
Editors: Tornetta, Paul; Einhorn, Thomas A.; Damron, Timothy A.
Title: Oncology and Basic Science, 7th Edition
Copyright ©2008 Lippincott Williams & Wilkins
> Table of Contents > Section
II – Specific Bone Neoplasms and Simulators > 7 – Congenital and
Inherited Bone Conditions > 7.4 – Retinoblastoma Syndrome
II – Specific Bone Neoplasms and Simulators > 7 – Congenital and
Inherited Bone Conditions > 7.4 – Retinoblastoma Syndrome
7.4
Retinoblastoma Syndrome
Timothy A. Damron
Retinoblastoma syndrome encompasses familial
retinoblastoma and a number of secondary malignancies that may develop
as a result of a “second hit” at the site of the RB1 gene, where a
somatic mutation added to the germline RB1 mutation inactivates the
tumor suppressor gene function. The synonym “retinoblastoma/osteogenic
sarcoma syndrome” reflects the fact that osteosarcoma is the most
common secondary tumor in these patients. Retinoblastoma is a
malignancy of the embryonic neural retina.
retinoblastoma and a number of secondary malignancies that may develop
as a result of a “second hit” at the site of the RB1 gene, where a
somatic mutation added to the germline RB1 mutation inactivates the
tumor suppressor gene function. The synonym “retinoblastoma/osteogenic
sarcoma syndrome” reflects the fact that osteosarcoma is the most
common secondary tumor in these patients. Retinoblastoma is a
malignancy of the embryonic neural retina.
Pathogenesis
Etiology
-
Prototypical example of “two-hit” theory for genetic predisposition to cancer
-
First hit: germline (familial form) or somatic (nonfamilial form) mutation of RB1 gene at 13q14.1
-
Second hit: somatic mutation in all cases at RB1 locus
-
Radiation increases the risk in a dose-dependent fashion above 5 Gy.
-
Resultant inactivation of RB1 tumor
suppressor gene function is associated with retinoblastoma,
post-retinoblastoma osteosarcomas, and other sarcomas (Box 7.4-1), and in some sarcomas not associated with retinoblastoma syndrome (breast and non–small-cell lung carcinoma).
-
-
-
Inheritance: autosomal dominant (AD) with almost full penetrance in familial form
Epidemiology
-
Retinoblastoma
-
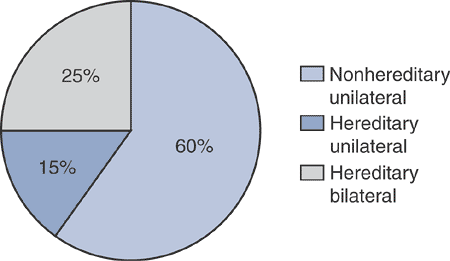
Frequency: 1/3,500 to 1/25,000 (Fig. 7.4-1)
-
Male:female equal
-
-
Secondary sarcoma
-
Relative risk compared to normal population: 30
-
Cumulative incidence over 50 years
-
Hereditary retinoblastoma: 51%
-
Nonhereditary retinoblastoma: 5%
-
-
Box 7.4-1 Secondary Tumors Associated with Retinoblastoma Syndrome
-
Osteosarcoma
-
Fibrosarcoma
-
Chondrosarcoma
-
Ewing sarcoma
-
Epithelial tumors
-
Leukemia
-
Lymphoma
-
Melanoma
-
Brain tumors, including pinealoblastoma
Pathophysiology
-
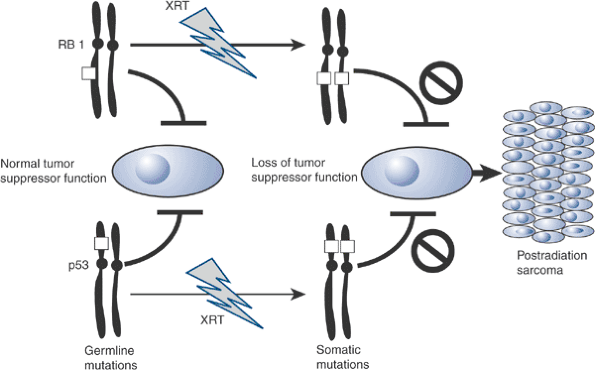
Known genetic defect (RB1) inactivates tumor suppressor gene, resulting in tumor formation (Fig. 7.4-2)
Diagnosis
History and Physical Examination
From an orthopaedic standpoint, patients with
retinoblastoma syndrome will have had retinoblastoma diagnosed earlier
in their life, so the key is to recognize that the history of
retinoblastoma predisposes to development of secondary tumors that may
either be musculoskeletal (osteosarcoma, chondrosarcoma, Ewing) or
present with musculoskeletal complaints (see Box 7.4-1).
retinoblastoma syndrome will have had retinoblastoma diagnosed earlier
in their life, so the key is to recognize that the history of
retinoblastoma predisposes to development of secondary tumors that may
either be musculoskeletal (osteosarcoma, chondrosarcoma, Ewing) or
present with musculoskeletal complaints (see Box 7.4-1).
History
-
Age at diagnosis of retinoblastoma: 90% at <3 years
-
Unilateral: 18 months
-
Bilateral: 12 months
-
-
Previous treatment for retinoblastoma: chemotherapy and/or radiotherapy
Physical Examination
-
Evidence of previous retinoblastoma: characteristic narrow facies, sometimes blindness
-
Secondary tumor: manifestations depend upon type of secondary tumor
 |
|
Figure 7.4-1 Frequency of various forms of retinoblastoma.
|
 |
|
Figure 7.4-2 Two-hit theory for retinoblastoma and p53 tumor suppressor genes.
|
Diagnostic Work-up
With the history of retinoblastoma, a high index of
suspicion for sarcoma must be maintained in any patient with pain
and/or a mass. The diagnostic work-up is the same as that for any other
bone or soft tissue mass, as discussed in earlier chapters.
suspicion for sarcoma must be maintained in any patient with pain
and/or a mass. The diagnostic work-up is the same as that for any other
bone or soft tissue mass, as discussed in earlier chapters.
Treatment
Treatment of the secondary tumors is the primary concern
for the orthopaedic surgeon. This treatment depends upon the specific
type of tumor, as discussed in each of the individual sections.
Aggressive treatment is indicated for most patients, although the
prognosis classically has been considered to be poor.
for the orthopaedic surgeon. This treatment depends upon the specific
type of tumor, as discussed in each of the individual sections.
Aggressive treatment is indicated for most patients, although the
prognosis classically has been considered to be poor.
Results and Outcome
Classically considered to have a dismal prognosis, the
latest results for aggressive treatment of postradiation osteosarcoma,
in particular, are encouraging.
latest results for aggressive treatment of postradiation osteosarcoma,
in particular, are encouraging.
-
Postradiation osteosarcoma treated with chemotherapy and surgery
-
5-year disease-specific survival: 71%
-
5-year overall survival: 68.3%
-
Suggested Reading
Aerts
I, Pacquement H, Doz F, et al. Outcome of second malignancies after
retinoblastoma: a retrospective analysis of 25 patients treated at the
Institut Curie. Eur J Cancer 2004;40(10):1522–1529.
I, Pacquement H, Doz F, et al. Outcome of second malignancies after
retinoblastoma: a retrospective analysis of 25 patients treated at the
Institut Curie. Eur J Cancer 2004;40(10):1522–1529.
Cavenee WK, Bogler O, Hadjistilianou T, et al. Retinoblastoma syndrome. In: Fletcher CDM, Unni KK, Mertens F, eds. Tumours of Soft Tissue and Bone: Pathology and Genetics. World Health Organization Classification of Tumours. Lyon: International Agency for Research on Cancer Press, 2002:363–364.
Koshy M, Paulino AC, Mai WY, et al. Radiation-induced osteosarcomas in the pediatric population. Int J Radiat Oncol Biol Phys 2005;63(4):1169–1174.
Smith LM, Donaldson SS. Incidence and management of secondary malignancies in patients with retinoblastoma and Ewing’s sarcoma. Oncology (Williston Park) 1991;5(5):135–141.
Wong FL, Boice JD Jr, Abramson DH, et al. Cancer incidence after retinoblastoma. Radiation dose and sarcoma risk. JAMA 1997;278(15):1262–1267.