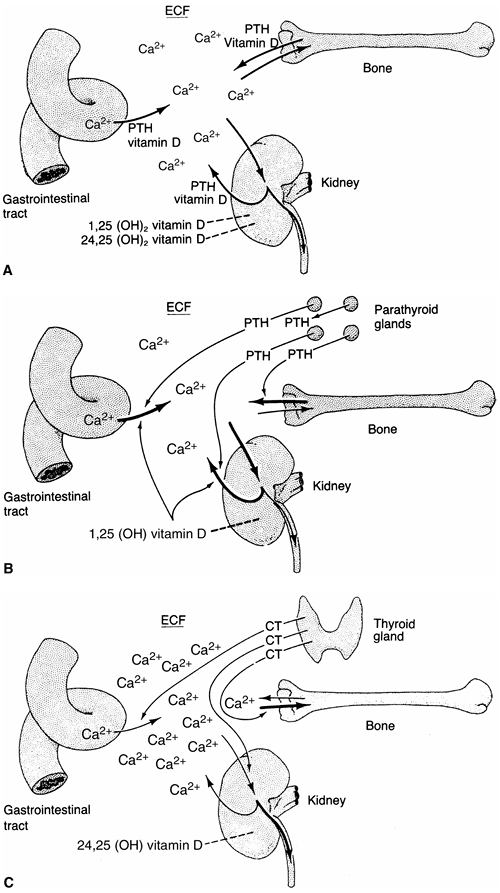
Figure 7.3
The roles of the bone, kidneys, gastrointestinal tract, parathyroid gland, and thyroid gland in calcium kinetics. These organs act to maintain calcium in the extracellular fluid (ECF) at the appropriate levels for normal cellular function. Vitamin D and parathyroid hormone (PTH) act to transport calcium ions across the gut wall and regulate renal excretion and, thereby, bone calcium content. Depending on the need for increased transport, 25-hydroxy vitamin D is converted to 24,25- or 1,25-dihydroxyvitamin D. A: In the normocalcemic state, an equilibrium between calcium intake and excretion is maintained by the various organs. B: In the hypocalcemic state, a reduced concentration of calcium signals the parathyroid glands to release more PTH, which acts at the levels of the gut cell, renal tubule, and bone to increase transport of calcium and rapidly replenish body fluids with it. An increase in PTH also favors the synthesis of 1,25-dihydroxyvitamin D in the kidney and acts to promote renal phosphate excretion by markedly diminishing the tubular reabsorption of phosphate. C: In the hypercalcemic state, low concentrations of calcium and PTH act independently to diminish the synthesis of 1,25-dihydroxyvitamin D and decrease transport of calcium in the gut cell, tubule, and bone. Increased concentrations of calcium also cause the release of calcitonin (CT) from the C-cells of the thyroid gland, thereby diminishing calcium concentration. This mechanism principally involves stabilizing the osteoclast and decreasing its action on the bone, but it is not very effective in humans. |
estrogen. Most of the clinical and experimental data on the role of
estrogen in bone have been generated from studies of postmenopausal
women. However, clinical data from children with deficiencies in sex
hormones, such as in Turner syndrome, also show that a lack of estrogen
in growing girls is responsible for profound loss of bone density. The
exact mechanism by which estrogen regulates bone formation and loss is
unknown. Estrogen receptors are present on both osteoblasts and
osteoclasts, yet the cellular mechanism by which estrogen alters the
behavior of
these
cells is not clear. Studies in animals suggest that estrogen exhibits
at least some of its effects through the regulation of pleuripotential
stromal cells in the bone marrow, a process that may be mediated by
interleukin-6. Estrogen also suppresses the activation of osteoclasts
by inhibiting the activation of RANK in the precursor cells (46,47).
mechanism is less well understood than it is in the case of estrogen.
Idiopathic hypogonadotropic hypogonadism is associated with decreased
bone mass, and there is an association between delayed puberty and low
bone mass in boys, suggesting a positive role for androgens in
regulating bone mass (48).
with nuclear proteins and DNA to increase the expression of a variety
of genes, ultimately regulating cell activity positively. Thyroid
hormone activates both osteoblasts and osteoclasts. The actual effect
on bone mass depends on the body’s balance between these two cell
types, and how well the normal control of the calcium level is able to
counteract their heightened activity. In general, the balance is in
favor of the osteoclast and, very often, increased thyroid hormone
levels lead to bone loss (49,50).
inhibit cellular activity in general, potentially decreasing the
ability of osteoblasts to lay down new bone. Corticosteroids also have
profound effects on the skeleton because of their effect on calcium
regulation in the kidney, where they increase calcium excretion; this
leads, secondarily, to elevated PTH levels, with consequent negative
effects on bone density (51,52).
CT causes inhibition of bone resorption by osteoclasts and osteoblasts,
and decreases reabsorption of calcium and phosphate in the kidneys in
animal models and cell cultures, it seems to play a minor role in
humans (53,54).
weightlessness (microgravity in outer space) or immobilization
(paralysis, prolonged bed rest, or application of casts) can cause
significant bone loss, whereas strenuous athletic activity can augment
certain bones (54,55).
This effect is important in pediatric orthopaedic patients, in whom
many of the neuromuscular disorders are associated with decreased
weight-bearing and associated osteoporosis. Bone remodels itself
according to the mechanical stresses applied, a phenomenon termed Wolff’s law.
It is well known that the mechanical environment alters cell behavior
and gene expression, and it is thought that such a mechanism, most
likely acting through osteocytes, is responsible for the effect of
weight-bearing on bone density, as well as for the changes attributable
to Wolff’s law (56,57).
General information about growth-plate development and its local
regulation is covered in the chapter on developmental biology. However,
it is apparent that growth-plate chondrocytes are affected, either
primarily or secondarily, by a variety of endocrine regulatory factors,
and so a brief review is warranted here.
growth plate reside in the resting zone; they begin to proliferate and
advance toward the metaphyseal side of the growth plate in the
proliferative zone; following this, they enter a prehypertrophic zone,
where they shift from proliferation to differentiation. It is also in
this prehypertrophic zone that important signals that regulate the
differentiation process, such as PTH-related protein and Indian
hedgehog, are present. Following this exposure, the cells undergo
hypertrophy, form columns in the hypertrophic zone, and then undergo
terminal differentiation and cell death. Blood vessels from the
metaphysis, adjacent to the terminally differentiated chondrocytes,
bring in new pleuripotential mesenchymal cells that differentiate into
osteoblasts, forming new bone on the scaffolding left behind by the
chondrocytes. This last region is sometimes called the zone of provisional calcification.
in favor of or against the differentiation process in these cells. In
addition, agents that alter normal bone formation by osteoblasts can
also alter the growth plate by preventing the normal replacement of the
terminally differentiating chondrocytes with new bone. This inhibition
of normal ossification results in the characteristic growth plate
changes in rickets, in which there is an increased zone of terminal
differentiation. Endocrinopathies can also alter size and matrix
components in the various zones of the growth plate. Such disorders
that affect terminal differentiation may make the growth plate
mechanically weaker in this region, predisposing the patient to
conditions such as slipped capital femoral epiphysis (SCFE). In a
similar manner, it may make the growth-plate chondrocytes easier to
deform with compressive pressure, causing deformities such as genu
varum; this explains the high frequency of these growth-plate
deformities in children with endocrine disorders. As in bone,
mechanical factors can also play a role in growth plate function. The
Hueter-Volkmann Principle states that growth plates exhibit increased
growth in response to tension and decreased growth in response to
compression (58). Therefore, an endocrinopathy can cause growth-plate deformities, which can then be
exacerbated by the effect of the changing mechanical axis in the affected limb.
hierarchal regulation of the growth plate, with endocrine factors
playing a dominant role over mechanical factors (3,59).
This fact is readily apparent in conditions such as rickets, where
surgery will not result in correction of the genu varum in the absence
of the correction of the underlying endocrinopathy in the growing
child. Therefore, it is important to avoid the temptation to undertake
surgical correction of a deformity in a growing child with an endocrine
disorder until the endocrinopathy is treated.
a role in regulating growth-plate function. In many cases, not much is
known about the intracellular signaling mechanisms utilized by these
factors. Growth hormone plays an important role in regulating the
proliferation of growth-plate chondrocytes, and the process is mediated
by somatomedins. In the absence of growth hormone, there is a slowing
of growth-plate maturation, as well as a slowing of the rate of long
bone growth. Thyroid hormone also plays a role in regulating
chondrocyte activity, by increasing the metabolic and proliferative
rates of growth-plate chondrocytes. PTH may alter chondrocyte
maturation in growth plates because the PTH receptor PTHR1 is expressed
in prehypertrophic chondrocytes and its stimulation results in an
inhibition of terminal differentiation. Nutrition and insulin also
regulate growth-plate chondrocytes, in a manner similar to that of
growth hormone, by regulating growth-plate chondrocyte proliferation. A
lack of dietary protein exerts a negative control over the
somatomedins. An excess of glucocorticoids also inhibits growth, partly
by an inhibitory effect on protein synthesis in cartilage, but also by
interference with somatomedin production and action (3).
Although all these factors play roles in regulating growth-plate
chondrocytes, we will likely learn more in the coming years about the
roles they play in a variety of growth-plate pathologies, including
disorders such as SCFE, where it is well known that a variety of
endocrinopathies are predisposing conditions.
clinical condition in which there is inadequate mineralization of
growing bone. Severe nutritional rickets was endemic in early
industrialized societies, particularly where sunlight was scarce.
Accordingly, severe rachitic deformities were commonly seen in the
early days of orthopaedics (60,61). In developed countries nutritional rickets is now a rarity, although it may present de novo to pediatric orthopaedists for diagnosis. Inherited forms of rickets are still commonly seen in the United States (62).
The surgeon should also be familiar with renal tubular abnormalities
that can result in rickets, as well as with the clinical entity of
renal osteodystrophy, which describes the bone disease associated with
end-stage renal disease and includes features of rickets as well as
secondary hyperparathyroidism.
similar, and therefore the clinical presentation will be covered first,
prior to a discussion of the various etiologies.
formed bone at long bone physes. The manifestations include changes in
growth-plate morphology, with decreased longitudinal growth and angular
deformities of the long bones. Osteomalacia, which is the failure of
mineralization of osteoid formed at cortical and trabecular surfaces,
often accompanies rickets in childhood. In the adult, osteomalacia is
the only result of the mechanisms that cause rickets in childhood.
early childhood and often before the age of 2 years. The child may have
a history consistent with hypocalcemia in infancy, including apneic
spells, convulsions, tetany, and stridor prior to the age of 6 months (63).
The child is often hypotonic with delayed motor activity milestones for
sitting, crawling, and walking. There is proximal muscle weakness and
sometimes profuse sweating. Cardiomyopathy and respiratory and GI
infections may accompany the clinical presentation (64,65,66,67,68,69).
wrists, elbows, and knees are thickened, and the long bones are short.
Genu varum or valgum may be present, as may coxa vara. Costochondral
enlargement leads to the characteristic rachitic rosary appearance of
the chest. Harrison’s sulcus is an indentation of the lower ribs caused
by indrawing against the soft bone. Kyphoscoliosis may be present.
Closure of the anterior fontanelle is delayed. Frontal and parietal
bossing of the skull is evident. Plagiocephaly may be related to the
effect of positioning on a soft skull. Delayed primary dentition is
common (70,71).
particularly with weight bearing. One should remember that crawling
children bear weight on their wrists, and this explains the thickened
wrists as well as knees. The metaphysis typically takes on a cupped and
splayed appearance. The long bones are short for age, and show evidence
of coxa vara and genu varum or valgum as described in the preceding
text. Radiography may reveal further evidence of osteomalacia. The
hallmark is Looser zones. These are
transverse
bands of unmineralized osteoid, which typically appear in the medial
aspect of the proximal femur and at the posterior aspect of the ribs.
These are described as pseudofractures and often have an osteosclerotic
reaction area around them. In an adult, they can progress to true
fractures. Acetabular protrusion and pathologic fractures complete the
radiographic signs of rickets (64,70,72,73).
 |
|
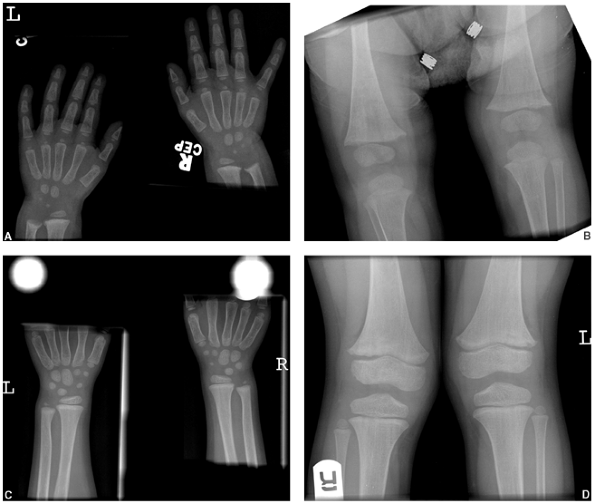
Figure 7.4 Rickets. Changes caused by rickets can be seen (A) at the wrist and (B)
at the knees of this 1-year-old child with X-linked hypophosphatemia. The growth plates are widened and the metaphyses are cupped, particularly at the ulna and femur. At 4 years of age (C, D) the changes have resolved with medical treatment. |
and phosphate in the presence of alkaline phosphatase enzyme. Calcium
and phosphate are maintained very close to their solubility coefficient
by a complex series of inhibitors. The control mechanisms in the
physiologic state have been discussed in earlier sections of this
chapter.
to consider those conditions that reduce the availability of calcium,
those conditions that reduce the availability of phosphate, and the
rare condition that reduces the availability of alkaline phosphatase at
the osteoblast-bone junction (Table 7.2).
Nutritional rickets and end organ insensitivity to calcitriol are
problems as far as calcium is concerned. X-linked hypophosphatemia is
the most common form of rickets seen today in the United States. It is
caused by renal tubular phosphate wasting in isolation (74).
Renal tubular abnormalities, including Fanconi syndrome, cause renal
wasting of phosphate, calcium, magnesium, and bicarbonate. Alkaline
phosphatase is deficient only in one rare recessive condition,
appropriately called hypophosphatasia.
|
TABLE 7.2 CLASSIFICATION OF RICKETS ACCORDING TO WHAT IS LACKING AT THE OSTEOBLAST-BONE INTERFACE
|
|
|---|---|
|
rickets, and appropriately so because many children with renal
osteodystrophy manifest signs of rickets. However, renal osteodystrophy
classically includes the changes of secondary hyperparathyroidism as
well as those of rickets.
northern industrialized societies in the 19th century. It has now
largely disappeared in developed countries. It remains a significant
clinical problem in the developing world; for example, a 66% prevalence
of clinical rickets was found in preschool children in Tibet in 2001 (75).
(cholecalciferol) can be produced in the skin by a process that
requires ultraviolet B radiation, or it can be ingested in the diet.
The peak age of presentation of nutritional rickets is between 3 and 18
months in children who have inadequate exposure to sunlight and no
vitamin D supplementation in the diet (76,77,78). Breast milk is poor in vitamin D, and prolonged breast-feeding is a risk factor (79,80). Vitamin D is supplemented in dairy foods in North America, and diets deficient in dairy foods are therefore a risk factor (80,81,82). Two hundred international units of vitamin D is the recommended daily amount for preventing rickets (37,62).
Exposure to sunlight also prevents rickets. Two hours per week of
summer sunshine at the latitude of Cincinnati (39 degrees north) is
sufficient to produce adequate vitamin D in the skin. However, during
the winter months in Edmonton (52 degrees north), there is insufficient
UVB exposure to allow for adequate intrinsic production of vitamin D (62).
nutritional rickets, it is possible to get rickets because of a
profoundly calcium-deficient diet, even in the presence of adequate
vitamin D intake (83). Probably much more
common is a subtle combination of calcium deficiency and vitamin D
deficiency interacting to produce dietary rickets (84). This interaction has been described among the modern Asian population in the United Kingdom (85,86) and black populations in the United States (71).
A diet that is low in calcium and high in phytate, oxalate, or citrate
(substances that bind calcium and are found in almost all fresh and
cooked vegetables) means that calcium availability in the diet is poor.
Vegetarians, especially those who avoid dairy products, are
particularly at risk. This lack of calcium produces an increase in PTH,
which in turn increases vitamin D catabolism. Vitamin D status may
already have been marginal because of low exposure to sunlight and poor
dietary intake. The increased catabolism of vitamin D, along with these
factors, results in vitamin D deficiency and the clinical presentation
of rickets. This combination of deficiencies of both calcium and
vitamin D is very prevalent among adolescents who present with rickets
in the United Kingdom and the United States.
provision of vitamin D. A dose of 5000 to 10,000 International Units
(IU) per day for 4 to 8 weeks should be prescribed, along with calcium
to the extent of 500 to 1000 mg per day in the diet (87).
Where daily dosing and compliance pose problems, much larger doses of
vitamin D (200,000 IU to 600,000 IU orally or intramuscularly) have
been given as single doses with good results (88).
rickets may include a low-normal or decreased calcium ion
concentration, a low serum phosphate level, a low serum
25-hydroxyvitamin D3 level, and a high alkaline phosphate level. Alkaline phosphate level drops to normal in response to successful therapy.
Gluten-sensitive enteropathy, Crohn disease, ulcerative colitis,
sarcoidosis, and short-gut syndromes have been implicated. If liver
disease interferes with the production of bile salts, then fat
accumulates in the GI tract and prevents the absorption of fat-soluble
vitamins including vitamin D. The resulting deficiencies of vitamin D
and calcium cause bone disease in the same way as nutritional
deficiencies do, but the treatments need to be aimed at rectifying the
underlying GI problem as well as at supplementing the deficient vitamin
and mineral.
It is an X-linked dominant disorder; this means the female-to-male
patient ratio is approximately 2:1, and there is no male-to-male
transmission. Approximately one-third
of the cases are sporadic (90).
Individuals in whom the condition has sporadically occurred do transmit
the defect to their offspring. The defect is in a gene called PHEX (91).
This gene product indirectly regulates the transport of renal
phosphates. The defect at the kidney is isolated renal phosphate
wasting, leading to hypophosphatemia. In addition, a low or normal
kidney production of 1,25-dihydroxyvitamin D3 is observed, which is inappropriate in the hypophosphatemic state.
Adults with the condition have osteomalacia accompanied by degenerative
joint disease, enthesopathies, dental abscesses, and short stature (95,96,97). The specific treatment for the condition is oral administration of phosphate and the active form of vitamin D3,
calcitriol, which is 1 α-hydroxylated. Treatment requires careful
metabolic monitoring. Hyperparathyroidism, soft tissue calcification,
and death caused by vitamin D intoxication have been problems with
medical therapy in the past. Calcitriol can be used in much lower doses
than the less-active vitamin D metabolites previously used, and is
thought to be a safer therapy (91,98,99). Angular deformities, particularly genu valgum, may persist after medical treatment, requiring osteotomy (100,101,102).
also develop hypophosphatemic rickets. This syndrome includes patients
with café au lait spots, precocious puberty, and fibrous dysplasia of
multiple long bones. The syndrome is caused by constitutional
activation of the cyclic adenosine monophosphate-protein kinase A
(AMP-PKA) signaling pathway, and is related to genetic defects in G
signaling proteins (103).
this was because the early patients were treated with very large doses
of vitamin D. It turns out that such patients have 1 α-hydroxylase
deficiency and they can be treated with much smaller quantities of the
biologically active 1 α-hydroxylated calcitriol (105).
Typically, the patients are less than 24 weeks of age with weakness,
pneumonia, seizures, bone pain, and the skeletal bone changes of
rickets. Serum findings include low calcium and phosphorus, high
alkaline phosphatase and PTH, with a normal level of 25-hydroxyvitamin D3 but a markedly decreased level of 1,25-dihydroxyvitamin D3. The patients are not able to convert the accumulated 25-hydroxyvitamin D3 to its biologically active form of 1,25-dihydroxyvitamin D3 and therefore develop clinical rickets. The autosomal recessive genetic pattern has been described (106) and the specific mutations were initially described in 1997 (107), since which time at least 31 distinct mutations in the 1 α-hydroxylase gene have been identified (105).
described two sisters with clinical rickets. The unusual clinical
feature was an exceedingly high circulating level of
1,25-dihydroxyvitamin D3. In patients having this condition, these levels can be 3- to 30-fold higher than normal (109).
A striking clinical finding is alopecia or near total loss of hair from
the head and body. These patients have an end organ insensitivity to
vitamin 3 (109). Treatment with
very high doses of vitamin D produces a variable but incomplete
clinical response. Intravenous high doses of calcium followed up with
oral calcium supplementation in large quantities has also been tried,
but as yet this rare form of rickets cannot be completely cured (105).
syndrome implies failure of tubular reabsorption of the many molecules
smaller than 50 Da. The kidneys lose phosphate, calcium, magnesium,
bicarbonate, sodium, potassium, glucose, uric acid, and small amino
acids. With this renal tubular abnormality, there are multiple
mechanisms by which bone mineral homeostasis is disrupted (89,110).
As a result, such patients are short, with rickets and delayed bone
age. The predominant cause of bone disease is hypophosphatemia from
renal phosphate wasting, very similar to that seen in X-linked
hypophosphatemic rickets. Other mechanisms include calcium and
magnesium loss, metabolic acidosis caused by bicarbonate loss, renal
osteodystrophy (if renal disease is sufficiently advanced that less
1,25-dihydroxyvitamin D3 is produced), and finally, decreased calcium and phosphate reabsorption.
hypophosphatemia: administration of oral phosphate and vitamin D.
Electrolyte imbalances from other causes need monitoring and treatment,
and the underlying renal disease should also be treated if possible.
overlap with rickets. Hypophosphatasia is caused by alkaline
phosphatase deficiency. Like most enzyme deficiencies, this is a
recessive condition with over 112 mutations described in the alkaline
phosphatase gene in chromosome 1 (111,112,113).
Clinically, alkaline phosphatase deficiency produces abnormal
mineralization of bone with a presentation of rickets in the child or
osteomalacia in the adult (114). Pathologic fractures can occur in children and in adults (115,116).
The fractures are accompanied by abnormal formation of dental cementum,
which causes loss of teeth. The primary teeth are lost early and with
minimal root resorption (117). Additional clinical manifestations may include failure to thrive, increased intracranial pressure, and craniosynostosis.
 |
|
Figure 7.5
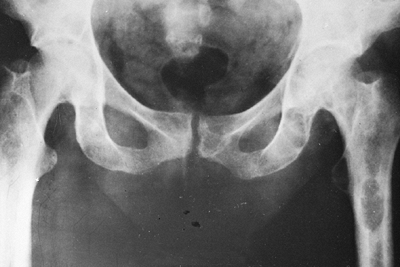
Radiograph of the pelvis of a patient with renal osteodystrophy shows the marked changes of secondary hyperparathyroidism. Several brown tumors are seen in the femoral shafts and ischial rami. These appear as expanded destructive lesions, resembling primary or metastatic bone tumors. |
There is a perinatal lethal form of the condition. A childhood form
presents with rickets at 2 or 3 years of age and remission of the
disease in adolescence. An adult form presents with mild osteomalacia
with pathologic fractures (111).
underlying defect. Bone marrow transplantation has been used
experimentally in severe cases, with the aim of repopulating the bone
marrow with osteoblasts capable of producing alkaline phosphatase (111).
Surgical treatment of femoral fractures and pseudofractures in the
adult has been reported, with rodding techniques found to be superior
to plating techniques in the abnormal bone (115).
accompanying end-stage renal disease; it is commonly seen in patients
on dialysis. The clinical presentation includes hyperparathyroidism as
well as rickets with osteomalacia in varying combinations (119,120,121,122).
from the blood once the renal function drops below 25% to 30% of
normal. The hyperphosphatemia drives the solubility equilibrium to
produce hypocalcemia, which signals the parathyroid glands to produce
PTH, causing secondary hyperparathyroidism. The bony changes of
hyperparathyroidism then become evident. These include subperiosteal
erosions and brown tumors (Fig. 7.5). The
subperiosteal erosions are described as classically appearing on the
radial margins of the middle phalanges of digits 2 and 3 of the hand in
adults. In children, they can also be seen at the lateral aspects of
the distal radius and ulna and at the medial aspect of the proximal
tibia (Fig. 7.6) (123).
Prolonged stimulation of the parathyroid glands can produce sufficient
hyperplasia to make the glands remain autonomous and maintain a
hyperparathyroid state, even if the end-stage renal disease is treated
by transplantation. In this case, the ongoing hyperparathyroidism is
described as tertiary rather than secondary.
there is inadequate renal mass to produce sufficient
1,25-dihydroxyvitamin D3 rickets (clinical and radiographic)
will accompany renal osteodystrophy. The clinical manifestations can
include varus or valgus deformities at the knees or ankles, and
radiographic evidence of widened and deformed growth plates and
rickets/osteomalacia such as Looser zones (Fig. 7.7).
-
Dietary phosphate restriction
-
Phosphate binding agents, especially those which are calcium-containing
-
Vitamin D, particularly calcitriol, to
decrease the secondary hyperparathyroidism as well as to treat clinical
rickets or osteomalacia -
Restoration of renal function by transplantation in order to improve the musculoskeletal manifestations
and occurs at a younger age, in children who are typically small
because of their chronic disease. Therefore, stabilization of the slip
should permit ongoing growth of the proximal femur if possible.
Unstable slips and avascular necrosis are rare in patients with renal
osteodystrophy, but avascular necrosis possibly associated with steroid use posttransplant has been reported (128).
If the child is young, the slip severe, and the bone disease has not
yet been treated medically, then traction plus medical treatment have
shown very good results. When considering surgical treatment of the
slipped epiphyses, the high incidence of bilaterality suggests that
both epiphyses should be stabilized. In young patients, a pinning
technique that allows for growth (smooth pins across the physis) can be
considered (127). Hardware cutout, including
pin protrusion into the joint, is more likely with the soft bone of
renal osteodystrophy, but has generally been associated with inadequate
medical control of hyperparathyroidism (127,128).
 |
|
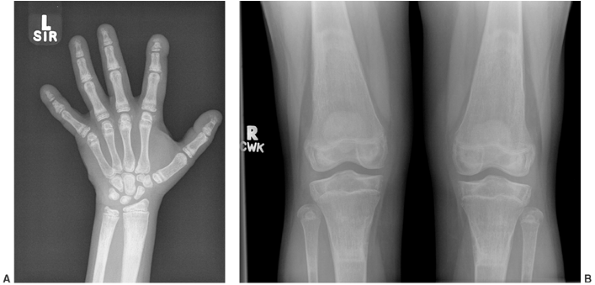
Figure 7.6 Renal osteodystrophy in an 8-year-old boy. A:
Radiographs of the hand show sclerosis, acroosteolysis, and soft tissue calcification around the metacarpal phalangeal (MCP) and proximal interphalangeal (PIP) joints. B: Radiographs of the knees show subperiosteal resorption at the medial border of the proximal tibia. |
 |
|
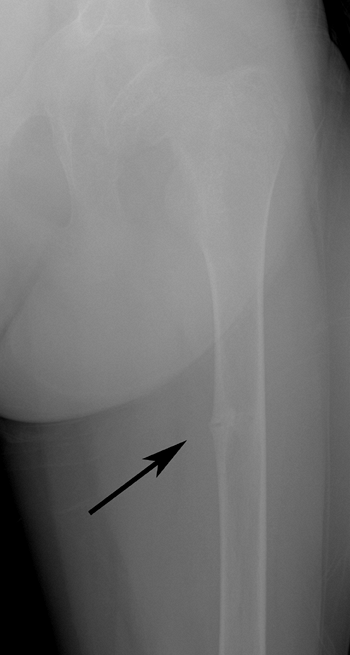
Figure 7.7 Renal osteodystrophy. A Looser zone is evident (arrow) in the medial femoral diaphysis.
|
(2000) has defined osteoporosis as “a skeletal disorder characterized
by compromised bone strength predisposing to an increasing risk of
fracture.” The Consensus Panel notes that bone strength includes both
bone density and bone quality. Childhood osteoporosis can come from
numerous primary and secondary etiologies, as summarized in Table 7.3.
with osteoporosis, and 18 million more with low bone mass are at risk
for osteoporosis (129). We associate
osteoporosis with senescence, and certainly most of the individuals
currently affected are old and not likely to be consulting pediatric
orthopaedists. However, the NIH emphasizes that “sub-optimal bone
growth in childhood and adolescence is as important as bone loss to the
development
of osteoporosis.” The recommended intake of calcium for children aged 9
to 18 is 1300 mg per day, and it is estimated that only 10% of girls
and 25% of boys meet this minimum requirement (129).
Whereas consumption of dairy-based beverages supplying calcium has
declined, consumption of carbonated beverages has increased (130).
Phosphoric acid is used in cola soft drinks to maintain carbonation,
and teenage girls who drink soft drinks are three to four times more
likely to report fractures than those who do not, the association being
strongest among active girls drinking cola (131).
The pediatric orthopaedic surgeon has at least three reasons to be
interested in osteoporosis. First, osteogenesis imperfecta (OI) is an
example of primary osteoporosis with profound implications for medical
and surgical management during childhood. The understanding of this
condition is advancing rapidly. Second, many children treated by
pediatric orthopaedists already have, or are at risk for, secondary
osteoporosis—especially nonambulatory children with neuromuscular
disorders. Finally, prevention of the most common osteoporosis
associated with aging, and its attendant morbidity, has to include a
focus on acquisition of adequate bone mineral strength in childhood and
adolescence—something pediatric orthopaedic surgeons are ideally
positioned to promote among their patients, in their community, and
nationally (132,133).
 |
|
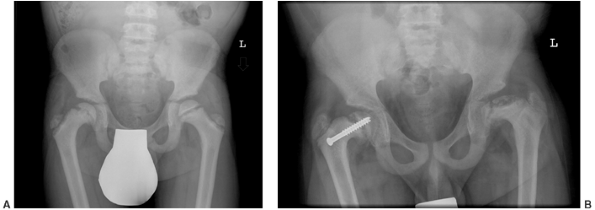
Figure 7.8 Renal osteodystrophy in a 12-year-old boy. A:
An anteroposterior pelvis x-ray reveals an early slipped capital femoral epiphysis on the right. Slipped capital femoral epiphysis is common in renal osteodystrophy and rare in rickets. B: Three years after fixation the right proximal femoral epiphysis remains open and stable; the left hip now shows signs of epiphyseal avascular necrosis and fragmentation. |
|
TABLE 7.3 CLASSIFICATION OF CHILDHOOD OSTEOPOROSIS
|
|
|---|---|
|
clinical disorders that share the symptom of abnormal bone fragility.
Most patients with OI have disorders of collagen production that affect
either the quantity or quality of collagen produced. Many of these
disorders can be traced to specific mutations in collagen genes, but
there are myriad such mutations. The phenotype of OI is quite variable.
It includes mildly affected individuals of normal stature without any
skeletal deformities and also those with extensive bony fragility, who
suffer dozens of fractures during childhood, are short in stature, and
have deformed, bowed
extremities and abnormal facial appearance. The most severe form of OI is fatal in the perinatal period.
management of the fractures and deformities that result from OI.
Diagnosis of milder forms of OI among children with frequent fractures
is easy if the sclerae are abnormal (blue or gray) but can be
challenging if they are normal (white). It can be particularly
difficult to distinguish mild OI from inflicted injury.
The hallmark of OI is brittle bones and the tendency to fracture, with
recurrent fractures occurring in childhood, in particular during the
preschool years. Bone pain is a feature in many patients. It is
described as chronic and unremitting and usually relates to old
fractures. Muscle weakness may be variously present. Ligamentous laxity
and joint hypermobility may be present. Wormian bones are present in
the skull in approximately 60% of patients. Abnormal collagen in the
eye leads to the blue or gray-blue sclerae classically associated with
OI. Abnormal collagen in the teeth leads to dentinogenesis imperfecta
(clinically small, deformed teeth which are “opalescent” due to a
higher ratio of transparent enamel to opaque dentin); this is present
in some, but not all, patients with OI (135–140).
a very wide spectrum. The clinical classification scheme that is most
commonly used is based on the one first proposed by Sillence et al. in
1979 (140). The Sillence classification has
stood the test of time and is a useful way of dividing the phenotype.
This classification has recently been modified (138,141) to incorporate genetic and biochemical abnormalities (Table 7.4). Greater genetic understanding has led to the addition of extra types to the four OI types initially described by Sillence.
low-normal height and do not have limb deformities; they share the
hallmark of bony fragility and often have multiple fractures during
childhood. Fractures become less common after puberty. Blue sclerae are
present in type I OI. Fifty percent of patients also have presenile
deafness (142,143); this presents typically in the third decade of life and therefore is not helpful for diagnosing children (144).
The deafness itself has a conductive component and a sensorineural
component, and is sometimes of sufficient severity to require surgery
for the ossicles of the ears, or cochlear implantation in very severe
cases (145,146).
Fractures which occur in type I OI include spiral and transverse
fractures of long bones, particularly lower extremity bones. In
addition, the avulsion type of fractures, such as olecranon fractures (147)
and patellar fractures, are common and are related to the decreased
tensile strength of the bone because of its underlying low collagen
content.
|
TABLE 7.4 CLASSIFICATION OF OSTEOGENESIS IMPERFECTA
|
|||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||||||||||||||||
with the condition die at or shortly after birth; they are born with
crumpled femora and crumpled ribs accompanied by pulmonary hypoplasia,
which usually leads to death. Central nervous system malformations and
hemorrhages are common due to the markedly abnormal collagen being
produced. Lethal OI can be diagnosed by prenatal ultrasonography.
Short, broad limbs are identified with low echogenicity and low
shadowing, and it is easier to see soft tissue features such as orbits
or arterial pulsations within the fetus. At present, it is not possible
to reliably distinguish on prenatal ultrasound between lethal type II
OI and severe but survivable OI type III described in the subsequent
text. Most patients with the lethal form of OI have blue sclerae,
although some are born with white sclerae (139,140,148,149).
These patients have relatively large skulls but undeveloped facial
bones, leading to a characteristic triangular appearance of the face.
The sclerae of patients with type III OI are described as pale blue at
birth, but become normal in color by puberty. Patients are short, with
severe limb deformities including bowing and coxa vara (Fig. 7.9). Multiple vertebral compression fractures lead to severe scoliosis, kyphosis, and rib
cage deformity. Many patients use wheelchairs for mobility, or require
walking aids. Radiographic characteristics include very osteopenic
bones with deformity related to previous fracturing. A characteristic
popcorn appearance of the epiphysis and metaphysis occurs in early
childhood. The pedicles of the vertebrae are elongated. The vertebrae
are wedged and may assume a codfishlike biconcave morphology. Posterior
rib fractures are seen. Additional clinical features may include
basilar invagination of the skull; this can present with headache,
lower cranial nerve palsy, dysphagia, limb hyperreflexia, nystagmus,
hearing loss, or quadriparesis. These patients often have multiple
fractures when they are born, but they do not have the severe thoracic
deformities seen in type II OI. Fractures heal at the normal rate but
recur frequently during childhood, particularly in the preschool years;
some patients with this form of OI have over 100 fractures (148,149).
 |
|
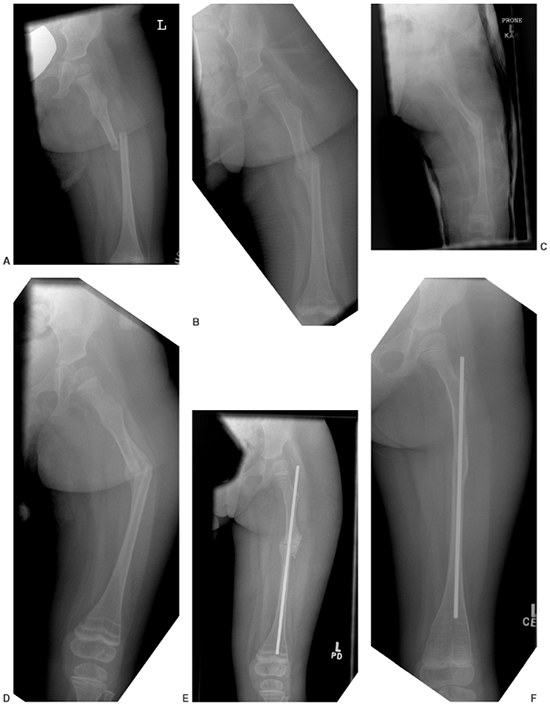
Figure 7.9 This female infant with severe osteogenesis imperfecta (OI) presented at 19 months of age with a left femoral fracture (A) which was treated in a spica cast and healed (B). A refracture was treated in a spica (C) with progressive varus. At age two, a second refracture occurred through the varus malunion (D) and was treated with open intramedullary (IM) Williams rodding (E).
Three years later the femur is intact and has grown distally, as evidenced by the position of the rod and by the transverse metaphyseal lines that occur with each pamidronate treatment cycle (F). |
presentation. Most have short stature and many have bowing and
vertebral fractures, although these are not as severe as in patients
with type III OI. Most of the patients are ambulatory, although some
use walking aids. There is a wide range of ages at the first fracture
and number of fractures in patients with type IV OI. Dentinogenesis
imperfecta may or may not be present in these patients. The sclerae are
typically white (139,140).
most of those who present with OI. There is some overlap in the
phenotypes that can make them difficult to distinguish—for instance,
type III and type IV OI. As the genetic defects are understood, at
least three more types of OI have been added, and these do not fit into
the scheme described in preceding text. Type V OI is described as a
hypercallus variety of OI (150). These patients develop profuse amounts of extraosseous callus following their fractures (Fig. 7.10),
and the presentation can be confused on clinical and radiographical
evidence with an osteosarcoma, although type V OI usually occurs in
much younger children. An additional clinical feature is the
ossification of interosseous membranes between the tibia and fibula,
and between the radius and ulna; this leads to the clinical sign of
diminished or absent pronation and supination of the forearm, which can
suggest the correct diagnosis. Type VI OI includes individuals who
phenotypically appear to have moderate or severe OI similar to a type
IV presentation (151). However, these patients
are known to have normal collagen, and they have a defect in new bone
mineralization without having any of the biochemical abnormalities or
growth-plate deformities associated with rickets. The exact etiology of
this condition remains uncertain. Type VII OI was initially described
in a cohort of First Nations individuals from Quebec, Canada (152).
These patients have rhizomelia, and the characteristic deformities are
coxa vara of the long bones. The bone is histologically similar to what
is seen in type I OI. Linkage analysis shows that the defect is on
chromosome 3, and therefore not in a collagen gene.
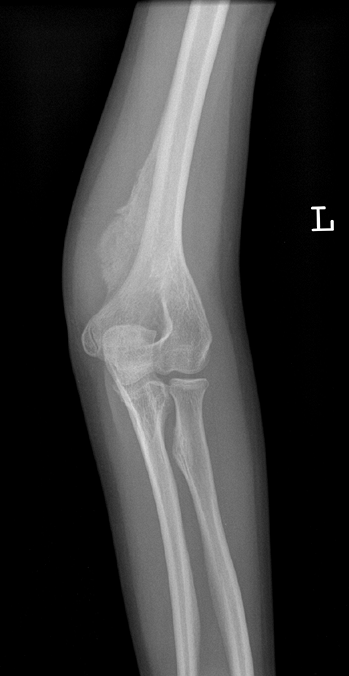 |
|
Figure 7.10
Osteogenesis imperfecta (OI). The excess callus formation around the distal humerus following injury (no displaced fracture was seen) is typical of type V OI. |
the collagen 1A1 gene found on chromosome 7q, or mutations in the
collagen 1A2 gene found on chromosome 17q. The complete list of
mutations found is kept up-to-date at the OI mutation database at http://www.le.ac.uk/genetics/collagen.
This is usually the result of a premature stop codon within the gene
and results in the production of nonsense messenger RNA instead of
proper messenger RNA coding for the preprocollagen of the collagen
molecule. The nonsense messenger RNA is detected and destroyed, and the
result is production of a diminished number of α 1 chains, and
consequently a decreased
quantity
of normal collagen being produced. With the decreased quantity of
collagen, the bone is weakened and becomes more susceptible to
microfractures. Bone can sense its mechanical environment, and
microfractures cause a new round of bone removal and reformation,
leading to constant activation of bone remodeling in OI. This process
demands an increased transcriptional activity of type I collagen and
induces more osteoclasts and an increase in the excretion of collagen
degradation of products. Bone turns over rapidly, but remains of poor
quality. When growth ceases at puberty, the transcriptional activity
demand is reduced and bone turnover can slow down; then the bone
strength and architecture become closer to normal, and the fracture
rate drops.
The collagen molecule is a triple helix formed by spontaneous
self-assembly of three long linear procollagen molecules. These
procollagen molecules have a typical repeating amino acid pattern of
glycine XY-glycine XY-glycine XY. Glycine appears at every third
position because it is the smallest amino acid and can be folded into
the interior of the triple helix. Glycine substitutions place a larger
amino acid where the glycine residue belongs, so that the collagen
triple helix cannot assemble appropriately. The collagen molecule
begins assembling at the C-terminal end and assembles toward the
N-terminal end. If the mutation substitutes for a glycine close to the
C-terminus at the beginning of the molecule, then a very short strand
of abnormal gene product is produced, and the corresponding clinical
diseases are severe. If the glycine substitution mutation is toward the
distal N-terminus end, then a longer partial collagen molecule can be
formed, and the clinical phenotype is less severe.
an abnormal quantity or quality of collagen. Mechanically the bone is
more ductile, rather than being more brittle (156).
Secondary osteoporosis caused by immobilization following fractures or
surgery, or by decreased physical activity and weight bearing with
severe deformity, adds to the complications of the primary defect.
Prevention of secondary osteoporosis is an important concept when
treating fractures, planning surgery, or recommending general care.
of OI. There is no single laboratory test that distinguishes
individuals with OI from those with normal bone. The clinical features
of severe OI type II and type III are distinct enough that physical
findings and plain radiography are usually sufficient for a diagnosis.
Patients with type I OI have blue or blue-gray sclerae and are readily
identified clinically. Normal babies may have blue sclerae until 1 year
of age, so this finding has diagnostic value only in the older child.
Patients with mild presentations of type IV OI are easy to diagnose if
they have dentinogenesis imperfecta, but those with normal teeth may
benefit from additional investigation, depending on the purpose of
making the diagnosis. Dual energy x-ray absorptiometry (DEXA) scanning
shows low levels of lumbar and femoral bone mineral density (BMD) in
patients with mild OI (157,158). Published values for BMD in healthy normal children are available for comparison (159).
Caution must be exercised in interpreting DEXA scans in children, and
overdiagnosis of osteoporosis is reported to be frequent (160);
this is because DEXA scanning reports BMD per square centimeter surface
area of bone, ignoring the third dimension (thickness of the bone in
the path of the photon), which is larger in adults, thereby leading to
a higher area density even if the true volumetric density were to
remain a constant.
collagen 1A1 and collagen 1A2 genes producing the main phenotypes of
type I, type II, type III, and type IV OI (138,148,149,154,161).
As such, many new patients often have new mutations specific to
themselves. Accordingly, a DNA-based genetic test for OI is not
currently clinically useful. An intermediate level of testing involves
culturing dermal fibroblasts and studying the collagen that they
produce. Quantitatively abnormal collagen production can be detected in
87% of individuals with known OI. Conversely, 13% of individuals with
known OI would be missed by a cultured dermal fibroblast test. One
common question is whether a patient has OI or inflicted trauma. OI is
very rare, and inflicted injury remains significantly more prevalent.
Clinical diagnosis remains a gold standard to distinguish these two
entities, and cultured dermal fibroblast testing is not considered
useful as a routine part of such investigation (162).
In cases with legal implications, positive findings in the past
history, family history, clinical examination, or properly interpreted
DEXA scan may assist in the diagnosis of osteoporosis or OI. OI cannot
be entirely ruled out in patients with negative findings, but is a
highly unlikely diagnosis in the presence of findings suggesting child
abuse (discussed elsewhere). Finally, a diagnosis of OI does not
exclude the possibility of child abuse.
OI, but cannot currently be recommended for mild OI. Bisphosphonates
are widely used drugs based on the pyrophosphate molecule, which is the
only natural inhibitor of bone resorption. The exact mechanism of the
drug is unclear, but its primary action is at the level of the
osteoclast. Glorieux et al. reported the effects of bisphosphonate
treatment in an uncontrolled observational study of 30 patients with
severe OI (163). The intravenous dose given was
3 mg per kg of pamidronate per cycle by slow intravenous infusion at
4-month intervals. All patients were given 800 to 1000 mg calcium and
400 international units of vitamin D per day. There was marked
improvement
in the clinical status of the patients. BMD increased at an average
rate of 42% per year. There was an increase in the cortical width of
the metacarpals and in the size of the vertebral bodies. The average
number of fractures dropped from 2.3 per year to 0.6 per year. No
nonunions or delayed unions of any fractures were noted. Patients
reported a marked reduction in bone pain 1 to 6 weeks following
initiation of treatment. The only adverse effect noted was the acute
phase reaction comprising fever, back pain, and limb pain on day two of
the first cycle. This adverse effect was treated with acetaminophen and
did not recur with subsequent infusion cycles. Mobility improved in 16
of the 30 patients treated, with no change in the other 14. Growth
rates increased. Patients younger than 3 years showed a faster and more
pronounced response to the bisphosphonate. The direct effect of the
bisphosphonate is decreasing resorption and turnover of bone. The
resulting decrease in bone pain and fractures led to increased weight
bearing and mobility. It is likely that the increased weight bearing
and mobility would have resulted in further strengthening of bone and
muscle. Bisphosphonate treatment has become a standard for severe OI.
It should be noted that this drug does not address the basic
abnormality underlying OI, but it does alter the natural course of the
disease. Radiographs of patients with OI treated with bisphosphonates
show characteristic dense sclerotic lines that form at the growth
plate, one per treatment cycle (164).
for bisphosphonate treatment to patients with milder forms of OI.
Reported clinical results apply only to patients with severe OI.
Osteopetrosis is a reported complication of bisphosphonates in humans (165). Animal studies suggest reduced longitudinal bone growth with these drugs (166,167,168).
Randomized controlled clinical trials are needed before routine
clinical use of bisphosphonates in milder forms of OI is considered.
specifically human growth hormone. Human growth hormone is an anabolic
agent, and therefore stimulates increased bone turnover, causing a
higher demand for collagen transcription and perhaps exacerbating the
underlying abnormality while attempting to ameliorate the decreased
stature. Because growth hormone may have both beneficial and negative
effects in OI, clinical research results are required before
indications can be stated. We suggest at present that growth hormone be
used in patients with OI only in the context of clinical research
studies.
modification of standard surgical treatments, whereas those with severe
(deforming) OI require multiple specialized surgical techniques and
implants.
such a way as to minimize disuse osteoporosis. Simple undisplaced
fractures can be treated with plaster cast or splints which should be
removed early to allow prompt return to function. Avulsion fractures of
the olecranon can be treated by tension band wiring and early motion
with good results (Fig. 7.11).
very fragile lower extremities can be supported with vacuum orthoses to
permit protected weight bearing (169).
Eventually, however, children with severe OI will be considered for IM
rodding of the long bones. This IM rodding is best carried out in
conjunction with medical treatment using bisphosphonates in an
interdisciplinary setting. Some centers propose that lower extremity
rodding be considered as soon as the patient stands and before walking
begins, at about the age of 18 months (148).
Upper extremity rodding is indicated if repeated long bone fractures
cause deformity which affects function, in particular, if the use of
crutches or a walker is compromised (148,170).
bowed long bones typical of severe forms of OI. Rodding allows for
correction of the deformity, puts the weight-bearing line along the
axis of the bone, is effective in soft bone, and allows for continued
growth.
fragmentation and rodding of the long bones in 1952 and reported the
results in 1959 (171). He described a long open
exposure with removal of the entire diaphysis of the bone, division
with multiple transverse osteotomies, and reassembly over a rod.
Williams (172) developed a threaded end rod
that can be attached to an insertor (which simplifies placement of the
rod in the bone) and then detached. Bailey and Dubow developed a rod
with overlapping male and female shafts allowing for expansion of the
rod during normal bone growth (Fig. 7.12). The
rod is anchored into the epiphysis using a t-piece at each end.
Complications of extensible rods are frequent; Nicholas and James (173)
reported 56 roddings in 16 patients, with 6 failed expansions, 12 t-end
loosenings, 4 rod migrations, 6 bent rods, and 5 fractures. Separation
of the rods after growth led to fracture and bowing in every case, and
close radiographic surveillance was proposed. Comparison of
nonelongating to elongating rods has shown slightly higher complication
rates with elongating rods, although crimping the t-piece may reduce
this complication rate. A newer elongating rod design, the Fassier
Duval rod, has cancellous screw threads at either end to provide stable
anchorage in the epiphysis or metaphysis (174).
In one series, the need for revision due to bone growth and rod
migration was reported as being higher for nonelongating rods (175), but in another series it was reported as being approximately equal to that seen with elongating rods (176). Methods of rod exchange through percutaneous techniques have been described (177,178), and a stereotactic device to assist this has been developed (178) but is not in wide clinical use.
rodding (180,181,182,183), but one emphasizes the possibility of ambulatory status worsening in a large number of patients (184).
Ultimately, the prognosis with regard to walking is much more strongly
influenced by the subtype of OI than by the treatment (185,186,187),
but modern combinations of medical and surgical treatment combined with
rapid advances in the understanding of the biology of the disease may
one day change this situation.
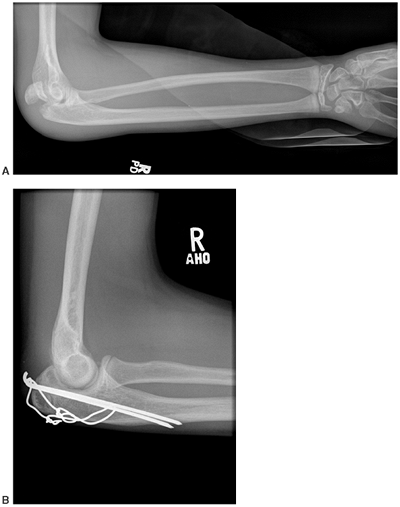 |
|
Figure 7.11
Osteogenesis imperfecta (OI). This olecranon avulsion in a 14-year-old boy with type 1 OI was treated with stable internal fixation and early return to motion. Olecranon avulsions are frequently associated with a diagnosis of OI. |
Patients with large progressive curves may suffer from pulmonary
compromise, and bracing can cause rib deformities that worsen it. It is
unknown whether operative management of the scoliosis leads to improved
pulmonary function, quality of life, and survival. Accordingly,
decision making regarding surgery must be individualized for each
patient. Spinal fusions have a higher complication rate in OI, with 20
patients out of 60 experiencing a total of 33 major complications (194).
The most common complications were blood loss greater than 2.5 L (nine
cases), intraoperative hook pullout (five cases), postoperative hook
pullout (five cases), and pseudarthrosis (five cases). Strategies to
prevent hook pullout include load sharing via segmental
instrumentation, supplementation of hook site bone with methyl
methacrylate, and consideration of fusion without instrumentation.
Preoperative medical treatment (bisphosphonate) to strengthen the bone
is logical but results are not yet reported. Progression of the curve
postoperatively, with or without pseudarthrosis, may occur (194).
The natural history, expectations, likelihood of complications, and
likelihood of success must be carefully assessed for each patient, and
decisions not to embark on surgical reconstruction are sometimes
correct.
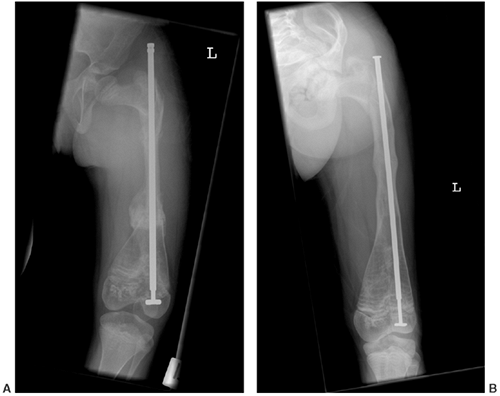 |
|
Figure 7.12 Osteogenesis imperfecta (OI). A: An extensible rod is used in this 7-year-old girl with severe OI. B: The rod has grown with the femur over a 3-year period.
|
or long bone fractures and bone pain. Fractures are typically
metaphyseal in location. Kyphosis, scoliosis, and pectus carinatum
deformities may be present. BMD is decreased 2.5 standard deviations
below age-appropriate norms. Idiopathic juvenile osteoporosis is a
diagnosis of exclusion; that is, other primary and secondary causes of
osteoporosis must be excluded (Table 7.3).
There are no blood test abnormalities specific to the diagnosis, and
the genetic cause is as yet unknown. Treatment includes optimizing the
intake of calcium and vitamin D and promoting physical activity,
including weight bearing and strength training but avoiding trauma.
Bracing can be used to control vertebral pain and to prevent
progression of kyphotic deformities (198).
Judicious use of bisphosphonate therapy may be considered in severe
cases. A remarkable remission of the condition at puberty is common.
there are several other primary causes of reduced BMD in children.
Children with Ehlers Danlos syndrome, Marfan syndrome, and
homocystinuria have reduced bone mass. Those who suffer fractures or
bone pain can also be considered to have a form of primary
osteoporosis, but fractures are not as typical a feature of these
conditions as they are of OI. Like OI, these conditions are based on
known deficiencies in the production of structural proteins. These
conditions are discussed in other chapters.
The phenotype is similar to OI, with thin bones, fractures, bone pain,
and blue or white sclerae. Joint contractures are a distinctive
clinical characteristic (199). The condition is a result of failure to cross-link collagen fibrils, and is specific to bone tissue (200).
autosomal recessive disorder phenotypically similar to OI but
accompanied by congenital blindness due to hyperplasia of the vitreous
humor (201,202). The mutation is in the low-density-lipoprotein-receptor–related protein 5 gene (203). Treatment with bisphosphonates has been successful (204).
Surgical management of fractures with intramedullary rodding techniques
has encountered complications related to severe fragility of the bone (205).
chapter. Interference with normal homeostatic mechanisms leads to
reduced bone mass—for example, many children have disuse osteopenia
following fracture treatment. Resumption of load bearing after healing
allows this to normalize. In conditions where weight bearing is
reduced, persistent low bone mass can lead to low-energy fractures and
a cascade of repeated fractures in the same extremity.
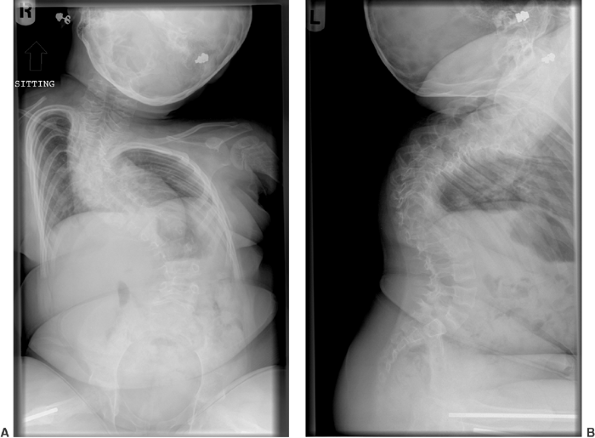 |
|
Figure 7.13 Osteogenesis imperfecta (OI). Marked spinal deformity in a 5-year-old girl with severe OI.
|
BMD. The fat-derived hormone leptin acts on hypothalamic neurons that
mediate bone mass via the sympathetic nerves (206,207,208,209).
The clinical significance of this recent finding has not been
elucidated, but it may become important in managing osteoporosis and
related regional conditions such as reflex sympathetic dystrophy. At
present, the management of neuromuscular osteoporosis focuses on the
downstream effects of the neuromuscular abnormalities on load bearing
and nutrition.
palsy (CP) (210–214). Typically, it is nonambulatory patients with the
lowest bone masses who are at risk for pathologic and low energy
fractures, but reduced bone mass has been observed in the affected
limbs of ambulatory hemiplegics also (215,216),
thereby suggesting that both weight bearing and muscle forces play
roles in the establishment and maintenance of bone mass. After absence
of ambulation, undernutrition is the second most important predictor of
low bone mass in patients with CP (214,217).
Fractures and a “fracture cascade” of increasing disuse osteoporosis
from treatment-related immobilization can cause significant problems
for many patients (218,219). Fracture prevalence among children and adolescents with moderate to severe CP was 26% in one study (214).
The orthopaedic surgeon should focus as much on the prevention as on
the treatment of osteoporosis in the CP population. Increasing weight
bearing, optimizing nutrition, and minimizing the extent and duration
of immobilization following surgery are important. A controlled trial
of weight bearing in patients with CP showed significant increases in
femoral neck bone mineral content (220). Medical treatments including vitamin D and calcium (221) or bisphosphonates (222)
have also been reported to increase BMD in patients with CP, although
the precise indications for their prescription are unclear.
Approximately one-third of children without CP suffer a fracture during
childhood, so the 26% fracture prevalence among children with CP does
not seem out of line. Accordingly, if weight bearing is easy to achieve
and enjoyable for the child it can be recommended; but if
extensive
surgery, elaborate standers, and many hours of professional care are
required, then perhaps it is not of benefit to the child.
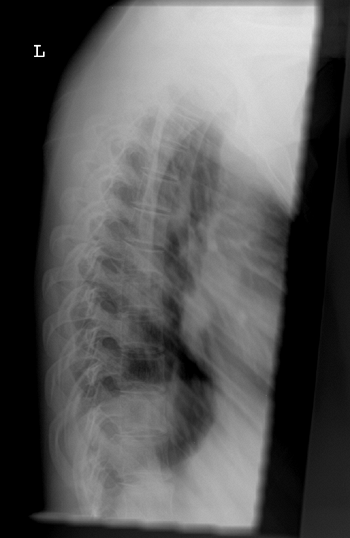 |
|
Figure 7.14
Juvenile idiopathic osteoporosis. This 14-year-old boy has marked osteoporosis evident on plain radiographs [dual energy x-ray absorptiometry (DEXA) scans indicated bone mineral densities 2.8 standard deviations below mean] and a healed compression fracture at T7. |
bone mass as the lower extremities weaken, with bone densities 1.6
standard deviations below the mean while they are still walking and 4
standard deviations below the mean once they become nonambulatory. Of
41 boys followed, 18 sustained fractures, 12 of which were lower
extremity fractures, and 4 of which caused loss of ambulation (223).
In boys with DMD, BMD has been reported as lower among those who were
treated with steroids (prednisone) than among untreated boys (224).
Deflazacort is a steroid medication which preserves muscle strength,
ambulation, and respiratory function as well as preventing the onset of
scoliosis. Despite being a glucocorticoid analog, it is reported to
have bone-sparing effects (225) and randomized
trial evidence (in juvenile arthritis) shows less bone loss among
patients treated with deflazacort compared with patients treated with
prednisone (226). Vertebral fractures have been reported in patients treated with deflazacort (227), whereas they are rare among untreated patients (223). No trials of bisphosphonates in DMD have been reported yet.
stature and low muscle mass. BMD is low in both adults and children,
and adults have a 2.7 times increased fracture rate compared with
normal controls (228). Treating the deficiency
with growth hormone improves longitudinal growth and muscle mass and
also improves calcium absorption, thereby exerting beneficial effects
on the osteoporosis both mechanically and biologically. Improved BMD
with growth hormone treatment has been documented in adults but
requires further study in children (229,230).
second most common endocrinologic cause of SCFE (after hypothyroidism).
Ninety-two percent of children with growth hormone deficiency and SCFE
present with the slip after growth hormone
treatment has been initiated, perhaps because the growth plate becomes
more active and potentially weaker, mechanically, relative to the
larger size of the body. The prevalence of bilaterality in slips with
endocrinopathies has been reported to be as high as 61%, and therefore
prophylactic pinning of the contralateral side has been suggested by
some specialists (231). Among 2922 children
followed prospectively while receiving growth hormone in Australia and
New Zealand, only 10 slipped epiphyses were reported by clinicians
performing active surveillance for complications (232).
bone turnover. T3 directly stimulates osteoblasts, resulting in linked
osteoclast activation and bone resorption. The increase in serum
calcium and phosphate suppresses PTH and 1,25-hydroxy vitamin D
production, which then decreases calcium and phosphate absorption and
increases calcium excretion. The net result is bone resorption in a
high turnover state (133). Treatment of the underlying disease to achieve a euthyroid state is the appropriate management for the osteoporosis.
during the pubertal years, and sex steroids are necessary for this
accelerated bone mineral accrual. Mineral accrual lags behind the
longitudinal growth spurt by a year or two, perhaps partially
explaining the increased fracture rate documented during and just after
the growth spurt (233). Over the last 30 years
there has been a statistically significant increase in the incidence of
distal forearm fractures in adolescents, but it is unclear whether this
relates to changes in diet or in physical activity (234).
bone mineral accrual. It can be treated with gonadotropin-releasing
hormone analogs to preserve epiphyseal function and allow attainment of
greater final height. Such treatment does not adversely affect peak
bone mass. Constitutionally delayed puberty in boys is associated with
lower BMD, but it is unclear whether testosterone treatment improves overall mineral acquisition (133).
for sport. Thirty-one percent of college level athletes who were not
using oral contraceptives reported athletic amenorrhea or
oligomenorrhea (235). Athletic amenorrhea plus disordered eating plus osteoporosis has been dubbed the “female athlete triad” (236,237). The full triad is rare among athletes (238) and was found to be nonexistent among female army recruits (239).
Not all sports are equal in propensity for bone loss. Gymnasts exhibit
higher bone mass than do runners despite similar prevalance of
amenorrhea (240); gymnasts have both higher muscle strength (241) and higher serum insulinlike growth factor 1 (IGF-1) (242)
than do runners, and these are both protective of bone mass. It must be
remembered that for the general population, more exercise means more
bone, as has been demonstrated in randomized trials of physical
activity in prepubescent and pubescent children (243,244).
The long-term prevalence of fractures amongst people with anorexia
nervosa is 57% at 40 years follow-up, 2.9 times higher than that in the
general population (246). Estrogen alone is insufficient to restore BMD, particularly if nutrition is compromised (247,248).
Restoration of body mass, provision of adequate calcium and vitamin D,
and correction of the hormonal environment should all occur.
hypothalamic–pituitary axis was associated with osteoporosis in 3 and
osteopenia in 10 of 19 girls (ages 16 to 18), compared with 0 among 20
controls with regular cycles (249).
and chronic medical conditions. Frequent short courses of oral
glucocorticoids (for asthma exacerbation) have been shown not to
adversely affect BMD in children (250), but
chronic glucocorticoid use is a well-established cause of osteoporosis
and is common among patients with juvenile arthritis, leukemia, and
organ transplantation. The underlying mechanism includes osteoblast
apoptosis and decreased intestinal calcium absorption and renal
reabsorption (133). Deflazacort is a bone-sparing glucocorticoid (225),
and randomized trial evidence (in juvenile arthritis) shows less bone
loss among patients treated with deflazacort than among patients
treated with prednisone (226). In adults using
glucocorticoids, there are now multiple trials demonstrating that
bisphosphonates are useful for prevention and treatment of bone loss
and for prevention of fractures (251,252,253,254). Case series in children have shown similar promising results (255).
Research is active in this area and it is likely that prevention and
treatment trials with children on long-term steroids will further
clarify the appropriate role for bisphosphonates, as well as for novel
agents such as subcutaneous PTH (256,257).
although the exact mechanism by which anticonvulsants interfere with
bone metabolism remains unknown. Induction of liver enzymes with
consequent increased vitamin D catabolism has been proposed (265,266), but other studies have reported vitamin D metabolism to be normal (267,268). There is laboratory evidence that phenytoin and carbamazepine exert a direct effect on bone cells (269).
Quantitative computerized tomographic studies of the distal radius in
patients using carbamazepine or valproic acid (for isolated epilepsy
without CP) showed decreased trabecular BMD but a compensatory increase
in cortical BMD in a high bone turnover state (270).
A controlled trial has shown that treatment with vitamin D and calcium
increases BMD in children with severe CP who are receiving
anticonvulsants (221).
osteoporosis in children include methotrexate, cyclosporine, and
heparin. Little information is available on prevention and treatment of
iatrogenic osteoporosis in children (133).
of the sclerosing bony dysplasias. All of these conditions are
characterized by an imbalance between the formation and the resorption
of bone, favoring formation. In osteopetrosis, it is decreased or
completely failed resorption of bone due to an osteoclast defect that
tips the balance in favor of formation.
The infantile or autosomal recessive form is the most severe and can be
fatal in the first decade of life. Patients present with pathologic
fractures or with an exceedingly dense radiographic appearance of the
bones (Fig. 7.15); they may have cranial nerve
problems including optic nerve compression and blindness, facial nerve
dysfunction, and sensorineural deafness due to narrowing of the skull
foramina; they may have bone pain related to fractures or stress
fractures; and they frequently have infections of the bones or
mandibles. Searching nystagmus is described. Patients are pale because
of anemia and hepatosplenomegaly caused by extramedullary
hematopoeisis. A prominent forehead and broad upper skull with
hypertelorism are common features.
and patients have mild degrees of short stature and prominent
foreheads. Other individuals with the autosomal dominant form are so
mildly affected that the diagnosis is made incidentally on the basis of
the very curious appearance of the bones.
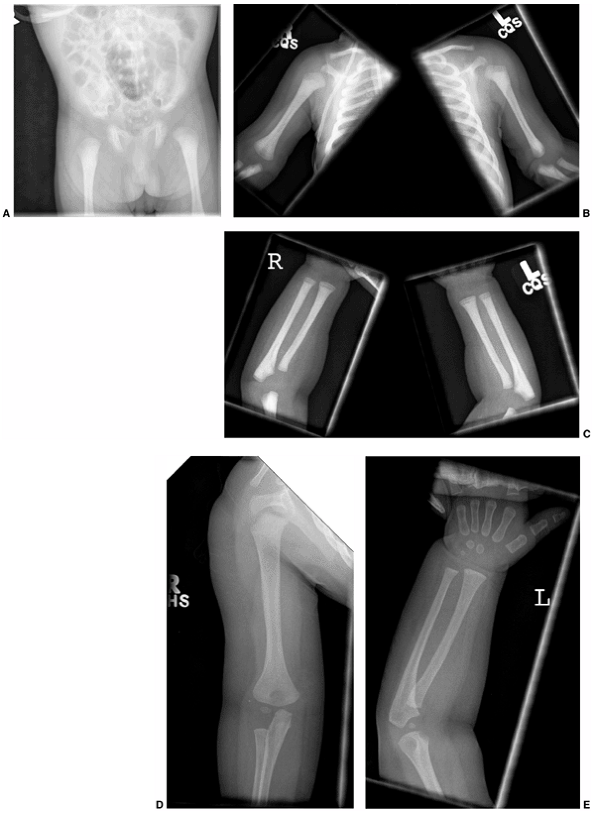 |
|
Figure 7.15 Six-month-old male infant with severe osteopetrosis and pancytopenia. A to E: Dense sclerotic bones at the pelvis (A), humerus (B), and forearm (C), without evident medullary cavities. D and E: After successful bone marrow transplant the bony architecture in the humerus (D) and forearm (E) were normalized.
|
described; it includes patients with more severe phenotypes who survive
into adulthood. Classifications based on the identification of genetic
defects have expanded the list to at least six types of osteopetrosis,
but the genetic classification is not currently comprehensive enough to
replace the clinical classification of the disease (272).
osteopetrosis is that the bones are dense white, without medullary
cavities, and appear marble-like. A bone within bone or endobone
appearance may be seen within the pelvis. Marked sclerosis of the end
plates of the vertebral bodies may give vertebrae a rugger jersey
appearance (Fig. 7.16). There may be
significant sclerosis of the base of the skull. Failure of metaphyseal
cutback by osteoclasts leads to an Erlenmeyer flask shape of the
epiphysis. Secondary deformities including progressive coxa vara or
apex lateral bowing of the femur may be present (271,273,274).
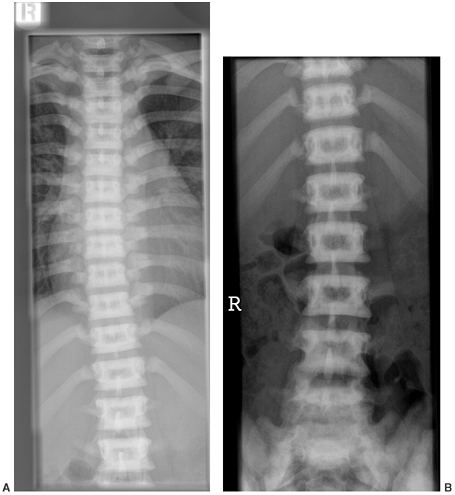 |
|
Figure 7.16 The classic rugger jersey appearance of the spine is seen in this 15-year-old girl with osteopetrosis.
|
that prevent the osteoclast from carrying out its normal function in
reabsorption of bone. Many specific defects have been identified. These
include carbonic anhydrase deficiency, which prevents the osteoclasts
from acidifying the extracellular space at the ruffled border. This is
associated with the milder clinical forms and may also be associated
with distal renal tubular acidosis and with intercranial calcifications
that are unique to this form (275,276,277). Fatal infantile forms of osteopetrosis have been associated with defects in the genes coding for proton pump or chloride
channel protein production (278). More recent case reports describe iatrogenic osteopetrosis as being the result of bisphosphonate treatment (165).
osteoclasts in the reabsorption of bone. The histologic hallmark of
osteopetrosis is remnants of primary spongiosa within the bone. These
are areas of calcified cartilage, which are precursors to enchondral
bone formation and are normally removed during the first pass
remodeling in fetal life. The ongoing failure to resolve and remodel
bone leads to the skeletal manifestations of the disease, limits the
narrow space available, contributes to pancytopenia and
hepatosplenomegaly, and is also responsible for the cranial nerve
compression at the base of the skull. The predisposition toward
musculoskeletal infections is thought to be caused by a combination of
the abnormal bony architecture and the insufficiency of white blood
cells.
Successful bone marrow transplantation corrects both the skeletal and
hematologic abnormalities associated with osteopetrosis. However, not
all bone marrow transplantations are successful and not all patients
have survived. Additional medical treatment can include very high doses
of calcitriol to stimulate osteoclastic activity and the administration
of interferon γ to stimulate superoxide production by osteoclasts.
Long-term therapy with interferon γ in patients with osteopetrosis
increases bone resorption and hematopoeisis and improves leukocyte
function (283).
The principal exceptions are fractures of the femoral neck and
intratrochanteric region, which can be extremely difficult to manage (Fig. 7.18). Armstrong’s survey of the experience of the Pediatric Orthopaedic Society of North America (284)
described a high incidence of nonunion and varus deformity of the femur
if femoral neck and intertrochanteric fractures were treated by closed
means. The results of early operative treatment of femoral neck and
intertrochanteric fractures were good, but technical difficulties
involved in obtaining fixation in the dense bone were noted. Worn-out
or broken drills and even drivers were reported. Subtrochanteric and
femoral shaft fractures did well with both closed and open management.
Most tibial fractures were managed closed. Multiple fractures of the
tibia were reported in some patients, but could be well managed with
casts.
posterior elements. These fractures are treated with immobilization.
Lumbar spondylolysis and listhesis have also been described (284,285)
and have responded well to nonoperative treatment in children and
adolescents, though occasionally requiring spinal fusion in adults.
valgus osteotomies fixed by conventional plates or screws, but with
technical difficulties similar to those encountered in treating femoral
fractures.
the hip and knee during midlife. These can be treated with total knee
arthroplasty and total hip arthroplasty, although difficulties with
reaming the canal and cutting the bone surfaces have been noted.
characteristically presents between the ages of 6 weeks and 6 months.
The clinical features are those of an irritable child, sometimes with
fever, and with tender soft tissue swelling over the affected bone.
Radiographically, there is abundant subperiosteal new bone and,
eventually, thickening of the cortex (Fig. 7.19).
The underlying cause remains unknown. The disease is usually
self-limiting and may recur episodically, but typically resolves by the
age of 2 years, and in most cases does not require any active
orthopaedic treatment. Laboratory investigation may show an increased
erythrocyte sedimentation rate, an increased white blood cell count, an
increased level of alkaline phosphatase, and iron deficiency anemia.
Differential diagnoses include child abuse, infection, and metastatic
neoplasms. John Patrick Caffey was a radiologist who initially
described the condition while working on the radiographic presentation
of child abuse. Table 7.5 presents a listing of other causes of periosteal reaction and cortical thickening in infancy and early childhood.
age group. Whether this remains part of the spectrum of Caffey disease
depends on the eventual elucidation of the underlying cause.
The rationale for treating Caffey disease with nonsteroidal
antiinflammatory drugs is supported by the clinical observation that a
bone lesion and hyperostosis similar to that in Caffey disease is also
seen in children who are treated with a prolonged course of
prostaglandin E for patent ductus arteriosus (169,290).
which normally degrades bone proteins during bone resorption (291,292,293,294).
This is an inherited autosomal recessive condition. Patients are
somewhat short, with deformities including pectus excavatum,
kyphoscoliosis, an oblique angle of the mandible, failure of fusion of
the sutures of the skull in adulthood, proptosis, oblique nose, and
frontal bossing (295,296,297). Growth hormone therapy has been used for improving stature (297). Orthopaedic management is similar to that for osteopetrosis.
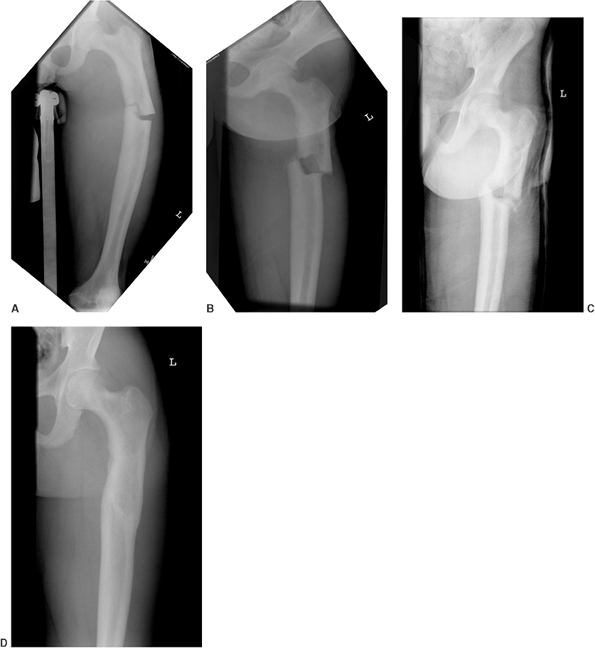 |
|
Figure 7.17 A to C: This 15-year-old boy with osteopetrosis sustained a fractured femoral shaft from a fall while running (A). He was successfully treated by traction (B) followed by spica casting (C). D: One-year follow-up shown.
|
overproduce bone because of abnormalities in regulation. These
conditions are caused by defects in the transforming growth factor β
superfamily of proteins, which are known to regulate bone growth. The
three clinical conditions are melorheostosis, Camurati-Engelmann
disease, and sclerosteosis.
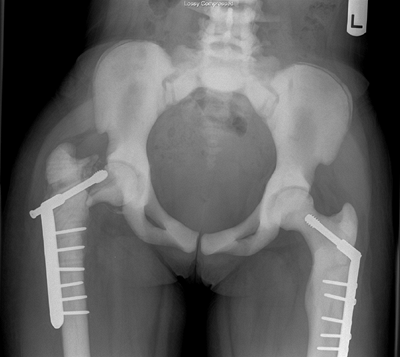 |
|
Figure 7.18
This 12-year-old girl with osteopetrosis has had bilateral femoral neck fractures treated with open reduction and internal fixation. The left has united but the right has had nonunion and cutout despite revision of hardware. The bone has been likened to chalk—dense but brittle—leading to a high rate of complications associated with internal fixation. |
which describes flowing wax on a burning candle. The condition is
characterized by new periosteal and endosteal bone that resembles
dripping wax radiographically. It typically affects one limb or one
side and produces irregularities of cortical bone, with a lesion or
rash overlying the skin. The etiology is believed to be down-regulation
of betaIG-H3 (298). Somatic mosaicism is thought to explain the usual single limb manifestation (299).
There may be stiffness in the soft tissues and shortening or
contracture of the affected extremity, and the lesion may present with
pain (300). Sarcomas arising in melorheostotic lesions have been reported (301,302,303). Ilizarov treatment has been useful in treating deformity and limb shortening (304,305), but complications relating to abnormal bone and soft tissue have been recorded (306).
Soft tissue contractures are often more problematic for the patient
than the bony deformity is, but they are difficult to treat
operatively, having an over 50% recurrence rate and associated
complications including distal ischemia (300).
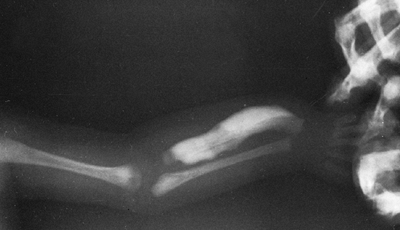 |
|
Figure 7.19 The ulna is the most frequently affected bone in the extremities of patients with Caffey disease.
|
condition characterized by hyperostosis, typically with accumulation of
bone within the medullary canal and the diaphyseal region, and also in
the skull. Clinical features include an enlarged head, proptosis, thin
limbs, weak proximal muscle sometimes resulting in a waddling gait, and
musculoskeletal pain (296,307).
Spontaneous improvement at puberty has been described. Cranial nerve
abnormalities and increased intracranial pressure are possible
sequelae. The disorder is caused by an excess of active TGF β1 which
leads to continuous stimulation of osteoblastic bone deposition (272,308,309).
|
TABLE 7.5 CAUSES OF CONDITIONS ASSOCIATED WITH PERIOSTEAL REACTION AND CORTICAL THICKENING IN INFANCY AND EARLY CHILDHOOD
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
It is characterized by skeletal overgrowth, particularly of the skull
and of the mandible. In addition, patients have sclerotic long bones
and gigantism. Bony syndactyly is a characteristic clinical marker (311,312,313,314,315,316).
The increased intracranial pressure associated with this condition may
lead to sudden death, so recognition of this condition is important (317). The underlying defect is loss of function and mutation in the SOST gene (318,319). No specific medical treatment is currently available.
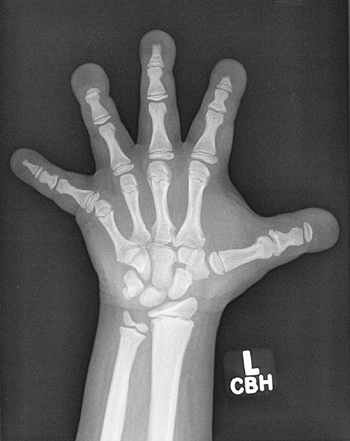 |
|
Figure 7.20 Pyknodysostosis. This 12-year-old boy has the dense bones and acroosteolysis characteristic of pyknodysostosis.
|
T, Robson H, Shalet SM, et al. Glucocorticoids, thyroid hormone and
growth hormone interactions: implications for the growth plate. Horm Res 2001;56:7–12.
HE, Harrison HC. The interaction of vitamin D and parathyroid hormone
on calcium, phosphorus and magnesium homeostasis in the rat. Metab Clin Exp 1964;13:952.
MF, Schnoes HK, DeLuca HF, et al. Isolation and identification of
24,25-hydroxycholecalciferol, a metabolite of D3 made in the kidney. Biochemistry 1972;11:4251.
MF, Schnoes HK, DeLuca HF, et al. Isolation and identification of
1,24-dihydroxycholecalciferol: a metabolite of vitamin D active in
intestine. Biochemistry 1971;10:2799.
IT, Gray RW, DeLuca HF. Regulation by calcium of in vivo synthesis of
1,25-dihydroxycholecalciferol and 21,25-dihydroxycholecalciferol. Proc Natl Acad Sci U S A 1971;68:2131.
LM, Greer FR. Section on Breastfeeding and Committee on Nutrition.
American Academy of Pediatrics. Prevention of rickets and vitamin D
deficiency: new guidelines for vitamin D intake. Pediatrics 2003;111(4 Pt 1):908–910.
A, Bravo G, Gauthier P. Meta-analysis of the effectiveness of physical
activity for the prevention of bone loss in postmenopausal women. Osteoporos Int 1997;7:331–337.
CT, Strafford B, Gross SB, et al. The proliferative and synthetic
response of isolated calvarial bone cells of rats to cyclic biaxial
mechanical strain. J Bone Joint Surg Am 1991;73:320.
rachitide sive marbo puerili qui vulgo The Rickets Dicitur Tracttatus.
Adscitis in operis societatem Georgio Bate et Ahasuero Regemortero. London: G Du-Gardi, 1650.
N, Pettifor JM, Cane RD, et al. Infantile apnoea due to profound
hypocalcaemia associated with vitamin D deficiency. A case report. S Afr Med J 1978;53:766–767.
NF, Kenney RD, Carrington PH, et al. Severe nutritional deficiencies in
toddlers resulting from health food milk alternatives. Pediatrics 2001;107:E46.
JB, Dunnigan MG, McIntosh WB, et al. Asian osteomalacia is determined
by dietary factors when exposure to ultraviolet radiation is
restricted: a risk factor model. Q J Med 1990;76:923–933.
JB, Dunnigan MG, McIntosh WB, et al. The importance of limited exposure
to ultraviolet radiation and dietary factors in the aetiology of Asian
rickets: a risk-factor model. Q J Med 1987;63:413–425.
CH, Dent CE, Harper C, et al. Vitamin D-resistant rickets: analysis of
twenty-four pedigrees with hereditary and sporadic cases. Am J Med 1964;36:222.
TO, Keller M, Schwartz D, et al. 24,25 dihydroxyvitamin D
supplementation corrects hyperparathyroidism and improves skeletal
abnormalities in X-linked hypophosphatemic rickets—a clinical research
center study. J Clin Endocrinol Metab 1996;81:2381–2388.
MT, Tylkowski C, Kriss VM, et al. The effect of osteotomy on bowing and
height in children with X-linked hypophosphatemia. J Pediatr Orthop 1999;19:114–118.
MD, Schwindinger WF, Levine MA. Clinical implications of genetic
defects in G proteins: the molecular basis of McCune-Albright syndrome
and Albright hereditary osteodystrophy. Medicine 1996;75:171.
A, Illig R, Heierli E. Eine besondere form des primare vitamin D
resistenten rachitis mit hypocalcemie und autosomal-dominanten erbgang:
die hereditare pseudomangelrachitis. Helv Paediatr Acta 1961;16:452–468.
AA, Miller WL. Rickets due to hereditary abnormalities of Vitamin D
synthesis or action. In: Glorieux F, Pettifor JM, Juppner H, eds. Pediatric bone: biology and diseases. San Diego, CA: Academic Press, 2003:583.
GK, Lin D, Zhang MY, et al. Cloning of human 25-hydroxyvitamin D-1
alpha-hydroxylase and mutations causing vitamin D-dependent rickets
type 1. Mol Endocrinol 1997;11:1961–1970.
MH, Bell NH, Love L, et al. Vitamin D-dependent rickets, type II:
resistance of target organs to 1,25-dihydroxy-vitamin D. N Engl J Med 1978;298:996.
BD, Salusky IB. Renal osteodystrophy: pathogenesis, diagnosis, and
treatment. In: Glorieux F, Pettifor JM, Juppner H, eds. Pediatric bone: biology and diseases. San Diego, CA: Academic Press, 2003:679.
Y, Yosipovitch Z, Licht A, et al. Bilateral slipped upper femoral
epiphysis: a rare manifestation of renal osteodystrophy. Case report
with discussion of its pathogenesis. Isr J Med Sci 1975;11:15.
R, Francis MJO, Bauze RJ. Osteogenesis imperfecta: a clinical and
biochemical study of a generalized connective tissue disorder. Q J Med 1975;44:555.
P, Vijayasekaran S, Lannigan F. Is it necessary to screen for hearing
loss in the paediatric population with osteogenesis imperfecta? Clin Otolaryngol 2003;28:199–202.
FH, Ward LM, Rauch F, et al. Osteogenesis imperfecta type VI: a form of
brittle bone disease with a mineralization defect. J Bone Miner Res 2002;17:30–38.
GS, David KE, Byers PH. Type I osteogenesis imperfecta: a nonfunctional
allele for pro alpha (I) chains for type I procollagen. Proc Natl Acad Sci U S A 1982;79:3838.
L, Palotie A, Prockop DJ. A defect in the structure of type I
procollagen in a patient who had osteogenesis imperfecta: excessive
mannose in the COOH-terminal peptide. Proc Natl Acad Sci U S A 1980;77:6179.
MS, Minch CM, Kruse RW, et al. The role of dual energy x-ray
absorptiometry in aiding the diagnosis of pediatric osteogenesis
imperfecta. Am J Orthop 1998;27:797–801.
RI, Baron J. Overdiagnosis of osteoporosis in children due to
misinterpretation of dual-energy x-ray absorptiometry (DEXA). J Pediatr 2004;144:253–257.
KD, Lau ST, Oberbauer AM, et al. Alendronate affects long bone length
and growth plate morphology in the OIM mouse model for osteogenesis
imperfecta. Bone 2003;32:268–274.
SC, Jee WS, Woodbury DM, et al. Effects of N, N, N’,
N’-ethylenediaminetetramethylene phosphonic acid and
1-hydroxyethylidene-1,1-bisphosphonic acid on calcium absorption,
plasma calcium, longitudinal bone growth, and bone histology in the
growing rat. Toxicol Appl Pharmacol 1985;77:230–239.
M, Monson R, Weber K. The prevention of recurrent fractures of the
lower extremities in severe osteogenesis imperfecta using vacuum pants:
a preliminary report in four patients. J Pediatr Orthop 1988;8:454.
HA, Millar EA. Fragmentation, realignment, and intramedullary rod
fixation of deformities of the long bones in children: a ten-year
appraisal. J Bone Joint Surg Am 1959;41:1371.
JG, Strudwick WJ, Rinsky LA, et al. Complications of intramedullary
rods in osteogenesis imperfecta: Bailey-Dubow rods versus nonelongating
rods. J Pediatr Orthop 1988;8:645.
S, Heller E, Seidman DS, et al. Functional results of operation in
osteogenesis imperfecta: elongating and nonelongating rods. J Pediatr Orthop 1991;11:200.
SJ, Sheridan JJ, Capelli AM, et al. Management of lower-extremity
deformities in osteogenesis imperfecta with extensible intramedullary
rod technique: a 20-year experience. J Pediatr Orthop 1998;18:88–94.
RH, Uiterwaal CS, Gulmans VA, et al. Osteogenesis imperfecta: profiles
of motor development as assessed by a postal questionnaire. Eur J Pediatr 2000;159:615–620.
RH, Beemer FA, van der Graaf Y, et al. Osteogenesis imperfecta in
childhood: impairment and disability—a follow-up study. Arch Phys Med Rehabil 1999;80:896–903.
RH, Uiterwaal CS, van der Hulst A, et al. Scoliosis in children with
osteogenesis imperfecta: influence of severity of disease and age of
reaching motor milestones. Eur Spine J 2003;12:130–134.
GJ, Finidori G, Engelbert RH, et al. Operative treatment of severe
scoliosis in osteogenesis imperfecta: results of 20 patients after halo
traction and posterior spondylodesis with instrumentation. Eur Spine J 2000;9:486–491.
RF, Bitan FD, Laplaza FJ, et al. Spinal deformity, pulmonary
compromise, and quality of life in osteogenesis imperfecta. Spine 1999;24:1673–1678.
RH, Gerver WJ, Breslau-Siderius LJ, et al. Spinal complications in
osteogenesis imperfecta: 47 patients 1–16 years of age. Acta Orthop Scand 1998;69:283–286.
DA, Winter RB, Lutter L, et al. Osteogenesis imperfecta. Radiographic
classification, natural history, and treatment of spinal deformities. J Bone Joint Surg Am 1992;74:598–616.
EJ, Engelbert RH, Pals G, et al. Bruck syndrome: a rare combination of
bone fragility and multiple congenital joint contractures. J Pediatr Orthop B 1998;7:35–38.
RA, Robins SP, Wijmenga C, et al. Defective collagen crosslinking in
bone, but not in ligament or cartilage, in Bruck syndrome: indications
for a bone-specific telopeptide lysyl hydroxylase on chromosome 17. Proc Natl Acad Sci U S A 1999;96:1054–1058.
RC, Lark RK, Gurka MJ, et al. Bone density and metabolism in children
and adolescents with moderate to severe cerebral palsy. Pediatrics 2002;110(1 Pt 1):E5.
DY, Yalcin E. Changes in skeletal maturation and mineralization in
children with cerebral palsy and evaluation of related factors. J Child Neurol 2001;16:425–430.
B, Barton LL, Lloyd J, et al. Dietary considerations in osteopenia in
tube-fed nonambulatory children with cerebral palsy. Clin Pediatr 1999;38:133–137.
F, Basu D, Pettifor JM. Pathological long-bone fractures in residents
with cerebral palsy in a long-term care facility in South Africa. Dev Med Child Neurol 2002;44:119–122.
KE, Bailey DA, McKay HA, et al. The effect of a weight-bearing physical
activity program on bone mineral content and estimated volumetric
density in children with spastic cerebral palsy. J Pediatr 1999;135:115–117.
M, Kocijancic A, Prezelj J. Effect of vitamin D and calcium on bone
mineral density in children with CP and epilepsy in full-time care. Dev Med Child Neurol 2000;42:403–405.
RC, Lark RK, Kecskemethy HH, et al. Bisphosphonates to treat osteopenia
in children with quadriplegic cerebral palsy: a randomized,
placebo-controlled clinical trial. J Pediatr 2002; 141:644–651.
J, Allen R, Hesp R, et al. Randomized, double-blind trial of
deflazacort versus prednisone in juvenile chronic (or rheumatoid)
arthritis: a relatively bone-sparing effect of deflazacort. Pediatrics 1991;88:428–436.
C, Seck T, Hinke V, et al. Effects of 6 years of growth hormone (GH)
treatment on bone mineral density in GH-deficient adults. Clin Endocrinol 2001;55:93–99.
DA, Wedge JH, McCulloch RG, et al. Epidemiology of fractures of the
distal end of the radius in children as associated with growth. J Bone Joint Surg Am 1989;71:1225–1231.
S, Melton LJ III, Dekutoski MB, et al. Incidence of childhood distal
forearm fractures over 30 years: a population-based study. JAMA 2003;290:1479–1485.
KM, Liu-Ambrose T, Sran MM, et al. New criteria for female athlete
triad syndrome? As osteoporosis is rare, should osteopenia be among the
criteria for defining the female athlete triad syndrome? Br J Sports Med 2002;36:10–13.
TL, Snow-Harter C, Taaffe DR, et al. Gymnasts exhibit higher bone mass
than runners despite similar prevalence of amenorrhea and
oligomenorrhea. J Bone Miner Res 1995;10:26–35.
RK, Bauer JJ, Snow CM. Jumping improves hip and lumbar spine bone mass
in prepubescent children: a randomized controlled trial. J Bone Miner Res 2001;16:148–156.
HA, Petit MA, Schutz RW, et al. Augmented trochanteric bone mineral
density after modified physical education classes: a randomized
school-based exercise intervention study in prepubescent and early
pubescent children. J Pediatr 2000;136: 156–162.
AR, Melton LJ III, Crowson CS, et al. Long-term fracture risk among
women with anorexia nervosa: a population-based cohort study. Mayo Clin Proc 1999;74:972–977.
E, Szmukler GI, Formica C, et al. Osteoporosis in anorexia nervosa: the
influence of peak bone density, bone loss, oral contraceptive use, and
exercise. J Bone Miner Res 1992;7: 1467–1474.
NH, Lanzkowsky L, Schebendach J, et al. The effect of
estrogen-progestin treatment on bone mineral density in anorexia
nervosa. J Pediatr Adolesc Gynecol 2002;15:135–143.
FM, Chabot G, Polychronakos C, et al. Safety profile of frequent short
courses of oral glucocorticoids in acute pediatric asthma: impact on
bone metabolism, bone density, and adrenal function. Pediatrics 2003;111:376–383.
JD, Roux C, Pitt PI, et al. A pooled data analysis on the use of
intermittent cyclical etidronate therapy for the prevention and
treatment of corticosteroid induced bone loss. J Rheumatol 2000;27:2424–2431.
DM, Hughes RA, Laan RF, et al. Efficacy and safety of daily risedronate
in the treatment of corticosteroid-induced osteoporosis in men and
women: a randomized trial. European Corticosteroid-induced Osteoporosis
Treatment Study. J Bone Miner Res 2000;15:1006–1013.
S, Cohen S, Reid DM, et al. Effects of risedronate treatment on bone
density and vertebral fracture in patients on corticosteroid therapy. Calcif Tissue Int 2000;67:277–285.
NE, Sanchez S, Modin GW, et al. Bone mass continues to increase at the
hip after parathyroid hormone treatment is discontinued in
glucocorticoid-induced osteoporosis: results of a randomized controlled
clinical trial. J Bone Miner Res 2000; 15:944–951.
NE, Sanchez S, Modin GW, et al. Parathyroid hormone treatment can
reverse corticosteroid-induced osteoporosis. Results of a randomized
controlled clinical trial. J Clin Invest 1998;102:1627–1633.
JL, Yeager H, Saunders RB, et al. Rickets in children receiving
anticonvulsant drugs: biochemical and hormonal markers. Am J Dis Child 1977;131:286.
RW, Cook SD, Thomas KA, et al. Effects of anticonvulsant drug therapy
on bone mineral density in a pediatric population. J Pediatr Orthop 1988;8:467.
TJ, Halstead LR. Anticonvulsant drug-induced osteomalacia: alterations
in mineral metabolism and response to vitamin D3 administration. Calcif Tissue Int 1979;27:13–18.
J, Becker A, Witte OW, et al. Long-term anticonvulsant therapy leads to
low bone mineral density—evidence for direct drug effects of phenytoin
and carbamazepine on human osteoblast-like cells. Exp Clin Endocrinol Diabetes 2000;108:37–43.
G, Tutlewski B, Stabrey A, et al. Analysis of the musculoskeletal
system in children and adolescents receiving anticonvulsant monotherapy
with valproic acid or carbamazepine. Pediatrics 2001;108:E107.
WS, Whyte MP, Sundaram V, et al. Carbonic anhydrase II deficiency in 12
families with the autosomal recessive syndrome of osteopetrosis with
renal tubular acidosis and cerebral calcification. N Engl J Med 1985;313:139–145.
WS, Hewett-Emmett D, Whyte MP, et al. Carbonic anhydrase II deficiency
identified as the primary defect in the autosomal recessive syndrome of
osteopetrosis with renal tubular acidosis and cerebral calcification. Proc Natl Acad Sci U S A 1983;80: 2752–2756.
E, Benichou O, Van Hul E, et al. Albers-Schonberg disease (autosomal
dominant osteopetrosis, type II) results from mutations in the ClCN7
chloride channel gene. Hum Mol Genet 2001;10:2861–2867.
FS, August CS, Fallon MD, et al. Successful treatment of infantile
malignant osteopetrosis by bone-marrow transplantation. J Bone Joint Surg Am 1988;70:617.
DG, Newfield JT, Gillespie R. Orthopedic management of osteopetrosis:
results of a survey and review of the literature. J Pediatr Orthop 1999;19:122–132.
K, Saito A, Nakano H, et al. Cortical hyperostosis following long-term
administration of prostaglandin E in infants with cyanotic congenital
heart disease. J Pediatr 1980;97:834.
V, Hou WS, Rialland X, et al. Cathepsin K deficiency in pycnodysostosis
results in accumulation of non-digested phagocytosed collagen in
fibroblasts. Calcif Tissue Int 2003;73:380–386.
Y, Nakata K, Yasui N, et al. Novel mutations of the cathepsin K gene in
patients with pycnodysostosis and their characterization. J Clin Endocrinol Metab 2000;85:425–431.
AT, Ramadan MA, Sherif A, et al. Pycnodysostosis: clinical, radiologic,
and endocrine evaluation and linear growth after growth hormone
therapy. Metab Clin Exp 2001;50:905–911.
JE, Kim EH, Han EH, et al. A TGF-beta-inducible cell adhesion molecule,
betaig-h3, is downregulated in melorheostosis and involved in
osteogenesis. J Cell Biochem 2000;77:169–178.
D, Lehman WB, Grant AD, et al. The Ilizarov apparatus for treatment of
melorheostosis. Case report and review of the literature. Clin Orthop Relat Res 1992;281:163–167.
K, ten Dijke P, Ralston SH, et al. Transforming growth factor-beta 1
mutations in Camurati-Engelmann disease lead to increased signaling by
altering either activation or secretion of the mutant protein. J Biol Chem 2003;278:7718–7724.
P, Durr L, Hamersma H. The clinical features of sclerosteosis. A review
of the manifestations in twenty-five affected individuals. Ann Intern Med 1976;84:393–397.
W, Ebeling M, Patel N, et al. Increased bone density in sclerosteosis
is due to the deficiency of a novel secreted protein (SOST). Hum Mol Genet 2001;10:537–543.
ME, Gardner JC, Van Ness J, et al. Bone dysplasia sclerosteosis results
from loss of the SOST gene product, a novel cystine knot-containing
protein. Am J Hum Genet 2001; 68:577–589.