
remodeling of bone and its cartilaginous precursor. The pathogenesis of
many of these conditions is slowly being worked out, teaching us about
the growth of the skeleton. In the preface to his classic text, Heritable Disorders of Connective Tissue,
Victor A. McKusick said, “Nature is nowhere more openly to display her
secret mysteries than in cases where she shows traces of her workings
apart from the beaten path” (1). It may even be
true that there is a mutation and a disorder representing nearly each
step of skeletal development. Most disorders result in short stature,
defined as height more than two standard deviations less than the mean
for the population at a given age. The term “dwarfing condition” is
used to refer to disproportionately short stature. The disproportion is
commonly referred to as “short trunk” or “short limb.” The short-limb
types are further subdivided into categories based on which segment of
the limb is short. Rhizomelic refers to shortening of the root (proximal) portion of the limb, mesomelic to the middle segment, and acromelic
to the distal segment. Achondroplasia is a classic example of
rhizomelic involvement, with the femora, especially the humeri, being
most affected by shortening. Some of these disorders are named after
the appearance of the skeleton (diastrophic means “to grow twisted,” camptomelic means “bent limbs,” and chondrodysplasia punctata refers to “stippled cartilage”). Eponyms are used to name other disorders (e.g., Kniest, Morquio, and McKusick).
skeletal dysplasia. Some dysplasias are due to an alteration in
transcription, or in intracellular or extracellular processing of
structural molecules of the skeleton. Others are caused by a defect in
a receptor or signal transduction in pathways of skeletal
differentiation and proliferation. These abnormalities tend to occur in
the pathway of cartilage differentiation, growth, and development.
a structural macromolecule may occur, as in type II collagen causing
spondyloepiphyseal dysplasia (SED). In some cases, the effect may be
magnified, a phenomenon termed a dominant-negative effect.
This occurs when the defective gene product binds to normal copies of
the product, leading to early destruction of both normal and defective
copies, as is seen in osteogenesis imperfecta type II.
Pseudoachondroplasia provides another example, with the abnormal
cartilage oligomeric matrix protein (COMP) accumulating in the rough
endoplasmic reticulum and causing secondary retention of type IX
collagen and other proteins. In contrast, models in which COMP is
completely knocked out and not expressed do not display any disease.
of structural molecules. One example of this pathway is the group of
conditions including diastrophic dysplasia (DD) and achondrogenesis
type 1, which are due to a defect in sulfate transport. Proteoglycan
assembly is thereby disturbed, leading to diffuse alterations in the
cartilage of the articular surface, growth plate, and other areas. An
example of receptors gone awry is the
family of disorders including achondroplasia, hypochondroplasia, and
thanatophoric dysplasia. These disorders occur because of varying
defects in
fibroblast
growth factor receptor protein. These mutations result in a
constitutively active receptor (gain-of-function). Because this
receptor downregulates endochondral growth, mutations result in
decreased endochondral growth. Another example is Jansen metaphyseal
dysplasia, which is due to a constitutively active mutation in the
parathyroid hormone receptor protein. This inhibits the expression of
Indian hedgehog, which, in turn, is needed for stimulating terminal
differentiation to hypertrophic chondrocytes and for producing normal
metaphyseal growth. Disorders of transcription
may also cause skeletal dysplasia—as seen in cleidocranial dysplasia, a
defect in core binding factor 1. This transcription factor stimulates
osteoblast differentiation. Therefore a defect in this factor leads to
a cartilage model that is well formed but not normally ossified.
been done according to the pattern of bone involvement, as in the
International Classification of Osteochondrodysplasias (2) (Table 8.1).
The newer trend, however, is to group them according to the specific
causative gene defect, in cases in which the defect is known (Table 8.2). A schematic representation of the effects of the known mutations on cartilage development is shown in Figure 8.1.
It is also useful for the orthopaedic surgeon to mentally classify the
dysplasias into those that are free from spinal deformity [e.g.,
hypochondroplasia and multiple epiphyseal dysplasia (MED) are rarely
associated with considerable spinal abnormalities] and those in which
spinal deformity is a frequent problem (e.g., SED, DD, and metatropic
dysplasia). Likewise, it is useful to consider which of the disorders
are free from epiphyseal involvement, and therefore from the risk of
degenerative joint disease down the road. Achondroplasia and
hypochondroplasia, cleidocranial dysplasia, and diaphyseal aclasia
rarely present these problems in adulthood, but SED, MED, DD, and
others commonly do.
|
TABLE 8.1 INTERNATIONAL CLASSIFICATION OF SKELETAL DYSPLASIAS, 1992 (A PARTIAL LIST)
|
|
|---|---|
|
Online Mendelian Inheritance in Man (OMIM). This Web-based compendium
is publicly available and readily accessible by name through the
National Library of Medicine. OMIM allows a user to search by physical
features or diagnosis, and provides a compilation of applicable
knowledge on each (3).
birth. When ultrasound shows a fetus with shortening of the skeleton,
femur length is the best biometric parameter for distinguishing among
the five most common possible conditions. Most commonly, fetuses with
femur length less than 40% of the mean for gestational age have
achondrogenesis, those with femur length between 40% and 60% have
thanatophoric dysplasia or osteogenesis imperfecta type II, and those
with femur length over 80% have achondroplasia or osteogenesis
imperfecta type III (4). Further testing may be performed, if indicated, by chorionic villous sampling and mutation analysis.
|
TABLE 8.2 CLASSIFICATION OF DYSPLASIAS BASED ON ETIOLOGY
|
|
|---|---|
|
 |
|
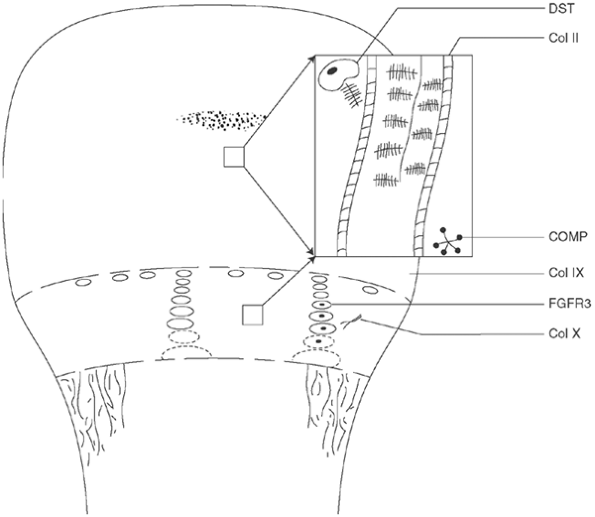
Figure 8.1
Schematic illustration of the sites and effects of the known cartilage defects associated with skeletal dysplasias. Section of cartilage matrix of physis and epiphysis is simplified and enlarged; genetic abnormalities often affect both regions. DST, diastrophic sulfate transporter, the deficiency of which leads to undersulfation of proteoglycans in epiphysis and physis of diastrophic dysplasia (DD) and achondrogenesis types 1B and 2; Col II, type II collagen, which is defective in Kniest syndrome and spondyloepiphyseal dysplasia; COMP, cartilage oligomeric matrix protein, abnormal pseudoachondroplasia, and some forms of multiple epiphyseal dysplasia (MED); Col IX, type IX collagen, which is closely linked to type II collagen and is abnormal in some forms of MED; FGFR3, fibroblast growth factor receptor 3, which inhibits chondrocyte proliferation in achondroplasia, hypochondroplasia, and thanatophoric dysplasia; Col X, type X collagen, which is synthesized only by the hypertrophic cells of the growth plate and is abnormal in Schmid-type metaphyseal chondrodysplasia. |
bone development for skeletal dysplasia, several aspects of the medical
history should be investigated for diagnosis and for coordination of
care. Length at birth should be recorded; this is decreased in
achondroplasia but not in pseudoachondroplasia. Head circumference
should also be noted; this is increased in achondroplasia. Respiratory
difficulty may occur in infancy because of restrictive problems in the
syndromes such as a small thorax, neurologic problems such as foramen
magnum stenosis in achondroplasia, or upper airway obstruction in
various conditions. A history of heart disease suggests the possibility
of chondroectodermal dysplasia, which may be associated with congenital
heart malformations; or of storage disorders such as Hurler or Morquio
syndromes, in which cardiac dysfunction may be acquired. A history of
immune deficiency or malabsorption is common in cartilage-hair
hypoplasia. Retinal detachment may occur with Kniest syndrome or SED.
The pertinent family history relating to short stature or dysmorphism
should be sought, as well as information about any prior skeletal
surgery the patient may have had.
malformations of the extremities should be noted. Height percentile for
age should be determined using standard charts. Most skeletal
dysplasias result in adult height of less than 152 cm. Measurement of
the upper:lower segment ratio of lengths may be helpful in
distinguishing disproportion early. This ratio can be determined by
measuring the distance from the top of the pubic symphysis to the sole
of the plantigrade foot, and by subtracting it from the overall length.
The normal ratio is 1.6 at birth (given that extremities develop later
than the trunk), and diminishes to 0.93 in teens and in adults. If
shortening of the extremities is noted, it is helpful to classify it as
rhizomelic (shortest in the humerus and femur), as in achondroplasia;
mesomelic (shortest in the forearms and legs); or acromelic (shortest
distally). The extremities should be examined for ligamentous laxity or
contracture. A thorough neurologic examination is needed because of the
frequent incidence of spinal compromise—at the upper cervical level in
SED, DD, Larsen syndrome, metatropic dysplasia, or at any level in
achondroplasia.
already available: a lateral film of skull and neck, and
anteroposterior views of the entire spine, pelvis, arms, hands, and
legs. Sometimes, pathognomonic features will be seen, such as the
caudal narrowing of the interpediculate distances in achondroplasia,
double-layered patella in MED, and the iliac horns in nail-patella
syndrome. Films of the cervical spine in flexion and extension should
be ordered if instability is suspected to be the cause of delay in
reaching milestones or loss of strength or endurance. In many
syndromes, such as SED, in which cervical instability is common, these
films should be ordered as a matter of course. Magnetic resonance
imaging (MRI) in flexion and extension may be helpful, in some cases,
in determining whether the instability is causing critical risk.
However, the limitation of this test is that it must often be done
under anesthesia or sedation, and the degree of cervical movement is
limited. If the plain radiographs show significant motion and a static
MRI shows signal changes at the same location, then flexion and
extension images are not needed.
calcium, phosphate, alkaline phosphatase, and protein to rule out
metabolic disorders such as hypophosphatemia or hypophosphatasia. The
urine should be screened for storage products (under the guidance of a
geneticist) if a progressive disorder is found. Serum thyroxine should
be measured if the fontanels in an infant are bulging and bone
development is delayed, to rule out hypothyroidism. DNA testing for
mutation analysis is currently done in the clinical setting for
skeletal dysplasias, once the differential diagnosis is clinically
focused. A geneticist should be consulted to help establish a diagnosis
and, thereby, a prognosis, and also to help deal with medical problems.
The geneticist sometimes functions as a primary physician for a patient
with a genetic disorder because geneticists have the best overview of
the medical issues facing the patient.
skeletal dysplasia should focus on several aspects: prevention of
future limitations, treatment of current deformity, and treatment of
pain. The patient’s parents should be counseled about the mode of
inheritance and risk of recurrence, so that they can make future family
plans appropriately. In most cases, it is advisable to see these
patients on a routine basis for surveillance, so that skeletal problems
can be detected at the optimum time for treatment. Weight management is
a continuing challenge for many, and reasonable attention should be
paid to this issue. Studies of quality of life in achondroplasia have
shown that, although many individuals are able to function at a high
level, as a group they achieve significantly lower scores in all
domains (5). Physical difficulties in an
environment that is not scaled for them was one of the most commonly
cited reasons, thereby indicating that treatments to increase stature
may have functional benefits.
dysplasia, special considerations apply. Anesthesia management is more
difficult if the dysplasia involves oropharyngeal malformations,
limited neck mobility, cervical instability, or stenosis (6).
Cervical instability is so common in the skeletal dysplasias that the
surgeon should make a point of ruling it out, based on knowledge of the
patient, knowledge of the condition and whether cervical instability is
associated with it, or viewing of special radiographs in flexion and
extension (7,8).
Problems relating to restriction in the airway accompany some
dysplasias, and laryngotracheomalacia affects many young diastrophic
children. Skeletal distortion may make deep venous access challenging
and, in some cases, a general surgeon should be consulted in advance.
Intraoperative positioning must accommodate both small stature and any
contractures that are present. Limb-lengthening is an option for many
patients who do not have a high risk of degenerative joint disease. In
the tibia, for instance, concomitant stabilization of the tibiofibular
joints during lengthening, as well as Achilles lengthening, can
decrease complications. However, achievement of a significant amount of
length in multiple extremities requires a major commitment of time.
individuals with skeletal dysplasia, because of contractures as well as
the abnormal shape and size of the joint. Extensive soft-tissue
releases may be necessary. However, pain and function scores improve
significantly as a result of surgery (9).
most of these patients have decreased ability to accommodate
postoperative immobilization, stiffness, or functional restrictions. In
some situations, postoperative
placement
in a rehabilitative setting may be most helpful to the patient and
family. The organization Little People of America may be a significant
resource for information and support.1
skeletal dysplasia, it is itself uncommon, with an incidence of
approximately 1 in 30,000 to 1 in 50,000 (10,11).
Achondroplasia is the form of skeletal dysplasia that physicians most
commonly encounter. The name of the condition is not strictly accurate
because cartilage does develop and grow, both at the physes and at
other locations. However, bone that is formed by endochondral means is
most underdeveloped in length, resulting in disproportionate, short
stature. The etiology of achondroplasia has been determined to be a
gain-of-function mutation in the gene that encodes for the
growth-suppressing fibroblast growth factor receptor 3 (FGFR-3), which further slows endochondral growth (12,13,14,15).
condition, and that information should be used while counseling the
family and the affected patient. However, in at least 80% of patients,
the disorder is because of a spontaneous mutation (16). The risk of having a child with achondroplasia increases with increasing paternal age.
which is part of the transmembrane domain of this receptor and is said
to be the single most mutable nucleotide in humans. The mutation causes
a change in a single amino acid, from arginine to glycine. Fibroblast
growth factor receptor is expressed in physeal cartilage and in the
central nervous system. FGFR3 seems to limit endochondral bone
formation in the proliferative zone of the physis, and this is a
mutation that actually increases that inhibition (a so-called gain-of-function
mutation). As discussed later in this chapter, this same receptor is
also the site of other mutations that cause hypochondroplasia and
thanatophoric dysplasia.
achondroplasia show a reduced hypertrophic cell zone and large collagen
fibrils (17). However, intramembranous and periosteal ossification processes are normal (18).
numerous features that are uniform and predictable. The facial
appearance is characterized by frontal bossing and midface hypoplasia (11,19). This hypoplasia develops because of the endochondral origin of the facial bones (12). The trunk length is within the lower range of normal, whereas the extremities are much shorter than normal (20), in a pattern that is termed rhizomelic (Fig. 8.2). The term rhizo–
means “root.” The proximal segments (roots) of the extremities—the
humeri and femora—are the most foreshortened. The fingertips usually
reach only to the tops of the greater trochanters, and this leads to
difficulties in personal care (19). The digits
of the hand have extra space between the third and the fourth rays, so
that the digits are separated into three groups, including the
thumb—the “trident hand.” There is usually a flexion contracture of the
elbows, and the radial heads may be subluxated. Neither of these
features causes significant functional
impairment.
Kyphosis at the thoracolumbar junction is common, especially in
infancy. The condition usually improves with increasing age of the
patient (21).
Lumbar lordosis increases. Ligamentous laxity is common at the knees
and ankles. The knees are most commonly in varus alignment, but in some
patients they may be straight or even in excessive valgus. The ankles
usually have varus alignment as well. Internal tibial torsion is
common. The joints are otherwise not directly affected by this
condition to any considerable degree. The limbs generally have a
muscular appearance. Intelligence is normal. Life expectancy is not
significantly diminished in this condition. Studies of health outcomes
have shown that although the physical component of the score is lower,
the overall functional health score is not dramatically lower in
achondroplasia than in the general population (22).
 |
|
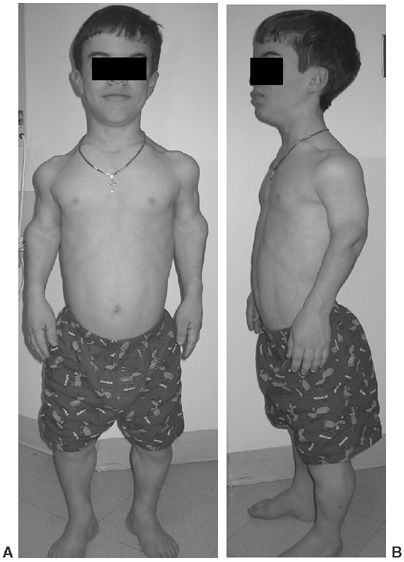
Figure 8.2 A 16-year-old boy with achondroplasia. A: Pronounced shortening of proximal limb segments (rhizomelic pattern). There is mild genu varum. The humeri are most affected. B: The elbows have a mild flexion contracture. He has had previous osteotomies of the tibias and fibulas for varus.
|
The stature of children with achondroplasia is diminished
proportionately throughout childhood, but the proportion declines
during the adolescent growth spurt (23). The predicted adult height is 132 cm for men, and 122 cm for women (16).
When the growth pattern of specific long bones is studied, it is found
that the growth of the femur deviates from the population mean even
more during the growth spurt, and the fibula overgrows the tibia (11). This latter fact is thought to explain the phenomenon of genu varum, which is seen in many children with achondroplasia.
much of the macrocephaly seen in children with achondroplasia. However,
although three fourths of the patients have ventriculomegaly, only a
small subset of them has clinically important hydrocephalus (24). Charts of head circumferences of children with achondroplasia are available and help assessment of these features (25).
Ventriculoperitoneal shunt surgery is indicated only for patients with
rapidly progressive head enlargement or signs of increased intracranial
pressure. Mental development is normal, but motor development is
delayed (26). Muscle tone is low in the trunk
and extremities in infancy. The most evident cause of this delay may be
neural compression at the foramen magnum. This neural compression
occurs most likely because of asynchronous growth between the neural
elements and the skull base, which is formed by endochondral bone (12). The foramen magnum is small for age in all infants with achondroplasia, although there is some “biologic variability” (27,28). Diminished foramen magnum measurements have been correlated with respiratory dysfunction and delayed motor development (24) (Fig. 8.3), although other studies have not shown a correlation between hypotonia and foramen magnum size (29).
The signs that are most predictive of severe stenosis of the foramen
magnum, which requires surgery, are the presence of clonus or
hyperreflexia, and central hypopnea, revealed by sleep study. Children
with achondroplasia meet developmental milestones later than
average-stature children. As an example, the mean age at which children
with achondroplasia walk unaided is 17 months. Normative tables for
standard milestones are available (26), and
these tables allow parents to detect delays. The development of spinal
stenosis is explained by the fact that the spinal canal forms through
endochondral ossification at the neurocentral synchondroses (Fig. 8.4).
These obliquely oriented growth plates contribute to the lengths of the
pedicles, and also to the distance between them. These dimensions are
decreased at all levels of the achondroplastic spine. It remains a
mystery
why the dimensions are most diminished in the distal lumbar spine.
 |
|
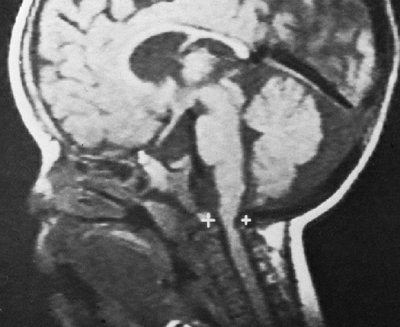
Figure 8.3
Magnetic resonance image (MRI) of a child with achondroplasia shows stenosis at the foramen magnum. (Courtesy of George S. Bassett, MD) |
 |
|
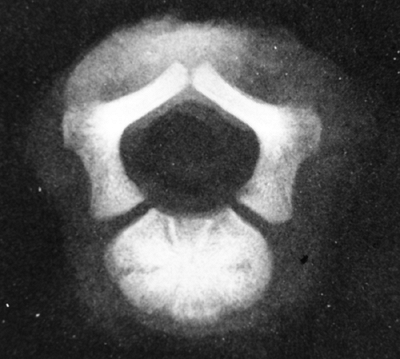
Figure 8.4
This specimen radiograph shows the obliquely oriented neurocentral synchondroses that contribute, by endochondral ossification, to both dimensions of the spinal canal. Because this process is impaired in achondroplasia, stenosis results. |
involve regions in which the growth and development occur primarily
through processes of endochondral ossification. In the skull,
therefore, the facial bones, skull base, and foramen magnum are
underdeveloped, whereas the cranial bones are normal in size and shape (11).
The growth curves relating to the foramen magnum demonstrate that the
dimensions of this aperture, as measured by computerized tomography
(CT) scan, are reduced at birth in achondroplasia and accelerate in
early childhood, but never quite reach normal (28). The spine displays central and foraminal stenosis, which becomes worse at progressively caudal levels (30,31).
Although stenosis may occur at any level, it is most common in the
lumbar spine; this is evident on plain films as a constant or
diminishing distance between pedicles, from the first to the fifth
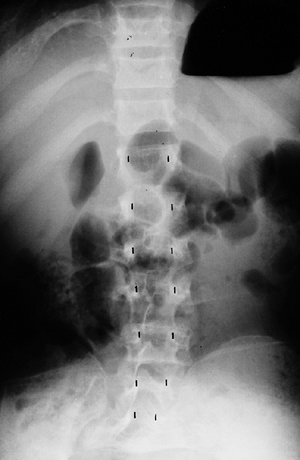
lumbar levels on the anteroposterior view (Fig. 8.5),
compared with the average-statured population, in whom 60% have
increasing interpedicular distance at more caudal levels, and 40% have
a constant distance. There is also decreased space between the
vertebral body and the lamina on the lateral view. The stenosis of the
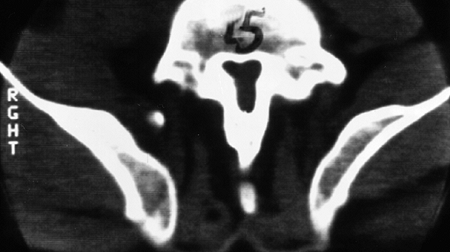
spinal canal is best visualized by CT scan, which graphically
illustrates the tapering of the spinal canal to a slitlike space at the
lumbosacral junction (Fig. 8.6). The vertebral bodies have a scalloped appearance (16).
If thoracolumbar kyphosis fails to resolve, the apical vertebrae
develop a progressively round or wedge shape. Lumbar lordosis
increases, and the sacrum may even become horizontal. Significant
scoliosis is rare. A point to remember is that cervical instability,
although so common in many forms of skeletal dysplasia, is not usually
seen in this, the most common type of dysplasia.
 |
|
Figure 8.5
This anteroposterior view of the entire spine shows the progressive narrowing of the interpedicular distance at more caudal levels of the lumbar spine; this is the opposite of the normal pattern. |
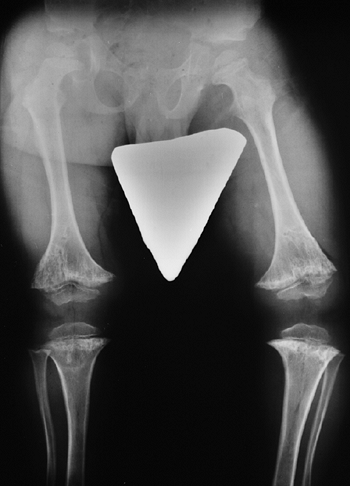
metaphyses of all long bones are flared in appearance. The diaphyses of
all long bones are thick, despite being short, owing to subperiosteal
bone apposition. Angulation at both the distal femoral and the proximal
tibial metaphyses contributes to abnormal knee alignment (Fig. 8.7).
The growth of the fibula is typically greater than that of the tibia,
and this also contributes to the varus in some cases. The shape of the
distal femoral physis is an exaggeration of the normal inverted “V” in
the midline, and the sites of major muscle insertions (such as the
tibial tubercle and the greater trochanter) are more prominent than
usual. The metacarpals and metatarsals are all of almost equal length.
In 50% of individuals with achondroplasia, there is increased space
between the third and fourth metacarpals, which is the basis for the
appearance of the “trident” hand (16). The
epiphyses throughout the skeleton are virtually normal in appearance
and development and, consequently, degenerative joint changes are rare.
in the first 2 years of life for signs of foramen magnum stenosis.
These signs may include severe developmental delay, sleep apnea,
persistent hypotonia, or spasticity. If stenosis is suspected, sleep
studies should be used for evaluating brain stem functions (32).
If the diagnosis of foramen magnum stenosis is made and the clinical
picture persists, decompression of the brain stem should be undertaken
by an experienced neurosurgeon. The decompression consists of
enlargement of the foramen magnum and, sometimes, the laminectomy of
the atlas. The clinical response is usually gratifying (27).
maintaining ideal body weight presents a continuous challenge. Obesity
is more common than in the general population, but standard curves of
weight-for-height or weight-for-age used in the general population are
not applicable to persons with achondroplasia. It is recommended that
these individuals be followed up using triceps skinfold thickness or
weight/height squared, a measure that is less sensitive to short
stature. Weight control measures should be instituted when these values
exceed 95% of those for the general population (33).
 |
|
Figure 8.6 Computed tomogram of the fifth lumbar vertebra in achondroplasia, showing the slitlike spinal canal.
|
hormone. Growth hormone has been used with limited success in this
disorder (14,34,35).
The most noticeable results seem to be in those patients with the
lowest growth velocities. Early data from the National Cooperative
Growth Study has shown that children with achondroplasia who were
treated with growth hormone gained a mean 0.3 standard deviations in
height after 2 years of treatment, but the ability to maintain this
over time is not known (14), nor is the effect on final height. One study (35)
showed a significant increase in the growth rate during the first year
of treatment, with a slower increase in the second year, as well as
variation in response between patients. Clinically, its use has not
been widespread up to now. Recent studies have suggested a decrease in
cellular expression of parathyroid hormone receptor protein and
increased tendency to chondrocyte apoptosis in achondroplasia; this
possibly explains the patient’s inability to respond to growth hormone (36).
 |
|
Figure 8.7
Radiograph of the lower extremities in a six-year-old with achondroplasia. The distal femoral physes have a pronounced inverted-“V” shape, and the knee is in varus. The acetabular roofs are horizontal. |
of underdevelopment of the midfacial skeletal structure. Maxillary
hypoplasia leads to dental crowding and malocclusion, which may require
orthodontic attention (37). The eustachian
tubes may not function normally, and this may lead to recurrent otitis
media. Given that the recurrent otitis media may result in hearing
loss, the physician should have a high index of suspicion, and early
hearing screening should be performed in all children with
achondroplasia (37). In one study, 60% of all the achondroplastic children who were screened had hearing loss (38).
Other causes of hearing deficit include ossicular chain abnormalities,
and neurologic causes resulting from brain stem compression.
Obstructive sleep apnea is found in three fourths of children with
achondroplasia when studied in the sleep laboratory (32,39).
Treatment, if necessary, begins with adenotonsillectomy, and may
progress to include more advanced procedures to enlarge the airway.
individuals with achondroplasia, not only because of upper airway
obstruction, but also because of decreased respiratory drive and
decreased pulmonary function. Early correction of brain stem
compression by decompression of critical foramen magnum stenosis may
help preserve ventilatory potential. Spirometry shows that patients
with achondroplasia also have decreased vital capacity (approximately
70% of the predicted value for height), although this is rarely a
clinically limiting factor (40,41).
individuals with achondroplasia. However, imaging studies have shown
that the cerebrospinal fluid dynamics are variable. Examples of true
megaloencephaly, dilated ventricles without hydrocephalus, and
communicating and
noncommunicating
forms of hydrocephalus have been identified. It is thought that
intracranial venous pressure increases because of jugular foraminal
stenosis, causing arrested hydrocephalus without elevated intracranial
cerebrospinal pressure. Treatment is not required. However,
occasionally there are patients who do appear to have clinical
hydrocephalus, and it is recommended that head circumference should be
measured throughout infancy and plotted against norms published for
individuals with achondroplasia (25).
Those with progressive head enlargement should be seen by a
neurosurgeon familiar with skeletal dysplasia, and they may benefit
from treatment with a ventriculoperitoneal shunt.
stable and healthy of those with skeletal dysplasias, nevertheless
mortality rates are elevated in all age groups. The most common causes
are sudden death in young infants, central nervous system events and
respiratory problems in older children and young adults, and
cardiovascular problems in older adults (33).
achondroplasia include angular deformities of the knees, thoracolumbar
kyphosis, and spinal stenosis. The former two are related to the
pathogenetic factors of ligamentous laxity and muscular hypotonia. Genu
varum is more common than valgus. Genu valgus almost never becomes
severe enough to require treatment. Varus may progress in some patients
and appear to cause pain and difficulty in walking. The fibula is long
compared with the tibia, and it has been proposed that this
differential growth between the long bones may be a cause of the
deformity, by causing laxity of the lateral collateral ligament at the
knee and by exerting a “push” on the talus distally (18,42). However, the deformity involves the distal femur, as well as the proximal and distal tibia (42).
Often, incomplete ossification of the epiphyses makes it impossible, on
plain films, to determine the joint line and to calculate the separate
contributions of the tibia and the femur to the deformity. An
arthrogram may be helpful in such cases. Along with the varus, there is
often tibial torsion. Decision making about treatment is clouded by the
fact that there are no natural history studies to suggest what extent
of varus in young children is likely to progress, and what extent of
varus, if any, is likely to cause degenerative problems in adulthood.
The clinician should rely on the patient or parents for a history of
walking difficulty or knee pain. Pain originating from the knee joint
should be differentiated from the leg pain of spinal stenosis. In
spinal stenosis, the aching is more diffuse and is relieved by
decreasing the lumbar lordosis by flexing the lumbar spine, or
“hunching over.”
involves surgery. There is no evidence of bracing being effective in
children with achondroplasia. Their short thighs make it difficult to
exert mechanical pressure unless the brace is extended to the waist.
Lax ligaments make it difficult to transmit any force to the growth
plates themselves. It does not seem wise to subject these children to
unproven therapies, in light of all of the other medical problems they
face. Tibial osteotomy may be done in any of several ways: opening or
closing, with internal or external fixation (43,44).
Usually, a decision for surgery is not made until the child is at least
4 years old. In patients who are skeletally immature, the osteotomy
should be performed below the tibial tubercle. If internal tibial
torsion exceeds 10 degrees to 20 degrees, that problem should be
corrected at the same time. Fibular shortening alone has been advocated
as a treatment for young children with genu varum (18),
but no long-term studies are available. Severe degenerative arthritis
of the knee is not often seen in adults with achondroplasia.
controversial, but is gradually gaining greater acceptance among
patients and physicians. In contrast to most other skeletal dysplasias,
conditions are favorable for extensive lengthening, in that the joints
are normal and the musculotendinous units and nerves have excellent
tolerance for stretch. Lengthening of 40% to 50% per segment has been
reproducibly achieved for the femur and the tibia (45,46) as well as for the humeri (47),
with lengthening indices of between 30 and 40 days per centimeter. Care
must be taken to minimize the complications of angular deformity and
joint stiffness. The expected benefits of limb lengthening include
increased function in the average-height world. After humeral
lengthening in particular, it may be easier to perform perineal care,
put on shoes and socks, and extend the reach (47).
Benefits of lower extremity lengthening include improved self-image,
and, possibly, decreased lumbar lordosis. The latter is purported to
occur if the hip flexors are lengthened and an extension osteotomy is
performed at the time of femoral lengthening (48,49).
This combination of steps has been claimed to produce a relative pull
of the pelvis into extension. However, most of these claims have not
yet been clinically validated by prospective study, and there are some
reports of increased symptoms of stenosis after lengthening (48).
Improved function has not been conclusively documented using standard
outcome measures, although such studies seem plausible. It is important
information because lengthening may affect muscle strength and joint
status. Because most achondroplasts would need approximately 25 to 30
cm of additional height to enter the range of average stature, some do
not quite achieve this goal. If the lower extremities are lengthened
significantly, the humeri should be lengthened also to facilitate
personal care. Six segments of major lengthening constitute a major
time commitment, even when opposite limbs are lengthened at the same
time. The total time required for such an undertaking may exceed 2
years, during a patient’s critical years of adolescence or young
adulthood. The effects on the lumbar
spine
require prospective study. The long-term effects of such lengthening in
this population also require study. In helping patients to come to a
decision about whether limb lengthening is personally appropriate,
several discussions should be held with the involved family members to
ensure that all the implications of the treatment plan have been
discussed, and that the information has been understood. Discussions
with knowledgeable counselors, as well as with others with
achondroplasia who have undergone limb lengthening, can be of help. In
the proper setting, patients may be gratified with the results of
lengthening.
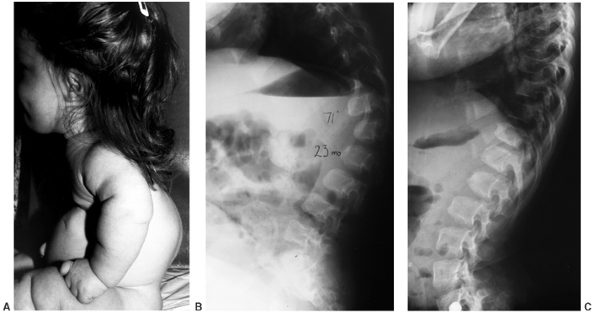
presumably because of low muscle tone, ligamentous laxity, and a large
cranium. The kyphosis is noncongenital and is centered at the twelfth
thoracic or first lumbar vertebra. These vertebrae become wedge shaped
anteriorly, although this is a reversible phenomenon (Fig. 8.8). Most patients improve by the second or third year of life after walking begins and muscle strength increases (11,21,50,51). However, in 10% to 15% of patients, kyphosis remains (Fig. 8.9),
and can increase the risk of symptomatic stenosis through pressure on
the conus, as well as through the secondary lumbar lordosis that it
induces. Therefore, treatment may be indicated at several phases of
life: in infancy, to prevent development of kyphosis; in childhood, to
assist in the correction of those kyphoses that do not correct with
time; and in adulthood, to correct surgically those kyphoses that
contribute to symptomatic spinal stenosis. Pauli et al. reported
favorable results from early intervention (21).
The authors recommend preventing children from sitting unsupported as
well as keeping them from sitting up beyond 60 degrees, even with
support, while kyphosis is present. We dispute the efficacy of this
latter recommendation, because the drive to sit is irrepressible, and
it seems to be more appropriate to provide earlier support, in the form
of a firm-backed chair, when sitting begins. Bracing is indicated if
the kyphosis is accompanied by significant and progressive vertebral
wedging, if it does not reduce below 30 degrees on prone hyperextension
radiographs, or if it does not resolve by the age of 3 years. We prefer
the use of a modified Knight brace, a double-upright thoracolumbosacral
orthosis that has an adjustable posterior pad under the apex of the
kyphosis and that does not constrain the thorax laterally. The modified
Knight brace should be worn full time during waking hours until
resolution of the vertebral wedging occurs, and a lateral film, taken
while the patient is out of the brace, shows no significant kyphosis;
then its use should be gradually tapered. For children in whom
treatment with a brace has failed, we have had some success with a
hyperextension cast incorporating the thighs, which is changed until
elimination of the kyphosis in cast is achieved, and worn for several
months, until the correction
is
maintained. The hyperextension cast has the advantage of not being
removable, and consequently a more sustained corrective force is
applied to the spine. For patients in whom this therapy also fails, two
options exist: prophylactic posterior fusion during childhood; or
observation, with stabilization or correction of kyphosis only in those
who require decompression for spinal stenosis. Although there are no
data directly comparing the two approaches, the former may be
preferable for those with severe kyphosis because of the difficulty and
risk of correcting a large kyphosis in these patients when they are
older (52).
 |
|
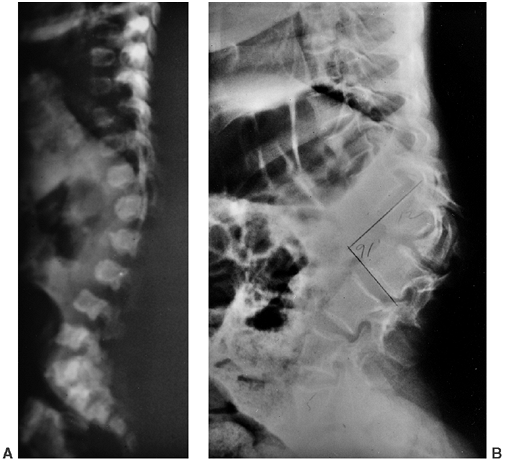
Figure 8.8 Thoracolumbar kyphosis in a 23-month-old achondroplastic child who has not walked yet. A: It is most pronounced in the sitting position. B: Radiograph shows hypoplasia of L1, with rounding-off of the anterior vertebral body corners. C: At 5 years of age, after a period of brace treatment, the shape of L1, as well as the overall kyphosis, has improved.
|
 |
|
Figure 8.9 Thoracolumbar kyphosis has progressed in this patient, who had no medical follow-up between age 6 months (A) and 12 years (B). Although (B) resembles a congenital kyphosis, (A) does not support that conclusion, suggesting that kyphosis was caused by compression over a period instead.
|
individuals with achondroplasia. Most patients present with symptoms of
neurogenic claudication. However, a small number develop muscle
weakness alone, some of which is detected only on routine physical
examination; this serves to emphasize the importance of periodic
neurologic screening. The neurologic deficit may involve upper or lower
motor neuron signs, or both. Symptomatic stenosis usually develops in
the third decade of life, although it has been noted as early as age 11
years. Diagnosis is best made by myelography performed through a
cervical puncture with CT scan; this is, in many cases, more sensitive
and specific than MRI in evaluating the site of blockage in a canal
that is diffusely narrow. Diagnosis of stenosis is an immediate
indication for spinal decompression to be carried out. This
decompression should extend from several levels above the blockage seen
on myelogram, down to the second sacral vertebra (53,54).
The laminectomy should be done by a surgeon experienced in this
procedure and should involve minimal use of instruments, such as
rongeurs or probes, inside the canal. The laminae to be removed should
be thinned with a high-speed burr. The dura often adheres to the
lamina, and the incidence of dural tearing is high. The nerve roots,
which are relatively long, often protrude through any hole in the dura.
Careful foraminotomy should be done if there are signs of root stenosis.
has phenotypic and genotypic similarities to achondroplasia;
nevertheless, the two disorders are distinct from each other. There are
no known instances of achondroplasia and hypochondroplasia existing in
the same family, except by marriage. The mutation that causes most of
the cases of hypochondroplasia has been found to be in the gene for FGFR-3 on the short arm of the fourth chromosome, just as in achondroplasia and thanatophoric dysplasia (12,55,56). However, the nucleotide change is in a different region (1,620, not 1,138) (57). In all these conditions, the mutation results in increased activation of factors that slow cell growth (58). In the case of hypochondroplasia, the mutation arises in a different portion of the gene
(the tyrosine kinase domain, in contrast to the transmembrane domain in
achondroplasia). However, there is more heterogeneity in this condition
than in achondroplasia: between 30% and 40% of patients with
hypochondroplasia have mutations in a different gene instead. As
mentioned earlier, almost all patients with achondroplasia have a
uniform mutation in the same gene. This finding probably accounts for
the clinical variability seen in hypochondroplasia. Increased paternal
age does not appear to play a role in the development of
hypochondroplasia in children.
skeletal dysplasias. The clinical abnormalities are rather mild, and
may go unnoticed until the pubertal growth spurt in some cases. The
eventual height ranges from 118 to 160 cm (16,59).
Head circumference is normal and frontal bossing is mild to absent.
Because of the absence of obvious midface hypoplasia, these patients do
not have a distinctive appearance. The limbs are not short in a
rhizomelic pattern, but rather in a mesomelic one. Body proportions are
closer to normal. The trident hand characteristic of achondroplasia is
not seen in hypochondroplasia. Thoracolumbar kyphosis is also not a
feature of this condition. Varus angulation of the knees is mild, and
may resolve with growth. Joint laxity is mild. Spinal stenosis has been
reported in about one third of patients (16),
but it is usually mild and does not require surgical treatment. Mental
retardation has been reported in some of these patients.
generally subtle. Hall and Spranger have proposed primary and secondary
radiographic criteria (16,60).
The primary criteria are narrowing of the lumbar pedicles; square iliac
crests; short, broad femoral necks; mild metaphyseal flaring; and
brachydactyly. Secondary criteria are shortening of the lumbar
pedicles, mild posterior scalloping of the vertebral bodies, and
elongation of the distal fibula and ulnar styloid. The pelvis has
sciatic notches that are of normal width, in contrast to the narrowed
notches seen in achondroplasia (61).
dysplasia, hypochondroplasia has more variation than achondroplasia in
its severity. In its more extreme form, hypochondroplasia may resemble
achondroplasia; conversely, it may be mistaken for constitutionally
short stature. Hypochondroplasia may also resemble Schmid metaphyseal
dysplasia in its mild short stature and mild genu varum.
Dyschondrosteosis also produces mild short stature, but it can be
distinguished by Madelung deformity and triangular carpal bones.
medical problems. The response to growth hormone administration in
pharmacologic doses has been shown to persist up to 4 years, although
decreasing over time. Studies with follow-up to maturity are still
needed, but this treatment remains an option (35,62,63).
Genetic counseling should be given about the pattern of transmission
for this autosomal dominant condition. If a patient becomes pregnant,
extra vigilance should be exercised for possible disproportion during
childbirth.
achondroplasia in achieving significant gains without undue risks,
because the joints are sound and the muscles tolerate lengthening (64).
Because these patients are generally approximately 20 cm taller than
patients with achondroplasia, limb lengthening may place them within
the normal range of stature. Choosing to undergo the procedure is a
personal decision for patients, who may benefit from talking to others
who have considered it or have undergone it. Long-term follow-up is
still needed to determine the effects on the joints.
or “changing form,” because patients with this condition appear to have
short-limb dwarfism early in life, but later develop a short-trunk
pattern as spinal length is lost during the development of kyphosis and
scoliosis. The condition has been likened to Morquio syndrome, because
of the enlarged appearance of the metaphyses and the contractures (65).
inherited in an autosomal dominant or recessive manner. The cause of
this dysplasia has not been elucidated. However, histologic
abnormalities of the growth plate have been studied and appear to be
characteristic, as shown in the study published by a group led by Boden
(66). The physis shows relatively normal
columns of proliferating chondrocytes. However, there is an abrupt
arrest of further development, with absence of a zone of hypertrophic
or degenerating chondrocytes. Instead, there is a mineralized seal of
bone over the metaphyseal end of the growth plate (Fig. 8.10).
The perichondral ring remains intact, and circumferential growth is
preserved. This uncoupling of endochondral and perichondral growth
appears to account for the characteristic “knobby” metaphyses. Further
understanding of the defects relating to this disorder will undoubtedly
shed light on the normal maturation of the physis.
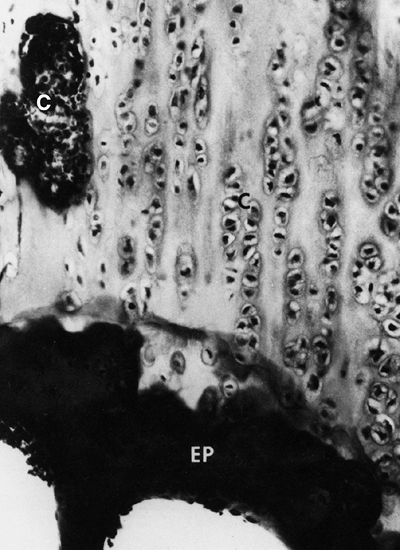 |
|
Figure 8.10 Histology of the growth plate in metatropic dysplasia, showing relatively normal columns of proliferating chondrocytes (C), but absence of the hypertrophic or degenerating zones, as well as a “seal,” or bony end plate (EP),
over the metaphysis. (From Boden SD, Kaplan FS, Fallon MD, et al. Metatropic dwarfism: uncoupling of endochondral and perichondral growth. J Bone Joint Surg Am 1987;69:174, with permission.) |
dysplasia is the presence of the “coccygeal tail,” a cartilaginous
prolongation of the coccyx that is not present in other dysplasias (Fig. 8.11).
The coccygeal tail is usually a few centimeters long and arises from
the gluteal fold. The facial appearance is not determined by the
condition, although there may be a high arched palate. The sternum may
display a pectus carinatum, and the limbs have flexion contractures of
as much as 30 degrees to 40 degrees from infancy, and may have
ligamentous laxity. They appear relatively short with respect to the
trunk. The metaphyses are enlarged, which, when combined with
underdeveloped musculature, gives a bulky appearance to the limbs.
Ventriculomegaly or hydrocephalus has been reported in up to 25% of
patients (67). Upper cervical spine instability develops in some patients. Scoliosis develops in early childhood and is progressive (68,69).
Inguinal hernias are common. Some restrictive lung disease is usually
present, which may cause death in infancy in the one third of patients
who are afflicted by the autosomal recessive form of the disease.
Others survive into adulthood, however, and adult height varies from
110 to 120 cm.
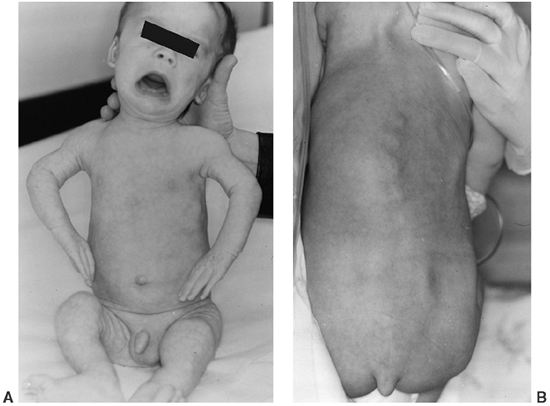 |
|
Figure 8.11 A 1-year-old infant with metatropic dysplasia, displaying knee-flexion contractures, “bulky” metaphyses (A), and a coccygeal tail (B).
|
may be possible in the first or second trimester of pregnancy, the
characteristics being significant dwarfism, narrow thorax, and enlarged
metaphyses (70,71).
Odontoid hypoplasia frequently exists in patients with this condition,
as it does in many patients with skeletal dysplasia. In infancy, the
vertebrae are markedly flattened throughout the spine, but normal in
width. Kyphosis and scoliosis are almost always seen. The ribs are
short and flared, with cupping at the costochondral junctions (Fig. 8.12).
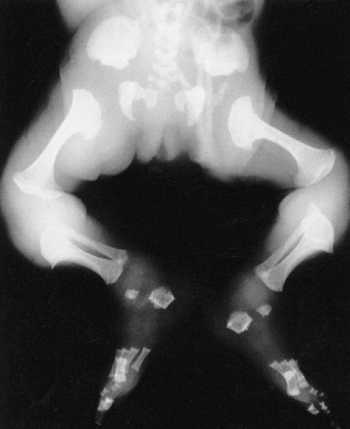
a dumbbell (Fig. 8.13).
The epiphyses have delayed and irregular ossification. Protrusio
acetabuli has been reported. Genu varum of mild to moderate degree
usually develops. Degenerative changes of major joints often occur in
adulthood.
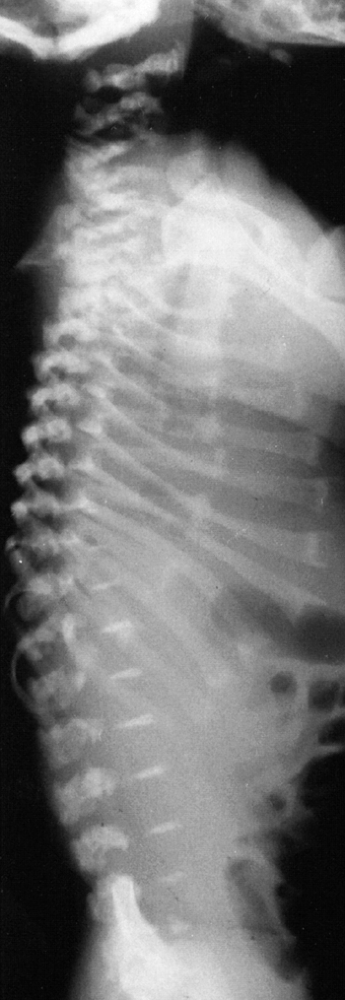 |
|
Figure 8.12
Newborn with metatropic dysplasia. Note platy-spondyly, with delayed vertebral ossification, and flared ribs. (Courtesy of Judy Hall, Vancouver, British Columbia.) |
 |
|
Figure 8.13
Newborn with metatropic dysplasia. The diaphyses are short and the metaphyses are broad and flared, with an appearance that has been likened to dumbbells. The iliac wings are flared, and the acetabulae are deep. (Courtesy of George S. Bassett, MD) |
often dominate infancy, and may be fatal. These problems result from
the smallness of the thorax and perhaps also, in part, from cervical
instability. These children need to be observed on a follow-up basis at
a center where pediatric pulmonary expertise is available. The neck
should be imaged early with lateral flexion–extension radiographs.
Because cervical quadriplegia has been reported after falls, fusion is
recommended if the translation is greater than approximately 8 mm, or
if neurologic compromise is present. If a patient has atlantoaxial
instability between 5 and 8 mm but is neurologically intact, MRIs
should be obtained in flexion and extension. Fusion should be
recommended if cord compromise is seen.
curvature. There is no documentation of the efficacy of brace treatment
for this condition. The treatment may be tried in small curves (less
than 45 degrees) in young patients or those who need support to sit,
but we do not recommend it for large curves, even if the patients are
young and still actively growing. Spinal fusion for scoliosis may be
advisable in patients with more severe curves. Deciding exactly when to
intervene is more of an informed judgment than a science. This author
recommends observation and accepting a larger curve threshold for
surgery in younger patients (under age 10) in order to document medical
health, and to allow the bone to attain a size adequate for
instrumentation. However, progressive sharp angular kyphosis with
paraparesis may occur in metatropic dysplasia, and should be treated
early with fusion if, in the surgeon’s estimation, neurologic
compromise is a risk. When surgery is undertaken, anterior as well as
posterior fusion is recommended, if the patient is able to tolerate it,
because of the high rate of pseudarthrosis in this condition (7).
Given that the curves are often rigid, one should aim to obtain only
the amount of correction that can be achieved safely. Halo-cast
immobilization is an option if patient size, stenosis, or poor bone
density make instrumentation inadvisable.
the mouth, teeth, limbs, and heart characterize the uncommon skeletal
dysplasia chondroectodermal dysplasia, also known as Ellis–van Creveld syndrome (72).
The syndrome is transmitted as an autosomal recessive condition, and is
therefore more common in closely knit populations, most notably in the
Pennsylvania Old Order Amish community. The basic defect is in a novel
gene, EVC, on the short arm of the fourth chromosome (73,74,75). It leads to a generalized defect of maturation of endochondral ossification.
the neonatal period (the most severely affected age group). Cardiac
defects are present in approximately half of patients, and most
commonly consist of atrial septal defects or single atrium. As the name
of this disorder would suggest, the teeth are also abnormal, appearing
early and also being lost early. The nails are hypoplastic. Urologic
features include hypospadias and epispadias. The skeletal features are
shortening of the middle and distal parts of the extremities
(acromesomelic) in combination with a normal spine (76). This distal shortening is the opposite of that seen in achondroplasia (77,78,79).
The chest is narrow. The ligaments are lax, and there is often
significant genu valgum. The proximal tibial epiphysis is often
markedly “wedged.” Rotational abnormalities often accompany this, such
as external rotation of the femur and internal rotation of the tibia.
The combination of these findings can give the limb an appearance of a
flexion contracture, when, in fact, the problem is really valgus.
Postaxial polydactyly is quite common in the hands, and occurs much
less commonly in the feet.
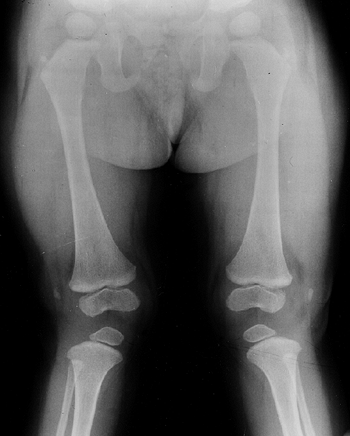
knee valgus is usually relatively symmetric. It results partially from
uneven growth of the proximal tibial epiphysis, with the lateral side
being underdeveloped (Fig. 8.14). An exostosis
may arise medially from the proximal tibial metaphy-sis. The acetabulae
have spike formations at the medial and lateral edges. The capital
femoral epiphyses ossify early, and the greater trochanteric apophyses
are pronounced. The wrists display fusion of the capitate and the
hamate, and sometimes of other bones. The carpal bones have delayed
maturation but the maturation of the phalanges is accelerated.
the cardiac status is stable, is usually successful. The angular and
rotational disturbance of the lower extremities is usually addressed
when it becomes clinically significant or is rapidly progressing,
usually at approximately 20 degrees of valgus (80).
Bracing seems to have little or no effect, and surgery remains the
mainstay of treatment. Careful preoperative planning is needed, taking
into account deformity at all locations from the proximal femur to the
ankle, and aiming to correct the mechanical axes and the malrotation
with as few procedures as possible (81).
Usually, external fixation is the most effective way of performing the
correction. If the deformity is one of simple valgus, medial
hemiepiphyseal stapling alone may be adequate. If the wedging of the
lateral proximal tibial epiphysis is severe, elevation of the lateral
plateau may be necessary in a manner similar to medial elevation for
Blount disease. Limb lengthening is possible in this condition with a
minimum of complications.
 |
|
Figure 8.14
Lower extremities of a 5-year-old child with chondroectodermal dysplasia, demonstrating the characteristic pronounced hypoplasia of the lateral proximal tibial epiphysis with marked genu valgus. |
with the most numerous, disparate, and severe skeletal abnormalities.
The term diastrophic comes from a Greek
root meaning “distorted,” which aptly describes the ears, spine, long
bones, and feet of patients with this condition. Before the current
level of understanding of the skeletal dysplasias was developed, an
early authority referred to this condition as “achondroplasia with
clubbed feet” (82,83). Certainly, the abnormalities are much more extensive than that.
population are carriers, and there are over 160 individuals known to be
affected because of an apparent founder effect. The defect is on
chromosome 5 in the gene that codes for a sulfate transporter protein
(aptly named diastrophic dysplasia sulfate transporter) (84,85).
This protein is expressed in virtually all cell types. It has been
demonstrated that patients with DD have a decreased sulfate content in
the cartilage (86). It is presumed that a
defect in this gene leads to undersulfation of proteoglycan in the
cartilage matrix. If one considers proteoglycans to be the “hydraulic
jacks” of cartilage at the ultrastructural level, it is understandable
that there should be such impairment of performance of physeal,
epiphyseal, and articular cartilage throughout the body.
Achondrogenesis types 1B and 2 are more serious disorders causing
mutations on the same gene.
Tracheal cartilage has some of the same abnormalities seen in other
cartilage types, but this still does not explain some of the focal,
specific malformations seen in DD, such as proximal interphalangeal
joint fusion in the hands, short first metacarpal causing hitchhiker
thumbs, and cervical spina bifida. Further work must be carried out on
the role of this sulfate transporter in skeletal growth and development
in order to explain these curious findings.
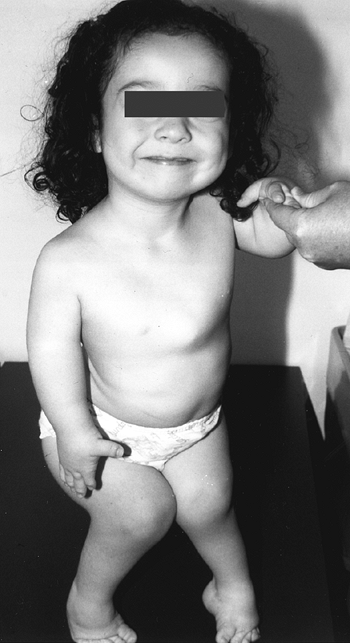
with this condition gave rise to the previously used name, “cherub
dwarf” (Fig. 8.15). The nasal bridge is flattened. Up to one half of patients have cleft palates, which may contribute to aspiration pneumonia (87).
The cartilage of the trachea is abnormally soft, and its diameter may
be narrowed. The ear is normal at birth, but develops a peculiar acute
swelling of the pinna at 3 to 6 weeks in 80% to 85% of cases. The
reason for this event and this timing is not known. The cartilage
hardens in a deformed shape—the “cauliflower ear,” which is one of the
pathognomonic features of this dysplasia.
(approximately 5%) perinatal mortality because of respiratory problems,
especially aspiration pneumonia and tracheomalacia. Motor milestones
are delayed: sitting occurs at a mean age of 8 months, pulling to stand
at 13 months, and walking at 24 months (89).
The posterior arches of the lower cervical spine are often bifid. There
are no external clues to this underlying abnormality, which is occult.
Cervical kyphosis is seen in one third to one half of patients (8,91);
this disorder may be present in infancy, and its course is variable.
Spontaneous resolution has been reported in a number of patients, even
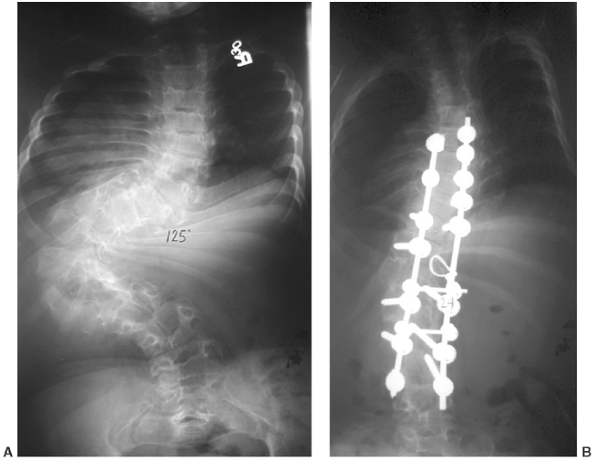
in those with curves of up to 80 degrees (92,93) (Fig. 8.16 A, B). However, other cases progress, and there are several reports of quadriparesis resulting from this deformity (8,94). Scoliosis develops in at least one third of patients (91),
but many curves do not exceed 50 degrees. Tolo and Kopits state that
the scoliosis may be one of two types: idiopathiclike or sharply
angular (95). The sharply angular type is
usually characterized by kyphosis at the same level as the scoliosis.
Spinal stenosis is not common, in contrast to achondroplasia. Most
patients have significant lumbar lordosis, which is likely to
compensate for the hip flexion contractures in diastrophism.
 |
|
Figure 8.15
A 5-year-old girl with diastrophic dysplasia. Note prominent cheeks, circumoral fullness, equinovarus feet, valgus knees with flexion contracture, and abducted or “hitchhiker” thumbs. |
shoulders may be subluxated, as may the radial heads (possibly because
of ulnar shortening). The hands are short, broad, and ulnarly deviated.
Hitchhiker thumb is due to a short, proximally placed, often
triangular, first metacarpal that may be hypermobile; this finding is
seen in up to 95% of diastrophic persons. The proximal interphalangeal
joints of the fingers are often fused (symphalangism).
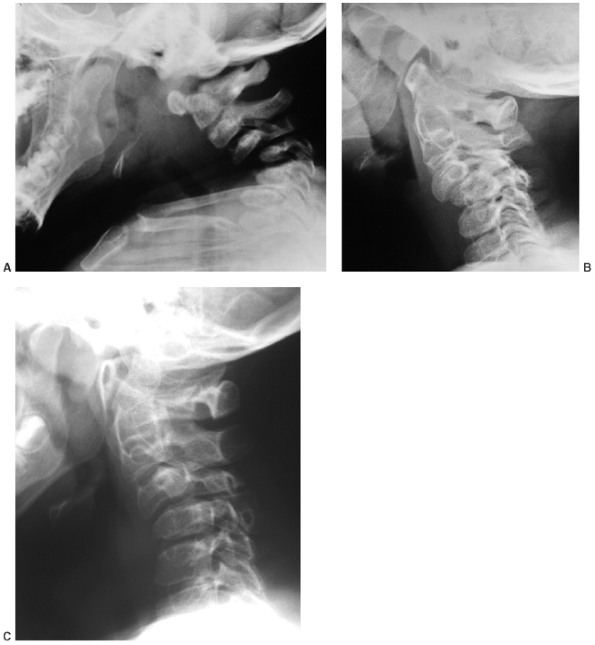
proximal femoral epiphyses progressively deform, and even subluxate in
some patients. Epiphyseal flattening and hinge abduction develop in
many patients (96). Arthritic changes develop
by early to middle adulthood. The knees usually have flexion
contractures that result from a combination
of ligamentous contracture and epiphyseal deformation (Fig. 8.17).
Excessive valgus is also common. As many as one fourth of these
patients have a dislocated patella. Degenerative joint disease of the
hips and knees develops in early to mid-adulthood.
 |
|
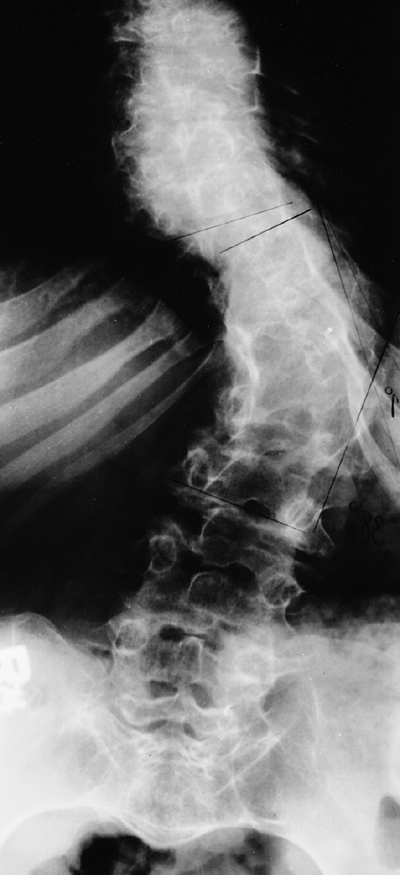
Figure 8.16 Cervical kyphosis in a 1-year-old child (A)
with diastrophic dysplasia is pronounced, with marked deformity of C4. The findings of neurologic examination are normal. Four years later, the condition is markedly improved without any intervention (B), and 7 years later the vertebral bodies have been restored to nearly normal shape, although the canal remains narrow (C). |
as being clubfeet, but many different variations exist. In the large
Finnish series of Ryoppy et al., the most common finding was adduction
and valgus (seen in 43%), followed in order of prevalence by
equinovarus (in 37%), and then by pure equinus (83).
The great toe may be in additional varus, beyond the degree commonly
seen in idiopathic clubfoot; this is analogous to the hitchhiker thumb.
The navicular may not be as medially displaced as in typical clubfoot.
The foot deformities are very stiff and involve bony malformations as
well as contracture and malalignment. These feet are as difficult to
correct as any type of clubfoot.
is related to overall severity of involvement, with taller people being
less severely affected (97,98).
These form part of the same spectrum of disorders. Growth curves
describing patients with DD are available in the literature (99). The median adult height of these patients is 136 cm for men and 129 cm for women (100).
Therefore, individuals with achondroplasia are shorter in stature, and
are approximately equal in height to those with pseudoachondroplasia
and
SED
congenita. The pubertal growth spurt is diminished or absent, so the
overall growth failure is progressive, thereby suggesting that the
physes are unable to respond to normal hormonal influences.
 |
|
Figure 8.17
The extremities as well as the feet are involved in diastrophic dysplasia. Joint contracture is accompanied by epiphyseal deformity, as this knee radiograph illustrates (A). A rigid, severe equinovarus foot is common (B). |
significantly reduced, except for the small number of patients
(approximately 8%) who die in infancy from respiratory causes, or in
childhood from cervical myelopathy. Patients with severe spinal
deformities are more prone to develop respiratory problems. Many
patients are able to lead productive work and family lives.
second trimester of pregnancy with demonstration of long-bone
measurements at least three standard deviations less than normal, as
well as clubfeet and adducted thumbs. In infancy, calcification
develops in the pinna of the ear, and later in the cranium and the
costal cartilages. The vertebrae are poorly ossified. The lower
cervical spine may demonstrate kyphosis in infancy and early childhood,
usually having an apex at approximately C4. The disorder tends to
decrease with time (101). MRI may be necessary
to judge the severity of this disorder in relation to the spinal cord.
Among patients with this condition, only one case of atlantoaxial
instability has been reported. The vertebral “wedging” decreases with
time in most patients (102). Spina bifida occulta is seen in over three fourths of the patients (102).
The interpediculate distances narrow only slightly at descending levels
of the lumbar spine, unlike in achondroplasia. Scoliosis may occur in
the form of either a sharp, angular curve or a gradual, idiopathiclike
one (Fig. 8.18).
findings. The first metacarpal is small, oval, and proximally placed.
Although the proximal interphalangeal joints of the digits are
ankylosed, a radiolucent space is present early on, which later fuses.
Both the ulna and the fibula are shortened, contributing to the valgus
of the knees and the radial head subluxation that is sometimes seen.
The diaphyses of the long bones are short and broad. The epiphyses of
both the proximal and the distal femur are delayed in appearing. The
capital femoral epiphyses may show signs of osteonecrosis well into
childhood. Arthrograms show flattening of both the proximal and the
distal femur, accounting for the stiffness observed clinically. The
proximal femur is usually in varus, but even so, hip dysplasia or
subluxation may develop progressively with time.
periodically on all children. A lateral cervical radiograph should be
performed during the first 2 years of life as well. If cervical
kyphosis is noted, the patient should be followed with clinical and
radiographic examinations every 6 months. The behavior of the kyphosis
appears to be related to the severity of the DD (101).
If the kyphosis is nonprogressive, and there is no neurologic deficit,
it should only be observed. Most kyphoses in this disorder will improve
with time and growth, probably because of strengthening of the extensor
muscles (96,97). However, if the kyphosis
progresses and there is no neurologic deficit, bracing may be employed.
Successful control of cervical kyphosis by full-time use of the
Milwaukee brace was reported by Bethem et al. (8,16).
If the curve continues to progress despite the brace, or if a
neurologic deficit occurs, posterior fusion should be performed. The
surgeon should be cognizant of the bifid lamina during the exposure. It
may not be technically possible to use instrumentation. If adequate
bone graft is not available from the iliac crests, it may be taken from
the proximal tibia(s) or other sources. Immobilization by a halo and
vest is needed for 2 to 4 months. The pins should be inserted at a
lower torque than in adults (4 inch-pounds), and the surgeon may elect
to use a slight distractive force and a slight posterior translation of
the head. A pad may be used behind the apex of the kyphosis to help
prevent it from increasing. If neurologic deficit is present along with
the curve, MRI in a neutral position and in extension will help
determine the degree of anterior compression and the type of procedure
required. If there is severe anterior cord compression, corpectomy and
strut graft may be indicated. Posterior fusion is indicated as well.
 |
|
Figure 8.18 Significant scoliosis may occur early in diastrophic dysplasia, as in this 125-degree curve in an 8-year-old child (A). After correction (B).
|
and follows one of three patterns, namely, early progressive,
idiopathiclike, and mild nonprogressive (103). It has been shown that it is unrelated to the type of mutation in diastrophic sulfate transporter (DTST) (102).
The success of bracing in preventing or slowing curve progression has
not been documented. It seems reasonable to offer bracing to patients
if the curve is less than 45 degrees, but to discontinue it in those
for whom there is no apparent benefit. Large curves often continue to
progress in adulthood (91). Surgery has a role
in preventing progression where the curve is more than 50 degrees.
Posterior fusion is the mainstay of treatment (95).
For younger patients, or those whose associated kyphosis is over 50
degrees, anterior fusion may be added as well. Instrumentation should
be used carefully, bearing in mind the short stature of the patient,
the stiffness of the spine, and the slightly diminished bone density.
Small hooks may be used if needed (95). Spinal
stenosis is seen much less commonly than in achondroplasia, but it may
occur if degenerative changes are superimposed on the baseline canal
size. Mild stenosis may be masked in some cases by the patients’
relative inactivity.
together. If they are significant (over 40 degrees), release may be
considered, provided an arthrogram shows no epiphyseal flattening and a
good potential for gaining range
of
motion. If there is epiphyseal flattening it is probably better to
avoid releases, given that recurrence is likely. Hip dysplasia is often
progressive because of deformation of the abnormal cartilage under
muscle forces and body weight. No long-term series has been carried out
in order to show the ability of surgery to arrest this process.
Therefore, the surgeon should use his or her own judgment as to whether
an acetabular augmentation or femoral osteotomy will help provide good
coverage without restricting range of motion or function. Conservative
treatment cannot be faulted in this condition.
of the main reasons for decreasing ability to walk in patients with
diastrophism. Hip joint arthroplasty is an option when the pain becomes
severe. Small or custom-made components are needed (9).
The femur often has an increased anterior bow, probably in compensation
for the hip flexion contracture. The isthmus of the femur is only 13 mm
on average. Femoral shortening osteotomy is often needed. Contracture
release (adductor, rectus, and sartorius) may be needed along with the
arthroplasty, but femoral nerve palsy may follow if it is done
extensively. Autograft augmentation of the acetabulum is often
necessary. The largest series of hip arthroplasty in this condition is
reported by Helenius et al., with 41 hip replacements in 24 patients
who had a mean age of 41 years (104).
Trochanteric transfer was performed in approximately half of the hip
replacements. Two had femoral palsies, which recovered. The range of
motion of the hip was increased slightly, and the Harris hip score
nearly doubled. At follow-up after a mean duration of 8 years, the
revision rate was 24%; all involved the acetabular side.
extension. Complete correction of knee flexion contractures is
prohibited by the shape of the condyles, which may be triangular,
creating a bony block to flexion, extension, or both. Residual
contracture at maturity may be diminished by distal femoral osteotomy.
Patellar subluxation is present in one fourth of diastrophic persons;
correcting these may help improve extensor power.
pain. Unique features of the procedure for diastrophic patients include
extensive lateral release with patellar relocation, use of constrained
prostheses whose stems must be shortened or bent, and femoral osteotomy
(105). The mean age at surgery is similar to
that for total hip arthroplasty (mid-forties). Pain and function are
improved, although many patients lose a slight amount of knee motion (105).
equinovarus, other types may be seen, including isolated equinus,
forefoot adduction, and valgus. The feet are rigid, and treatment with
a cast is usually futile. A plantigrade foot is the goal of treatment.
Surgical treatment should be deferred until the feet of the patient are
large enough to work on (usually after 1 year of age), and the neck is
safe to handle. If soft-tissue release is performed, it should be as
extensive as is needed to correct the deformity.
tibiofibular ligament to bring the dome of the talus into the mortise
is required to achieve the intended extent of the release. Partial
recurrence of deformity is common (83), and salvage procedures include talectomy, talocalcaneal decancellation, or arthrodesis (in the older child).
profound effects. The syndrome is characterized by typical facial
features and large, stiff joints with contractures (106,107).
The syndrome has been likened by some to metatropic dysplasia because
of the enlarged stiff joints, and to SED because of the generalized
disorder of both spinal and epiphyseal growth. The syndrome is now
known to be due to a defect in type II collagen, the predominant
protein of cartilage. Most mutations occur between exons 12 and 24 of
the COL2A1 gene. Although numerous
different mutations have been described, their phenotypic similarity is
because of the fact that they are all in this region, and that they
tend to occur at splice sites, resulting in exon skipping and,
therefore, in shorter type II collagen monomers (108,109).
These type II collagen monomers combine with normal-length monomers
from both ends to form heterotrimers with the missing segment excluded
from the helix (110). This combination allows
the mutation to express autosomal dominant behavior, in that one copy
of the mutant allele disrupts the structure of the entire cartilage
matrix. In most patients, it is a new mutation that causes the disorder.
Scanning electron microscopy of the cartilage demonstrates deficiency
and disorganization of cartilage fibrils, and large, open cystlike
spaces.
condition too have a somewhat characteristic facial appearance, with
prominent eyes and forehead and a depressed midface. Many patients have
cleft palates. The sternum may be depressed, and the trunk is broad,
unlike the findings in metatropic dysplasia. However, as with that
condition, the joints appear enlarged because of broad metaphyses of
the long bones, and they are also stiff, often lacking both extension
and full flexion. This stiffness affects the hands as well as the large
joints. Motor development may be delayed
because of contractures or myelopathy. Intellectual development, however, is normal. Inguinal and abdominal hernias are common.
which is made more likely by cleft palate or tracheomalacia. As with
many other patients with skeletal dysplasia, otitis media may be a
recurrent problem and may even contribute to hearing impairment. Myopia
is common, and retinal detachment and glaucoma may cause severe visual
loss, as in Kniest’s original patient, who became blind during
adolescence (107). Adult height ranges from 106 to 145 cm.
perhaps as a result of disuse. All regions of the spine are affected
with problems, from atlantoaxial instability (due to odontoid
hypoplasia) to hypoplasia of the cervical vertebrae and flattening of
all vertebrae (112). The vertebral bodies have vertical clefts (113). There is kyphosis, and often mild scoliosis (Fig. 8.19),
in the thoracolumbar spine. The femoral necks, as with all metaphyses,
are short and broad. There are irregular calcifications in the
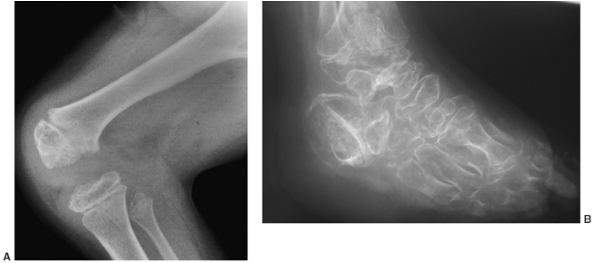
epiphyseal and metaphyseal regions. Valgus deformities often develop in
the distal femur or proximal tibia. The epiphyses are flattened and
irregular (Fig. 8.20). Degenerative arthritis of the major weight-bearing joints develops early, even in the second decade of life.
 |
|
Figure 8.19 Scoliosis is common in Kniest syndrome, but rarely severe enough to require intervention.
|
the diagnosis is first made, when intubation is planned, or with any
missed milestones or loss of of strength or coordination (114).
Kyphoscoliosis should be monitored, but the efficacy of brace treatment
has never been studied, and remains doubtful. Surgery for spinal
deformity is not often needed. Physical therapy has been recommended to
increase joint mobility, but the efficacy of this too has not been
proven.
be helpful in certain circumstances. Osteotomy of the proximal femur
may help improve joint congruity, if hinge abduction is developing. An
arthrogram in different positions may aid in making this decision. Any
flexion deformity can be corrected at the same time by incorporating an
extension component into the osteotomy, as long as adequate range of
flexion will remain for sitting (at least 80 to 90 degrees). Osteotomy
of the distal femur or proximal tibia is indicated if knee valgus is
excessive. Equinovarus of the foot may be treated initially with casts,
but surgery is often indicated. If the stiffness of the first
metatarsophalangeal joint produces hallux rigidus, traditional measures
such as cheilectomy or arthrodesis may help.
its key features include significant spinal and epiphyseal involvement,
without metaphyseal enlargement or contractures of other joints (115).
SED congenita is heritable in an autosomal dominant form, but most
patients acquire the disease because of a new mutation. The genetic
defect has been characterized as a defect in type II collagen, the gene
for which is located on chromosome 1293. Type II collagen is the
predominant protein of cartilage, and mutations have been observed in
the α1 chain, resulting in alteration in length (116).
As in many other skeletal dysplasias, electron microscopy has
demonstrated intracellular inclusions, which are probably due to
intracellular retention of procollagen (117).
appearance is characteristic—the face is taut and the mouth small.
Cleft palate is common. The trunk and extremities are
shortened, although the extremities are more shortened proximally because of the coxa vara (Fig. 8.21).
There is pectus carinatum, possibly because the rib growth outpaces the
increase in trunk height. There are many similarities to Morquio
syndrome, but an absence of visceral involvement. Scoliosis and
kyphosis usually develop before the teen years. Back pain is also
common by this time.
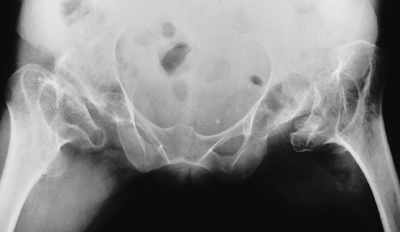 |
|
Figure 8.20
Like most epiphyses in Kniest syndrome, the femoral heads are flattened and irregular. Also note the short, broad femoral necks. |
variable. The degree of varus has been felt to be the best marker for
the severity of the disease (118). If the varus is severe, it
is often accompanied by a significant hip flexion contracture. Patients
often walk with the trunk and head held back to compensate for this
contracture. The knees are often in mild varus, and external rotation
of the femora and internal rotation of the tibiae often coexist.
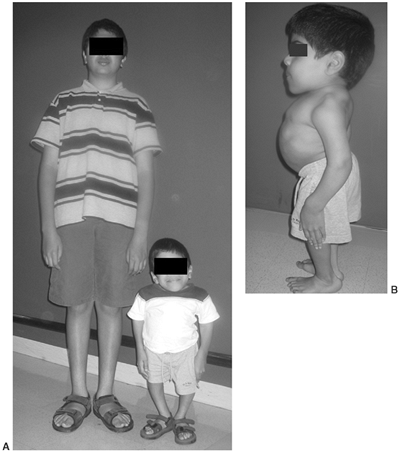 |
|
Figure 8.21
Spondyloepiphyseal dysplasia congenita produces the most extreme short stature. This 12-year-old boy is with his 14-year-old brother (A). Note extreme spinal shortening, increased lumbar lordosis, and hip flexion contracture (B). |
stiffness is not nearly as much as that in DD. Growth curves of
patients with this condition are available in the literature (118). Adult height varies from 90 to 125 cm.
There is often odontoid hypoplasia or os odontoideum. Flattened
vertebral ossification centers with posterior wedging give the vertebra
a “pear-shaped” appearance when viewed laterally. If scoliosis is
present, it is often sharply angulated over a few vertebrae (Fig. 8.22).
Disc spaces become narrow and irregular before maturity. Ossification
of the pubis is delayed. The proximal femora are in varus with short
necks, but the degree of this involvement varies. The proximal femur
may not ossify for up to 9 years of age (118). Often, the varus is progressive (Fig. 8.23).
There is possible progressive extrusion of the femoral head. An
arthrogram may be necessary to visualize the extrusion clearly. The
distal femoral metaphyses are flared. Genu valgum is more common than
genu varum. Early osteoarthritis is likely in the hips, even more so
than in the knee. The carpal bones are delayed in ossification, but the
tubular bones of the hands are near normal.
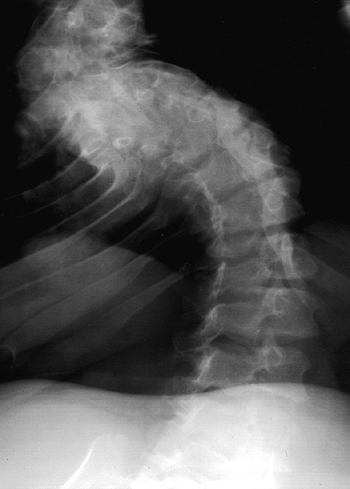 |
|
Figure 8.22
Scoliosis, with a sharp apex concentrated over a limited number of vertebrae, is characteristic of spondyloepiphyseal dysplasia congenita. |
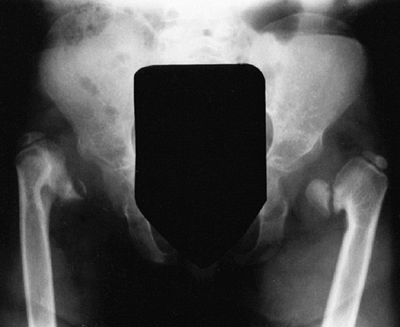 |
|
Figure 8.23
The hips of this patient with spondyloepiphyseal dysplasia congenita show severe coxa vara, with delayed ossification of the capital femoral epiphyses. (Courtesy of George S. Bassett, MD) |
because of a small thorax. The most common disabling problem in this
syndrome involves the eyes: retinal detachment is frequent and is
reported to occur especially during the adolescent growth spurt.
Regular ophthalmologic examinations are recommended. Hearing impairment
is noted in a few patients.
can involve neck instability. Os odontoideum, odontoid hypoplasia, or
aplasia may cause instability and, potentially, myelopathy (Fig. 8.24 A,B,C). Numerous cases have been reported (7).
Careful neurologic examination should be done at each clinic visit.
Flexion–extension radiographs should be performed approximately every 3
years if an upper cervical anomaly is identified. If the odontoid is
difficult to see, one can use CT scan or MRI. Stenosis often coexists
and makes subluxation more critical. It is recommended that the
atlantoaxial interval should be fused if instability exceeds 8 mm, or
if symptoms develop. If severe stenosis exists, or if a fixed
subluxation cannot be reduced, it may be necessary to perform a
decompression of the atlas and, consequently, fusion to the occiput (121).
Transarticular screw fixation is often possible after the age of
approximately 6 to 8 years. However, in children younger than this,
lack of bone strength or insufficient size of the neural arches may
make rigid internal fixation impractical; in these cases, bone graft
and halo-cast immobilization are usually successful. Scoliosis is
present in more than one half of patients with SED congenita, and may
become severe. Control with a brace may be attempted
if
the curve is less than 40 degrees. However, the long-term efficacy of
bracing has not been demonstrated. Fusion may be necessary if the curve
is progressive. Thoracolumbar stenosis is not as severe as in
achondroplasia. Instrumentation is not contraindicated but should be
used judiciously. If internal stabilization is not judged to be strong,
the use of a halo brace for immobilization postoperatively should be
considered. Correction is usually modest [17% in one series (7)].
Anterior surgery should be used if the patient is young (younger than
11 years) or the curve is rigid (correcting to less than approximately
45 degrees). Kyphosis is also common; use of a Milwaukee brace has been
shown to be effective if it can be worn until maturity (7).
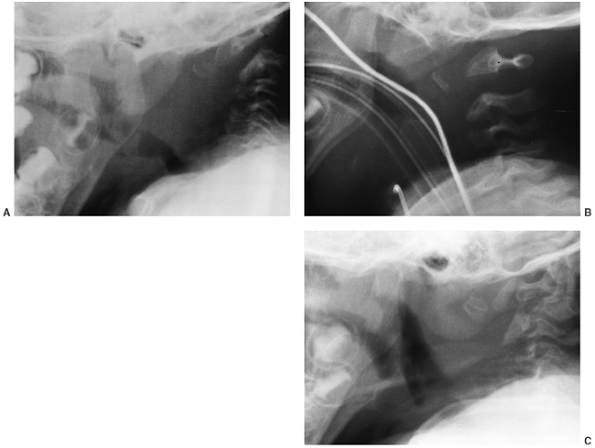 |
|
Figure 8.24
Atlantoaxial instability is common in spondyloepiphyseal dysplasia congenita. This 2-year-old boy experienced delayed motor milestones. The upright lateral film (A) of the cervical spine demonstrates odontoid hypoplasia with marked atlantoaxial subluxation. Less evident is the stenosis of the ring of the atlas. When supine in a neutral position (B), the alignment improved. Following decompression of the atlas and fusion of occiput to C2 (C), he gained the ability to walk. |
less than 100 degrees. Insufficient correction makes recurrence more
likely. It is helpful to correct any flexion contracture at the same
time if enough flexion will remain. Malrotation should be corrected as
well. If a patient is experiencing painful hinge abduction, a valgus
osteotomy may improve the symptoms. An arthrogram may help in planning
the surgical procedure.
of femoral and iliac osteotomies. When doing any procedure on the hip,
the knee alignment should be assessed at the same time and corrected if
necessary. The clinician should also consider the effect that
correction of the knee angle will have on the hip. For instance,
correction of severe knee valgus deformity has the same effect on hip
congruity as does a varus osteotomy of the proximal femur.
the hip is stiff, custom-made components are often needed, and
concomitant osteotomy is sometimes necessary (9).
Foot deformities can usually be treated according to standard
principles for the treatment of clubfoot. If the foot is stiff, an
osteotomy or decancellation of the talus, calcaneus, and/or cuboid may
be needed.
appear in later childhood, or even in adulthood. The spine and only the
larger joints are affected. Several genetic patterns of transmission
have been reported (122,123,124).
The most common SED tarda is X-linked, in which male patients are more
commonly or more severely affected and female patients may show milder
(or no) manifestations. SED tarda occurs because of a defect in the
gene SEDL (125), whose function is not yet known. A recessive form has also been reported. SED tarda is one of several conditions (termed the COL2A1 group or the SED family), which may result from a mutation in type II collagen (10,55).
The mechanism by which the particular mutation for this condition
produces the mildest phenotype in this family will doubtlessly be
elucidated in the near future.
clinical attention when the child is approximately 4 years old, at the
earliest. Stature is mildly shortened. Armspan is significantly longer
than height. The condition may be first diagnosed as bilateral Perthes
syndrome (126). Back pain and hip or knee pain
may be present in childhood. The range of motion of the joints is
minimally limited, if at all. Varus or valgus deformities are rare.
Degenerative changes may occur in the hip or the knee by young
adulthood. Adult height may be 152 cm or more (124).
involved. The hips manifest varying degrees of coxa magna, flattening,
or epiphyseal extrusion, differing markedly even within the same family
(Fig. 8.25). A few patients present with
bilateral coxa vara. Odontoid hypoplasia or os odontoideum may cause
atlantoaxial instability. Spinal involvement ranges from mild
platyspondyly (Fig. 8.26), with ax-like
configuration of the vertebral bodies in the lateral view, to isolated
disc-space narrowing. Mild-to-moderate scoliosis develops in a few
cases.
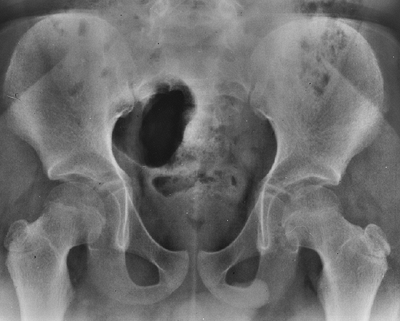 |
|
Figure 8.25 The pelvis in this patient with spondyloepiphyseal dysplasia tarda shows somewhat small, flattened epiphyses.
|
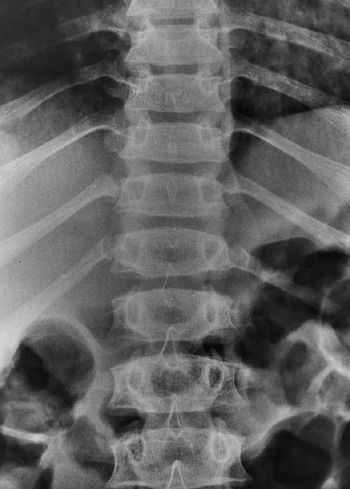 |
|
Figure 8.26
The spine in this patient with spondyloepiphyseal dysplasia tarda shows typical mild flattening of the vertebral bodies, but no scoliosis. |
with this dysplasia varies widely, even within a family. There are
undoubtedly many affected individuals whose problems are so mild that
no diagnosis is ever made. One large family was reported in which only
4 of the 31 affected members requested any orthopaedic treatment (127).
SED is one condition to consider whenever spine, hip, and/or knee pains
run in a family, and the radiographs seem to be just a little atypical.
Bracing may be recommended if scoliosis exceeds 30 degrees in the
skeletally immature patient. Surgery should be offered for the rare
patient in whom it exceeds 50 degrees. All patients should be screened
for atlantoaxial instability. Fusion should be recommended if the spine
is unstable in either flexion or extension, according to criteria given
earlier for SED congenita. A role for procedures to increase coverage
of the dysplastic, extruded femoral head by the acetabulum during the
childhood years has not been well documented. However, it may be
helpful in the rare young patient with increasing extrusion and
persistent pain, in whom the hip contact surface is markedly
compromised. If hip pain becomes a problem after the femoral heads are
mature or nearly mature, osteotomy may help increase congruity or
decrease hinge abduction. Usually, a valgus or valgus-extension
osteotomy is most appropriate, so long as there is reasonable joint
space, and adequate contact remains. A preoperative arthrogram is
helpful
in the younger patient to see the full outline of the articular
surface. Osteotomies of knees or ankles are rarely needed. Total joint
replacement is often needed for the hips or knees, at an age much
younger than in the general population.
it was later recognized as being distinct from SED because of
late-onset physical findings and milder spinal involvement.
Pseudoachondroplasia involves the metaphyses, as well as the spine and
epiphyses. The significant features are ligamentous laxity and
“windswept” knees. With a prevalence of approximately four per million,
it is one of the more common skeletal dysplasias. Early histologic
studies demonstrated that the chondrocytes of persons with
pseudoachondroplasia contained lamellar inclusions within the
endoplasmic reticulum (68,129).
It has since been demonstrated that pseudoachondroplasia results from a
mutation in COMP, the same protein that is disordered in MED (130,131).
These two conditions have been classified as a family of dysplasias,
with pseudoachondroplasia on the severe end and MEDs on the milder and
more heterogeneous end. COMP is normally a large extracellular matrix
glycoprotein that is found in the territorial matrix surrounding
chondrocytes. COMP is a member of the thrombospondin family and has a
pentameric structure and is also found in the extracellular matrix of
ligament and tendon tissues. A number of different mutations have been
found in this gene (130). Some of the mutations
involve a GAC trinucleotide repeat within the gene; it has been found
that either expansions or deletions in this region may cause the
disease. Normal growth and development occur in mice whose COMP
gene has been deleted, illustrating that it is accumulation of an
abnormal form of COMP, not its absence, which causes
pseudoachondroplasia. Mutant COMP accumulates in the rough endoplasmic
reticulum of chondrocytes and tenocytes in persons with this disorder,
and may cause apoptosis (132) (Fig. 8.27).
Abnormal COMP also makes its way into the extracellular matrix. It is
thought that with accumulation of the mutant COMP, the abnormal matrix
does not help maintain the shape of the cells, and allows them to
become flatter (133,134).
Three separate families have been reported who have somatic/germline
mosaicism, which allows this dominant condition to behave like a
recessive disorder, with up to a 50% recurrence risk even from two
parents with normal stature (131).
recognized as having a skeletal dysplasia at birth. The mean length at
birth is 49 cm, which is within normal limits. Growth tapers soon after
this, however, so that the height falls to less than the fifth
percentile by the age of 2 years (135). This
pattern of progressive involvement is typical of storage disorders,
which is essentially the nature of this condition. Usually, the
diagnosis is made by the age of 2 to 4 years.
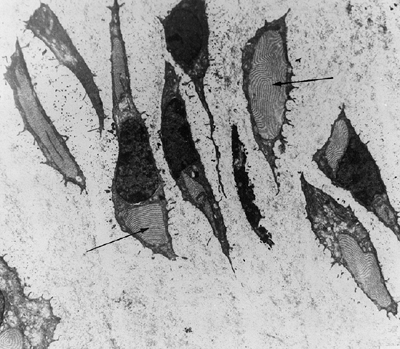 |
|
Figure 8.27
Abnormal lamellar inclusion bodies in endoplasmic reticulum of growth-plate chondrocytes of a patient with pseudoachondroplasia. (From Cooper RR, Ponseti IV, Maynard JA. Pseudoachondroplastic dwarfism. A rough-surfaced endoplasmic reticulum storage disorder. J Bone Joint Surg Am 1973;55:475, with permission.) |
Mild scoliosis often occurs, but few cases become severe. The pattern
of shortening of the extremities is rhizomelic and progresses with
time. This finding helps explain why early writers often confused this
disorder with achondroplasia. However, the hands are not
trident-shaped. In almost all cases, the hips are dysplastic and the
patient exhibits a waddling gait. Knees are most commonly in excessive
valgus or windswept (one in varus, one in valgus), because of lax
ligaments, as well as having epiphyseal and metaphyseal abnormalities.
The joints may have flexion contractures or recurvatum. The mean adult
height is 119 cm (range, 106 to 130 cm) (135).
No changes outside the skeletal system have been noted as part of the
disorder. Patients have normal intelligence, and a normal life
expectancy (138).
ways to make an early diagnosis because affected babies are outwardly normal (Fig. 8.29).
This flattening may be discovered if a routine chest radiograph is
ordered, for instance. Almost one half have odontoid hypoplasia or
aplasia. Therefore, flexion–extension radiographs are advised at the
initial evaluation; these should be repeated at intervals because the
degree of instability may increase with time. In the lumbar spine, the
pedicles are not narrower caudally, as in achondroplasia, but are short
in the sagittal plane. Sternal deformities may appear (carinatum or
excavatum).
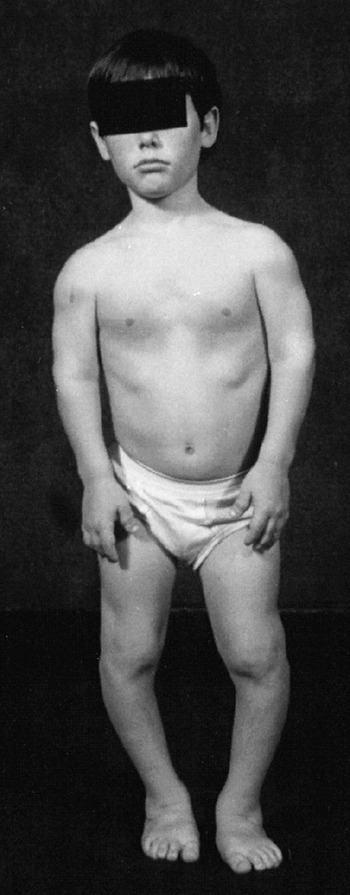 |
|
Figure 8.28
A 12-year-old patient with pseudoachondroplastic dysplasia. The head and trunk are normal, but there is rhizomelic shortening of the extremities. The hands and feet are short and broad. (Courtesy of George S. Bassett, MD) |
ends, and flared at the edges. The epiphyses ossify late, and are
irregular in appearance. There is progressively irregular ossification
of epiphyses. Delayed maturation of triradiate cartilage is more common
than in any other dysplasia. The pubic rami are delayed in closing, and
the greater trochanteric apophysis is delayed also (139). The femoral head is enlarged, and undergoes progressive subluxation (Fig. 8.30). The metaphyses all show irregularity and beaking. The height of the epiphysis of the distal femur decreases (Fig. 8.31).
An arthrogram may be helpful if operative intervention is planned, in
order to visualize the surface of the joint and determine the location
of the deformity(ies). Because epiphyseal ossification is delayed, this
can be difficult to visualize. The tibial plateau may be depressed on
one side, and the fibula may be relatively long. Delayed maturation of
carpal bones makes it difficult to predict bone age or skeletal
maturity.
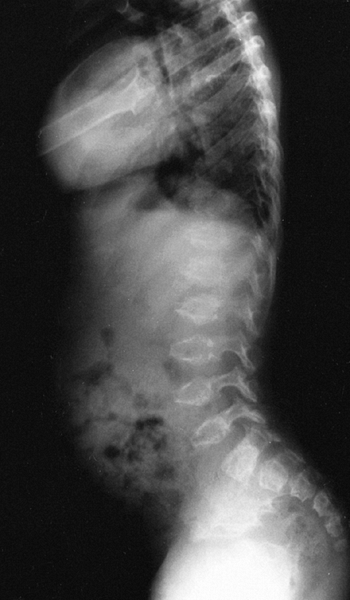 |
|
Figure 8.29
A 3-year-old child with pseudoachondroplastic dysplasia has platyspondyly with anterior beaking. (Courtesy of George S. Basset, MD) |
according to guidelines given earlier in this chapter. Myelopathy has
been reported in several series. Posterior cervical fusion may be
indicated if translation of the atlas
on the axis exceeds approximately 8 mm, or if neurologic signs are present.
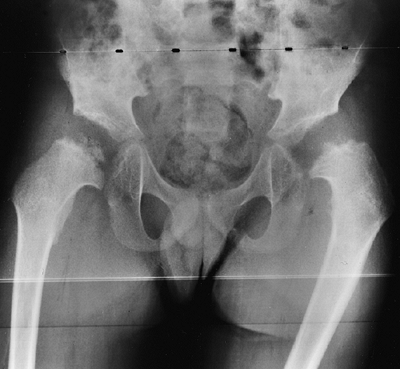 |
|
Figure 8.30 Deformation and delay in the ossification of the epiphyses are frequently seen in pseudoachondroplasia.
|
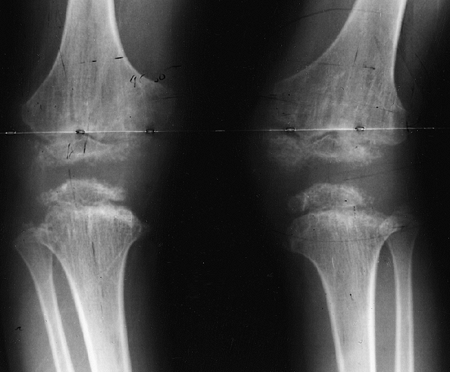 |
|
Figure 8.31 Epiphyseal flattening and a “windswept” alignment are characteristic of the knees in pseudoachondroplasia.
|
45 degrees in a skeletally immature patient, a trial of brace is
warranted. Larger curves (over 50 degrees) may require surgery. Spinal
stenosis is not a clinical problem, so the surgeon can use laminar
fixation, if needed, to supplement other fixation methods.
deformation, to a hip adduction contracture, or to valgus of the knee.
It is recommended that the orthopaedist try to arrest this process. A
femoral osteotomy, as well as an iliac osteotomy or augmentation, may
be needed. Preoperatively, it is wise to obtain radiographs with the
limb in the corrected position, to see whether the hip will be
congruous in this position. If the hip is aspherical or incongruous,
the patient may need an acetabular augmentation to achieve coverage.
Acetabular augmentation is generally a more versatile procedure than a
rotational iliac osteotomy in the person with skeletal dysplasia. At
the same time, the surgeon should look for and correct flexion and
rotation contractures as well.
correction. It is up to the discretion of the patient and the surgeon
to decide when to proceed with the corrective procedure. The objective
of the procedure is to obtain a horizontal joint surface with a
well-aligned knee. Both tibial and femoral procedures may be necessary.
The risk of recurrence is high, even with a well-done procedure, so it
is wise to educate patients about this risk. Early onset of
osteoarthritis often occurs—about one half of adults in one long-term
study had undergone at least one arthroplasty (9,133,138).
commonly occurring skeletal dysplasias. MED is usually dominantly
inherited, although recessive cases have recently been described as due
to mutations in transport protein (140). This
type of dysplasia affects many epiphyses, produces symptoms mainly in
those bones with significant load bearing, and causes few changes in
the physes or metaphyses. Historically, it was described as occurring
in two separate forms, with eponyms that are still used today: Ribbing
dysplasia, having mild involvement, and Fairbank dysplasia, a more
severe type (141,142,143).
With current understanding of the genetic basis of these conditions,
this may not be an absolute distinction, and a wide variability is
recognized.
that are similar to, but not as severe as, those seen in
pseudoachondroplasia. Growth plate organization is still noticeably
abnormal, despite the minimal changes seen in the metaphyses. The
genetic basis for this disorder is now reasonably well understood. MED
is a heterogeneous disorder. In many patients, mutations have been
found in the gene for COMP on chromosome 19, as in
pseudoachondroplasia. However, in other cases of MED, abnormalities
have been found in the α22 fibers of collagen type IX
(COL9A2). Collagen type IX is normally a trimer that is found on the
surface of type II collagen in cartilage; it may form a macromolecular
bridge between type II collagen fibrils and other matrix components,
and may therefore be important for the adhesive properties of
cartilage. A COL9A2 mutation has been described in one large family
with peripheral joint involvement only (144). In addition, dominant MED in some cases is caused by defects in matrilin-3, another oligomeric extracellular matrix protein (140,145). Rarer, recessive forms of MED are caused by mutations in transporters (140).
It therefore appears that MED is a very heterogeneous disorder, which
may help us to better understand some of the more common cartilage
disorders such as Perthes disease, osteochondritis dissecans, and
osteoarthritis (140).
of several reasons. They may be referred for joint pain in the lower
extremities, decreased range of motion, gait disturbance, or angular
deformities of the knees (146). There may be
flexion contractures of knees or elbows. Symptoms may develop as late
as adulthood. These patients have somewhat short stature, ranging from
145 to 170 cm (57 to 67 in.) (142). The face and spine are normal. There is no visceral involvement.
ossification centers may be delayed in appearing. There are occasional
irregularities of streaking in the metaphyses, but they are minor. The
appearances of the epiphyses in the immature and in the mature patient
are different
and characteristic (147). In the growing patient, the epiphyses are fragmented and small in size (Fig. 8.32).
The epiphyseal ossification centers eventually coalesce, but the
overall shape of the epiphysis is smaller. An arthrogram may be helpful
when it is necessary to assess the shape of the joint surface. The more
fragmentation there is in the capital femoral epiphysis, the earlier is
the onset of osteoarthritis (148). Coxa vara
occurs in some patients. After maturity, there is some degree of
flattening of the major load-bearing epiphyses—flattening of the
femoral condyles, an ovoid femoral head, decreased sphericity of the
humeral head, and squaring of the talus. In adulthood, major joints
develop premature osteoarthritis, which is most common and most severe
in the hips.
occurs in about one half of the femoral heads. It can be recognized by
the presence of a crescent sign, resorption of bone that had already
been formed and, sometimes, by the presence of metaphyseal cysts (149). MRI at this time may show loss of signal in a portion of the femoral head. A “sagging rope sign” may develop later (138).
Several radiographic clues may be helpful. In MED, abnormalities in the
acetabulum are primary, and are more pronounced. The radiographic
changes are symmetric and fairly synchronous. It is also helpful to
obtain radiographs of the knees, ankles, shoulders, and wrists.
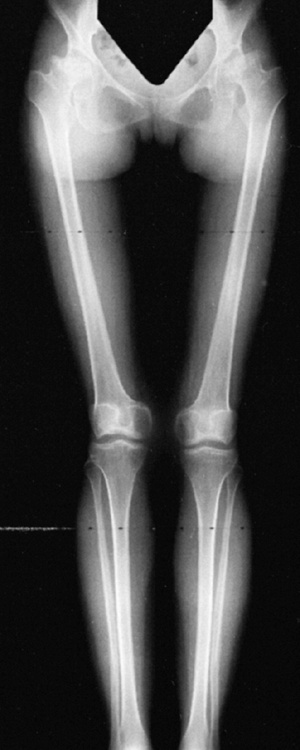 |
|
Figure 8.32 Multiple epiphyseal dysplasia.
|
are flattened, and may be in valgus. There may be irregular
ossification, just as in the hip. The condyles appear somewhat squared
in the lateral view. Osteochondritis dissecans may be superimposed.
Some patients with MED also show a double-layered patella in the
lateral view (150). This is a complete or partial double radiodensity, which is rarely seen in other conditions.
more in the talus than in the distal tibia. Upper extremity involvement
is less severe; there may be irregularities in the proximal and distal
humerus and radius. The humeral head involvement in adulthood, termed hatchet head,
results from diminished growth of the head and neck. The hatchet head
occurs in those children who are the most severely affected with MED.
Radial ray hypoplasia may occur sporadically (151). The carpal ossification centers are delayed in appearing. The hand and wrist involvement may predict future stature (152).
The spine may be normal, or may have slight endplate irregularities or
ossification defects on the anterior margins of the vertebrae (153).
of the patient with MED in either of two periods. There is a small role
for realignment procedures in the early period during which the hip is
deformed, provided there is progressive subluxation or pain. Pain is
more likely to occur in cases in which avascular necrosis has
supervened (149). Although the principle of
coverage is the same as that used in Perthes disease, there is often a
degree of coxa vara preexisting in the hips of patients with MED, which
contraindicates use of a femoral osteotomy. Acetabular shelf
augmentation is a worthwhile procedure in these instances (154,155).
can be helped. Significant deformities may be corrected near maturity,
either in the femur or tibia, depending on the site of the abnormality.
Patients having a double-layered patella may have symptoms because of
the relative movement of one over the other. This may be treated by
excision of one or fusion of the two segments, as appropriate (156).
Degenerative joint disease is the biggest problem, and it occurs in the
second or third decade of life. The disease results not so much from
malalignment of the joints as from an intrinsic defect in cartilage,
and it produces stiffness from an early age and pain, leading to a
total joint arthroplasty. The shoulder too is commonly affected by
degeneration, and shoulder arthroplasty may be necessary (133).
Key features include depressed nasal bridge and multiple punctate
calcifications in infancy, which are best visualized on the newborn’s
radiographs (157).
There are many different types, the most common being an X-linked
dominant type (Conradi-Hünermann syndrome). There is also an autosomal
recessive rhizomelic type, which is usually lethal in infancy, and a
rare X-linked recessive type. Four other types have been described that
are even more rare (158). Although the
appearance of neonatal epiphyseal calcification is striking, it is not
very specific. Wulfsberg has listed various other conditions that may
present with the same phenomenon: Zellweger (cerebrohepatorenal)
syndrome, gangliosidosis, rubella, trisomy 18 or 21, vitamin K
deficiency, hypothyroidism, and fetal alcohol or hydantoin syndromes (158,159,160,161,162,163).
Rhizomelic chondrodysplasia punctata is a peroxisomal deficiency of
dihydroxyacetone-phosphate acyltransferase; it is often (but not
always) fatal in the first year of life (164,165).
The genetic defect and pathogenesis of the Conradi-Hünermann syndrome
has not been elucidated. The final common pathway of the various types
is a defect in cholesterol synthesis (166). Histologic examination shows perilacunar calcifications throughout the cartilage matrix (167).
In addition, many have alopecia, congenital heart and/or renal
malformations, and mental retardation. In rhizomelic chondrodysplasia
punctata, findings include microcephaly, a high incidence of congenital
cataracts, growth retardation, and a well-formed nasal bridge (172,173,174,175).
Some have feeding difficulties, and most succumb to respiratory death
or seizures in the first year of life. Diagnosis may be made by
amniocentesis, by measuring plasmalogen biosynthesis and phytanic acid
oxidation.
disappear by the age of 1 year. These calcifications involve the
epiphyses, carpal bones, and pelvis (179) (Fig. 8.33).
Extraskeletal sites include the trachea and larynx. The appearance is
of small flecks of calcium, “which appear as if paint had been flecked
on by a brush” (180). The centers of
ossification themselves may be delayed in appearing. Coxa vara may
affect one or both hips, or it may be absent (181).
The fibula often overgrows the tibia significantly. Spine radiographs
may have the appearance of a hemivertebra or a congenital bar.
Calcification of the intervertebral discs may develop (Fig. 8.33B). The occurrence of odontoid hypoplasia and os odontoideum has been reported (178).
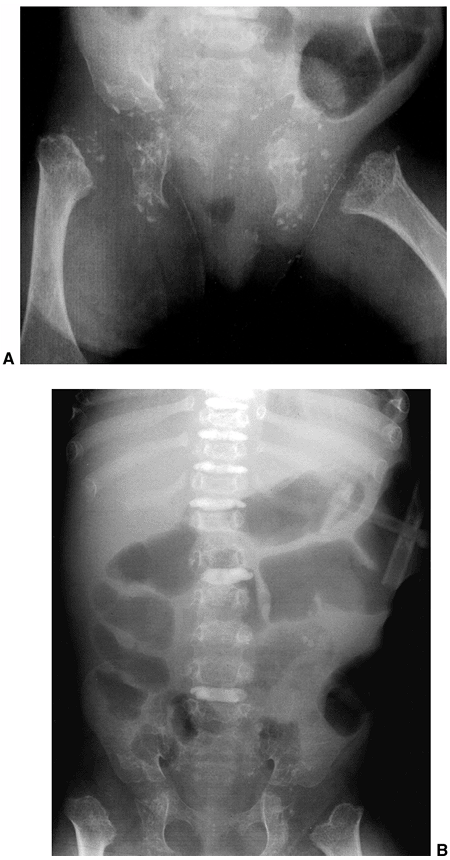 |
|
Figure 8.33 A: Diffuse punctate epiphyseal calcifications in infancy are the hallmark which gave chondrodysplasia punctata its name. B: At age 2½ years, the epiphyseal calcifications are mostly resolved, but calcification of the intervertebral discs persists.
|
each patient should have a lateral cervical radiograph and, if
instability appears possible, a flexion–extension view. Scoliosis may
occur early because of secondary congenital anomalies. It may require
early fusion if progression is documented and the patient, medically,
is a suitable candidate.
Because
of the dysplastic vertebrae, hemiepiphyseodesis and anterior
morsellized grafts are not recommended. Anterior structural grafting,
followed by posterior fusion and cast immobilization, has the highest
rate of success (166).
Coxa vara should be treated if the neck-shaft angle is less than 100
degrees. Lower limb-length inequality should be monitored and treated
appropriately.
of disorders characterized by metaphyseal irregularity and deformity,
but with preservation of epiphyseal structure. This, of course, is in
contrast to almost all the dysplasias previously described in this
chapter (181,182,183,184).
The name may be a bit misleading because it refers to the end
result—changes in the metaphyses as revealed in radiographs. Of course,
logically, the real defect is in the growth plate itself, resulting in
failure of uniform ossification of the cartilage columns, with
persistence of cartilage islands, underdevelopment, and deformity as
the sequelae (185,186).
There are many different named disorders that come under the heading of
metaphyseal chondrodysplasia. We discuss here the commonest
types—McKusick, Schmid, and Jansen types, as well as Kozlowski-type
spondylometaphyseal dysplasia, which is associated with mild changes in
the vertebral bodies.
common in the Amish community of Lancaster County, Pennsylvania, as
well as in Finland. The condition also occurs sporadically throughout
the world; it is also known by the term cartilage-hair hypoplasia. The condition is autosomal recessive and maps to chromosome 9 (187). The defect is in the RMRP gene, which encodes a mitochondrial RNA-processing enzyme (188).
This should serve as a clue to the more important medical problems that
this group of patients may have. An alteration in T-cell immunity
causes an increased risk of viral infection (especially Varicella zoster,
which may be more severe in these persons). Continued antibiotic
prophylaxis in the first 6 months of life has been recommended (189,190). Anemia may develop, and 16% of these patients require a blood transfusion (191).
Bone marrow transplant has been reported in one infant with severe
recurrent infections; it corrected the immune problem, but did not
improve skeletal growth (192). Hematologic problems have a tendency to become less
severe after childhood. Hirschsprung disease, intestinal malabsorption,
and megacolon may also develop. There is an increased risk of
malignancy, such as lymphoma, sarcoma, and skin cancer. Life expectancy
is decreased. In the largest series reported, 8% of the patients had
malignancies (189,193).
Clearly, then, these patients need medical surveillance into adulthood,
more than would most patients with skeletal dysplasia.
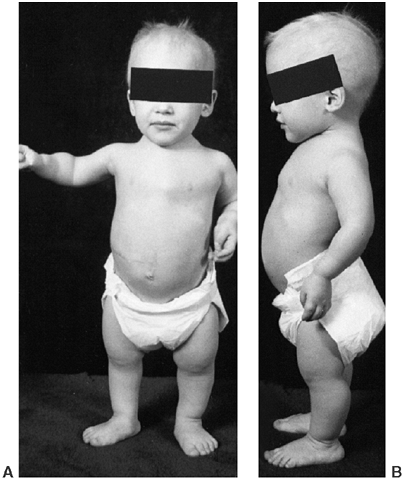 |
|
Figure 8.34 A:
An 18-month-old child with McKusick-type metaphyseal chondrodysplasia. Notice the characteristic light, sparse hair, disproportionate short stature, pectus excavatum, and varus deformities of the lower extremities. B: Mildly increased lumbar lordosis, flexion contractures of the elbows, and expansion of the wrists are also part of the deformity. (Courtesy of George S. Bassett, MD) |
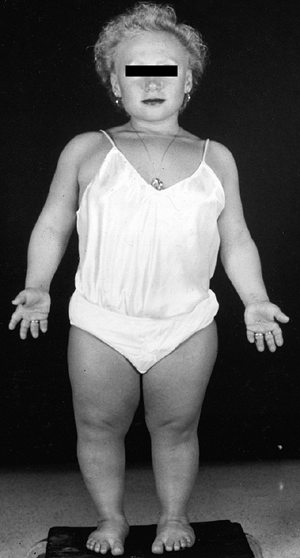 |
|
Figure 8.35
A 22-year-old woman with metaphyseal chondrodysplasia, McKusick type (cartilage-hair hypoplasia). Note disproportionately short stature, fine light hair, and genu varum. |
ligamentous laxity, but the elbows actually have flexion contractures.
There is mild genu varum, which may bring them to an orthopaedist.
Pectus excavatum or carinatum may be observed. The adult height is 106
to 147 cm (42 to 58 in.).
there is more shortening and less varus of the long bones than is seen
in the Schmid type. The metaphyseal involvement is more evenly
distributed, not all on the medial side of the knee. There is distal
fibular overgrowth, perhaps because this bone is less inhibited by the
forces of weight bearing. Atlantoaxial instability has been reported.
The sternum is angulated so that the distal end is anterior (194).
The thoracolumbar spine shows some minimal changes, which are not of
much clinical importance—columnization (increased height) of the
vertebrae and increased lumbar lordosis.
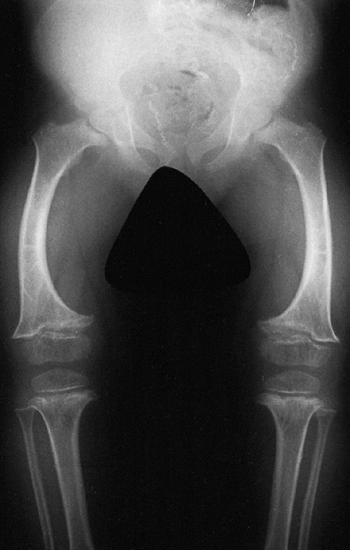 |
|
Figure 8.36
A 4-year-old patient with McKusick-type metaphyseal chondrodysplasia. The pelvis is normal, apart from silver clips from surgical treatment of megacolon. Mild coxa vara, bowing of the femurs and tibias with metaphyseal expansion, and irregular zones of provisional calcification are evident. (Courtesy of George S. Bassett, MD) |
atlantoaxial instability, at least on the patient’s first visit. MRI in
flexion and extension may be helpful if the plain films seem equivocal
diagnostically. Posterior spine fusion should be performed if there is
more than 8 mm of translation, or if any signs of cord compression are
present. Congenital hip dislocation has been reported in 3% of
patients, and if it is detected early, successful closed reduction may
be performed (189). In some patients (15%), the varus at knee or ankle should be corrected, if severe.
Type X is a highly specialized extracellular matrix component, the
synthesis of which is restricted to hypertrophic chondrocytes in the
calcifying zones of the growth plate and in zones of secondary
ossification. Type X is a homotrimer of three α1 chains, which has been
implicated in morphogenetic events of endochondral ossification,
including calcification of hypertrophic cartilage prior to its
replacement by bone. Many mutations described involve the C-terminal
end, where joining of the three individual chains starts and prevents
the abnormal chains from forming trimeric structures (198).
On histologic examination, one sees cartilage islands that extend into
the metaphyses. The differential diagnosis for this and other forms of
metaphyseal dysplasia, most importantly, includes various types of
rickets and hypophosphatasia.
clinical abnormalities. They are normal at birth. The facial appearance
is normal. They may present to the orthopaedic surgeon with leg pains,
varus knees and ankles, short stature, or a waddling gait. The adult
height is minimally shortened, the average being 150 cm (59 in.).
and may have cysts. The physes are slightly widened. Weight bearing may
play a role in these changes: they have been reported to improve after
rest or cast immobilization, and to recur after resumption of loading (195). There is a varus deformity of the knees. Atlantoaxial instability has been reported, but is rare.
degenerative changes. The orthopaedic surgeon may be called upon to
correct bowing of the knees if it becomes severe. Otherwise, there is
little need for orthopaedic intervention in these patients.
dysplasia. It is an autosomal dominantly inherited disorder, which has
been linked to a defect in the receptor for parathyroid hormone and
parathyroid hormone-related protein (199). This
provides an interesting link between the skeletal dysplasias and the
metabolic bone diseases. Patients with Jansen syndrome may have
hypercalcemia, and they have more severe metaphyseal changes than do
patients with McKusick or Schmid dysplasia.
dominant disorder characterized by spinal, as well as metaphyseal,
changes. The disorder is recognized in preschool-aged children by the
findings of short stature and mildly increased kyphosis. There may be
slight limitation of joint movement, a Trendelenburg gait, and early
osteoarthritis. Adult height reaches approximately 150 cm.
disorders just described. The bone age of the carpals and tarsals is
retarded. The metaphyseal chondrodysplasia is most pronounced in the
proximal femur.
dysplasias, it is clear that patients with multiple exostoses have a
generalized disturbance of skeletal growth. Diaphyseal aclasia has been
localized to three different chromosomal locations—sites on chromosomes
8, 11, and 19. These sites are referred to as EXT
1, 2, and 3, respectively. The differing locations of mutation may
account for the phenotypic variability of the condition. Theories
include defects in tumor suppression versus signaling for fibroblast
growth factor and Indian hedgehog (200). Most
cases are transmitted as autosomal dominant, but a large number of
patients acquire it by spontaneous mutation. The metaphysis of a person
with multiple osteochondromas is characterized by thinning of the
cortex, innumerable small bumps, and cartilage rests extending into the
trabecular bone.
years, when the first exostoses are noted and other features develop.
The features become progressively more pronounced until maturity, at
which point bony prominences should cease to grow. Affected persons are
at the low end of normal for stature. The metaphyses are
circumferentially enlarged throughout the body, not only in regions
where there are obvious exostoses. This gives a rather “stocky”
appearance, which is then further exaggerated by the appearance of the
exostoses. They may cause soreness when they arise under tendons or in
an area vulnerable to bumping, such as the proximal humerus. The
exostoses tend to steal from the longitudinal growth of the long bones.
The categories of problems caused by this condition are fourfold:
-
Localized pressure on tendons and nerves,
among other sites. Peroneal palsy may arise from a lateral exostosis,
and it may occur in such a way as to cause brachial plexus or spinal
cord compression. -
Angular growth of two-bone segments—the
arms and forearms. Usually the thinner of these two bones is more
inhibited in its growth than the wider one, thereby tethering the
growth of the latter. Valgus may develop at the wrist, knee, and ankle.
The radial head may subluxate or dislocate. -
Limb-length inequality. Often one limb is
more involved than the other with exostoses, and it may show shortfall
in growth by as much as 4 cm. -
Malignant degeneration. Transformation to chondrosarcoma occurs in approximately 1% of patients after maturity (200).
Such a change may be signaled by increased growth of an exostosis, or
pain over an exostosis. Bone scans every 2 years in adulthood have been
advocated as one way to detect this change. Self-examination and
periodic pelvic examination, with MRI in the case of an apparent
change, is another way to follow these patients in adulthood (200).
irregularities can be seen. The exostoses may be sessile or
pedunculated, and have continuity with the main cortex, as solitary
exostoses do. Exostoses on the undersurface of the scapula may be
identified on plain films, but are best evaluated by CT scan. The
femoral necks are usually wide and in valgus (Fig. 8.37). Valgus is much more common than varus at the knee (Fig. 8.38), and the distal tibial epiphysis may be triangular if the fibula is pulling the ankle into valgus.
Radial head subluxation may occur with ulnar shortening, and the
resultant carpal subluxation can readily be identified in wrist films.
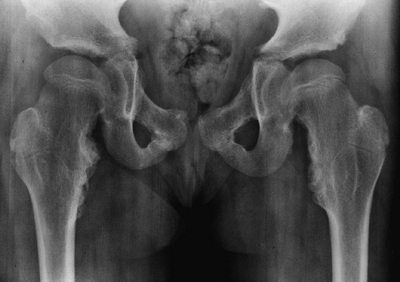 |
|
Figure 8.37
The hips in diaphyseal aclasia are characterized by broad, irregular femoral necks that are usually in valgus. There are osteochondromas and irregularities in the formation of the pelvis also, and these can be difficult to monitor over time. |
clinical examination because all bones are affected, and the lesions
are too numerous to image routinely. A brief neurologic examination
should be followed by a check of the range of motion of the joints. The
knee, ankle, elbow, and wrist angulations and the limb lengths should
be measured. Any exostoses that are causing significant symptoms should
be removed, but the patient should be warned that the metaphyseal
widening will persist, and so the effect on appearance may not match
expectations. Removal of lesions impairing radioulnar motion may result
in a slight increase in range of motion, but not a dramatic
improvement. Ulnar lengthening may help avert radial head subluxation.
Hemiepiphysiodesis is a minimally invasive way of correcting angulation
at the wrist, knee, and ankle. Valgus angulation at the distal tibia is
correlated with degenerative changes in the long term (201).
If the patient is near maturity and needs the angulation to be
corrected, osteotomy may be indicated. Limb-length inequality can be
corrected by the standard algorithm because the growth remains
proportionate. After maturity, patients should be taught to examine
themselves for signs of growth because this may signal malignant
degeneration. Bone scan may be a helpful adjunct if a problem is
suspected.
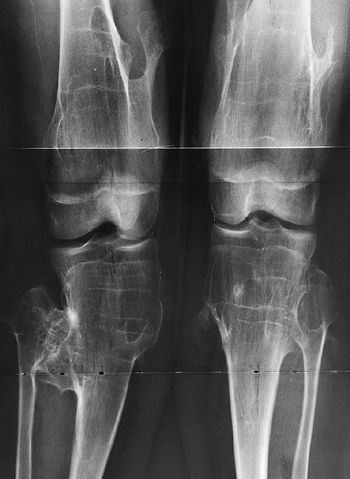 |
|
Figure 8.38
This figure of the knees in a patient with diaphyseal aclasia best illustrates that this defect is a systemic abnormality of bone formation, rather than a series of discrete tumors. The metaphyses are broad and irregular in the region where the exostoses are located. The knees are developing a valgus alignment, because of the short fibulae, as are the ankles. |
is characterized by mild mesomelic short stature, the middle segments
being the shortest. The growth disturbance of the middle segments is
most notable in the distal radius, which usually develops a Madelung
deformity (202,203,204). The condition is inherited in an autosomal dominant fashion, with about 50% penetrance (203,205).
The expression is more severe in female patients than in male patients.
It has been demonstrated to involve a mutation or deletion in the
short-stature homeobox-containing gene SHOX (206).
stature, disproportion or deformity of the forearms, or pain or
deformity of the wrist (207). The deformity of
the distal forearm, or Madelung deformity, is characterized by a
deficiency of growth of the volar–ulnar portion of the radius. The
differential diagnosis of this phenomenon includes Turner syndrome,
trauma, Ollier disease, and multiple hereditary exostoses. Most
patients begin to experience pain in the wrist during adolescence, as
well as limitation of pronation and supination. A variation on this
theme, seen in some patients with dyschondrosteosis, is shortening of
both radius and ulna together, without angulation. The mesomelic
shortening also involves the lower extremities, specifically the
tibiofibular segments. Here, however, there is not so much angular
deformity—usually only a mild genu varum or ankle valgus. Short stature
is usually, but not always, a feature; adult height ranges from 135 to
170 cm. In one series of patients, deficiency in growth hormone was
found, and stature was increased by growth hormone supplementation (208).
volar–ulnar part of the distal radial epiphysis. The distal radial
epiphysis develops a triangular appearance and a tilt of the surface of
the joint (207) (Fig. 8.39). A physeal bar may be seen on CT scan at the lunate facet (205).
The ulna is subluxated or dislocated dorsally. The ulna is as long as,
or longer than, the radius, in contrast to other causes of Madelung
deformity mentioned in the differential diagnosis (208).
The tibia and fibula are short, with the fibula longer than the tibia
at the ankle and/or the knee. There may be some degree of genu varum or
ankle valgus. Cubitus valgus, hypoplasia of the humeral head, and coxa
valga have all been noted, but rarely do all occur in the same patient.
response, and patients who are concerned about their short stature may
be referred to an endocrinologist for discussion of this treatment (208,209).
Patients who experience wrist pain may be treated initially with a
wrist splint and antiinflammatory agents. If symptoms persist, a
reconstruction with a double osteotomy of the distal radius and an
ulnar recession provides good results (210).
The reconstruction has reportedly produced improvement in symptoms and
clinical appearance, but lunate subluxation, grip strength, and range
of motion were only minimally influenced (204).
Although it has been described, it is unclear whether bar resection can
allow normal growth to occur. Osteotomy of the tibia is occasionally
indicated for correction of genu varum (203).
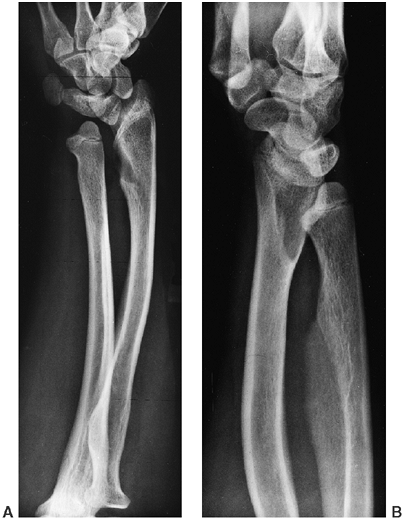 |
|
Figure 8.39 A: Madelung deformity in the forearm of a patient with dyschondrosteosis. B: The distal radial epiphysis has a markedly triangular epiphysis, and the ulna is dorsally subluxated.
|
because it affects the growth of many bones in all parts of the
skeleton, primarily those of membranous origin. The classic features
include a widening of the cranium, and dysplasia of the clavicle and
the pelvis (211,212). The incidence is estimated at 1:200,000 (213). This dysplasia is transmitted as an autosomal dominant condition, and the defect is in the RUNX2/CFBA1 gene on chromosome 6, which encodes an osteoblast-specific transcription factor required for osteoblast differentiation (214,215,216).
affected, there are numerous abnormalities. The patients have mildly to
moderately diminished stature, with most male patients and some female
patients less than the fifth percentile for age. The mean adult height
attained by male patients is 162 cm. There is bossing in the frontal
parietal and occipital regions. The maxillary region is underdeveloped,
giving apparent exophthalmos and maxillary micrognathism. Cleft palate
and dental abnormalities are common (217,218,219,220).
 |
|
Figure 8.40
The clavicles are completely absent in this patient with cleidocranial dysplasia, although in many patients they are merely hypoplastic. There is also a characteristic mild scoliosis and an occult bifid lamina of T2. |
complete absence is seen only 10% of the time. The absence of clavicles
causes the shoulders to drop and the neck to appear longer. The classic
diagnostic feature is that the shoulders can be approximated, an
ability that helped one college wrestler to escape holds (220).
The pelvis is narrow. The hips are occasionally unstable at birth. Coxa
vara may occur, causing limitation of abduction and a Trendelenburg
gait. There is an increased incidence of scoliosis, and often a double
thoracic curve. Syringomyelia has been reported in several patients,
along with cleidocranial dysplasia and scoliosis (221,222,223). It is recommended that MRI be performed in patients with this dysplasia who have progressive scoliosis.
If there is uncertainty, molecular prenatal diagnosis may be performed.
If a portion of the clavicle is present, it is usually the medial end.
The skull of a newborn with this disorder has the maturation of a
20-week fetus (217). Wormian bones are present in the skull. The anterior fontanel may be open in adulthood (Fig. 8.41). In the vertebral column, spina bifida occulta and spondylolysis are common (225).
The pelvis is narrow, and shows widening of the triradiate cartilage,
delay in pubic ossification, and progressive deformation of the base of
the femoral neck into varus (Fig. 8.42), with a triangular metaphyseal fragment typical of coxa vara (226). Skeletal maturation may be delayed.
vara may be treated by valgus-rotational osteotomy if the neck-shaft
angle is less than 100 degrees and the patient has a Trendelenburg gait
(227). If there is acetabular dysplasia, this
should be corrected first. Scoliosis should be treated according to the
usual guidelines. MRI should be performed if the curve is progressive,
because of the increased risk of syringomyelia.
dysplasia. Craniofacial surgery may be helpful in correcting the skull
defects and many dental problems. Pregnant women may have cephalopelvic
disproportion, especially if the fetus has the same disorder, because
of the mother’s narrow pelvis and the fetus’ enlarged cranium.
 |
|
Figure 8.41
The skull in this teenager with cleidocranial dysplasia shows an enlarged cranium, widened sutures, and a persistent anterior fontanel. |
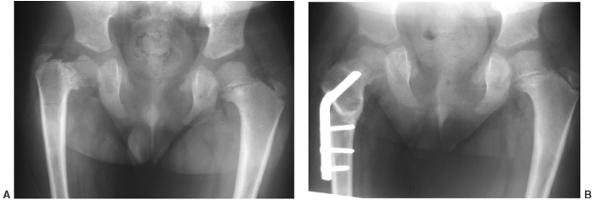 |
|
Figure 8.42
The pelvis in cleidocranial dysplasia is narrow. The symphysis pubis is widened, the ischiopubic synchondrosis is unossified, and there may be coxa vara [(A) preoperative, (B) postoperative view]. |
cases were described with the unique combination of hypertelorism,
multiple joint dislocations, and focal bone deformities (228).
This syndrome has been reported in both autosomal dominant and
recessive patterns. The gene is on chromosome 3 near, but distinct
from, COL7A1 locus 4207. The dominant form has been associated with a mutation in filamin B, a cytoskeletal structural protein (229).
depressed nasal bridge, and a prominent forehead. Cleft palate is
common. The thumb has a wide distal phalanx and the fingers do not
taper distally. Hypotonia has also been reported, but this may result
from cervical compression (230). Sudden death has been reported (228,231),
most likely caused by exacerbation of this compression. Dislocations
most commonly involve the elbows (or radial heads), hips, and knees (Fig. 8.43)
and, less frequently, the midfoot and shoulders. Characteristic foot
deformities involve equinovarus or equinovalgus. Atrial and ventricular
septal defects have been reported. Within the range of abnormalities
just described, every patient with this syndrome is unique in his or
her pattern of associated problems.
findings in this syndrome. Virtually every patient described, however,
has some abnormality in some part of the spine. The cervical spine is
the most commonly and severely affected. Spina bifida is very common in
the cervical spine. Perhaps because of this, the cervical vertebrae may
develop a progressive kyphosis (Fig. 8.44). The
vertebral bodies in this situation are very hypoplastic, especially C4
and C5. It is not clear whether this vertebral hypoplasia is a result
of pressure from the kyphosis, or whether it is a separate,
coincidental phenomenon that coexists with the posterior element
deficiency. The incidence of cervical kyphosis in different series
ranges from none to 60% (230,232,233). Other cervical problems that may occur include atlantoaxial or subaxial instability (232)
and spondylolisthesis of vertebrae. The thoracic spine may also
manifest spina bifida; scoliosis is seen in many patients, but it is
usually mild and rarely requires treatment (232). In the lumbar spine, spondylolysis, kyphosis, scoliosis, and back pain
may occur (232). Sacral spina bifida is common, but no neurologic compromise is reported.
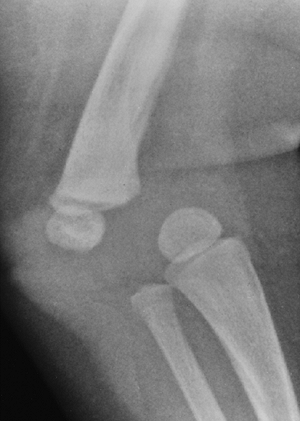 |
|
Figure 8.43 Congenital anterior knee dislocation is common in Larsen syndrome.
|
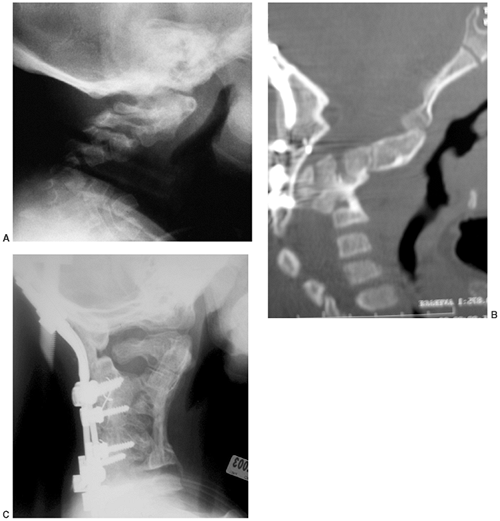 |
|
Figure 8.44 Cervical kyphosis occurs in many patients with Larsen syndrome, in association with spina bifida occulta of this region (A). The disorder is usually progressive (B), and may require decompression and fusion if myelopathy from focal kyphosis occurs (C).
|
findings in Larsen syndrome is the presence of accessory calcaneal or
carpal ossification centers (Fig. 8.45). Shortened metacarpals are also noted.
must rule out the cervical kyphosis that may accompany this syndrome,
because of the catastrophic complications that have been reported to be
associated with it. It may be easy to ascribe any developmental delay
to the many other skeletal problems these children have, when in fact
the cervical kyphosis may cause a neurologic basis for it. Because
spontaneous improvement has not been reported in this kyphosis, as it
has been in DD, it should be fused posteriorly if it exceeds
approximately 35 degrees to 45 degrees. At this level, posterior fusion
alone over the involved segments may be successful, and may result in
spontaneous correction with growth by acting as a posterior tether. If
the
kyphosis
progresses to the point of myelopathy, an anterior corpectomy and
fusion may be needed, and anterior growth will not occur. If enough
iliac crest bone is not available for fusion, tibial bone may be used.
After surgery, the patient should be in a brace or cast for 4 to 6
months (230). Laryngotracheomalacia may complicate the induction of anesthesia.
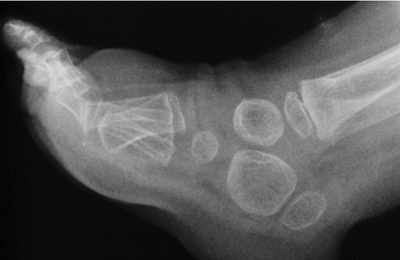 |
|
Figure 8.45
The feet in patients with Larsen syndrome are usually in equinovarus, and show a characteristic accessory calcaneal ossification center. |
sequence beginning with the feet and the knees, and then the hips.
Treatment of clubfeet may be started early, inasmuch as some respond to
manipulation and cast treatment with tenotomy. Recurrence is common,
and should then be treated with complete subtalar release and
shortening osteotomy or decancellation, as necessary. Knees that are
hyperextended or subluxatable may be treated with casts as well, but
this is unlikely to succeed in cases of complete dislocation. Such
cases usually require open reduction with VY quadricepsplasty, anterior
capsulotomy, and release of the anterior portions of the collateral
ligaments. If cruciate deficiency leads to persistent anterior
instability, reconstruction using parapatellar fascia is usually
successful.
remains controversial. Some series report that they are resistant to
treatment (232,233), whereas others report some successful results (231,233).
After failed treatment, a dislocated hip is less functional than one
left untreated. A reasonable approach is to consider treatment of hips
in which the dislocation is not too high and the acetabulum is not too
shallow, in patients with otherwise good prospects for activity. The
medial approach may be used in infants, but in older children or those
with a shallow acetabulum, an anterolateral approach is preferred, with
osteotomy or augmentation. If the hip subluxates easily or has a narrow
safe zone, the clinician should not hesitate to perform a femoral
shortening and derotation. Our preference is to begin cast treatment
for the feet and knees together, then operate on the knees if they are
resistant, and then on the feet if they are resistant. By that time,
the surgeon will have a better idea of the patient’s potential and can
decide on the most appropriate treatment for the hips.
the orthopaedic surgeon may be questioned about some of the lethal
dysplasias that would not otherwise be encountered in practice.
Perinatal lethal skeletal dysplasias are mentioned here to provide
passing familiarity. The combined incidence of lethal dysplasias has
been estimated at 15 per 100,000 births. The natural history of these
conditions should be considered carefully if one is facing a decision
about whether to provide respiratory support.
disproportionately small limbs, normal trunk length, a protuberant
abdomen, and a large head with frontal bossing. The chest is narrow and
the lungs hypoplastic. The femora are bowed, and their appearance has
been likened to old-style telephone receivers. There is phenotypic
resemblance to homozygous achondroplasia, and in fact, this condition
results from a mutation in the same gene, FGFR3.
Only a few children with this disorder have been reported to survive
past the age of 2 years, even with full respiratory support.
head, distended abdomen, and severely underdeveloped limbs. The
condition has been subclassified into four types. Achondrogenesis may
be autosomal dominant or recessive. In fact, achondrogenesis type I
results from a mutation in the DD sulfate transporter. Survival beyond
birth is very rare. No treatment is available to prolong the life span
at present.
characterized by dislocations of large joints and, in some cases, by
clubfeet. Midface hypoplasia, micrognathism, and a narrow chest are
also seen. At least one of the two types results from a mutation in the
DD sulfate transporter.
and is characterized by polydactyly, which is classically postaxial but
may be preaxial, short horizontal ribs, and defects in the kidneys and
lungs.
dysplasia. Because of the poor prognosis, some of these children have
been treated with bone marrow transplants, with reports of prolonged
survival.
the neonatal period include achondroplasia (homozygous form),
rhizomelic chondrodysplasia punctata, camptomelic dysplasia, and a
congenital form of hypophosphatasia.
patient is older than 2 years; decompress symptomatic stenosis; C-spine
is stable; osteotomies for genu varum if symptomatic
“cauliflower” ears; contractures of joints, cervical kyphosis;
scoliosis; degenerative disease of joints, equinovarus feet
normal epiphyses; genu varum, mild short stature, fine sparse hair;
immune and GI disorders in McKusick type
instability; correct genu varum if severe; monitor medical problems in
McKusick type
tendons and nerves, angular deformities, limb length in equality,
malignant degeneration
remove symptomatic exostoses, educate about signs of slight malignant
degeneration
SE, Thompson RE, Gooding HC, et al. Living with achondroplasia in an
average-sized world: an assessment of quality of life. Am J Med Genet 2003;120A:447–458.
R, Thompson LM, Zhu Y-Z, et al. Mutations in the transmembrane domain
of FGFR3 cause the most common genetic form of dwarfism,
achondroplasia. Cell 1994;78:335.
RM, Horton VK, Glinske LP, et al. Prospective assessment of risks for
cervicomedullary-junction compression in infants with achondroplasia. Am J Human Genet 1995;56:732.
JT, Nelson FW, Butler IJ, et al. Computerized tomography of the foramen
magnum: achondroplastic values compared to normal standards. Am J Med Genet 1985;20:355.
KA, Everett FM, Sillence DO, et al. Treatment of obstructive sleep
apnea in achondroplasia; evaluation of sleep, breathing and
somatosensory-evoked potentials. Am J Med Genet 1995;59:460.
H, Kubo T, Yamate T, et al. Effect of growth hormone therapy in
children with achondroplasia: growth pattern, hypothalamic-pituitary
function and genotype. Eur J Endocrinol 1998; 138:275.
R. Distraction osteogenesis for lengthening of the tibia in patients
who have limb-length discrepancy of short stature. J Bone Joint Surg Am 1999;81:624.
JM, Ginebreda I, Jimeno E. Lengthening of the lower limbs and
correction of lumbar hyperlordosis in achondroplasia. Clin Orthop 190;250:143.
S, Hurko O. The subarachnoid fluid space in achondroplastic spinal
stenosis: the surgical implications. In: Nicoletti B, ed. Achondroplasia: human achondroplasia, a multidisciplinary approach. New York: Plenum, 1986:275.
WC, Kitawaga M, Xue N, et al. Activation of Stat1 by mutant fibroblast
growth-factor receptor in thanatophoric dysplasia type II dwarfism. Nature 1997;386:288.
F, Danks DM, Jansen F, et al. Achondroplasia and hypochondroplasia.
Comments on frequency, mutation rate, and radiological features in
skull and spine. J Med Genet 1979;16:140.
RWB, van Creveld S. A syndrome characterized by ectodermal dysplasia,
polydactyly, chondrodysplasia and congenital morbis cordia. Arch Dis Child 1940;15:65.
SE, Ortiz de Luna RI, Francomano CA, et al. Exclusion of the MAX1
homeobox gene as the gene for the Ellis-van Creveld syndrome in the
Amish. Hum Genet 1996;98:572.
F, Jacques SM, Evans MI, et al. Skeletal histopathology in fetuses with
chondroectodermal dysplasia (Ellis-van Creveld syndrome). Am J Med Genet 1996;3:471.
D, Tetsworth K. Mechanical axis deviation of the lower limbs:
preoperative planning of uniapical frontal plane angular and bowing
deformities of the femur or tibia. Clin Orthop 1992;280:65.
A, Rossi A, Steinmann B, et al. Achondrodysplasia family produced by
mutations in the diastrophic dysplasia sulfate transporter gene:
genotype/phenotype correlations. Am J Med Genet 1996;63:144.
WA, Hall JG, Scott CL, et al. Growth curves for height for diastrophic
dysplasia, spondyloepiphyseal dysplasia congenita and
pseudoachondroplasia. Am J Dis Child 1982;136:316.
VM, Marttinen EJ, Poussa M, et al. Cervical spine in patients with
diastrophic dysplasia—radiographic findings in 122 patients. Pediatr Radiol 2002;32:621–628.
AR, Pidoux I, Reiner A, et al. Kniest dysplasia is characterized by an
apparent abnormal processing of the C-propeptide of type II collagen
resulting in imperfect fibril assembly. J Clin Invest 1988;81:579.
WG, Hall RK, Rogers JG. The clinical features of spondyloepiphyseal
dysplasia congenita resulting from the substitution of glycine 997 by
serin in the alpha 1 (II) chain of type II collagen. J Med Genet 1993;30:27.
GE, Weis MA, Polumbo PA, et al. An RNA-splicing mutation in the type II
collagen gene in a family with spondyloepiphyseal dysplasia congenita. Am Hum Genet 1995;56:388.
G, Cottefava F, Senes FM. Spondyloepiphyseal dysplasia tarda: linkage
with genetic markers from the distal short arm of the X chromosome. Hum Genet 1988;81:61.
JF, Wynne-Davies R, Fulford GE. Bilateral failure of the capital
femoral epiphysis: bilateral Perthes disease, multiple epiphyseal
dysplasia, pseudoachondroplasia, and spondyloepiphyseal dysplasia. Pediatr Orthop 1986;8:197.
M, Sanford T, Francomano CA, et al. Identification of nine novel
mutations in COMP in patients with pseudoachondroplasia and multiple
epiphyseal dysplasia. Am J Med Genet 1999;85:486.
JT, Montufar-Solis D, Decker G, et al. Retention of COMP and cell death
in redifferentiated pseudoachondroplasia chondrocytes. Matrix Biol 1998;17:33.
WA, Hall JG, Scott CI, et al. Growth curves for height for diastrophic
dysplasia, spondyloepiphyseal dysplasia congenita and
pseudoachondroplasia. Am J Dis Child 1982;136:316.
R, Hall CM, Young ID. Pseudoachondroplasia: clinical diagnosis at
different ages and comparison of autosomal dominant and recessive
types. A review of 32 patients. J Med Genet 1986;23:425.
MD, Chapman KL. Pseudoachondroplasia and multiple epihyseal dysplasia:
mutation review, molecular interactions and genotype to phenotype
correlations. Human Mutat 2002; 19:465–478.
T, Stanescu V, Muriel ME, et al. Multiple epiphyseal dysplasia,
Fairbank type: morphologic and biochemical study of cartilage. Am J Med Genet 1993;45:501.
O, Mortier GR, Czarny-Rataczak M, et al. Clinical and radiographic
findings in multiple epiphyseal dysplasia caused by MATN3 mutations:
description of 12 patients. Am J Med Genet 2004;125A:278–284.
AE, Poznanski AK, Pudlowski RM, et al. Distal femoral epiphyses: normal
standards for thickness and application to bone dysplasias. Radiology 1968;159:515.
NJ, Jensen FO, Bankier A, et al. Development of the hip in multiple
epiphyseal dysplasia. Natural history and susceptibility to premature
osteoarthritis. J Bone Joint Surg Br 1990; 72(6):1061–1064.
EA, Curtis J, Jayne CH. Chondrodysplasia punctata: a boy linked with
X-linked recessive chondrodysplasia punctata due to an inherited x-y
translocation with a current classification of these disorders. Am J Med Genet 1992;43:823.
RM, Lian JB, Mosher DF, et al. Association of congenital deficiency of
multiple vitamin K-dependent coagulation factors and the phenotype of
the warfarin embryopathy: clues to the mechanism of teratogenicity of
coumarin derivatives. Am J Med Genet 1987;41:566.
MG, Applegarth DA, Dunn HG, et al. Congenital rubella syndrome
associated with calcific epiphyseal stippling and peroxismal
dysfunction. J Pediatr 1990;116:88.
CJR, Magenis RE, Brown M, et al. Inherited chondrodysplasia punctata
due to deletion of the terminal short arm of X-chromosome. N Engl J Med 1984;311:1010.
TD, Pagon RA, Powell BR, et al. Rhizomelic chondrodysplasia punctata
and survival beyond one year: a review of the literature and five case
reports. Clin Genet 1990;38:84.
EF, Opitz JM, Spranger JW, et al. Chondrodysplasia punctata—rhizomelic
form, pathologic and radiographic studies of three infants. Eur J Pediatr 1976;123:89.
MC, Luzzatti L, Silverman FN. Clinical and genetic aspects of
Conradi-Hünermann disease: a report of three familial cases and review
of the literature. J Pediatr 1980;97:911.
JA, Ippolito EG, Ponseti IV, et al. Histochemistry and ultrastructure
of the growth plate in metaphyseal dystosis: further observations on
the structure of the cartilage matrix. J Pediatr Orthop 1981;1:161.
GE, Polumbo PA, Weis MA, et al. Dominant mutations in the type II
collagen gene, COL2A1, produce spondyloepimetaphyseal dysplasia,
Strudwick type. Nat Genet 1995;11:87.
M, Jain P, McKusick VA, et al. The major mutation in the RMRP gene
causing CHH among the Amish is the same as that found in most Finnish
cases. Am J Med Genet 2003;121C:81–83.
E, Makitie O, Makipernaa E, et al. Defective in-vitro colony formation
of haematopoietic progenitors in patients with cartilage-hair
hypoplasia and history of anemia. Eur J Pediatr 1995;154:30.
der Burgt I, Haraldsson A, Oosterwijk JC, et al. Cartilage hair
hypoplasia: description of seven patients and review of the literature.
Am J Med Genet 1991;41:371.
EP, Jacenko O, Olsen B, et al. Fourier transform infrared
microspectroscopic analysis identifies alterations in mineral
properties in bones from mice transgenic for type X collagen. Bone 1996;19:151.
GA, Rash B, Sykes B, et al. Mutations within the gene encoding the
alpha-1 (X) chain of type X collagen cause metaphyseal chondrodysplasia
type Schmid but not several other forms.J Med Genet 1996;33:450.
E, Jensen GS, Pincus J, et al. Constitutive activation of the cyclic
adenosine monophosphate signaling pathway by parathyroid hormone
receptors mutated at the two loci for Jansen’s metaphyseal
chondrodysplasia. Mol Endocrinol 1997;11:851.
KJ, Feinberg JR, Levanda A, et al. Natural history of multiple
hereditary osteochondromatosis of the lower extremity and ankle. J Pediatr Orthop 2002;22:120–124.
DJ, Vassal HJ, Goodman FR, et al. Mutation and deletion of the
pseudoautosomal gene SHOX causes Leri-Weill dyschondrosteosis. Nat Genet 1998;19:70.
PA, Yu JS, Wiand W, et al. Madelung deformity in skeletally immature
patients: morphologic assessment using radiography, CT and MRI. J Comput Assist Tomogr 1996;20:505.
D, Nielsen G. Madelung deformity: surgical prophylaxis (physiolysis)
during the late growth period by resection of the dyschondrosteosis
lesion. J Hand Surg Br 1992;17:401.
ML, Herranz I, Salvador Al. Prevalence of dominant mutations in Spain:
effect of changes in maternal age distribution. Am J Med Genet 1988;31:645.
V, Corral DA, Ahou L, et al. Genomic organization, expression of the
human CBFA1 gene, and evidence for an alternative splicing event
affecting protein function. Mamm Genome 1999;9:54.
B, Thirunavukkarasu K, Zhou L, et al. Missense mutations abolishing DNA
binding of the osteoblast-specific transcription factor OSF2/CBFA1 in
cleidocranial dysplasia. Nat Genet 1997;16:307.
F, Thornell AP, Crompton T. cfbal, a candidate gene for cleidocranial
dysplasia syndrome, is essential for osteoblast differentiation and
bone development. Cell 1997;89:765.
HT, Chambers HG, Mubarak SJ, et al. Congenital coxa vara: computed
tomographic analysis of femoral retroversion and the triangular
metaphyseal fragment. J Pediatr Orthop 2000; 20:551–556.
D, Robertson SP, King LM, et al. Mutations in the gene encoding filamin
B disrupt vertebral segmentation, joint formation, and skeletogenesis. Nat Genet 2004;36(4):323–324.