
Editors: Tornetta, Paul; Einhorn, Thomas A.; Damron, Timothy A.
Title: Oncology and Basic Science, 7th Edition
Copyright ©2008 Lippincott Williams & Wilkins
> Table of Contents > Section
II – Specific Bone Neoplasms and Simulators > 7 – Congenital and
Inherited Bone Conditions > 7.6 – Werner Syndrome
II – Specific Bone Neoplasms and Simulators > 7 – Congenital and
Inherited Bone Conditions > 7.6 – Werner Syndrome
7.6
Werner Syndrome
Kathryn Palomino
Werner syndrome is a rare hereditary systemic disease,
also known as adult progeria. It is characterized by signs of premature
aging (gray hair, skin changes, and cataracts) in patients after the
onset of puberty. Atherosclerosis, diabetes, and neoplastic diseases
are also associated with the syndrome. Life expectancy is around 50
years, with cardiovascular, atherosclerotic disease and cancers being
the most common causes of death.
also known as adult progeria. It is characterized by signs of premature
aging (gray hair, skin changes, and cataracts) in patients after the
onset of puberty. Atherosclerosis, diabetes, and neoplastic diseases
are also associated with the syndrome. Life expectancy is around 50
years, with cardiovascular, atherosclerotic disease and cancers being
the most common causes of death.
Etiology
-
Werner syndrome gene (WRN) is located on chromosome 8 (8p11-12).
-
WRN protein encodes DNA helicase and exonuclease activities and is involved in response to DNA damage.
-
Inheritance: autosomal recessive
Epidemiology
-
Prevalence: ranges from 1/22,000 to 1/106
-
More common in Japanese and Caucasian populations but has been described throughout the world
-
Frequency is higher in populations with consanguineous marriage.
-
Male:female equal
-
Age: diagnosis is usually not made until adulthood
Pathophysiology
-
Precise mechanism for the phenotype of Werner syndrome is unknown.
-
Defective DNA metabolism is involved, leading to altered bioavailability of growth factors.
-
-
RecQ helicase deficiency syndrome leads to heritable cancer predisposition (10% to 30% increased risk).
-
Numerous types of tumors are seen (Fig. 7.6-1).
-
Diagnosis
Nonorthopaedic Clinical Features
-
Develop at onset of puberty
-
Dermatologic changes resembling scleroderma
-
Short stature (mean height: male 157 cm; female 147 cm)
-
Premature graying and alopecia
-
Nonsenile cataracts
-
Weak, high-pitched voice
-
Calcific deposits on heart valves
-
Atherosclerosis
-
Diabetes mellitus
-
Hypogonadism
-
Neoplasm (nonorthopaedic)
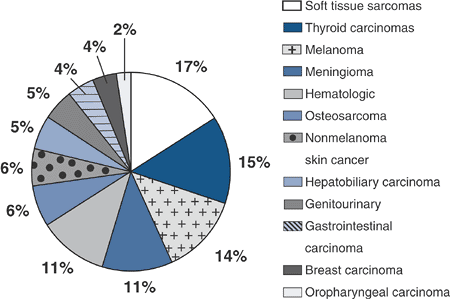 |
|
Figure 7.6-1 Tumors associated with Wermer syndrome.
|
Orthopaedic Clinical Features
-
Osteoporosis, usually presenting with vertebral abnormalities or insufficiency fractures
-
Muscle wasting
-
Calcific deposits in ligaments and tendons
-
Pes planus
-
Neoplasms (orthopaedic)
-
Soft tissue sarcomas: most common malignancy
-
Osteosarcoma
-
Diagnostic Algorithm
-
Presence of all consistent clinical features with at least two of the following:
-
Loss of WRN protein from peripheral lymphocytes or fibroblasts
-
Elevated 24-hour urinary hyaluronic acid level
-
Detection of mutation of WRN gene on chromosome 8 by Western blot
-
Treatment
-
There is no specific therapy for Werner syndrome.
-
Management involves symptomatic medical management of conditions as they arise.
-
Management of orthopaedic conditions
-
Osteoporosis: human insulin-like growth factor has been used
-
Fractures: heal poorly regardless of method of treatment
-
Pes planus: best managed with orthotics
-
Surgical incisions heal poorly.
-
-
Tumor management
-
Early diagnosis of Werner syndrome and
subsequent hypervigilance in investigating skin of soft tissue
abnormalities or bone pain are paramount in decreasing death rate from
malignancy in the Werner syndrome patient. -
Unique features of malignancy in Werner syndrome patients
-
Develop at early age
-
Develop in unusual sites
-
May develop multiple primaries (synchronously or metachronously)
-
One of most common causes of death
-
-
Suggested Reading
Monnat RJ Jr. Werner syndrome. In: Fletcher CDM, Unni KK, Mertens F, eds. Tumours of Soft Tissue and Bone: Pathology and Genetics. Lyon: World Health Organization Classification of Tumours. International Agency for Research on Cancer Press, 2002:366–367.
Walton NP, Brammer TJ, Coleman NP. The musculoskeletal manifestations of Werner’s syndrome. J Bone Joint Surg [Br] 2000;(82):885–888.