
sequence of the human genome by government-funded and privately funded
research groups is an amazing accomplishment. The task was completed
ahead of schedule as a result of the cooperative efforts of many
international research groups. These groups used high-throughput
automated DNA sequencing and advanced bioinformatics. The human genome
was shown to contain only 30,000 rather than the expected 100,000
genes. The momentum continues, with the sequencing of the genomes of
many other organisms.
accompanied by an equally impressive effort to convert the raw data
into usable genetic information. Both the raw and annotated data are
available in online databases. Bioinformatic tools enable complex
searches to be undertaken of nucleotide, protein, and disease databases.
resulted in a rapid acceleration in the identification of new disease
genes of importance in orthopaedics. Such advances result in more
precise diagnoses and yield insights into the pathogenesis,
classification, prognosis, and potential treatments of genetic
disorders of the musculoskeletal system. This chapter summarizes the
principles of genetics as they apply to orthopaedic conditions and
highlights the advances in knowledge in this field. The information
provided, however, will need to be updated periodically because
knowledge in the field of genetics is advancing rapidly. Online access
to the National Library of Medicine Web sites provides an entry point
to a massive amount of genetic information.
diseases mentioned in this chapter can be found in the “Online
Mendelian Inheritance in Man” (OMIM) database (http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM&itool=toolbar). References 1 to 5, cited at the end of the chapter, provide comprehensive background material on human genetics.
The chromosomes contain genes, which are the DNA units of genetic
information. They are linearly arranged along the chromosomes, and each
gene occupies a particular position or locus.
Twenty-two of them are autosomes that occur in men and women. The
remaining pair, the sex chromosomes, are designated XX in women and XY
in men. The members of a pair of autosomes and a pair of X chromosomes
contain matching genetic information.
of two chromatids joined at the centromere, which is the primary
constriction of the chromosome. The centromere divides the chromosome
into the short p arm and the long q arm. Cytogenetic techniques divide
the arms into banded regions that are used for indicating the sites of
chromosomal rearrangements and the loci of genes. For example, the COL1A1
gene for one of the type I collagen protein chains is located on
chromosome 17 at locus q21.3-22. The latter notation indicates that the
gene is located on the q arm of chromosome 17 at the band 21.3-22 (Fig. 6.2).
repair by the process of mitosis. The daughter cells contain genetic
profiles identical to those of the parent cells. Germline cells undergo
meiosis during gametogenesis. In this process, the diploid number of 46
chromosomes is reduced to the haploid number of 23, including one of
each of the autosomes and either an X or a Y chromosome. The random
assortment of each of the chromosome pairs during meiosis is central to
the Mendelian inheritance pattern of single-gene disorders and some
forms of chromosomal rearrangements.
genome of approximately 7 billion base pairs of DNA. Genes are made up
of linearly aligned nucleotides. Each unit or nucleotide of DNA
consists of a deoxyribose sugar, a purine or pyrimidine base, and a
phosphate group. There are two purine bases, adenine (A) and guanine
(G), and two pyrimidine bases, thymine (T) and cytosine (C). The
nucleotides form long polynucleotide chains.
in which the component polynucleotide chains run in opposite directions
and contain complementary sequences. Central to the Watson and Crick
model of the DNA double helix are the complementary sequences of the
chains, which are held together by hydrogen bonding between
complementary pairs of nucleotide bases. An A of one chain pairs with a
T of the other, and a G of one chain pairs with a C of the other.
codons that encode specific amino acids. Each codon contains three
nucleotides. Exons often delimit functional domains within the protein.
The 5′, or upstream, end of the gene contains promotor sequences that
regulate the expression of the gene. The promotor immediately precedes
the start site of transcription of messenger RNA (mRNA).
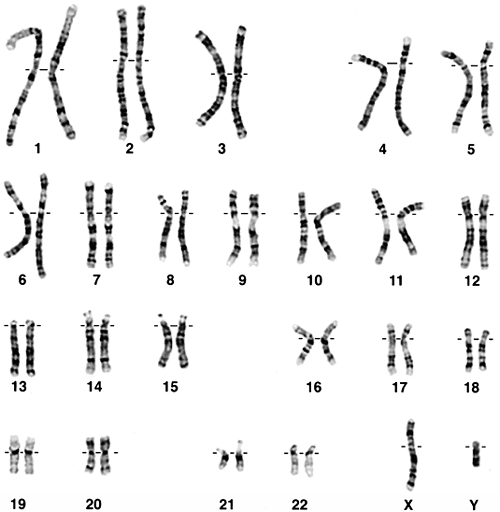 |
|
Figure 6.1 Normal 46, XY karyotype.
|
is synthesized from the start site of transcription to the 3′
untranslated region. The pre-mRNA undergoes several modifications to
form mRNA, which is transported from the nucleus to the cytoplasm and
ribosomes. After the introns are spliced out, the remaining exons form
a continuous coding sequence. The coding region is flanked by a 5′
untranslated region that contains sequences essential for ribosomal
binding and translation. The 3′ untranslated region contains sequences
that are important for mRNA stability. The polyadenylation signal
contains sequences that result in the addition of a polyA nucleotide
tail, a polyadenosine sequence that characterizes most mRNAs.
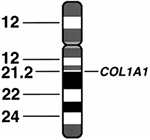 |
|
Figure 6.2 Diagram of chromosome 17 showing its banded structure and the location of the COL1A1 gene encoding the pro-α1(I) chain of type I collagen.
|
amino acid code of the corresponding protein, is achieved on the
ribosomes. The key to the genetic code is the codon, which is a group
of three bases. Because each codon contains three of the four
nucleotide bases, there are 64 possible triplet combinations. In
humans, there are only 20 relevant amino acids, and most of them are
encoded by more than one codon. Three of the codons are called stop or nonsense codons because they designate the site of termination of translation.
the translational reading frame, ensuring that the correct amino acids
are added sequentially to the growing polypeptide chain. Addition of
the appropriate amino acids is achieved by specific transfer RNAs
(tRNAs) for each amino acid. They contain the anticodon sequences that
recognize the complementary codon sequences of the mRNA. As an amino
acid is added to the carboxyl end of the polypeptide chain, the mRNA
slides exactly one codon length along the ribosome and brings the next
codon into line for interaction with its specific tRNA. The proteins
are synthesized from the amino terminus to the carboxyl terminus,
corresponding to translation of the mRNA from 5′ to 3′ Translation
ceases at the first stop codon. The completed polypeptide is released
from the ribosome.
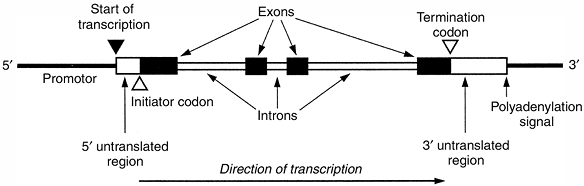 |
|
Figure 6.3 General structure of a typical human gene, showing the main functional domains. (From Thompson MW, McInnes RR, Willard HF. Genetics in medicine, 5th ed. Toronto: WB Saunders, 1991, with permission.)
|
modifications in the rough endoplasmic reticulum, Golgi apparatus, and
outside the cell. For example, the core proteins of collagen and
glycosaminoglycans undergo extensive enzymatic modification. Many
pre-proproteins have terminal extensions that are removed to convert
these pre-proproteins into functional proteins. The functional proteins
are assembled into complex polymers.
arrangement of DNA. It can occur in somatic cells, as is observed in
many cancers, but, when it occurs in germline cells, the mutation can
be transmitted to subsequent generations. Permanent changes in DNA
sequences are rarely deleterious but add to the genetic diversity among
individuals. Loci that have many alternative forms, called alleles,
are polymorphic. The Human Genome Project and related projects have
identified many thousands of single nucleotide polymorphisms, called SNPs.
that involve misaggregation of chromosomes to chromosome mutations that
involve chromosome rearrangements and specific gene mutations.
-
missense mutations that alter the amino acid sequence
-
nonsense mutations that introduce a premature termination codon
-
alteration of promotor sequences
-
mRNA splicing mutations that result in exon loss.
randomly, there are mutational hot spots in the genome, commonly at CG
dinucleotides, and mutations tend to recur at such sites. Transitions,
which exchange one pyrimidine for the other or one purine for the
other, are more common than transversions, which exchange a purine for
a pyrimidine or vice versa. Transitions and transversions are
responsible for most of the mutations of the type I collagen genes in
osteogenesis imperfecta and of the type II collagen gene in the
spondyloepiphyseal dysplasias.
sporadic point mutations and is referred to as the paternal age effect
on new mutations (6). It is common in
achondroplasia. Germline mosaicism for the new mutation also occurs in
achondroplasia and other skeletal dysplasias. It accounts for the birth
of affected siblings from clinically normal parents (7).
The paternal age effect and germline mosaicism are explained by
differences in gametogenesis in men and women. Spermatogonia go through
a few mitotic divisions before embarking on the meiotic divisions that
lead to mature sperm (8). Some of the products
of the mitotic divisions are returned to the “cell bank” to replenish
the supply of spermatogonia. Mutations that occur during DNA
replication can accumulate, providing a basis for the paternal age
effect and for germline mosaicism.
substitution alters the sense of a codon and a different amino acid is
added to the elongating polypeptide. Mutations of this kind are common
in many structural proteins, such as the collagens in osteogenesis
imperfecta and in some of the chondrodysplasias.
substitution converts a codon for an amino acid into a termination
codon. The introduction of a termination codon into the sequence
results in the premature termination of translation and a truncated
protein. Such proteins are rarely functional because they lack the
carboxyl-terminal domains that are usually required for protein
assembly. The mRNAs containing a premature translational termination
codon are often retained within the nucleus. Because the mutant allele
is essentially functionless, it produces a state of haploid
insufficiency. This type of mutation produces the common mild form of
osteogenesis imperfecta.
transcription of the gene. They have been identified in the β-globin
gene and in the factor IX gene in hemophilia B. Few other mutations of
this type have been identified in humans.
altered transcription or instability of mRNA, thereby reducing the
production of the relevant protein. Such mutations have been identified
in the β-globin gene, but not in the genes that produce musculoskeletal
diseases.
that contain numerous exons and introns. Commonly, the point mutations
occur in the consensus sequences at the exon–intron boundaries. The
adjoining exon is usually spliced out, resulting in a shortened protein
chain. If the exon normally starts and finishes with complete codons,
the normal translational reading frame is retained, and the amino acid
sequence is normal beyond the spliced-out exon. The resulting protein
functions abnormally because it is shorter than normal, and because it
lacks the functional domain encoded by the lost exon. If the exon
contains split codons at its ends, the translational reading frame
beyond the spliced-out exon is abnormal, and the amino acid sequence is
incorrect. A frequently encountered premature translational termination
codon results in the synthesis of a truncated protein.
mutations that create a new or cryptic splice site. The consequences of
such mutations are often complex because splicing may remove part of an
exon and include intron sequences. Lethal forms of osteogenesis
imperfecta and spondyloepiphyseal dysplasia frequently result from such
mutations of the type I and type II collagen genes, respectively.
changes in gene structure and in the transcript. These genetic
variations result from several types of molecular alterations:
-
frameshift mutations caused by partial codon deletions or insertions
-
complete codon deletions or insertions
-
gene deletions and duplications
-
insertion of duplicated elements.
produced. The protein may be partially functional, as observed with the
shortened forms of dystrophin produced by deletions in the DMD gene in patients who have the Becker form of muscular dystrophy.
These disorders occur in approximately 0.7% of live births, in 2% of
all pregnancies of women older than 35 years of age, and in 50% of all
spontaneous first-trimester abortions; they are being recognized with
increasing frequency because of improvements in cytogenetic techniques.
Chromosome abnormalities of number or structure can involve autosomes
or sex chromosomes.
occurs in approximately 4% of pregnancies. Most patients with
aneuploidy are trisomic; they have three, instead of the normal pair,
of a particular chromosome. Monosomy, which is the loss of one member
of a pair, occurs less commonly. The most common trisomies of an entire
autosome compatible with postnatal survival are trisomy 21 (i.e., Down
Syndrome), trisomy 18, and trisomy 13. All the trisomies produce growth
retardation, mental retardation, and multiple congenital anomalies. It
is likely that the additional dosage of the specific genes on the extra
chromosome is responsible for the abnormal phenotype (10).
and the frequency is higher among pregnancies of mothers older than 35
years.
phenotype but is important for counseling. In 95% of patients, trisomy
21 results from meiotic nondisjunction of the chromosome 21 pair (Fig. 6.4).
The recurrence risk increases with maternal age, particularly in women
older than 30 years of age. Nondisjunction usually occurs during
maternal meiosis I, and occasionally during paternal meiosis I. The
cause of nondisjunction is uncertain.
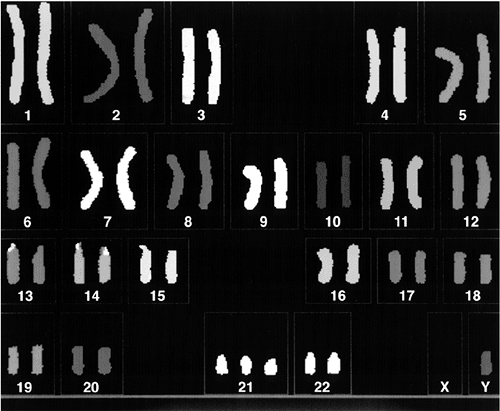 |
|
Figure 6.4
Karyotype in Down syndrome attributable to meiotic nondisjunction of the chromosome 21 pair. There are three copies of chromosome 21. |
which is a translocation between chromosome 21q and the long arm of
chromosome 14 or 22. The resulting karyotype for a Robertsonian
translocation between chromosome 14 and 21 is 46, XX or XY, – 14,
+t(14q21q), with a loss of chromosome 14 (designated -14), and a new
hybrid form of the 14q21q chromosome, designated +t(14q21q)(9).
This karyotype produces a trisomy 21 state. The translocation forms of
Down syndrome are not related to maternal age, but there is a high
recurrence risk, particularly when the mother is a carrier of the
translocation. A carrier involving chromosomes 14 and 21 has only 45
chromosomes because one of each of these chromosomes is missing and is
replaced by the translocation chromosome t(14q2lq). Down syndrome is
produced when the fetus inherits a normal chromosome 21 from one parent
and an unbalanced complement of chromosomes, including a normal
chromosome 21 and the translocation chromosome, from the other parent.
The unbalanced chromosome complement appears in 15% of the progeny of
carrier mothers, which is less than the expected proportion, and it
rarely appears in the progeny of carrier fathers.
inheritance of a translocation chromosome t(21q21q), made up of two
chromosome 21 long arms from one parent and a normal chromosome 21 from
the other parent. Carriers of this translocation chromosome usually
only have children with Down syndrome.
for the trisomy state. There is wide variability in the severity of the
phenotype, probably because of the variable proportion of trisomic and
euploid cells. Germline mosaicism may account for the
higher-than-expected risk of recurrence in young mothers.
anomalies of chromosome number. These anomalies are balanced if the
chromosome set has the normal complement of DNA and unbalanced if there
is additional or missing DNA.
information, and commonly produce abnormal phenotypes. Duplication of
part of a chromosome produces a partial trisomy, and deletion leads to
a partial monosomy. Increasingly, small deletions and insertions are
being detected by cytogenetic techniques. The phenotypes of
some
of the deletion syndromes can be readily explained by the loss of
contiguous genes. For example, in the Langer-Giedion syndrome, deletion
of chromosome 8q24.11-q24.13 produces mental retardation,
trichorhinophalangeal syndrome, and osteochondromas. The
osteochondromas occur because the deletion includes the EXT1
locus, which is abnormal in some patients with autosomal dominant
multiple exostoses. The trichorhinophalangeal syndrome is produced by
the deletion of the TRPS1 gene.
effect because all of the genetic information is present, although it
is arranged differently. Occasionally, such rearrangements do disrupt a
gene at the site of chromosome break. Balanced rearrangements increase
the risk of unbalanced rearrangements in progeny.
associated with abnormally tall and short statures. The 47, XXY
chromosome constitution, called Klinefelter syndrome,
and the 47, XYY constitution produce abnormally tall stature in men.
Trisomy X (47, XXX) occurs in women and is the female counterpart of
Klinefelter syndrome, producing tall stature, whereas 45, X and its
variants (e.g., Turner syndrome) are associated with short stature.
defects are not detectable by current cytogenetic methods. Single-gene
defects alter one or both copies of a gene. Many genes are polymorphic
as they have many alleles that contain nonpathologic changes of DNA
sequence. Mutant alleles contain changes in DNA sequence that can
produce single-gene disorders.
Single-gene disorders are produced by a specific allele at a single
locus of one or both members of a chromosome pair. If the alleles are
identical, the individual is homozygous for that trait; if they are
dissimilar, the individual is heterozygous; and if they have two
different mutant alleles, the individual is a compound heterozygote.
Men are hemizygous for X-linked genes because they only have one X
chromosome.
determined by pedigree analysis. They may involve genes on autosomes
(i.e., autosomal inheritance) or genes on the X chromosome (i.e.,
X-linked inheritance) (11). If the disease is expressed when just one out of the pair of chromosomes carries a mutant allele, the phenotypes are termed dominant; if the disease is expressed only when both chromosomes carry the mutant allele, the phenotype is termed recessive.
For many genetic diseases, there is little detailed knowledge of the
critical factors that link the genotype and the phenotype. Many other
genetic and environmental factors modify the expression of the
genotype; some affected individuals show minimal or no clinical
anomalies, but others show severe changes.
probability that a gene defect will have any phenotypic expression at
all. In pedigrees, particularly autosomal dominant pedigrees, some
affected individuals fail to express the genotype. The penetrance of a
gene can be defined as the proportion of individuals with the
appropriate genotype who express it.
to different severities of the phenotype among individuals who have the
same genotype. Many autosomal dominant disorders show variable
expressivity; for example, patients with Marfan syndrome may have few
or all of the classic features of the condition.
which refers to the apparent worsening of the disease in successive
generations. This form is a feature of pedigrees of myotonic dystrophy,
Huntington disease, and fragile X mental retardation, and it is caused
by variable and unstable expansions of DNA. Myotonic dystrophy type 1,
for example, is caused by the unstable expansion of a CTG trinucleotide
repeat located in the 3′ untranslated region of a gene on chromosome 19
that encodes a protein kinase (12). Severity
varies with the number of repeats: normal individuals have from 5 to 30
repeat copies; mildly affected individuals, from 50 to 80; and severely
affected individuals, 2,000 or more copies. Amplification is frequently
observed after parent-to-child transmission, but extreme amplifications
are not transmitted through the male line. This explains the
anticipation and the occurrence of the severe congenital form almost
exclusively in the offspring of affected women.
of onset of the phenotype. Some single-gene disorders, such as
achondroplasia, are evident at birth, and are therefore congenital.
Others, such as pseudoachondroplasia, are not apparent at birth but
become so after the patient is 2 to 3 years of age, when growth
retardation and dysmorphism appear.
For example, the pleiotropic musculoskeletal, ocular, and
cardiovascular manifestations of Marfan syndrome are causally linked by
fibrillin1, the microfibrillar protein at fault in this syndrome, which
is distributed throughout all of the affected tissues (13).
autosomal dominant traits. Affected individuals are heterozygous for
the mutation; they have one normal and one mutant allele of the gene.
However, the product of the
normal
allele is unable to compensate for the abnormality produced by the
mutant allele. The mating of two heterozygous individuals can produce
homozygous autosomal dominant traits. The homozygotes are usually much
more severely affected than heterozygotes, often resulting in perinatal
death.
musculoskeletal anomalies. These disorders include many of the
chondrodysplasias, osteogenesis imperfecta, Marfan syndrome,
Ehlers-Danlos syndrome, acrocephalosyndactyly syndromes, absent tibial
syndromes, Charcot-Marie-Tooth disease types IA and IB, and
neurofibromatosis 1.
transmitted from generation to generation by affected individuals, who
transmit the mutant gene to about half of their offspring (Fig. 6.5).
Men and women are equally affected, and unaffected individuals do not
carry or transmit the mutant gene. Typical multigeneration autosomal
dominant pedigrees (Fig. 6.5) are common in
families with neurofibromatosis, osteogenesis imperfecta type I, and
Marfan syndrome. However, there is wide variability of penetrance and
expression of the genotype in such families. For example, in families
with the common type I form of osteogenesis imperfecta, some members
have gray–blue scleras, a characteristic feature of the disease, and
severe osteoporosis with multiple fractures, whereas others have
gray–blue scleras without clinical evidence of bone fragility. Similar
variability is observed in families with neurofibromatosis 1 and Marfan
syndrome when the clinical manifestations are correlated with the
inheritance of the mutant allele. Many of the individuals shown to
carry the mutant allele lack the major clinical features required for a
firm clinical diagnosis and are unaware that they have the disease. The
latter observation applies particularly to young individuals, who are
likely to develop more obvious features with age.
dominant mutations. About half of the individuals with osteogenesis
imperfecta or Marfan syndrome, and most individuals with
achondroplasia, have new autosomal dominant mutations. The mutation
occurs in the ovum, or in the sperm involved in the formation of the
fertilized ovum, for the first affected individual in the family. New
dominant mutations are often associated with increased paternal age,
presumably as a result of an increased level of mutagenesis during
spermatogenesis in older men. The affected individuals transmit the
trait to half of their offspring, which is typical of an autosomal
dominant inheritance pattern.
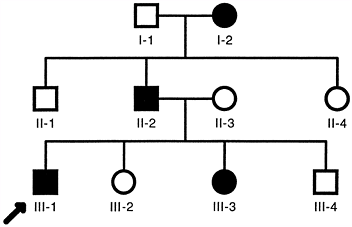 |
|
Figure 6.5
Typical autosomal dominant pedigree. Each individual is identified by a generation number and the position within each generation. Men are indicated by squares and women by circles. Filled symbols indicate clinically affected individuals. The proband (arrow) is the family member through whom the family history was ascertained. |
syndrome show an apparently autosomal recessive form of inheritance,
with clinically normal parents and multiple affected offspring. In most
instances, genetic testing has shown that one parent is mosaic for the
dominant mutation, and transmits the trait to multiple children.
Presumably, a spontaneous mutation occurred early in the embryogenesis
of the mosaic parent, and some of the somatic cells and gametes carry
the mutation. Mosaic parents may show some minor clinical features of
the disease. Genetic testing of dermal fibroblasts, hair follicles, and
leukocytes reveals the proportion of cells carrying the mutant allele.
The sperm can be similarly tested. Rapid progress is being made in
identifying mutant genes in autosomal dominant disorders that produce
musculoskeletal anomalies. Important principles of autosomal dominant
disorders are discussed in the following sections.
diseases are illustrated by recent findings in osteogenesis imperfecta.
In most instances, autosomal dominant diseases are either inherited as
autosomal dominant traits or occur from new autosomal dominant
mutations. The mutations usually involve one of the two genes that
encode the chains of type I collagen, the principal collagen of the
tissues affected by the disease. The COL1A1 gene on chromosome 17 encodes the pro-α1(I) chain, and the COL1A2 gene on chromosome 7 encodes the pro-α2(I) chain. Each type I collagen molecule contains two α1(I) chains and one α2(I) chain.
The common type IA form, with gray-blue scleras, osteoporosis, mild
bone fragility, normal teeth, ligament laxity, and premature deafness,
is caused by mutations of the COL1A1 gene,
in which the mutant allele is functionless. The mutant allele usually
produces an mRNA containing a premature stop codon that would be
expected to produce a truncated and functionless α1(I) collagen chain.
However, the nucleus retains most of the mutant mRNA, and the cytoplasm
contains predominantly normal α1(I) mRNA, although in 50% of the normal
amount. The type I collagen produced in osteogenesis imperfecta type IA
cells is normal, but the amount is about
half
of the normal amount. Each family has been shown to have its own unique
mutation, leading to premature stop codons at different sites of the
mRNA. Despite this genetic heterogeneity, there is a final common
pathway of type I collagen deficiency that accounts for this type of
osteogenesis imperfecta. Nonetheless, because the severity of the
disease varies between and within families, it is likely that modifying
genes and epigenetic factors also play a role in the pathogenesis of
the disease.
gene. These mutations result in the production of a mixture of normal
and mutant collagen chains and type I collagen molecules. A registry of
type I collagen mutations is available at http://www.le.ac.uk/genetics/collagen/.
The most common mutation involves the substitution of a glycine residue
in one of the 338 glycine-X-Y triplets, the mandatory repetitive
triplet sequence required for triple helix formation. Proline is often
in the X position and hydroxyproline in the Y position of the triplets.
Abnormal helix formation occurs after substitution of glycine, the
smallest amino acid, with the larger amino acids, alanine, valine,
cysteine, arginine, aspartic acid, and glutamic acid. Collagen α chains
carrying these substitutions are able to combine with normal chains to
produce type I collagen molecules. In cases of COL1A1
mutations, half of the α1(I) chains are expected to be mutant and half
are expected to be normal. Because type I collagen molecules contain
two α1(I) chains, it is expected that approximately 25% of the
molecules will be normal and 75% will contain one or two mutant α1(I)
chains. The particular α1(I) chain composition of the type I collagen
molecules enhances the impact of the heterozygous COL1A1 mutation.
mutations, approximately half of the α2(I) chains will be normal and
half will be mutant. Because type I collagen molecules contain only one
α2(I) chain, approximately half of the molecules will be normal and
half will contain the mutant α2(I) chain. The mutant molecules, whether
containing the mutant α1(I) or α2(I) chain, are more susceptible to
degradation and are poorly secreted. Once secreted, they interfere with
the formation of the extracellular matrix of bone and other tissues
containing type I collagen. These mutations act in a dominant-negative
fashion because the mutant collagen chains impair the function of the
normal α chains.
mutations, as shown for the perinatal lethal forms of osteogenesis.
There are a few examples of unrelated families with the same mutation.
Variability in the severity of the disease has also been observed in
such families, indicating that modifying genes and epigenetic factors
contribute to the pathogenesis of the dominant-negative forms of
osteogenesis imperfecta.
determining the clinical severity of the disease resulting from
dominant-negative mutations of the type I collagen genes. However, most
of the perinatal lethal cases result from mutations that involve the
carboxyl-terminal half of the collagen chains. Substitutions of glycine
by cysteine yield a gradient of severity, with lethal cases at the
carboxyl terminus, moderately severe cases in the middle, and milder
cases at the amino terminus of the α chains.
autosomal recessive form of inheritance. As a result, the empiric risk
of recurrence in a family with a sporadic form of osteogenesis
imperfecta is approximately 6%. The risk can be better assessed by
genetic testing of the parents, but it is still only a rough estimate
because the proportion of affected gametes is usually unknown.
Intrauterine DNA testing for osteogenesis imperfecta is available at
specialized centers.
determined diseases that affect the structure and function of
cartilage. Spranger grouped the disorders with similar features into
families (16). One family consists of a
heterogeneous group of spondyloepiphyseal dysplasias. The severity of
these disorders varies markedly among the lethal forms of
achondrogenesis type II and hypochondrogenesis, the severely dwarfing
forms of spondyloepiphyseal dysplasia congenita and Kniest syndrome,
the marfanoid form of Stickler syndrome or hereditary
arthroophthalmopathy, and mild forms with premature osteoarthritis.
Heterozygous mutations of type II collagen (the principal collagen of
cartilage) or type XI collagen (a minor collagen of cartilage) are
found in this family of spondyloepiphyseal dysplasias. The general
categories of mutations found in osteogenesis imperfecta are also found
in this family of dysplasias.
gene in patients with osteogenesis imperfecta type IA. In both of these
diseases, the mutant alleles of the respective genes are functionless
and lead to the production of normal collagen, although in about half
of the normal amount. Other individuals with Stickler syndrome have
mutations of the COL11A1 gene on chromosome 1p21, which encodes the α1(XI) chain, or of the COL11A2 gene on chromosome 6p21.3, which encodes the α2(XI) chain of type XI collagen.
dysplasias are caused by heterozygous mutations that alter the
structure of the triple helical domain of type II collagen (15,17).
Unlike the marfanoid habitus of individuals with Stickler syndrome,
these individuals are often severely dwarfed. The dominant-negative
effects of the mutations are severe because type II collagen molecules
contain three α1(II) chains. Approximately 12.5% of the
molecules
contain three normal chains, and 87.5% of them contain one, two, or
three mutant chains. As in osteogenesis imperfecta, the mutant
molecules are poorly secreted, are more susceptible to degradation, and
impair normal formation of the extracellular matrix.
dwarfism. It is inherited as an autosomal dominant trait with complete
penetrance. Approximately 87% of cases are caused by new mutations.
There is a considerable reduction in the effective reproductive fitness
of patients with achondroplasia.
heterogeneity than occurs in other skeletal dysplasias, such as
osteogenesis imperfecta and the type II collagen family of
spondyloepiphyseal dysplasias. The clinical and radiographic features
are remarkably constant, and the growth plates are histologically
normal, despite the severe retardation of longitudinal growth. The
similarity of phenotype between unrelated patients can be explained by
the molecular defects in achondroplasia.
at locus p16.3 by linkage analysis, and mutations were identified in
the gene for fibroblast growth factor receptor 3 (FGFR3) (18,19,20,21).
Transcripts of this gene are most abundant in the nervous system, and
may account for the megaloencephaly of some patients. Outside the
nervous system, the highest levels are found in the cartilage anlage of
all bones and in the resting chondrocytes of the growth plates (22).
All patients have missense mutations that change glycine residue 380 to
arginine or, less often, that change a nearby amino acid residue (18,23).
The codon for amino acid residue 380 includes a CG dinucleotide, which
is a “hot spot” for mutations. These mutations are expected to alter
the structure of the transmembrane domain of the receptor and to
produce similar functional abnormalities, accounting for the relatively
invariant phenotype of achondroplasia.
gene. Thanatophoric dwarfism, a lethal chondrodysplasia that shares
some phenotypic features with achondroplasia, is also caused by
mutations of FGFR3 (20).
neurofibromatosis 1 shows complete penetrance, in that all individuals
who carry the mutation express the mutation. However, the expression is
highly variable, and some individuals within affected families have
extremely severe disease, whereas others may have café-au-lait spots as
their only manifestation of neurofibromatosis 1.
Approximately 80% of these mutations potentially encode a truncated
protein because of premature termination of translation. The disease
expression is probably the result of haploid insufficiency because the
truncated proteins are likely to be functionless. The normal allele
produces a reduced amount of normal neurofibromin, insufficient for
normal development and functioning of the tissues that express the NF1 gene.
This phenomenon probably reflects genomic imprinting (still a poorly
understood process) that alters the relative expression of the
paternally and maternally derived genes.
from a new mutation that is not inherited from either parent. The
spontaneous mutation rate is approximately 1 in 10,000 gametes, which
is one of the highest levels in humans. This high rate presumably
reflects the large size of the gene and its resulting susceptibility to
deletions, insertions, point mutations, and major rearrangements. In
most cases, the new mutation occurs in the paternally derived gene.
This finding suggests that the mutation may occur during mitotic
division, which takes place in male gametogenesis but not in female
gametogenesis. Because there is little or no evidence of the
accumulation of mutations, reflected by the absence of a paternal age
effect, the mutations may accumulate in cells that are not involved in
the process of replenishment of the germ cell bank.
heterozygotes, also called carriers.
Carrier frequency varies considerably but, for common autosomal
recessive disorders such as cystic fibrosis, it is approximately 1 in
45 individuals. The mutant alleles in a population occur much more
frequently in carriers than in affected individuals. For example,
approximately 98% of the cystic fibrosis alleles are present in
asymptomatic carriers, and only 2% are present in homozygous patients.
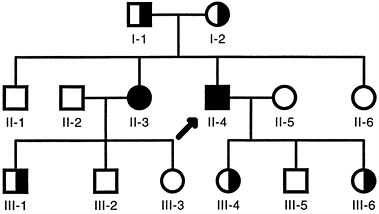 |
|
Figure 6.6 Typical autosomal recessive pedigree. Homozygous affected individuals are indicated by filled symbols. Asymptomatic carriers, who are heterozygotes, are indicated by half-filled symbols. The proband is indicated by the arrow.
|
Autosomal recessive traits are more frequent in consanguineous
marriages, particularly if the mutant gene is rare.
They result from deficiencies of specific enzymes that lead to a block
in a normal metabolic pathway, with accumulation of the substrate and a
deficiency of the product. Because most enzymes are normally present in
vast excess, a major reduction in their activity is required before a
metabolic pathway is blocked. As a result, carriers rarely express
inborn errors of metabolism because the activity of the enzyme produced
by the normal allele is sufficient to ensure normal metabolic activity.
In the homozygous state, the activity of the specific enzyme is often
reduced to approximately 5% or less of normal values. Reductions of
this magnitude are usually required before a metabolic pathway is
blocked.
accumulation of its substrate, the deficiency of its product, or both.
Substrates may be readily diffusible and are found in excessive amounts
in all body fluids and in all tissues. An example is phenylalanine,
which accumulates in phenylketonuria, the classic example of an
autosomal recessive disease. In diseases of this kind, the widespread
accumulation of the substrate may result in pathologic changes in
tissues that are not normally involved in the particular metabolic
pathway. Damage to the developing nervous system in phenylketonuria
results from this mechanism. Most of the inborn errors of amino acid
metabolism produce types of changes similar to those observed in
phenylketonuria. Homocystinuria is one of the few inborn errors of
amino acid metabolism that produce musculoskeletal anomalies. Affected
individuals have a marfanoid appearance.
that are normally involved in the metabolic process. Cell function
deteriorates, eventually producing cell death as the substrate
progressively accumulates intracellularly. Diseases caused by this
abnormality are often referred to as storage diseases
because the affected tissues progressively enlarge. Typical examples
include lysosomal storage diseases, such as Gaucher disease, and the
mucopolysaccharidoses. The lysosomal enzymes are responsible for the
degradation of macromolecules, such as the mucopolysaccharides of the
extracellular matrix. Deficiencies of the lysosomal enzymes involved in
the degradative cascade of the mucopolysaccharides produce a
heterogeneous group of diseases, some of which manifest severe skeletal
anomalies. This group includes the following syndromes: Hurler, Scheie,
Sanfilippo A to D, Morquio A and B, Maroteaux-Lamy, and Sly.
syndromes, can occur with different enzyme deficiencies, a phenomenon
referred to as locus heterogeneity. Partial and complete deficiencies of the enzymes can also alter the severity of the phenotype, which is referred to as clinical heterogeneity.
These syndromes may also show wide variation in clinical severity as a
result of allelic heterogeneity, in which different defects occur in
the same gene.
are caused by a deficiency of the normal product rather than an
accumulation of the substrate. For example, some forms of congenital
hypothyroidism result from enzyme defects in the synthesis of thyroxine.
mechanisms, including defects in receptor proteins, membrane transport,
and cell organelles. Cystic fibrosis is caused by mutations of a
protein called cystic fibrosis transmembrane conductance regulator.
Disorders of peroxisomes, which are subcellular organelles, produce a
variety of diseases, including rhizomelic chondrodysplasia punctata.
Men are hemizygous for X-linked genes because they have only one X
chromosome. Women are homozygous unaffected, homozygous affected, or
heterozygous because they have a pair of X chromosomes.
disorders because of the normal random inactivation of one of the X
chromosomes in their somatic cells. The random inactivation of the X
chromosome is called the Lyon hypothesis,
which accounts for the similar levels of expression of one allele in
men and a pair of alleles in women. This process is also referred to as
dosage compensation; the level
of expression of one dose of an X-linked gene in a man is equivalent to that of two doses of an X-linked gene in a woman.
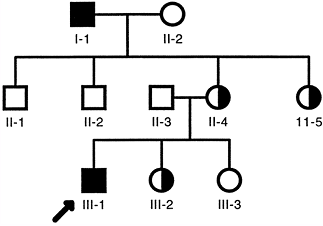 |
|
Figure 6.7 Typical X-linked recessive pedigree. Affected hemizygous men are indicated by filled squares. Asymptomatic female carriers are indicated by half-filled circles.
|
inactivation of the paternal or maternal X chromosome occurs in each
somatic cell. The descendants of each cell have the same inactive X
chromosome. As a result, the somatic cells of women are mosaic, with
some cells expressing one X chromosome and the remainder expressing the
other. The inactive X chromosome is condensed and, with the exception
of the pseudoautosomal region, its genes are not expressed. Because
heterozygous women have various proportions of cells expressing either
of the X-linked alleles, there is marked variability in the expression
and clinical phenotypes. Some women appear normal, whereas others,
referred to as manifesting heterozygotes, have the typical phenotype
displayed by hemizygous men.
expressed in heterozygous women. A characteristic feature of such
pedigrees is that all of the daughters of the affected fathers are
themselves affected, but none of the sons is affected. The affected
women transmit the mutation in a manner similar to an autosomal
dominant trait because they have a pair of X chromosomes. As a result,
affected women transmit the mutation to half of their children,
regardless of gender. Affected women are usually less severely affected
than affected men because of random inactivation of one of the X
chromosomes. The expression depends on the ratio of cells that express
the mutant allele to cells that express the normal allele.
manifestations include X-linked hypophosphatemic rickets and Rett
syndrome. Rett syndrome is lethal in male babies at birth, whereas
heterozygous affected girls have severe mental retardation.
It resembles metaphyseal chondrodysplasia–type Schmid, which results
from mutations of type X collagen. This collagen is specific to the
hypertrophic zone of the growth plate. These disorders are
differentiated by the low serum inorganic phosphorus levels in children
with hypophosphatemic rickets.
are hemizygous, with only one X chromosome. Variable expression occurs
in heterozygous girls because of random inactivation of the paternal
and maternal X chromosomes (32). Mutations of the PHEX gene (phosphate-regulating gene with homologies to endopeptidases on the X chromosome) cause the disease.
is expressed in all boys, but only in homozygous girls. The latter
situation is rare because expression in girls is usually limited to the
manifesting heterozygotes, in whom the normal X chromosome has by
chance been inactivated in most somatic cells.
from an affected father through all his daughters. Consequently, a
daughter’s sons have a 50% chance of inheriting the gene. Fathers do
not transmit the gene directly to their sons.
A, which produces a deficiency of factor VIII, and Duchenne muscular
dystrophy, which produces a deficiency of dystrophin.
gene that encodes the protein dystrophin, a normal component of the
muscle membrane. Approximately one third of cases are new mutations,
and the remainder are inherited from female carriers. Most of the
mutations are deletions (33). Affected men
reproduce only infrequently, and the disease is transmitted by female
carriers who are usually clinically unaffected. Some mutations produce
Becker muscular dystrophy, which has a milder phenotype.
fall into the typical dominant or recessive pattern. Fragile X syndrome
is an example of a disorder with an atypical X-linked inheritance
pattern.
common cause of mental retardation in boys. Girls too can be affected,
although the phenotype is usually milder, and is characterized by
learning disabilities or mild mental retardation. Postpubertal boys
have a
marfanoid
appearance, macroorchidism, and mental retardation; they also have lax
joints, resembling milder forms of Ehlers-Danlos syndrome.
characteristic cytogenetic anomaly. The chromatin in the fragile site
at position Xq27.3 fails to condense during mitosis. The molecular
defect is attributable to an amplification of a region containing a
variable CGG trinucleotide repeat in the 5′ untranslated region of the FMR1 gene (34). Expression of the FMR1
gene is deficient in affected men, although normal individuals, women
carriers, and men with the premutation all show normal expression (35).
Allele sizes vary from 6 to 54 repeats in normal individuals, from 52
to 200 repeats in individuals with premutations, and from 200 to more
than 1,000 repeats in affected individuals (34).
Expansion of premutations to full mutations occurs only after passage
through the maternal germline. Fathers can pass on the premutation for
this condition to their daughters, but it is only after female
gametogenesis that sufficient trinucleotide expansion occurs to silence
the FMR1 gene and give rise to the clinical manifestations found in grandsons of the premutation men (36).
unstable expansion of trinucleotide repeats in other genes. These
disorders, like fragile X syndrome, also have a parental sex bias in
the transmission of the mutation with respect to the age of onset and
clinical expression (37).
with Mendelian principles. However, alternative modes of inheritance
have been identified in humans. For example, some neuromuscular and
ocular diseases are caused by mutations of mitochondrial, rather than
nuclear, DNA. They are inherited from the mother because mitochondria
are transmitted in the ovum, but not in sperm. As a result, women
transmit their mitochondrial DNA to all their children, but men do not
transmit their mitochondrial DNA to any of their children.
identical nuclear DNA derived from a single zygote. However, mutations
can produce cell clones that are genetically different from the
original zygote (37). Such individuals are said to be mosaic. Mosaicism can be somatic, gonadal, or both.
both somatic and gonadal mosaicism; later in embryogenesis or in
postnatal life, mutations are limited to producing somatic mosaicism.
Some unusual clinical manifestations and inheritance patterns have been
observed. Asymmetrical Marfan syndrome affects one side of the body,
and segmental neurofibromatosis 1 affects one segment of the body.
These mutations appear to arise early in embryogenesis, and produce
somatic and gonadal mosaicism, with transmission of the typical disease
to the offspring. Many mutations, however, occur later in embryogenesis
and are limited to somatic cells. Examples include somatic mutations in
McCune-Albright syndrome (i.e., polyostotic fibrous dysplasia,
café-au-lait spots, and endocrinopathies); benign and malignant
neoplasms; as well as in overgrowth disorders including Proteus
syndrome and hemihypertrophy.
spots, sexual precocity, and other dysfunctional endocrinopathies.
Activating missense mutations in the gene for the α-subunit of Gs (the
G protein that stimulates cyclic adenosine monophosphate formation)
have been identified in these patients (38).
The mutations are found in variable abundance in different affected
endocrine and nonendocrine cells, including osteoblast precursors,
consistent with the mosaic distribution of abnormal cells generated by
somatic cell mutation early in embryogenesis. However, because these
mutations are not transmitted to the offspring, the mutations
presumably occur after cells are committed to form gametes.
disorders that probably arise through a similar mechanism of somatic
mosaicism include Proteus syndrome, other hemihypertrophy and local
gigantism syndromes, and Ollier disease.
Using retinoblastoma, which can be associated with osteosarcomas, as a
model, the inherited types can be explained by a germline mutation of
the RB1 gene, followed by a somatic
mutation of the remaining normal allele in a given cell. In the
sporadic form, the two mutations are somatic in origin, affecting both
copies of the normal allele of the RB1
gene in the same cells. A similar mechanism applies to the development
of malignant tumors in individuals with neurofibromatosis 1. However,
more complex arrangements occur, with combinations of somatic mutations
and chromosomal rearrangements. The chromosomal rearrangements in
tumors, such as the t(11;12)(q24;q12) translocation in Ewing sarcoma,
alter the structure or regulation of cellular oncogenes or
tumor-suppressor genes (40). Mutations involving the tumor-suppressor gene P53 are common in many malignant tumors.
dominant diseases, such as osteogenesis imperfecta and Marfan syndrome,
and in X-linked disorders. In affected families, multiple affected
children
can
be shown by genetic testing to be heterozygous for the mutation,
although the parents are clinically normal. Such pedigrees were
previously considered to show autosomal recessive inheritance of the
trait, with the resulting prediction that 25% of offspring would be
homozygous for the mutation and would therefore be clinically affected.
The predicted recurrence risk may be greater, depending on the
proportion of germline cells that contain the mutant gene. If there is
only one affected child, the prediction of recurrence risk is
difficult. If neither parent is mosaic for the mutation, the recurrence
risk is equal to the spontaneous occurrence rate of the disease in that
ethnic group, which is usually low. However, the recurrence rate is
significantly higher if either parent has germline mosaicism. In the
absence of genetic testing of germline cells, the empiric recurrence
risk of autosomal dominant or X-linked disorders for phenotypically
normal parents is approximately 6%. The affected heterozygous children
will transmit the mutation to half of their offspring.
genes are marked, or imprinted, in such a way that they are expressed
differently when they are inherited from the mother than when they are
inherited from the father (37). The process of
imprinting often involves differences in DNA methylation that alter the
transcriptional regulation of the paternally derived and the maternally
derived genes.
diseases, including familial cancers, chromosomal deletion syndromes,
and single-gene disorders such as retinoblastoma, neurofibromatosis 1,
Beckwith-Wiedemann syndrome, Huntington disease, and myotonic
dystrophy. More severe forms of myotonic dystrophy and
neurofibromatosis 1 occur when the mutant gene is inherited from the
mother. More severe forms of Huntington disease and autosomal dominant
spinocerebellar ataxia occur when the mutant gene is inherited from the
father.
syndrome. Hemihypertrophy, Wilms tumors, and other tumors are common in
affected individuals. Cytogenetic duplication of band p15 of chromosome
11 occurs in these patients, and it is paternal in origin (41). There is increased expression of the insulinlike growth factor type 2 gene (IGF2), which maps to this band. The maternal IGF2
allele is normally repressed, but is activated in some maternally
inherited forms of the syndrome. These women carry chromosomal
rearrangements involving chromosome 11 at locus p15, which appear to
activate the IGF2 gene. The syndrome results from increased expression of IGF2 by paternal duplication or maternal activation of the gene.
Angelman syndromes, further highlight the importance of genomic
imprinting and the parental origin of genetic material (37,39).
Prader-Willi syndrome produces hypotonia, obesity with hyperphagia,
hypogonadism, mental retardation, short stature, and small hands and
feet. Angelman syndrome is clinically distinct. Individuals with this
syndrome have a happy disposition, mental retardation, repetitive
ataxic movements, abnormal facies with a large mouth and protruding
tongue, and an unusual type of seizure. Despite their clinical
dissimilarity, some patients with these syndromes share the same
cytogenetic deletion of chromosome 15 (15qll-ql3). In Prader-Willi
syndrome, the deletion is inherited from the father, and in Angelman
syndrome, it is inherited from the mother.
contain two chromosomes of a particular type that have been inherited
from only one parent (37,39).
Isodisomy exists when one chromosome is duplicated, and heterodisomy
exists when both homologs have been inherited from one parent. Examples
include patients with Prader-Willi syndrome lacking cytogenetic
anomalies, in whom both copies of chromosome 15 had been inherited from
the mother. Conversely, some cases of Angelman syndrome lacking
cytogenetic anomalies result from the inheritance of both copies of
chromosome 15 from the father. Some of these individuals carry two
identical copies of the same chromosome 15, and have uniparental
isodisomy, whereas others carry two different copies of chromosome 15
from one parent and have uniparental heterodisomy. These findings
suggest that the lack of the q11-13 region of the paternal chromosome
15 leads to Prader-Willi syndrome, and that the lack of the equivalent
region of the maternal chromosome 15 produces Angelman syndrome. These
observations also indicate that both parental chromosome contributions
serve necessary and complementary functions in normal growth and
development.
with cystic fibrosis who had unexplained short stature at birth. It is
unclear whether there is a higher frequency of uniparental disomy in
patients with intrauterine growth retardation syndromes, such as
Russell-Silver syndrome, which is also associated with limb-length
discrepancy.
For example, a boy with hemophilia A inherited both sex chromosomes
from his father, with no contribution of sex chromosomes from his
mother. Although such events occur rarely, they add to the difficulties
of predicting recurrence risks.
Neural tube defects, congenital talipes equinovarus, and developmental
dislocation of the hip are examples of such disorders. It is likely
that
environmental factors play important roles in the development of the
phenotypes. Little is known about the genetic or environmental factors
involved in the pathogenesis of clubfeet or developmental dislocation
of the hip. However, folic acid intake during pregnancy appears to be
an important nutritional factor in the pathogenesis of neural tube
defects.
There appears to be an underlying continuous variation in
susceptibility to each multifactorial disease that has to exceed a
threshold before the abnormal phenotype appears.
The disorders are familial, but do not show the inheritance patterns
typical of single-gene defect disorders. The risk to first-degree
relatives is approximately the square root of the population risk, but
the risk is much lower for second-degree relatives. For example, the
risk of congenital talipes equinovarus in the general population is
approximately 0.001, but it is 25 times higher in first-degree
relatives, only 5 times higher in second-degree relatives, and only
twice as common in third-degree relatives. If the disorder is more
common in one sex, the recurrence risk is higher for relatives of the
less susceptible sex. The recurrence risk is higher when there is more
than one affected family member, and when the malformation is more
severe. The recurrence risk is also increased when the parents are
consanguineous.
includes information about empiric risk, which is the recurrence risk
observed in similar families. It may not be accurate for a given
family. Progress in defining the genes at fault can be expected to
improve the accuracy of the risk estimates. Preventative measures, such
as taking folic acid during the periconception period, may diminish the
risk of neural tube defects. The pregnancy can also be monitored using
α-fetoprotein levels in maternal serum and amniotic fluid, and by
ultrasonography of the fetus.
emphasis of many research groups is on identifying all the genes that
are involved in the development of diseases of the musculoskeletal
system. Studies of this kind are complementary to equally ambitious
projects that aim to identify all the genes that are involved in the
development, maintenance, repair, and aging of the musculoskeletal
system. Considerable progress is being made toward achieving these
objectives, as is outlined in this and other chapters.
several approaches. In some diseases, candidate genes are selected and
tested for their association with the disease. For example, the type I
collagen genes were the candidate genes in osteogenesis imperfecta
because type I collagen is found in all of the major tissues affected
by the disease. The candidate genes can be directly studied for
mutations in affected individuals. Alternatively, linkage analysis is
used for determining whether genetic markers or polymorphisms, either
within or flanking the candidate gene, are coinherited with the disease
phenotype in families. Mutational analyses can then be undertaken to
determine the genotypes and the genotype/phenotype relations.
a list of candidate genes cannot be prepared. The chromosome, and
region of the chromosome, containing the disease gene may be revealed
by cytogenetic analysis. Translocations may disrupt a gene and can
therefore produce the disease, and a microdeletion may indicate loss of
contiguous genes. Translocations, which are common in many tumors, may
interrupt and inactivate a gene or may result in the fusion of two
genes, which then produce a new fusion protein. The study of contiguous
gene deletion syndromes has enabled researchers to associate these
genes with specific phenotypes. An example is the deletion of the EXT1
gene that produces multiple exostoses in children with the contiguous
gene deletion Langer-Giedion syndrome. The identification of the EXT1
gene led to the identification of other gene family members that reside
in other parts of the genome. At least one of the other EXT genes is also involved in producing multiple and solitary exostoses.
used for identifying a disease gene in humans when no likely candidate
genes can be postulated or where candidate gene screening has not
revealed any anomalies. Large families are usually needed for such
studies, and careful evaluation is needed in classifying individuals as
phenotypically affected or unaffected. Phenotypic ascertainment can be
straightforward, as in classical Ehlers-Danlos syndrome type I, in
which symptoms include skin scars, skin laxity, and generalized joint
instability. The syndrome is fully penetrant, in that all individuals
bearing the mutant allele show the clinical phenotype. The skin and
joints are obviously abnormal at all ages and in both sexes although
the severity of the skin scarring worsens with age. In contrast, it may
be difficult in the case of other genetic disorders to clinically
determine whether asymptomatic individuals bear the mutant allele or
not. This difficulty may be due to low penetrance, variable
expressivity, age, and gender. Such difficulties are likely to account
for the lack of progress in identifying genes for common conditions
such as idiopathic scoliosis and developmental dysplasia of the hip.
families from closed communities. The inheritance of common alleles in
such communities has enabled more rapid progress to be made, often with
relatively small families. For example, the disease genes for autosomal
recessive diastrophic dysplasia and cartilage hair hypoplasia were
identified in Finland where many apparently healthy
individuals carry a single copy of one or both of these disease-gene alleles.
had a major positive impact on the rate of progress, from the
identification of a region of the genome containing a disease gene of
interest to the identification of the disease gene itself and its
mutations. Previously, many years of hard work would commonly follow
the identification of the region of the genome containing the disease
gene. Currently, once a disease-gene locus has been determined, the
information from the Human Genome Project provides the complete DNA
sequence of the region as well as an ordered list of its known and
tentative genes. Additional databases provide information about the
expression patterns of the genes in the linked region. Consequently, it
is possible to select genes that are known to be expressed in the
tissue or tissues of interest. In this way, even large linked regions
containing several hundred genes can often be reduced to a small number
of candidate genes. “DNA chips” that contain arrays of short DNA
sequences from all human genes can also be used to narrow the list of
candidate genes that are expressed in the tissues of interest.
identification of disease genes from among a list of candidate genes.
Previously, laborious mutation detection techniques were used in order
to localize mutations for DNA sequencing. Although such technology has
improved considerably, it is often quicker to proceed directly to DNA
sequencing as the definitive means of identifying the disease gene.
numerous normal DNA sequence variants and variants producing pathologic
changes. Premature termination codons, splicing mutations, and other
major rearrangements are often pathological. However, missense
mutations that alter a single amino acid often require functional
studies to demonstrate that the observed change was responsible for the
disease.
successful identification of disease genes in humans. There are many
examples of mouse models of human diseases and even fruit fly models of
human diseases. Some of these models were the result of spontaneous
mutations, whereas others were produced by targeted mutations or
inactivation of genes of interest. Examples include the identification
of RUNX2 as the disease gene for cleidocranial dysplasia and LMX1B
as the gene for the nail-patella syndrome. A very exciting development
is the use of random mutagenesis techniques to generate musculoskeletal
phenotypes in mice. For example, random mutagenesis is being used to
gain new insights into the genetics of osteoporosis and skeletal
development. The mutagenized mice are screened for abnormal bone
density and for anomalies of skeletal development. Genetic analyses are
undertaken to identify the genes and the mutations giving rise to the
phenotype of interest. The mutagenesis techniques randomly introduce a
single-nucleotide point mutation into a single gene of each mouse. The
mice are bred to yield dominant and recessive phenotypes.
establishing whether a putative missense mutation identified in humans
is a cause of a given phenotype. Such studies were, until recently,
undertaken by random insertion of mutated complementary DNA (transgene)
into the mouse genome. Valuable phenotypic information was often
obtained from these transgenic mice. However, the phenotypes often
varied widely between different transgenic mouse lines because of
differences in the integration sites of the transgene, and in the
number of copies of the transgene that were integrated into the mouse
genome. In addition, the expression of the transgene was usually
regulated by promotors other than the normal endogenous promotor for
the gene. Most of these shortcomings have been overcome by homologous
recombination technology in which the normal gene sequence is exchanged
for the mutant DNA sequence. Another major advance involves the
restricted expression of the mutated gene in specific tissues such as
cartilage or bone. Such studies are often resorted to when normal,
unrestricted expression of the mutation yields a lethal phenotype.
identifying the genes that cause many of the single-gene disorders of
the musculoskeletal system, little progress has been made in
identifying the variables that modify their clinical severity. For
example, in a family with the mild form of osteogenesis imperfecta
there are likely to be individuals with grey-blue sclera and no
fractures, as well as other individuals with grey-blue sclera and
numerous fractures. Although the type I collagen gene mutation shared
by affected individuals is the major variable causing the phenotype,
the severity of the phenotype is likely to depend upon other genes as
well as on environmental factors. Little information is currently
available concerning the network of interacting genes and environmental
factors that determines the severity of the phenotype of skeletal
disorders that are usually considered to be single-gene disorders.
Complex studies in mouse models will be needed in order to delineate
the genetic and environmental variables.
genes involved in multi-gene or multifactorial disorders of the
musculoskeletal system, for example, in degenerative arthritis,
intervertebral disk disease, and osteoporosis. The latter studies
provide new insights into the etiologies of these disorders and show
that some of the multifactorial disorders are part of larger disease
families. For example, some mutations of the type IX collagen genes
cause multiple epiphyseal dysplasia, whereas other sequence variants
predispose adults to the development of degenerative intervertebral
disk disease. It is likely that close links will be established between
many of the rare single-gene disorders and the common multi-gene
disorders of the musculoskeletal system.
During blastocyst formation, teratogens usually result in fetal death
and spontaneous abortion. During the period of organogenesis, 18 to 60
days after conception, the fetus is most vulnerable to the effects of
teratogens. Easily recognizable structural defects are the usual
result. Later in pregnancy, teratogens may produce only subtle changes
or no anomaly at all.
processes. These teratogens may act on cell membranes or on the
metabolic machinery of cells. The final common pathway of these various
levels of action is cell death or a failure of replication, migration,
or fusion of cells. These changes often involve specific organs, but
can produce more general changes in the fetus.
does not appear to play a significant role in the development of birth
defects. Most agents that interfere with the DNA of sperm produce
sterility rather than teratogenic effects in the fetus.
inadequate. Interspecies differences in sensitivity are common. For
example, thalidomide is teratogenic in rabbits, but not in rats and
mice. Many agents known to be teratogenic in animals, such as
glucocorticoids in rats, do not produce any detectable anomalies in
humans.
identified from clinical observations of unexpected outbreaks of
malformations. In most instances, however, unexpected clusters of cases
result from natural fluctuations in the frequency of specific birth
defects, as shown by birth defect registers. Epidemiologists who deal
with birth defect registers play an important role in assessing whether
apparent outbreaks of malformations are potentially significant.
-
androgens
-
aminopterin
-
chlorobiphenyls
-
warfarin (Coumadin)
-
cyclophosphamide
-
diethylstilbestrol
-
D-penicillamine
-
goitrogens and antithyroid drugs
-
isoretinoin
-
methyl mercury
-
phenytoin
-
tetracyclines
-
thalidomide
-
valproic acid
-
infections
-
cytomegalovirus
-
rubella
-
syphilis
-
toxoplasmosis
-
maternal metabolic imbalance
-
alcoholism
-
diabetes mellitus
-
phenylketonuria
-
virilizing tumors
-
ionizing radiation
effects of drugs and environmental factors on the developing fetus.
However, relatively few agents have proven to be teratogenic.
in Australia reported an increased frequency of limb-deficient babies
born to mothers who used thalidomide as a sedative during pregnancy. In
clinical studies, the agent was shown to produce its major effects
during the period of limb formation.
warfarin between 6 and 9 weeks of gestation. Stippling of the epiphyses
is one of the characteristic changes. Exposure during the second and
third trimesters produces severe neural anomalies.
cystic acne. Women who receive retinoic acid are often of childbearing
age, and their fetuses are at risk from the potent teratogenic effects
of this agent. It produces craniofacial, cardiac, thymic, and central
nervous system defects. Megadoses of vitamin A are also teratogenic.
Vitamin A, retinoic acid, and its analogs should be avoided during
pregnancy. If women of childbearing age use these agents, pregnancies
should be avoided by the use of contraception.
The regular intake of two alcoholic drinks each day during pregnancy
results in a slightly reduced birth weight for the baby. Chronic intake
of 8 to 10 drinks each day by women during pregnancy is likely to cause
low birth weights, craniofacial anomalies, mental retardation,
incoordination, short stature, and increased propensity for congenital
heart disease in their babies. A gradient of severity of these effects
is seen with intermediate levels of alcohol intake. Alcohol should be
avoided by the mother during pregnancy.
radiographs and isotopes. Doses in excess of 1 Gy should be avoided,
and doses in excess of 10 Gy will cause microcephaly, growth
retardation, and mental retardation. A woman of childbearing age should
not be exposed to unnecessary radiation if she is pregnant or if there
is a possibility that she is pregnant.
deleterious effects on the fetus can be prevented by routine testing of
pregnant women and by providing treatment when necessary. The virus
that causes acquired immunodeficiency syndrome (AIDS) has emerged as a
major teratogen. Rubella embryopathy is preventable by vaccination of
young girls. When the fetus is exposed to the virus in the first
trimester, blindness, deafness, cataracts, microphthalmos, congenital
heart disease, limb deficiencies, and mental retardation may occur.
Cytomegalovirus infection and toxoplasmosis also produce birth defects.
mothers with diabetes, particularly if their diabetes is poorly
controlled in the first trimester of the pregnancy. For example,
cardiac malformations occur three to four times more often in babies of
mothers with diabetes than of healthy mothers, and anencephaly and
myelomeningocele occur in 1% to 10% of babies born to mothers with
diabetes. Caudal regression syndrome, with sacral hypoplasia and fusion
of the legs, is a rare disorder, but it is more common in babies of
mothers with diabetes.
information for an individual or a couple to make an informed decision
about future pregnancies and to assist them in coming to terms with the
issues they face (47).
child being born with a birth defect, formal genetic counseling is
called for. Appropriate genetic counseling requires diagnostic
precision and knowledge of the recurrence risk, the burden of the
disorder, and the reproductive options. There are several indications
for genetic counseling (31,47):
-
The couple has had a stillbirth or multiple miscarriages.
-
The couple already has a child with a birth defect.
-
The couple already has a child with mental retardation.
-
There is a family history of any of the problems mentioned in preceding text.
-
The couple has relatives with known genetically transmittable diseases, such as muscular dystrophy.
-
The mother has been exposed to radiation, drugs, or infections during pregnancy.
-
The mother is of an advanced age.
-
The parents are consanguineously related.
-
There are chromosomal translocations.
the correct diagnosis. A precise diagnosis cannot be made for about
half of children who present with mental retardation or dysmorphic
features. However, there is a large amount of empiric data that can be
used for counseling in this group.
recurrence risk is made. This is a numeric estimate of the likelihood
of a particular disorder occurring in subsequent children, such as a 1
in 4 risk of an autosomal recessive disorder and a 1 in 2 risk of an
autosomal dominant disorder. The recurrence risk for multifactorial
disorders, after a single affected child, is approximately 3% to 5%.
need a careful discussion to give meaning to any risk estimate. For
example, a 1 in 4 risk applies to each pregnancy, but many families
believe that they can have three more children without worry, if they
already have one abnormal child.
for major birth defects. Approximately 1 in 25 children is born with a
major defect. In this setting, risks of 1 in 2 and 1 in 4 are high, and
risks of 1 in 100 are low.
counseling. Clinodactyly is a common autosomal dominant condition with
a high recurrence risk of 1 in 2, although it has minimal or no burden
to those who have the condition. Clubfeet and congenital dislocation of
the hip are multifactorial diseases with lower risks of recurrence. The
potential burden of these conditions is minimized by early diagnosis
and treatment. In contrast, the burden of additional children with
Duchenne muscular dystrophy, severe osteogenesis imperfecta, or severe
chondrodysplasia is considerable because there are no curative
treatments available.
family’s concept of burden. Some families are prepared to accept a 1 in
4 risk of a perinatally lethal disorder, knowing that the child will
die at birth or soon afterward, or be normal. Other families may not be
willing to accept the burden of recurrent deformities such as clubfeet,
despite the lower risk and the availability of treatment.
metabolism, such as phenylketonuria and hypothyroidism, have been
highly successful. The severe consequences of these diseases have been
prevented by early diagnosis and treatment.
offered to an increasing number of families as the number of diseases
that can be detected in early pregnancy increases. The most common
indication is a maternal age of 35 years or older. The indications for
prenatal diagnosis are shown in the following list:
-
The mother is of advanced age (older than 35 years).
-
There is a known chromosomal anomaly in one parent or in a previous pregnancy.
-
There has previously been a neural tube
defect, or there is a high serum level of α-fetoprotein or a neural
tube defect is suspected from ultrasound results. -
There is a family history of disorders
detectable by biochemical or DNA technology; these include Duchenne and
Becker muscular dystrophy, myotonic dystrophy, hemoglobinopathies,
hemophilia A or B, Huntington disease, cystic fibrosis, and other rare
detectable genetic diseases.
abnormality, thereby providing reassurance to the parents. Because of
the availability of prenatal diagnosis, more families are willing to
have children, instead of refraining from having them for fear of birth
defects.
α-fetoprotein reaches a peak in fetal serum at approximately 13 weeks
of gestation and decreases thereafter. Amniotic levels of this protein
are high in the fetus with a lesion that is not covered by skin, such
as open spina bifida, anencephaly, and exomphalos. The protein leaks
into the amniotic fluid and into the maternal circulation. An increased
maternal serum level of α-fetoprotein is not diagnostic of open spina
bifida but is an indication for further investigation. Abnormally high
levels also occur in cases of fetal death, cystic hygroma, polycystic
kidneys, and Turner syndrome.
fetus and fetal movements. Ultrasonography is commonly undertaken to
determine gestational age. However, more extensive studies by
experienced ultrasonographers are required when examining for fetal
abnormalities in at-risk pregnancies. Such examinations are
increasingly being undertaken as screening investigations in all
pregnancies.
procedure when undertaken at 16 weeks of gestation. The risk of fetal
loss is approximately 0.5% to 1%. The amniotic fluid is most often used
for determination of α-fetoprotein levels. The amniotic cells are used
for karyotype analysis for the determination of enzyme levels in cases
of inborn errors of metabolism and for DNA diagnosis using direct
detection of a previously defined mutation, or indirect detection using
polymorphisms. Chorionic villus sampling can be undertaken between 9
and 11 weeks of gestation; it allows earlier diagnosis of many genetic
diseases and offers the option of first-trimester termination of
pregnancy. The risk of fetal loss because of the procedure itself is
approximately 4%.
should seek counseling before the mother becomes pregnant. This
approach ensures that there is sufficient time to establish the
diagnosis, recurrence risk, burden of the disorder, reproductive
alternatives, and suitability for prenatal diagnosis. These options may
be limited when counseling is sought during the pregnancy. Parents must
be fully informed about the risks of investigational procedures and
anticipated delays in receiving test results, and they must be given
all test results and appropriate explanations about the implications of
those results. Although genetic testing may reveal that the fetus bears
a disease mutation, it is difficult to predict the severity of the
phenotype. Parents require much support at this difficult time, and
they alone are responsible for the decision about whether to terminate
a pregnancy. Parents may not wish to terminate a pregnancy but may use
the prenatal diagnostic results to plan the most appropriate method of
delivery of the baby. For example, prenatal diagnosis of mild
osteogenesis imperfecta (type IA) may be sought in order to determine
whether to use vaginal or cesarian delivery. If the fetus is affected,
the parents may choose cesarian delivery in order to reduce the
likelihood of a birth fracture from a vaginal delivery.
hypothyroidism has effectively prevented mental retardation and other
consequences of these diseases. However, these instances are the
exceptions because treatment is not available for most genetic
diseases, and, when available, it is relatively ineffective. Table 6.1 lists the various levels at which intervention is possible.
manipulations and protein replacement, occur beyond the level of the
gene. However, bone marrow transplantation has been used successfully
to cure or ameliorate diseases such as congenital immune deficiencies,
infantile malignant osteopetrosis, thalassemia, lysosomal storage
diseases, infantile agranulocytosis, and chronic granulomatous disease.
The
normal
genes of the transplanted cells produce the protein, usually an enzyme,
which corrects the metabolic defect. The stem cells within the donor
marrow continue to replicate, providing a continuing source of the
normal protein.
|
TABLE 6.1 LEVELS OF TREATMENT OF GENETIC DISEASES
|
||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||||||||
that specific subsets of stem cells within the bone marrow, peripheral
blood, or other tissues will be used to target specific organs. For
example, stem cells that can develop into osteoblasts, fibroblasts, or
chondrocytes may be used as a form of cell therapy to correct genetic
defects of the appropriate connective tissues. Growth factors will be
needed to direct the cell differentiation pathways. An alternative
approach to cell therapy is to use embryonic stem cells, which are
pluripotential. Despite the great potential of this treatment, many
ethical and practical issues need to be resolved before embryonic stem
cells are used in clinical practice.
become available for many genetic diseases. Gene transfer is used for
replacing a gene that is nonfunctional, such as the DMD
gene in boys with Duchenne muscular dystrophy. In the case of other
diseases, the adverse effects of the mutant allele are blocked by
specific gene therapy directed to the mutant sequence. Therapy of the
latter kind is most applicable to diseases in which many affected
individuals share the same mutation. Designing gene therapy is more
difficult for diseases in which most individuals have their own
mutations, as occurs in osteogenesis imperfecta.
effective somatic gene therapy. One of the challenges is to target the
therapy to specific cells. Transplantation of modified autologous bone
marrow cells is likely to be suitable for hematologic diseases and for
some bone diseases. Specific cell surface receptor–mediated gene
transfer is likely to be more suitable for other diseases, as in
transferring a replacement gene into the skeletal and cardiac muscle
cells of young children with Duchenne muscular dystrophy.
patients with autosomal dominant disorders that give rise to haploid
insufficiency. In such patients, the protein produced is qualitatively
normal, but is reduced in amount because only the normal allele is
functional. In this situation, the amount of normal protein produced
from the normal allele can be increased by specific pharmacologic
modulation of gene expression. Therapy of this kind is potentially
curative for half of all patients with osteogenesis imperfecta and
Marfan syndrome.
JB, Fenwick SK, Fu TH, et al. Relationship between parental
trinucleotide GCT repeat length and severity of myotonic dystrophy in
offspring. JAMA 1993;269:1960.
HC, Cutting GR, Pyeritz RE, et al. Marfan syndrome caused by a
recurrent de novo missense mutation in the fibrillin1 gene. Nature 1991;352:337.
R, Thompson LM, Zhu Y-Z, et al. Mutations in the transmembrane domain
of FGFR3 cause the most common genetic form of dwarfism,
achondroplasia. Cell 1994;78:335.
CA, Ortiz de Luna RI, Hefferon TW, et al. Localization of the
achondroplasia gene to the distal 2.5 Mb of human chromosome 4p. Hum Mol Genet 1994;3:787.
A, Eich G, Bucher HU, et al. A glycine 375- to cysteine substitution in
the transmembrane domain of the fibroblast growth factor receptor-3 in
a newborn with achondroplasia. Eur J Pediatr 1995;154:215.
E, Marchuk DA, Collins FS, et al. Somatic deletion of the
neurofibromatosis type 1 gene in a neurofibrosarcoma supports a tumour
suppressor gene hypothesis. Nat Genet 1993;3:122.
M, Nagamine M, Ohtoshi A, et al. Suppression of oncogenic Ras by mutant
neurofibromatosis type 1 genes with single amino acid substitutions. Proc Natl Acad Sci U S A 1993;90:6706.
KM, O’Connell P, Martin GA, et al. Loss of the normal NF1 allele from
the bone marrow of children with type 1 neurofibromatosis and malignant
myeloid disorders. N Engl J Med 1994;330:597.
EF, Chamberlain JS, Murphy EG, et al. Molecular and phenotypic analysis
of patients with deletions within the deletion-rich region of the
Duchenne muscular dystrophy (DMD) gene. Am J Hum Genet 1989;45:507.
Y, Kuhl DP, Pizzuti A, et al. Variation of the CGG repeat at the
fragile X site results in genetic instability: resolution of the
Sherman paradox. Cell 1991;67:1047.
A, Weinstein LS, Sweet DE, et al. An activating Gs α mutation is
present in fibrous dysplasia of bone in the McCune-Albright syndrome. J Clin Endocrinol Metab 1994;79:750.
O, Zucman J, Melot T, et al. The Ewing family of tumors: a subgroup of
small round-cell tumors defined by specific chimeric transcripts. N Engl J Med 1994;331:294.
RL. Environmental causes of human congenital malformations: the
pediatrician’s role in dealing with these complex clinical problems
caused by a multiplicity of environmental and genetic factors. Pediatrics 2004;113(Suppl. 4):957.