
Editors: Tornetta, Paul; Einhorn, Thomas A.; Damron, Timothy A.
Title: Oncology and Basic Science, 7th Edition
Copyright ©2008 Lippincott Williams & Wilkins
> Table of Contents > Section IV – Basic Science > 15 – Genetic Basis of Musculoskeletal Disorders
15
Genetic Basis of Musculoskeletal Disorders
Sathappan S. Sathappan
Kirill Ilalov
Paul E. Di Cesare
The orthopaedic genome
consists of all genes involved in the development and functioning of
the musculoskeletal system. The Human Genome Project (1990–2003) has
facilitated an in-depth understanding of the orthopaedic genome as well
as the molecular biology of musculoskeletal conditions. Current and
future molecular interventional modalities are expected to represent a
paradigm shift in the management of orthopaedic disorders.
consists of all genes involved in the development and functioning of
the musculoskeletal system. The Human Genome Project (1990–2003) has
facilitated an in-depth understanding of the orthopaedic genome as well
as the molecular biology of musculoskeletal conditions. Current and
future molecular interventional modalities are expected to represent a
paradigm shift in the management of orthopaedic disorders.
Pathogenesis
Etiology
-
Human DNA consists of 46 chromosomes (22
pairs of autosomes and 1 pair of sex chromosomes), comprising
approximately 30,000 genes. -
Each gene represents a chain of nucleotides (Fig. 15-1) characterized by:
-
Promoter and other regulatory elements that control gene expression
-
A coding region, consisting of introns
and exons, that defines the unique order of amino acids and hence
determines protein form and function -
Gene expression: the transfer of genetic
information (DNA) to messenger RNA by the process of transcription,
followed by the translation of mRNA into a specific protein (Table 15-1 gives definitions of terms used in this chapter)
-
-
Genetic abnormalities
-
Can involve individual chromosomal
aberrations, such as deletions (as in Turner syndrome), additions (as
in Down syndrome), translocations (where portions of two chromosomes
are exchanged, as seen in the 11:22 translocation associated with Ewing
sarcoma), and nucleotide repeat expansions (as in trinucleotide
disorders) -
Can affect autosomes and sex chromosomes
-
Inheritance may be autosomal dominant or recessive.
-
X-linked mode of inheritance typically is
recessive (e.g., Duchenne’s muscular dystrophy, hemophilia) but rarely
may be dominant (e.g., X-linked hypophosphatemic rickets). -
Many diseases exhibit more than one pattern, depending on the mutation.
-
Can be inherited or spontaneous (new mutations)
-
Can follow a simple or complex pattern of inheritance
-
Single-gene disorders follow Mendelian genetics.
-
Polygenic disorders result from interaction of multiple genes (e.g., congenital hip dislocation
P.348
or neural tube defects, such as myelomeningocele).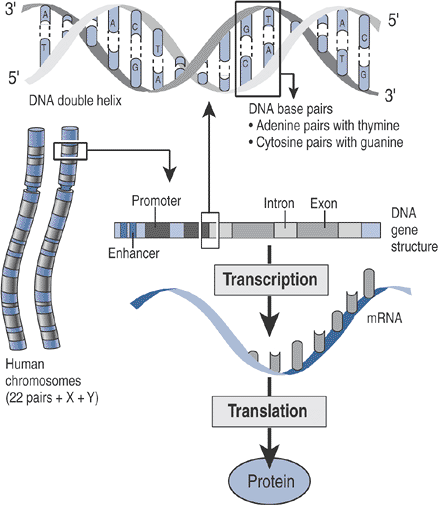 Figure 15-1 Outline of the human genome.
Figure 15-1 Outline of the human genome. -
Mosaicism refers to an expression of two cell lines in one individual (e.g., X chromosome inactivation in female).
-
-
May have variable penetrance (percentage
of individuals with the mutation expressing the phenotype) and
expressivity (degree to which the individuals with the mutation express
the phenotype)
-
-
The underlying basis for various musculoskeletal disorders has been attributed to disorders in genotype.
-
The disease manifestations of abnormalities in the orthopaedic genome (Fig. 15-2) can be broadly classified as:
-
Skeletal dysplasias
-
Connective tissue disorders
-
Neuromuscular disorders
-
Trinucleotide disorders
-
Other genetic disorders
-
Epidemiology
The incidence of many musculoskeletal conditions is
increasing due to longer life expectancies. Estimating incidence rates
for inheritable musculoskeletal diseases is difficult, since new
detection methods, changing classification schemes, and more complete
understanding of the disease mechanisms constantly shape our perception
of these disorders.
increasing due to longer life expectancies. Estimating incidence rates
for inheritable musculoskeletal diseases is difficult, since new
detection methods, changing classification schemes, and more complete
understanding of the disease mechanisms constantly shape our perception
of these disorders.
Diagnosis (Algorithm 15-1)
Physical Examination
-
Age at presentation
-
Most genetic diseases present early.
-
Autoimmune disorders with genetic contribution may present in adulthood only.
-
-
Presence of characteristic facial features (e.g., Down syndrome and achondroplasia)
-
Stature: lesser- or greater-than-normal arm spanto—height ratio
-
Skin lesions (e.g., cafe-au-lait spots)
-
Neurologic assessment: weakness in myotomes can be either neural or muscle related
-
Limb configuration and range of motion of joints (e.g., joint contractures)
Laboratory Tests
-
Hematology (e.g., erythrocyte sedimentation rate elevated in rheumatoid arthritis)
-
Biochemistry (e.g., creatine phosphokinase [CPK] in Duchenne’s muscular dystrophy)
-
Molecular diagnostic tests
-
Gene product analysis using immunoblotting techniques
-
DNA mutation analysis with polymerase chain reaction (PCR)
-
Chromosomal studies
-
Indications include multiple orthopaedic
abnormalities, two or more siblings with the same condition, multiple
miscarriages in the mother, multiple organ system abnormalities, mental
retardation.
-
-
Treatment
-
Varies with specific disease
-
Supportive
-
Orthotics to address joint and spinal
deformities (e.g., ankle-foot orthosis [AFO] in Charcot-Marie-Tooth
disease associated with peroneal muscle weakness) -
Physical therapy to condition and maintain tone of functioning musculature (e.g., in spinal muscular atrophy)
-
-
Medical treatment
-
Medications to suppress or curb disease activity (e.g., anti-inflammatory agents and steroids in autoimmune disorders)
-
-
Surgical procedures
-
Fusion procedures
-
Spinal instability and scoliosis (e.g., for severe atlantoaxial instability in Down syndrome)
-
-
Realignment osteotomies
-
Sofield osteotomies in osteogenesis imperfecta
-
-
Lengthening procedures have been considered for short stature in selective cases of skeletal dysplasia.
P.349P.350Table 15-1 Definitions of Terms Associated With Genetic DisordersTerm Definition Alleles Alternate forms of a gene found at the same locus Anticipation Phenomenon that refers to a
progressive increase in the number of nucleotide triplets down the
successive familial generation with worsening in disease presentationArthrogryposis Congenital nonprogressive limitation of joint movement attributable to soft tissue contractures that involve at least two joints Autoimmune disorders A group of clinical conditions
that have a predilection for accumulation of selective antibodies, the
detection of which facilitates clinical diagnosis and categorizationAutosomal dominant Condition that occurs when a
mutation of only one of the alleles is enough to cause the disease, as
in achondroplasia, Marfan syndrome, and neurofibromatosisAutosomal recessive Condition that requires mutations be present in both alleles of a gene, as in spinal muscular atrophy Cartilage oligomeric matrix protein A member of the thrombospondin family of proteins; expressed in high levels in the chondrocyte matrix Charcot-Marie-Tooth disease Type 1 or 2 hereditary sensory
and motor neuropathy; an autosomal-dominant disease that presents with
motor and sensory demyelinating neuropathyCoding region (of a gene) Consists of introns and exons; defines the unique order of amino acids and thus determines protein production Dwarfism Pathological decrease in
stature; can be divided as proportionate (midget) versus
disproportionate (short limb or trunk); the short-limb type can be
defined by location of maximal shortening as rhizomelic (proximal
portion of limb), mesomelic (middle portion of limb), or acromelic
(distal portion)Dysplasia Intrinsic skeletal developmental abnormality Ehlers-Danlos syndrome A group of the most common
inheritable connective tissue disorders, consisting of six major
subtypes; all have features of skin and joint hypermobility.Fibroblast growth factors A group of polypeptide growth factors involved in chondrocyte development and wound healing Gene A chain of nucleotides that contain a promoter and a coding region Hereditary sensory and motor neuropathy A group of disorders
characterized by a slow neural conduction, with muscle biopsy revealing
uniformly small-diameter fibers and nerve biopsy revealing demyelinationMendelian disorders Disorders involving a single gene Mosaicism Expression of two cell lines in the same individual Muscular dystrophies A group of inherited, noninflammatory disorders that cause progressive muscular weakness Neuromuscular disorders A group of conditions that may
have genetic anomalies leading to abnormalities in signal transmission
in one of three regions: neural junction, neuromuscular junction, or
muscleOrthopaedic genome The genes involved in the genesis and functioning of the musculoskeletal system Osteogenesis imperfecta A disorder associated with mutation in type I collagen, primarily characterized by bone fragility and long bone deformities Pleiotropic A single genotype producing multiple phenotypic effects Promoter region The region of the DNA that regulates gene expression Rheumatoid arthritis An autoimmune disorder characterized by symmetrical inflammatory arthropathy with varied extra-articular manifestation Scleroderma A chronic disease of unknown etiology, characterized by skin fibrosis Seronegative spondyloarthropathies A group of conditions that are
negative for rheumatoid factor and are associated with inflammatory
arthritis and various typical extra-articular manifestationsSkeletal dysplasias A spectrum of disorders that
are caused by abnormalities in bone and cartilage metabolism and
phenotypically identified by short statureSpinal muscular atrophy An autosomal-recessive disease
characterized by loss of anterior horn cells in the spinal cord and
presenting with progressive weaknessTranscription A process of transferring genetic information found in DNA to mRNA Transduction The insertion of genetic material into a host cell by use of a viral vector, most commonly an adenovirus Translation The process of converting RNA to protein Trinucleotide disorders A group of conditions associated with an increased number of nucleotide triplets in DNA -
-
Gene therapy
-
Gene therapy is a potential therapeutic
option when a single functional gene product is absent and treatment is
affected by transfer of a wild-type gene; possible orthopaedic examples
are Duchenne’s muscular dystrophy, osteogenesis imperfecta, familial
osteoarthritis. -
Gene transfer into cell lines requires the aid of vectors, which can either be viral (recombinant viruses) or nonviral.
-
Key issues concerning gene therapy use in orthopaedics
-
Orthopaedic genetic diseases have an early onset, which necessitates administration of gene therapy at a very early age.
-
In heterozygous individuals with
selective genetic conditions, half of the genes are abnormal and can
behave as mutations producing inhibitory protein products that
interfere with normal gene products. A successful gene therapy outcome
therefore requires eliminating this endogenous mutant allele. -
Longevity of gene expression
-
Economics of gene therapy
-
-
Current status of orthopaedic applications for gene therapy
-
Genetic diseases
-
Osteogenesis imperfecta
-
Trials ongoing with implantation of precursors of normal bone (stem cells) into osteogenesis imperfecta mice models
-
-
Duchenne muscular dystrophy
-
In animal models, dystrophin gene delivery success has been hampered by immu-nogenicity and rejection issues.
-
-
-
Nongenetic diseases
-
Arthritis was the first orthopaedic condition to be targeted by gene therapy.
-
Involves delivery of antiarthritic genes into synovial linings of diseased joints (rheumatoid arthritis, osteoarthritis)
-
Phase I trials in human have suggested
efficacy in transfer of genes (interleukin-1 receptor antagonists) in
patients with rheumatoid arthritis.
-
-
Current challenge is to sustain longevity of gene expression.
-
-
Osteoporosis
-
Osteoporotic mice models injected with
vector carrying transgenes to block interleukin-1, an osteoclastic
cytokine, showed decreased bone loss and in some cases bone mass was
restored to normal.
-
-
Tissue repair
-
Gene expression is required for a limited time period until healing is complete.
-
Bone healing can be enhanced in animal models using an adenovirus that carries the BMP-2 gene.
-
In experimental models, cartilage defects
have been treated using “gene plugs”—bone marrow aspirates combined
with a vector carrying a localized “healing” transgene that allows the
stem cells to differentiate into chondrocytes and thus repair the
cartilage defect.
-
-
-
Skeletal Dysplasias
Definition
Skeletal dysplasias, which are mostly heritable
disorders, are caused by abnormalities in bone and cartilage
development and phenotypically characterized by a variably short
stature.
disorders, are caused by abnormalities in bone and cartilage
development and phenotypically characterized by a variably short
stature.
Epidemiology
The incidence of skeletal dysplasias is 1:3,000 to 5,000 births depending on disease subtype and geographic area.
P.351
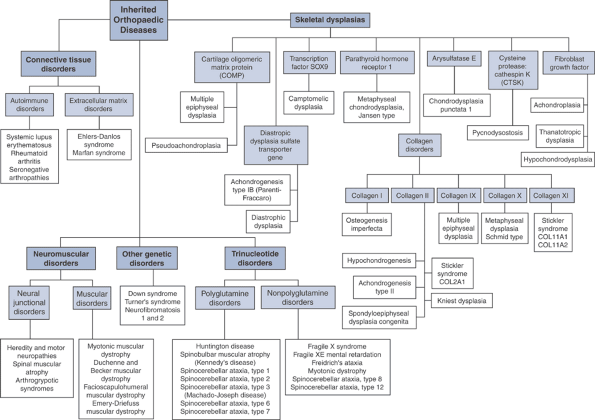 |
|
Figure 15-2 Flowchart summary of genetic basis for musculoskeletal disorders.
|
P.352
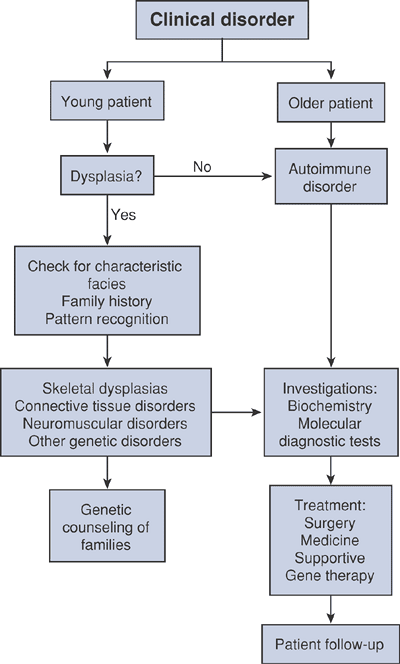 |
|
Algorithm 15-1 Diagnostic work-up of musculoskeletal disorders.
|
Classification
The clinical classification of dysplasias is based on
whether the dwarfism is proportionate or disproportionate, and, if
disproportionate, whether it is predominantly short-trunk or
short-limb. It is also possible to define these disorders in terms of
their genetic basis (Fig. 15-3), as in the section that follows.
whether the dwarfism is proportionate or disproportionate, and, if
disproportionate, whether it is predominantly short-trunk or
short-limb. It is also possible to define these disorders in terms of
their genetic basis (Fig. 15-3), as in the section that follows.
Skeletal Dysplasias by Genetic Basis
Skeletal Dysplasias Due to Mutations in Fibroblast Growth Factor Receptor Genes
-
Fibroblast growth factors (FGF) are a
subtype of polypeptides that control cell differentiation and
proliferation and are critical in chondrocyte development and wound
healing. Mutation in FGF receptor 3 genes leads to:-
Abnormal chondrocyte maturation
-
Impeded endochondral bone growth with
rhizomelic shortening (femur and humerus are the most affected as they
are the bones with the largest amount of endochondral growth)
-
-
Three main disease subtypes
-
Achondroplasia
-
Hypochondroplasia
-
Thanatrophic dysplasia
-
Achondroplasia
Achondroplasia, an autosomal-dominant disorder, is the
most common type of short-limb, disproportionate dwarfism and occurs
due to a mutation of guanine to adenine in FGF receptor type 3 (FGFR3)
on chromosome 4. Although endochondral ossification is affected,
intramembranous ossification is essentially spared. The genetic defect
affects the proliferative zone of the physis. The disorder is
associated with increased paternal age.
most common type of short-limb, disproportionate dwarfism and occurs
due to a mutation of guanine to adenine in FGF receptor type 3 (FGFR3)
on chromosome 4. Although endochondral ossification is affected,
intramembranous ossification is essentially spared. The genetic defect
affects the proliferative zone of the physis. The disorder is
associated with increased paternal age.
Clinical Features
-
Frontal bossing, low nasal bridge, midface hypoplasia
-
Rhizomelic shortening of the extremities (humerus and femur) (Fig. 15-4)
-
Trident hand: increased space between the middle and ring finger and inability to approximate them
-
Thoracolumbar or lumbar kyphosis in infancy that usually resolves with the onset of ambulation
-
Infants are often hypotonic at birth:
this can be due to foramen magnum and upper cervical stenosis, which
can present with apnea and lead to hydrocephalus. When this is
suspected, magnetic resonance imaging (MRI) is indicated. -
Radiographs in older children and adults
often reveal increased lumbar lordosis with short pedicles and a
decreased interpedicular distance, know as “champagne glass” pelvis
(width > depth). -
The most serious clinical problem is lumbar spinal stenosis, which can be evaluated with computed tomography (CT) or MRI.
Treatment
-
Fusion for significant cervical instability and decompression for spinal stenosis
-
Osteotomies for genu varum
-
Limb-lengthening procedures are a controversial treatment option.
Hypochondroplasia
-
An autosomal-dominant disorder that results from a cytosine-to-adenine substitution in FGFR3 gene on chromosome 4
-
The condition, in which mild achondroplasia-type features become apparent at age 2 to 3 years, is characterized by:
-
Rhizomelic shortening of extremities
-
Facial features less distinctive than in achondroplasia patients
-
Lack of trident hand seen in achondroplasia
-
Neurologic complications such as lumbar
stenosis can occur in individuals with hypochondroplasia, but they
occur less frequently than in patients with achondroplasia.
-
P.353
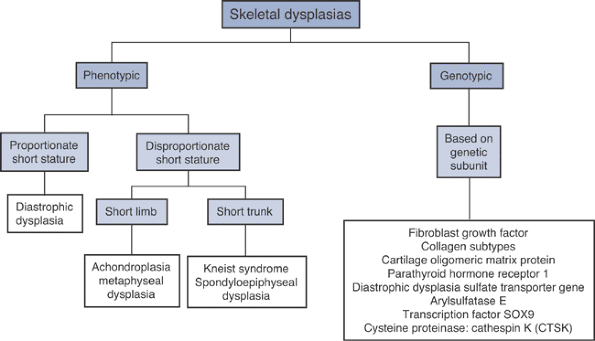 |
|
Figure 15-3 Classification of skeletal dysplasias.
|
 |
|
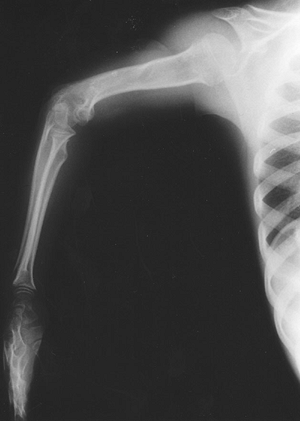
Figure 15-4
Radiograph of 10-year-old girl with achondroplasia. Note features of rhizomelic shortening of the upper extremity. (From Baitner AC, Maurer SG, Gruen MB, et al. The genetic basis of the osteochondrodysplasias. J Pediatr Orthop 2000;20:594–605.) |
Thanatophoric Dysplasia
-
Lethal bone dysplasia
-
New mutations in FGFR3 gene result in new
cysteine residues, which trigger the formation of abnormal disulfide
bonds between the mutant FGFR receptors. -
FGFR receptor complexes then inhibit endochondral ossification.
-
Clinical Features
-
Rhizomelic dwarf with large head and normal trunk
-
Generalized platyspondyly (flattening of the vertebral bodies)
-
Extreme rib shortening
Skeletal Dysplasias due to Mutations in Collagen Genes
Mutations in Type I Collagen: Osteogenesis Imperfecta
Etiology
-
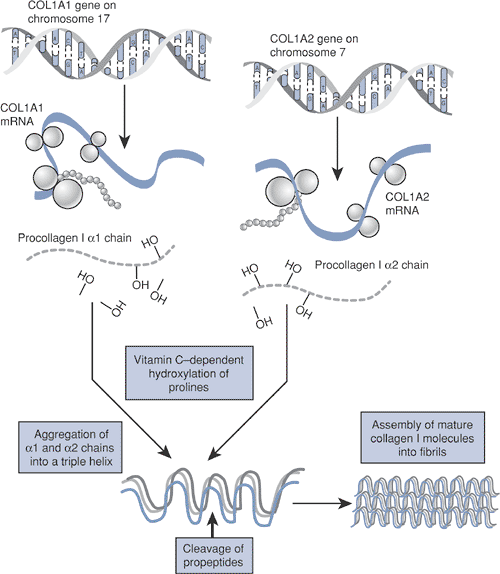
Type I collagen (Fig. 15-5) is a major structural protein in bone, skin, and tendons.
-
A heterotrimer of two identical a-1 chains and one α-2 chain folded into a triple helix configuration
-
The α-1 and α-2 chains consist of amino acid repeats (Gly-X-Y).
-
-
COL1A1 gene, coding for the α-1 chain, is on chromosome 17, whereas the a-2 subunit is coded by the COL1A2 gene on chromosome 7.
-
COL1A1/A2 gene mutations disrupt the synthesis of type I collagen.P.354
![]() Figure 15-5 Type I collagen synthesis.
Figure 15-5 Type I collagen synthesis. -
Affect all bone and connective tissue containing type I collagen as their major structural protein
-
Classical findings: bone fragility, long bone deformities (Fig. 15-6)
-
May also have blue sclerae, dentinogenesis imper-fecta, and scoliosis
Clinical Features and Classification
-
Most patients are categorized among the
four distinct types using the Sillence classification; the rest are
classified recently under V, VI, VII types (Table 15-2). -
May be autosomal dominant or autosomal recessive. New mutations are rare.
Radiographic Features
-
Osteoporosis
-
Multiple fractures
-
Bowing of long bones (often described as “gracile”)
-
Deficient ossification of the skull
Diagnosis and Work-up
-
Prenatal ultrasound and DNA analysis (chorionic villi sampling)
-
Collagen synthesis analysis of dermal fibroblasts (from skin biopsy)
-
Osteogenesis imperfecta should be considered in the differential diagnosis of child abuse.
Treatment
-
Medical treatment options aim to improve bone mass.
-
Bisphosphonates (pamidronate most common)
-
Bone marrow transplantation has been used with some success in severe cases but is not standard treatment.
-
Gene therapy (discussed in detail in section on treatment below)
-
-
Bracing may be used for fracture prevention (not effective in management of scoliosis).
-
Surgical treatment options include Sofield osteotomies
P.355
to correct long bone deformities, decompression of basilar impression, and spine fusion for significant scoliosis.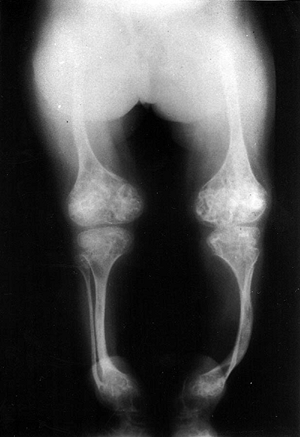 Figure 15-6
Figure 15-6
Radiograph of a young patient with osteogenesis imperfecta. (From
Baitner AC, Maurer SG, Gruen MB, et al. The genetic basis of the
osteochondrodysplasias. J Pediatr Orthop 2000; 20:594–605.) -
Genetic linkage studies: for risk assessment and genetic counseling
Mutations in Type II Collagen
Etiology
-
Type II collagen: found in large quantities in cartilage, tectorial membrane, nucleolus pulposus, and ocular vitreous humor
-
Polypeptide chains are encoded by COL2A1 gene on chromosome 12.
-
Mutations involving the gene lead to several connective tissue disorders (Table 15-3).
Mutations in Type IX Collagen—Multiple Epiphyseal Dysplasia Type II (Ribbing’s/Milder Form)
Etiology
-
Type IX collagen: essential for cartilage fibril formation and found only in cartilage
-
Encoded by three genes: COL9A1 (on chromosome 6), COL9A2 (on chromosome 1), and COL9A3 (on chromosome 20)
-
Mutations of these genes result in multiple epiphyseal dysplasia type II (Ribbing’s).
Clinical Features
-
Present at 2.5 to 6 years of age with knee and hip pain, worse after exercise
-
Mildly short stature, limbs affected more than the trunk
-
Waddling gait
-
Occasional angular deformities
-
The course is progressive pain and joint deformity leading to early-onset osteoarthritis.
-
Radiologic features: small, flattened epiphyses in many joints (severe in knees)
Mutations in Type X Collagen
Etiology
-
Type X collagen: produced by the COL10A1 gene
-
Gene expression is essential in
chondrocyte hypertrophic stage in the physis, which controls
endochondral ossification and facilitates long bone development. -
Gene mutations result in an inability of
nascent alpha chain to be incorporated in the collagen X triple helix,
which decreases the quantity of collagen X in the zone of endochondral
ossification (metaphyses affected, with normal epiphyses).
Clinical Features
-
Metaphyseal chondrodysplasia, Schmid type (MCDS), is the mildest and most common of the metaphyseal chondrodysplasias.
-
Patients present in childhood with small stature and bowed legs.
Radiologic Features
-
Varus deformities of the hips and knees
-
Flared metaphyses
-
Physes widened
Mutations in Type XI Collagen
-
Type XI collagen: found primarily in physeal cartilage
-
Three encoding genes: COL11A1 (chromosome
1), COL11A2 (chromosome 6), and COL2A1 (the gene on chromosome 12 that
also encodes type II collagen)-
Most type XI collagen abnormalities are associated with mutations in these genes and exhibit an autosomal-dominant pattern.
-
COL2A1 gene mutations are the most common
and result in Stickler syndrome type I (classified as type II collagen
disorder; see section on type II collagen). -
Mutations of the COL11A1/A2 genes are more rare but result in distinct phenotypes.
-
Stickler syndrome, COL11A1 or type 2
-
Referred to as vitreoretinal type because of characteristic myopia, glaucoma, and retinal detachment
-
Skeletal changes affect the joints and manifest similar to arthritis on x-rays.
-
P.356Table 15-2 Sillence Classification For Osteogenesis ImperfectaType Inheritance and Incidence Genetic defect Clinical Features/Comments I Autosomal dominant, 1:30,000 - “Functional null alleles” of COL1A1 or COL1A2 genes that lead to 50% reduction in type I collagen (mild disease)
- Bone fragility and blue sclera throughout life
- Susceptibility to multiple fractures with minimal trauma; though
initial bone healing is normal, there is a lack of bone remodeling phase - Life expectancy: normal or slightly reduced
II Autosomal dominant (new mutation or parental gonadal mosaicism) 1:60,000; autosomal recessive (rare) - Various mutations in COL1A1 gene leading to the disruption of Gly-X-Y repeat in the α-1 chain
- Disruption in triple helix patter
- Perinatal lethal form
- Present with numerous fractures at birth
- Soft skull, beaded ribs, dark sclera
- Death within first year of life
III Autosomal dominant (new mutation) or autosomal recessive, 1:70,000 - Deletions in α-1 and α-2 chains leading to abnormal collagen synthesis
- In certain mutations amino acid glycine is replaced by a different amino acid during procollagen synthesis
- Present in utero or neonatal period with multiple fractures
- Progressive limb deformities throughout life
- Shortened life span due to respiratory infections and skull fractures
IV Autosomal dominant, rare - COL1A1/A2 mutation
- Results in shortened pro-α1 chain affecting all chain structures in triple helix
- Similar to type I, except patients have normal hearing and scleral hue
- Life expectancy may be slightly reduced.
-
-
Stickler syndrome, COL11A2 or type 3
-
Referred to as nonocular because of the absence of ocular pathology (unlike types I and II)
-
Patients exhibit marfanoid habitus, hearing loss, and facial features similar to Stickler, COL2A1, or type I.
-
Skeletal Dysplasias due to Mutations in the Cartilage Oligomeric Matrix Protein (COMP) Gene
A member of the thrombospondin family of proteins,
cartilage oligomeric matrix protein (COMP) is expressed in high levels
in the chondrocyte matrix. Two autosomal-dominant disorders have been
mapped to the region of the COMP gene on chromosome 19 and are
described below.
cartilage oligomeric matrix protein (COMP) is expressed in high levels
in the chondrocyte matrix. Two autosomal-dominant disorders have been
mapped to the region of the COMP gene on chromosome 19 and are
described below.
Multiple Epiphyseal Dysplasia (MED) Type I (Fairbanks’s/ Severe Form)
-
MED has been linked to the mutations in
the COMP gene (MED, Fairbanktype 1) and collagen type IX genes (MED,
type II; see section on collagen IX). -
Presentation and prognosis of MED type I
-
Dependent on the type of the COMP mutation
-
Selective mutation types present with a milder phenotype that is similar to COL9 mutations.
-
Short stature, genu valgum and
ossification abnormalities about the knees (femoral condyle flattening,
“double-layer” patella), shortened metatarsals and metacarpals -
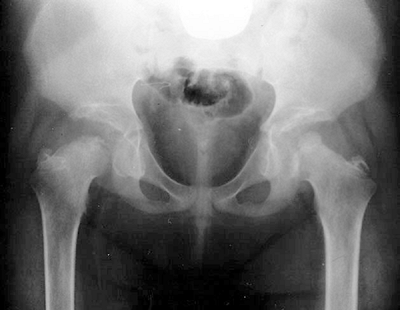
Severe disease entity presents with major involvement of the capital femoral epiphyses and irregular acetabuli (Fig. 15-7). This can be mistaken for bilateral Perthes disease, but Perthes does not appear as symmetric.
-
Pseudoachondroplasia
-
Infants appear normal at birth; growth retardation becomes apparent only at age 2 years or later.
-
Form of short-limb dwarfism, similar to achondroplasia
-
Head and face are normal; fingers, although short, do not exhibit the trident hand of achondroplasia
-
Cervical instability, scoliosis, increased lumbar lordosis
-
Genu varum or valgum
-
-
Radiographically, all tubular bones are short, with uneven, splayed metaphyses and irregularly developed epiphyses.
Skeletal Dysplasias due to Mutations in the Parathyroid Hormone Receptor 1 Gene
Etiology
-
Parathyroid hormone receptor 1 (PTHR1) is
a G-protein receptor and is highly expressed in the growth plate and
involved in chondrocyte proliferation and differentiation. -
An autosomal-dominant mutation of the
PTHR1 gene in 3p22 results in a severe form of metaphyseal
chondro-dysplasia called the Jansen’s type.
Clinical Features
-
Large skull
-
Widely spaced and exophthalmic eyesP.357Table 15-3 Diseases Associated With Mutation of the Col2A1 Gene
Disease Inheritance Genetic Defect Clinical Features/Comments Stickler syndrome, type I (hereditary arthro-ophthalmopathy) Autosomal dominant - Single base pair mutations (stop codons)
- Result in the absence of protein synthesis and inability of the healthy allele to compensate (haploinsufficiency)
- Craniofacial abnormalities (midface hypoplasia, cleft palate)
- Hearing loss
- Premature joint degeneration, abnormal epiphyseal development, marfanoid hypermobility, may have scoliosis or kyphosis
- Ocular abnormalities (congenital myopia, vitreoretinal degeneration, retinal detachment)
Achondrogenesis, type II (Langer-Saldino dysplasia) Autosomal dominant (new mutation) - Glycine-to-serine substitutions in COL2A1
- Inability to form the triple helix
- Neonatal dwarfism, micromelia, short trunk, fetal hydrops
- Early death in utero or after birth
Hypochondrogenesis Autosomal dominant - Different glycine-to-serine substitutions
- Milder allelic variant of type II achondrogenesis
- Infants: oval facies and widely spaced eyes
- Neonatal death from respiratory infections
Spondyloepiphyseal dysplasia congenita Autosomal dominant - Single base pair mutations or entire exon deletions and duplications
- Abnormal protein produced that interferes with function of trimer
- Short stature (140 cm or less)
- Barrel-chested trunk, more shortened than extremities
- Cleft palate, myopia
Kniest dysplasia Autosomal dominant - Single amino acid mutations or deletions of entire exons
- Secretion of type II procollagen that lacks a c-propeptide
- Results in abnormal fibril formation
- Short-trunk dwarfism
- Severe kyphoscoliosis
- Flattened facies, retinal detachment, deafness
- Physical therapy for joint contractures and reconstructive surgery for hip osteoarthritis
![]() Figure 15-7
Figure 15-7
Radiograph of a 10-year-old girl with multiple epiphyseal dysplasia
(Fairbanks type). (From Baitner AC, Maurer SG, Gruen MB, et al. The
genetic basis of the osteochondrodysplasias. J Pediatr Orthop 2000;20:594–605.) -
Severe limb shortening
-
Angular deformities at the diaphyseal—metaphyseal junction
-
Radiographs: metaphyseal fragmentation, normal epiphysis
-
Differential diagnosis: hyperparathyroidism
-
Both disease entities are characterized by hypercalcemia, hypophosphatemia, and increased renal phosphate secretion.
-
Abnormal parathyroid hormone (PTH) levels: only in hyperparathyroidism
-
Skeletal Dysplasias due to Mutations in the Diastrophic Dysplasia Sulfate Transporter Gene
Etiology
-
The diastrophic dysplasia sulfate
transporter (DTDST) gene is found on chromosome 5 and encodes a Na +
-independent membrane transporter of sulfate.-
DTDST found in human cartilage and intestines
-
Probable role in endochondral bone formation
-
-
Mutations are associated with recessively inherited osteochondrodysplasias.
P.358
Diastrophic Dysplasia
-
Cartilage of diastrophic dysplasia (DTD)
patients is associated with decreased sulfation of glycosaminoglycans,
which leads to severe limb distortion (“twisted” dwarf).-
A rare form of micromelic dwarfism
-
Spinal malalignment (cervical kyphosis and scoliosis)
-
Cleft palate and irregular calcifications of the pinnae of the ears (“cauliflower ears”)
-
Rigid foot deformities (e.g., talipes equinovarus)
-
“Hitchhiker’s thumb”
-
Achondrogenesis Type IB (Parenti-Fraccaro Type Achondrogenesis)
-
Autosomal-recessive DTDST mutation leading to impaired sulfation of proteoglycan
-
A lethal neonatal dwarfism associated with severe micromelia, large head, and fetal hydrops
-
Distinguished from type II
achondrogenesis, which results from a mutation leading to abnormal type
II collagen and has a normal cranial vault
Skeletal Dysplasias due to Mutations in the Arylsulfatase E Gene
-
Arylsulfatase E (ARSE): a steroid sulfatase, a family that includes enzymes that convert sulfated steroids to free steroids
-
The gene for ARSE is located on the X chromosome.
-
These sulfatases are active in osteoblast cell lines and play an important role in bone and cartilage formation.
-
-
Mutations of the ARSE gene have been implicated in at least one type of osteochondrodysplasia: chondrodys-plasia punctata.
-
Similar to diastrophic dysplasia and type I achondroplasia (mutation in sulfate transport)
-
Chondrodysplasia punctata is due to an inherited deficiency of the sulfatase enzyme.
-
Causes a high degree of sulfation of certain components within the cartilage matrix (exact disease etiology unclear)
-
-
Chondrodysplasia Punctata 1 X-Linked Recessive Type
-
Characterized by asymmetry of extremities
-
Hypoplasia of distal phalanges
-
Patients present in infancy with failure
to thrive, atypical facies (commonly nasal hypoplasia), ichthyosis
(pig-mented hyperkeratosis of the skin), and apparent mental
retardation. -
The name refers to the punctate epiphyseal calcifications that are prominent on the radiographs.
Skeletal Dysplasias due to Mutations in the SOX9 Gene
-
SOX9: a tissue-specific transcription
factor that is highly expressed in cartilaginous tissue and is involved
in testicular development -
The gene for SOX9 is on chromosome 17.
-
It codes for a transcription factor that results in the production of specific regulatory proteins.
-
These regulatory proteins bind to the
first intron on COL2A1 gene, thus regulating type II collagen
expression and chondrogenesis. -
Camptomelic dysplasia is caused by an autosomal-dominant SOX9 mutation.
-
Clinical Features of Camptomelic Dysplasia
-
Neonatal dwarfism with congenital angulation and bowing of long bones; the tubular bones are thickened
-
Other skeletal abnormalities include talipes equinovarus and hypoplastic scapulae.
-
Cleft palate, micrognathia, flat face
-
Most individuals with XY karyotype have a partial or full female phenotype since testicular development is affected.
-
Most patients do not survive past the neonatal period because of respiratory problems (tracheobronchial hypoplasia).
-
Radiologic features
-
Long bones are shortened and bowed.
-
Iliac bones, scapulae, and cervical spine are often hypoplastic.
-
Skeletal Dysplasias due to Mutations in the Cysteine Proteinase Cathepsin K Gene
-
The cysteine proteinase cathepsin K (CTSK) gene is located on chromosome 1q21 and produces an endopro-tease, which belongs to the cysteine proteinase family.
-
Vital role in the degradation of bone matrix, collagen, and osteonectin
-
Selectively expressed in osteoclasts and thus controls bone resorption
-
Associated with rheumatoid arthritis
-
-
CTSK gene
mutation may result in pyknodysostosis, a form of dwarfism
characterized by osteosclerosis on radiographs and bone fragility.
Connective Tissue Disorders
Connective tissue disorders can be classified as:
-
Extracellular matrix disorders
-
Autoimmune disorders
Extracellular Matrix Disorders
Pleiotropic mutations in genes responsible for
extracellular matrix production can lead to various abnormalities of
bone, vascular system and viscera. (“Pleiotropic” refers to a single
gene influencing several distinct and seemingly unrelated phenotypic
traits.)
extracellular matrix production can lead to various abnormalities of
bone, vascular system and viscera. (“Pleiotropic” refers to a single
gene influencing several distinct and seemingly unrelated phenotypic
traits.)
P.359
-
Ehlers-Danlos syndrome
-
Marfan syndrome
-
Osteogenesis imperfecta and other collagen disorders
Ehlers-Danlos Syndrome
-
Most common heritable connective tissue disorders, with six major subtypes, all characterized by skin and joint hypermobility (Table 15-4).
All are autosomal-dominant disorders except for kyphoscoliosis and the
dermatosparaxis subtypes, which are autosomal recessive. The classic,
hypermobility, and vascular subtypes are the most common.
Marfan Syndrome
-
An autosomal-dominant disorder with pleiotropic mutation of FBN1 gene on chromosome 15q21 that affects fibrillin production
-
Fibrillin is a major protein component of microfi-brils.
-
Fibrillin serves as a scaffold for deposits of newly synthesized elastin.
-
Microfibrils are seen in large quantities
in the eye (zonule of Zinn: the suspensory ligament of the eye that
holds the lens in place), skin, cartilage, and viscera (heart, lung,
and kidney).
-
-
Marfan syndrome has a high penetrance and variability
P.360
in clinical expression both within and between families (25% of cases arise from new mutations).Table 15-4 Subtypes of Ehlers-Danlos Syndrome According to the Villefranche ClassificationSubtype Inheritance Genetic Defect Clinical Features/Comments Classic Autosomal dominant Type V collagen abnormality due to mutations in COL5A1 or A2 genes - Hypermobility of skin
- Joint hypermobility
- Tower vertebra (high vertebral height), scoliosis (30%)
- Aortic root dilation (30%)
- Tissue fragility
- >50% of patients may present with chronic pain.
Hypermobility Autosomal dominant Genetic mutation uncertain - Both small and large
joint hypermobility (multidirectional instability of shoulder, patellar
subluxation, chronic ankle instability) - Soft, lax skin
- Most common subtype associated with orthopaedic intervention due to its debilitating presentation
Vascular Autosomal dominant Genetic mutation involving COL3A1 with abnormalities in type III collagen synthesis - Hypermobility of joints
- Clubfoot
- Laxity of viscera
- Vascular abnormalities predisposing to rupture of large arteries
- 75% of patients have aortic root dilatation; 25% of affected women are at risk of death due to uterine rupture
Kyphoscoliosis Autosomal recessive Procollagen-lysine,
2-oxoglutarate 5 dioxygenase-1 (PLOD) gene mutations resulting in
deficient production of lysyl hydroxylase, an enzyme involved in the
modification of collagen- Hypotonia at birth
- Progressive infantile scoliosis (double thoracic pattern)
- Hypermobility of joints
- Ocular complications: fragile sclera with globe rupture
- Patients with this disease have similar phenotypic presentation to Marfan syndrome.
Arthrochalasis Autosomal dominant COL1A1/A2 genetic abnormality with absence of pro-α 1 or 2 collagen type I chains at their N-terminal end - Hypermobility of joints
- Excessive laxity of skin
- Bilateral developmental dysplasia of hip with a high recurrence rate following surgical intervention
Dermatosparaxis Autosomal recessive Lack of production of procollagen I N-terminal peptidase - Fragile redundant skin
- Easy bruising
-
Diagnosis of Marfan syndrome is based on the characteristic phenotypic presentation and is confirmed by genetic studies.
-
Skeletal abnormalities: dolichostenomelia
(disproportionately long limbs), arachnodactyly, scoliosis (60% of
patients), chest wall deformity (pectus carinatum or excavatum), tall
body habitus, ligamentous laxity, abnormal joint mobility, and
protrusio acetabula -
Patients have an arm span-to-height ratio of more than 1.05.
-
Most common extraskeletal manifestations
are superior dislocation of lens (ectopia lentis, often bilateral) and
abnormalities in cardiovascular system (aortic root dilation/aortic
regurgitation and aortic aneurysms).
-
-
Treatment options include beta-blockers (to help reduce stress on aorta) and scoliosis treatment with bracing or surgery.
Autoimmune Disorders
Autoimmune disorders are a group of clinical conditions
characterized by an accumulation of selective antibodies.
Identification of disease subtypes is facilitated by tests for these
antibodies.
characterized by an accumulation of selective antibodies.
Identification of disease subtypes is facilitated by tests for these
antibodies.
-
Common examples
-
Systemic lupus erythematosus
-
Rheumatoid arthritis
-
Seronegative spondyloarthropathy
-
Systemic Lupus Erythematosus
-
Characterized by microvascular inflammation and generation of autoantibodies affecting multiple organ systems
-
Prevalence of 1:2,000; greater in premenopausal women and African Americans
-
Genetic basis for system lupus erythematosus: high rate of concordance in monozygotic twins (57%)
-
Associated with 10 different genetic loci—in particular, alleles of the human leukocyte antigens: HLA-DR2 and HLA-DR3
-
-
Musculoskeletal symptoms
-
Arthralgia and myalgia
-
Early morning joint stiffness and pain
-
In systemic lupus erythematosus arthritis is migratory and polyarticular (small joints of the hands, wrist, and knee joints).
-
10% of patients develop Jaccoud’s
arthropathy of the hand (bony erosion of metacarpal heads), resembling
rheumatoid arthritis disease pattern (Fig. 15-8).
-
-
Presence of any four of the following clinical findings confirms the diagnosis:
-
Arthritis
-
Malar/discoid rash
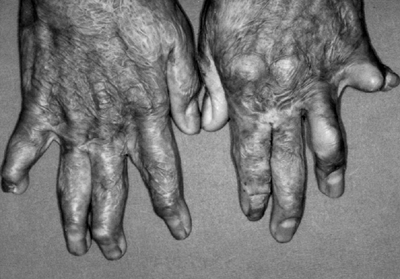 Figure 15-8 A 49-year-old woman with systemic lupus erythematosus and Jaccoud’s arthropathy of the hands.
Figure 15-8 A 49-year-old woman with systemic lupus erythematosus and Jaccoud’s arthropathy of the hands. -
Photosensitivity
-
Oral ulcers
-
Serositis
-
Renal disorder
-
Neurologic disorder
-
Hematologic disorder
-
Immunologic disorder
-
Antinuclear antibody
-
-
Treatment for systemic lupus erythematosus arthritis
-
Supportive: nonsteroidal
anti-inflammatories (NSAIDs), hydroxychloroquine (antimalarial drug
effective in refractory myalgias and arthralgias), cor-ticosteroid
therapy for renal and neuropsychiatric symptoms (can cause osteoporosis) -
Joint reconstruction/replacement for severely affected joints (e.g., avascular necrosis of the hips)
-
Rheumatoid Arthritis
-
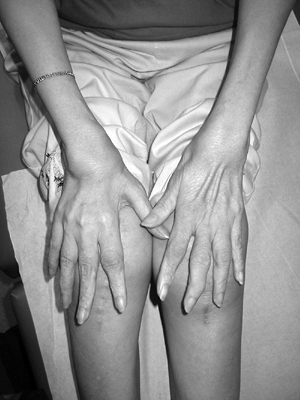
Autoimmune disorder characterized by symmetrical inflammatory arthropathy (Fig. 15-9), positive rheumatoid factor, and a varied extra-articular manifestation (e.g., rheumatoid nodules and neuropathy)
-
Recent evidence suggests that in at least
50% of patients genetic susceptibility is attributable to major
histocom-patibility complex class II loci (DR4(3 chains), with the
strongest associations with rheumatoid arthritis being DRB*0401 and
DRB*0404. -
Treatment principles: similar to those for systemic lupus erythematosus
Seronegative Spondyloarthropathies
-
Group of conditions negative for
rheumatoid factor and associated with inflammatory arthritis and
various typical extra-articular manifestations -
Four main conditions in this group:
ankylosing spondylitis, psoriatic arthropathy, reactive arthritis, and
inflammatory bowel disease -
All have their etiology associated with HLA-B27 (class
P.361
I major histocompatibility antigen), which can facilitate diagnosis (Fig. 15-10):![]() Figure 15-9 A 40-year-old woman with bilateral, symmetrical proximal polyarthropathy.
Figure 15-9 A 40-year-old woman with bilateral, symmetrical proximal polyarthropathy.-
About 50% to 95% of patients with ankylosing spon-dylitis test positive for HLA-B27.
-
However, about 5% of the normal population also test positive.
-
Neuromuscular Disorders
Neuromuscular disorders are a group of conditions in
which genetic defects cause abnormalities in signal transmission in one
of three regions: the neural junction (synapse), neu-romuscular
junction, or muscle.
which genetic defects cause abnormalities in signal transmission in one
of three regions: the neural junction (synapse), neu-romuscular
junction, or muscle.
Neural Junction Disorders
Hereditary Sensory and Motor Neuropathy
-
Hereditary sensory and motor neuropathy
(HSMN) is characterized by slow neural conduction identified by nerve
conduction studies and also associated with abnormal electromyographic
results.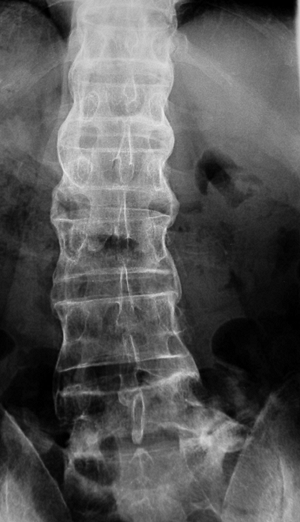 Figure 15-10
Figure 15-10
Radiograph of a 51-year-old man with a longstanding history of
ankylosing spondylitis. Note “bamboo spine” characterized by bridging
syndesmophytes between vertebrae.-
The histologic hallmark is demyelination on nerve biopsy.
-
Muscle biopsy is characterized by uniformly small-diameter fibers.
-
-
HSMN has been associated with 10 different genetic mutations.
-
The most common mutation involves overexpression of the peripheral myelin protein 22 gene on chromosome 17.
-
Other associated genes: myelin protein zero gene and connexin-32 gene
-
-
Patients with HSMN present to the
orthopaedist with hip dysplasia, scoliosis (10%), and cavovarus foot
deformity (most common orthopaedic manifestation).-
Approximately two thirds of patients have marked hand involvement characterized by intrinsic muscle weakness.
-
Six main types: the three main types encountered by orthopaedic surgeons are types 1,2, and 3 (Table 15-5)
P.362Table 15-5 The Seven Subtypes of Hereditary Sensory and Motor NeuropathiesType Inheritance Genetic Defect Clinical Features/Comments I Autosomal dominant Duplication of a portion of
chromosome 17p or a point mutation that results in an abnormality in
the peripheral myelin protein-22- Neuropathy marked by demyelination
- Includes disorders referred to as peroneal atrophy,
Charcot-Marie-Tooth disease type IA (hypertrophic form), or Roussy-Levy
syndrome
II Autosomal dominant (majority) Can have a variable inheritance pattern - Normal reflexes, sensory, and motor nerve conduction times
III Autosomal recessive Genetic mutation of PMP (peripheral myelin protein) 22 - More severe than type
I or II with clinical features of delayed ambulation, pes cavus, and
“glove-and-stocking” dysesthesia; most patients confined to a
wheelchair by third or fourth decade - Synonymous with Dejerine-Sottas disease
IV Autosomal recessive Mutation of peroxin 7 (PEX7)
gene or phytanoyl-CoA hydroxylase (PAHX), which controls the production
of the enzyme phytanoyl-CoA, resulting in accumulation of phytanic acid
in tissues and fluids- Synonymous with Refsum disease
V Autosomal dominant Undetermined genetic mutation - Inherited spastic paraplegia with distal weakness in the limbs
VI Autosomal recessive Undetermined genetic mutation - Optic atrophy in association with peroneal muscular atrophy
VII Autosomal dominant Undetermined genetic mutation - Retinitis pigmentosa associated with distal muscle weakness
-
-
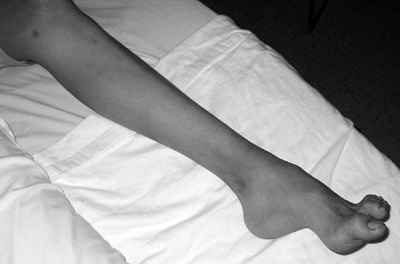
Charcot-Marie-Tooth disease refers to
HSMN type 1 or 2 disease, an autosomal-dominant condition characterized
by motor and sensory demyelinating neuropathy-
Can present either in the second decade
of life as the hypertrophic form or, more classically, in the third or
fourth decades as the axonal form -
Distinguished by extensive foot involvement, with the phenotypic appearance of “upside-down champagne bottle” (Fig. 15-11)
-
Other features include cavovarus feet, hammer-toes, peroneal weakness, and intrinsic muscle wasting.
-
May require orthotics, surgical releases, and/or muscle transfers for the foot
-
Spinal Muscular Atrophy
-
An autosomal-recessive disease that
manifests as a loss of anterior horn cells in the spinal cord; the most
common diagnosis in girls presenting with progressive muscle weakness-
Incidence: 1:200,000 births
-
Classified into three disease types based on age at presentation (Table 15-6)
-
-
Pathology is consistent with muscle denervation, and nerve conduction is unaffected.
-
Survival motor neuron (SMN) gene mutation
on chromosome 5q has been detected in a significant number of patients;
this gene plays a critical role in RNA metabolism and is a mediator of
apoptosis. -
No cerebral dysfunction; patients have a normal intellect
-
The disease presents with:
-
Progressive scoliosis
![]() Figure 15-11
Figure 15-11
A 32-year-old man with Charcot-Marie-Tooth disease. Note typical
extensive lower limb and foot involvement (“upside-down champagne
bottle”).P.363Table 15-6 Types of Spinal Muscular Atrophy Based on Age At PresentationType Name Genetic Defect Clinical Features/Comments 1 Acute Werdnig Hoffman disease Defective gene(s) for all three types located on chromosome 5q, but appear to be different mutations at the same locus - Presents at birth and in infants
- Short life span
2 Chronic Werdnig Hoffman disease - Presents between age 6 months and 2 years
3 Kugelberg-Welander disease - Presents in children after 2 years
- Typically long life span
-
Symmetric paresis and weakness predominating in the proximal musculature of both lower limbs
-
Hip dysplasia
-
Arthrogrypotic Syndromes
-
Arthrogryposis refers to a congenital,
nonprogressive limitation of joint movement attributable to soft tissue
contractures involving two or more joints. -
150 different genetic manifestations have been linked to the disease.
-
Decreased intrauterine movement a common etiology in all types
-
Pathophysiology due to the replacement of muscle in affected areas with fat and fibrous tissue
-
-
A severe disease entity that may be
managed by a pediatric orthopaedic surgeon is called arthrogryposis
multiplex congenita or amyoplasia. Arthrogryposis multiplex congenita
has the following disease features:-
Three types: myopathic, neuropathic, and mixed
-
Loss of anterior horn cells from the spinal cord
-
Multiple rigid joints with muscle wasting
-
Normal facies and intellect
-
Muscular Disorders (Inherited Myopathies)
-
A group of nine inherited myopathies that cause progressive muscular weakness
-
>30,000 people in the United States with inheritable myopathies
-
Clinical diagnosis based on family
history, physical examination, laboratory findings (CPK, aldolase),
electro-physiologic studies (electromyography, nerve conduction
studies), muscle biopsy, and molecular diagnostic tests -
CPK is an enzyme found in high levels in skeletal muscle and is closely associated with the myopathic disorders.
-
Levels normal in neuropathic disorders
-
Normal or mildly elevated levels in congenital myopathies and spinal muscular atrophies
-
High levels seen when there is significant myocyte destruction
-
Following this phase when the muscle has been replaced with fat and fibrous tissue, CPK levels return to normal.
-
-
Myotonic Muscular Dystrophy
-
The most common muscular dystrophy, with an incidence of 1:8,000
-
Marked phenotypic variability
-
Inherited as an autosomal-dominant trait, it is caused by a trinucleotide expansion in the myotonic dystrophy protein kinase (DMPK) gene on chromosome 19q13.2–q13.3.
-
Patients present with hypotonia in
infancy, myotonia in childhood, weakness, cataracts, hypogonadism,
frontal balding, mental retardation, and electrocardiographic changes.
Duchenne’s and Becker Muscular Dystrophies
-
Duchenne’s muscular dystrophy and the
milder Becker muscular dystrophy are two disorders that result from
different mutations in the dystrophin gene on the X chromosome (Xp21) (Table 15-7). -
Special diagnostic tests include:
-
Muscle biopsy
-
Necrotic foci
-
Fatty infiltration of muscle
-
Dystrophin immunoblotting: lack of detection of dystrophin with an antibody in a muscle biopsy
-
-
DNA mutation analysis with PCR or restriction fragment length polymorphism
-
Dystrophin RNA testing (requires a blood sample only)
-
Facioscapulohumeral Muscular Dystrophy
-
The third most common muscular dystrophy
(after Duchenne’s and myotonic muscular dystrophies),
facioscapulohumeral muscular dystrophy (FSHMD) is an autosomal-dominant
disorder associated with the gene on chromosome 4q. -
Patients present in late childhood or
early adulthood with progressive muscular weakness of the face,
shoulder girdle (scapular winging), and arm muscles (Fig. 15-12). -
Patients generally have normal life expectancy.
Emery-Dreifuss Muscular Dystrophy
-
A rare muscular dystrophy: an X-linked
disorder that affects males and is caused by a mutation in the emerin
gene on chromosome Xq28 (dystrophin is normal)P.364Table 15-7 Two Forms of Muscular Dystrophy That Arise From Dystrophin MutationParameter Duchenne’s Becker Incidence 1:3,500 1:30,000 Inheritance X-linked recessive Protein Dystrophin (found in skeletal, smooth, and cardiac muscle and the brain) Mutations Disrupt the translational
reading frame or promoter region, leading to truncated/ unstable
dystrophin with absence of functional dystrophinUsually associated with a less
disruptive type of mutation, resulting in a smaller and/or
semifunctional protein (nonsense mutation)Creatine phosphokinase Highly elevated (100 times normal) Moderately to highly elevated (50 to 100 x normal) Dystrophin immunoblotting Complete absence of dystrophin Dystrophin decreased in amount, size, or both Onset Between 3 and 6 years of age Later in childhood Characteristics Proximal muscle weakness (Gowers maneuver) Proximal muscle weakness (less severe) Skeletal manifestations Joint contractures, scoliosis, equinovarus Similar to Duchenne’s but less severe Other manifestations - Cardiac involvement
with sinus tachycardia and right ventricular hypertrophy with
arrhythmias and congestive heart failure in 10% of patients - Progressive loss of respiratory capacity
- Gastric dysmotility
- Risk of malignant hyperthermia
- Cardiomyopathy can occur, but complications such as congestive heart failure and arrhythmias are infrequent.
- Risk of malignant hyperthermia
Course - Progressive weakness of axial and appendicular muscles
- Able to run as children, preserve ability to walk sometimes into adulthood
- Death from respiratory problems in late 20s
- More variable, protracted course
- Walking until age 10 years
- Fewer respiratory problems
- Cardiac involvement
-
Slow progression, with initial
nonspecific features (e.g., muscle weakness in the first few years of
life, awkward gate, tendency to toe-walk)-
Differential diagnosis: Duchenne’s and Becker muscular dystrophy
-
The early diagnosis is confirmed by electromyography and muscle biopsy.
-
Definitive clinical features are noted in the second decade:
-
Fixed equinus deformities
-
Elbow flexion contractures
-
Neck extension contractures (limit neck flexion)
-
Cardiac abnormalities (risk of sudden cardiac death)
-
-
Trinucleotide Disorders
-
A group of conditions associated with an increased number of nucleotide triplets in the DNA
-
There is a progressive increase in
trinucleotide number down the successive familial generation, with
worsening in disease presentation (phenomenon called anticipation). -
The repeat expansions are associated with
a “loss-of-function” mechanism: these expansions prevent the expression
of normal protein.
-
-
The genetic abnormality may be located on
various chromosomes, and some disease entities share a characteristic
increase in CAG repeat.-
CAG trinucleotide codes for amino acid glutamine.
-
-
To date, 14 trinucleotide disorders have been documented.
-
Trinucleotide disorders may be classified as polyglutam-ine or nonpolyglutamine disorders.
-
Most polyglutamine disorders are rare and not associated with any major orthopaedic issues.
-
-
Friedrich’s ataxia is an autosomal
recessive nonpolyglutamine trinucleotide disorder characterized by
spinocer-ebellar involvement.-
Findings include a classic triad of ataxia, loss of deep tendon reflexes (areflexia), and an extensor Babinski response.P.365
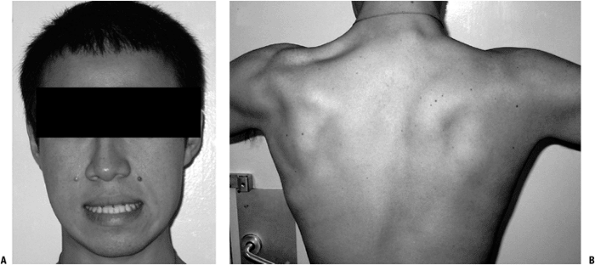 Figure 15-12
Figure 15-12
A 22-year-old man with facioscapulohumeral muscular dystrophy. Note
loss of facial muscle contracture (patient is attempting to say “E”)
(A) and winging of scapulae (B). -
Patients typically present to an
orthopedic surgeon because of scoliosis (which occurs in approximately
80%) and/or cavovarus feet. -
Average age of onset is 12 years; patients often die by age 25 years due to cardiomyopathy-related heart failure.
-
Molecular basis linked to the presence of
expanded guanine-adenosine-adenosine trinucleotide repeats in the gene
in chromosome 9 involved in the production of the mitochondrial protein
frataxin, which plays an essential role in iron metabolism and
oxida-tive stress-
~66 to 1,500 abnormal repeats, the number correlating to disease severity (normal patients have up to 32 abnormal repeats)
-
Presence of this genetic defect can lead
to accumulation of free radicals (produced when excess iron reacts with
oxygen), which then cause irreversible damage in the nervous system,
the heart, and the pancreas. -
Knowledge of the genetic basis of the
disease has given physicians the opportunity to extend the lives of
patients at risk for cardiac failure (secondary to accumulation of
bioactive radicals) by administration of antioxidants.
-
-
Other Genetic Conditions
Down Syndrome (Trisomy 21)
-
Duplication of long arm of chromosome 21
-
Increasing incidence with increasing maternal age
-
Clinical features: ligamentous laxity, mental impairment, heart disease, endocrine disorders, early aging
-
Orthopedic implications: atlantoaxial
instability, scoliosis, hip instability, slipped capital femoral
epiphyses, patella dislocation, metatarsus primus varus, plano-valgus
Turner’s Syndrome (XO)
-
Single X chromosome instead of the normal XX or XY configuration
-
Clinical features: short stature, sexual infantilism, web neck, renal anomalies in 66%, cardiac anomalies in 33%
-
Orthopedic issues: short stature,
scoliosis, genu valgum, shortened fourth and fifth metacarpals, risk of
malignant hyperthermia following administration of anesthesia
Neurofibromatosis
-
Neurofibromatosis is an autosomal-dominant disorder of the bones, soft tissues, skin, and nervous system. Two distinct subtypes:
-
Neurofibromatosis-1 (NF-1 or von Recklinghausen disease)
-
Neurofibromatosis-2 (NF-2)
-
-
NF-1, the more common peripheral subtype,
is characterized by a mutation in the neurofibromin tumor-suppressor
gene on chromosome 17.-
Incidence: 1:3,000 births (highest incidence of all single gene disorders)
-
Half of cases can be new mutations.
-
Affects multiple organ systems with varying severity; this can help in making the diagnosis
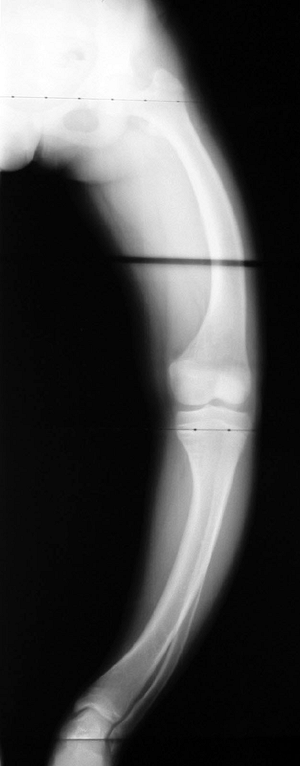
![]() Figure 15-13 Radiograph of a 16-year-old girl with a diagnosis of neurofibromatosis. Note anterolateral bowing of tibia.
Figure 15-13 Radiograph of a 16-year-old girl with a diagnosis of neurofibromatosis. Note anterolateral bowing of tibia. -
Two or more of these criteria are necessary for diagnosis:
-
At least two neurofibromas (tumors of Schwann cells) or one plexiform neurofi-broma
-
At least six cafe-au-lait spots (irregular brown spots on skin) that measure at least 5 mm in children, at least 15 mm in adults
-
Axillary or inguinal freckling
-
Optic glioma
-
At least two iris hamartomas (Lisch nodules)
-
Distinctive bony lesion
-
Sphenoid dysplasia or thinning of the cortex of the long bones
-
Congenital pseudarthrosis or anterolateral bowing of tibia (Fig. 15-13)
-
Dystrophic scoliosis (short, sharp curve
with or without thinning of ribs, enlargement of neural foramina,
vertebral body scalloping)
-
-
First-degree relative with NF-1
-
-
Treatment
-
Excision of enlarging or symptomatic neurofibromas
-
Treatment of scoliosis and pseudoarthrosis of the tibia
-
Resection of malignant peripheral nerve sheath tumors
-
-
-
NF-2 is characterized by various central
nervous system tumors. Its incidence is 1:40,000, and it has been
localized to chromosome 22 (half are de novo mutations).-
Patients present with unilateral/bilateral vestibular schwannomas (may be associated with various tumors).
-
Treatment: excision of acoustic neuromas causing tinnitus, vertigo, epilepsy
-
Suggested Reading
Baitner AC, Maurer SG, Gruen MB, et al. The genetic basis of the osteochondrodysplasias. J Pediatr Orthop 2000;20:594–605.
Beighton P, Giedion ZA, Gorlin R, et al. International classification of osteochondrodysplasias. AmJ Med Genet 1992;44:223–229.
Dietz FR, Mathews KD. Update on the genetic basis of disorders with orthopaedic manifestations. J Bone Joint Surg [Am] 1996;78(10): 1583–1598.
Evans CH, Ghivizzani SC, Robbins PD. Orthopaedic gene therapy. Clin Orthop Relat Res 2004;(429):316–329.
Mackenzie WG, et al. Genetic diseases and skeletal dysplasias. In: Vaccaro AR, ed. Orthopaedic Knowledge Update, Home Study Syllabus 2005:663–675.
Online
Mendelian Inheritance in Man (OMIM) Database [Internet]. Bethesda, MD:
National Center for Biotechnology Information for the National
Institute of Health. Available from http:// www.ncbi.nlm.nih.gov/Omim/
Mendelian Inheritance in Man (OMIM) Database [Internet]. Bethesda, MD:
National Center for Biotechnology Information for the National
Institute of Health. Available from http:// www.ncbi.nlm.nih.gov/Omim/
Sollazzo V, Bertolani G, Calzolari E, et al. A two-locus model for non-syndromic congenital dysplasia of the hip (CDH). Ann Hum Genet 2000;64:51–59.
Wilkinson JA. Etiologic factors in congenital displacement of the hip and myelodysplasia. Clin Orthop Relat Res 1992;(281):75–83.