
Editors: Tornetta, Paul; Einhorn, Thomas A.; Damron, Timothy A.
Title: Oncology and Basic Science, 7th Edition
Copyright ©2008 Lippincott Williams & Wilkins
> Table of Contents > Section
II – Specific Bone Neoplasms and Simulators > 7 – Congenital and
Inherited Bone Conditions > 7.3 – Skeletal Dysplasias
II – Specific Bone Neoplasms and Simulators > 7 – Congenital and
Inherited Bone Conditions > 7.3 – Skeletal Dysplasias
7.3
Skeletal Dysplasias
Danielle A. Katz
Polyostotic Fibrous Dysplasia and Albright’s Syndrome
Fibrous dysplasia is a condition in which there is
fibro-osseous tissue in place of normal lamellar bone. Most often (85%)
this process is monostotic (affecting a single bone), but it may be
polyostotic (involving multiple bones). Cutaneous markings and
endocrinopathies may accompany the polyostotic form, which usually has
more severe skeletal involvement. This section will focus on the
polyostotic form.
fibro-osseous tissue in place of normal lamellar bone. Most often (85%)
this process is monostotic (affecting a single bone), but it may be
polyostotic (involving multiple bones). Cutaneous markings and
endocrinopathies may accompany the polyostotic form, which usually has
more severe skeletal involvement. This section will focus on the
polyostotic form.
Pathogenesis
Etiology
-
Not inherited
-
May be due to failure of woven bone to mature to lamellar bone
-
G-protein gene mutations in both
monostotic and polyostotic fibrous dysplasia, Albright’s syndrome, and
solitary pituitary adenoma-
Activating (gain of function) mutations in GNAS1 (encodes alpha subunit of stimulatory G protein)
-
Epidemiology
-
Female predominance
-
Diagnosis usually made in late childhood or early adolescence
-
30% to 50% of patients with polyostotic fibrous dysplasia have Albright’s syndrome.
Pathophysiology
-
Fibro-osseous tissue within bone, most often metaphyseal
-
One side more involved than other
Classification
-
Monostotic or polyostotic
-
McCune-Albright (Albright’s) syndrome is:
-
Polyostotic fibrous dysplasia
-
Café-au-lait spots (“coast of Maine” irregular border)
-
Precocious puberty or other endocrine abnormalities (Box 7.3-1)
-
-
Mazabraud’s syndromeP.216
-
Fibrous dysplasia
-
Soft tissue myxomas
-
Box 7.3-1 Conditions Associated with Mccune-Albright Syndrome
-
Sexual precocity
-
Pituitary adenoma
-
Hyperthyroidism
-
Gastrointestinal polyps
-
Thymus hyperplasia
-
Splenic hyperplasia
-
Pancreatic islet cell hyperplasia
-
Hepatobiliary disease
-
Cardiac disease
-
Failure to thrive
-
Metabolic acidosis
-
Abnormalities in serum electrolytes, glucose, or insulin levels
-
Hyperphosphaturic hypophosphatemia
-
Osteosarcoma
-
Developmental delay
-
Microcephaly
-
Sudden or premature death
Diagnosis
Clinical Features
Polyostotic Fibrous Dysplasia
-
May be asymptomatic, although that is less common in polyostotic form
-
Pain
-
Limp (can be from pain, leg-length discrepancy, or Trendelenburg gait from “shepherd’s crook” deformity of proximal femur)
-
Swelling (if bone in subcutaneous location)
-
Angular deformity
-
Leg-length discrepancy
-
50% with craniofacial manifestations
McCune-Albright Syndrome (features in addition to the above)
-
Cutaneous macular pigmentation similar to café-au-lait spots of neurofibromatosis
-
Irregular “coast of Maine” border (unlike smooth “coast of California” border seen in neurofibromatosis) (Fig. 7.3-1)
-
Tend to cluster centrally, especially on the back
-
Most frequent extraskeletal manifestation (approximately one third)
-
Unusual in monostotic fibrous dysplasia
-
-
Precocious puberty or endocrinopathy
-
Precocious puberty more common (20%)
-
Females > males
-
Female presentation: vaginal bleeding, premature development of sexual organs, premature secondary sex characteristics
-
Male presentation: enlarged genitals, premature secondary sex characteristics
-
-
Endocrinopathy may include
hyperparathyroidism, hyperthyroidism, Cushing syndrome, acromegaly,
diabetes, rickets, osteomalacia, hyperprolactinemia.
-
 |
|
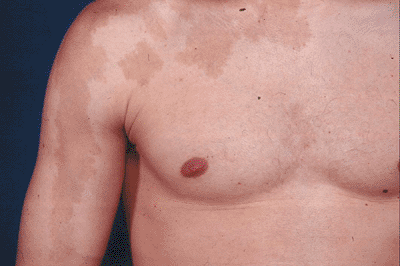
Figure 7.3-1
Irregular “coast of Maine” border in the pigmented skin lesion associated with polyostotic fibrous dysplasia. This differs from the smooth “coast of California” border seen typically in type I peripheral neurofibromatosis and in Jaffe-Campanacci syndrome (multiple nonossifying fibromas and café-au-lait spots). |
Mazabraud’s Syndrome
-
Myxomas with fibrous dysplasia bone lesions
-
Usually polyostotic (86%) over solitary fibrous dysplasia, rarely with McCune-Albright’s
Radiologic Features
-
Diaphyseal or metaphyseal
-
Epiphyseal involvement rare
-
Involvement of flat bones (skull, jaw, ribs) common
-
Spinal involvement uncommon
-
-
Lucent or “ground glass” appearance (Fig. 7.3-2)
-
May have calcifications
-
Sclerotic rim typical
-
May expand cortex (usually does not break cortex)
-
May have angular deformities (e.g., “shepherd’s crook” deformity) (Fig. 7.3-3)
-
Increased uptake on bone scan
Histologic Features
-
Fibrous stroma with spindle cells
-
Spicules of osteoid or woven bone that
have the appearance of “alphabet soup” (often described as resembling
the letters C, O, J, and Y) or “Chinese characters” (Fig. 7.3-4) -
Few osteoblasts (lacking osteoblastic rimming)
-
Lack of osteoblastic rimming
distinguishes fibrous dysplasia from osteofibrous dysplasia, more
common in the tibia, which is characterized by osteoblastic rimming.
-
-
Cartilaginous foci may be present.
Treatment
Observe if asymptomatic. Endocrinologist should manage precocious puberty or other endocrinopathies.
P.217
 |
|
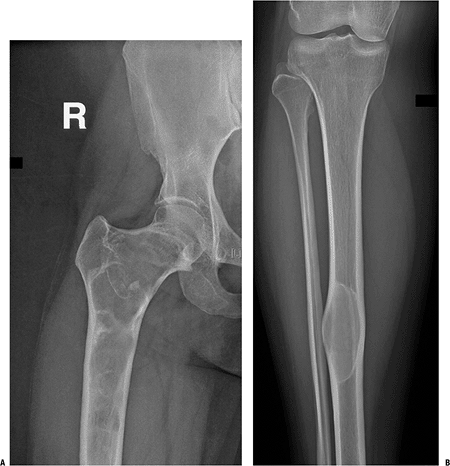
Figure 7.3-2 Radiographs from a patient with polyostotic fibrous dysplasia show extensive involvement of the proximal femur (A) and a single lesion in the ipsilateral tibia (B).
Note the characteristic loss of normal trabeculation within the lesion in the tibia, which has been referred to as a “ground glass” mineralization pattern. This less organized appearance reflects the histology, which shows a more random “Chinese character” pattern of mineralization. |
Indications for Surgery
-
Biopsy indicated if diagnosis in question
-
Fracture through lesion
-
If alignment unacceptable or high-risk location (e.g., proximal femur)
-
In children may be able to treat pathologic fractures with casting
-
-
Progressive deformity
-
If causing functional disability, unacceptable disfigurement, significantly increased risk of pathologic fracture
-
-
Pain
Surgical Technique
-
In adults, curettage and bone grafting has better success.
-
If open reduction and internal fixation (ORIF) is performed:
-
Must have good alignment of bones (may need osteotomies to accomplish this)
-
Screws may not have good purchase in abnormal bone.
-
Allograft cortical struts may provide additional stability and be less likely to be resorbed.
-
 |
|
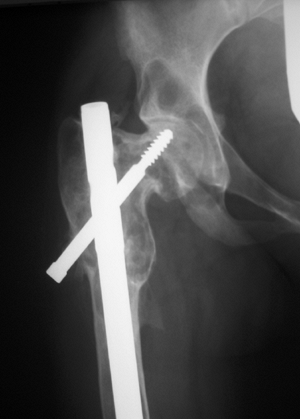
Figure 7.3-3
A classic “shepherd’s crook” deformity of the proximal femur consisting of coxa vara due to fibrous dysplasia is shown on this hip radiograph. In this case, the patient has undergone previous internal fixation. |
 |
|
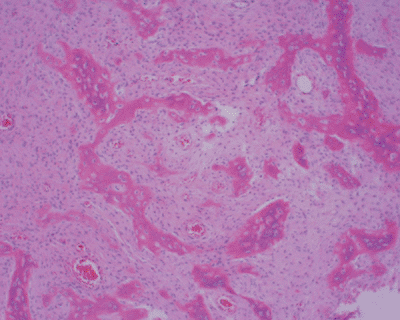
Figure 7.3-4
Typical histology of fibrous dysplasia, with multiple “alphabet soup” or “Chinese character” bone formation surrounded by a fibrous tissue background and lacking osteoblastic rimming. |
Results and Outcome
Surgical treatment may be challenging because of
tendency for recurrence and pathological nature of bone. Malignant
transformation of lesions is rare and usually is associated with a
history of radiation therapy. For this reason radiation therapy is not indicated for this disease. When malignancy develops, the prognosis is poor.
tendency for recurrence and pathological nature of bone. Malignant
transformation of lesions is rare and usually is associated with a
history of radiation therapy. For this reason radiation therapy is not indicated for this disease. When malignancy develops, the prognosis is poor.
Ollier’s Disease and Maffucci’s Syndrome
Solitary enchondromas are relatively common benign
tumors of bone. Multiple enchondromatosis is far less common and has
been given the eponym “Ollier’s disease.” Maffucci’s syndrome consists
of multiple soft tissue hemangiomas in the presence of multiple
enchondromatosis. This section will focus on Ollier’s disease and
Maffucci’s syndrome.
tumors of bone. Multiple enchondromatosis is far less common and has
been given the eponym “Ollier’s disease.” Maffucci’s syndrome consists
of multiple soft tissue hemangiomas in the presence of multiple
enchondromatosis. This section will focus on Ollier’s disease and
Maffucci’s syndrome.
Pathogenesis
Etiology
-
Unknown
-
Mutations in parathyroid hormone receptor
1 (PTHR1), the receptor for parathyroid hormone (PTH) and parathyroid
hormone-related protein (PTHRP), identified in some patients with
Ollier’s disease -
Ollier’s disease usually sporadic but affected families reported
-
Maffucci’s syndrome sporadic
Epidemiology
-
Uncommon
-
Typically diagnosed in childhood
Pathophysiology
Cartilaginous rests remain within normal bone, possibly
due to delay in differentiation resulting from constitutive activation
of Ihh (Indian Hedgehog) signaling secondary to mutant PTHR1. In
Ollier’s disease there is up to a 20% to 33% chance of malignant
degeneration to chondrosarcoma. In Maffucci’s syndrome the risk is
higher; some authors report 100%. Patients with Maffucci’s syndrome are
also at increased
due to delay in differentiation resulting from constitutive activation
of Ihh (Indian Hedgehog) signaling secondary to mutant PTHR1. In
Ollier’s disease there is up to a 20% to 33% chance of malignant
degeneration to chondrosarcoma. In Maffucci’s syndrome the risk is
higher; some authors report 100%. Patients with Maffucci’s syndrome are
also at increased
P.219
risk for the development of acute lymphocytic leukemia (ALL), astrocytoma, and malignancies of the gastrointestinal system.
Classification
-
Ollier’s disease: multiple enchondromatosis
-
Maffucci’s syndrome: multiple enchondromatosis with multiple soft tissue hemangiomas
Diagnosis
Clinical Features
-
Multiple enchondromas (Fig. 7.3-5), frequently unilateral predominance
-
Enlargement of affected bones
-
Deformity (bowing)
-
Especially genu valgum in lower extremities
-
May lead to decreased forearm rotation
-
-
Leg-length discrepancy possible (may be 10 to 25 cm at maturity)
-
In Maffucci’s syndrome, can also see:
-
Cutaneous hemangiomas
-
Pigmented nevi (may be multiple)
-
Vitiligo
-
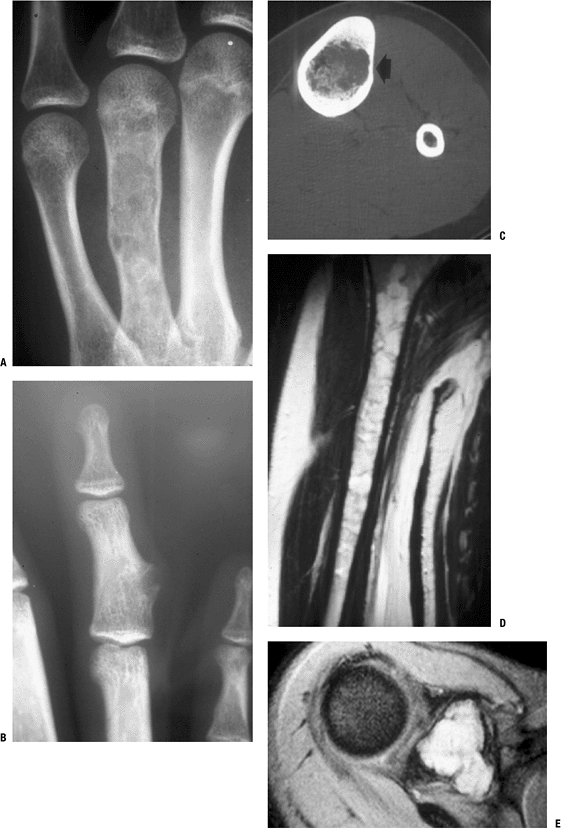
Radiologic Features (see Fig. 7.3-5)
-
Typically metaphyseal (may extend into diaphysis) (see Fig. 7.3-5D)
-
Thinning of cortex
-
Endosteal scalloping
-
May have longitudinal radiolucent “streaks”; “fan-like” septation of metaphyses
-
Usually have hyaline cartilage matrix calcifications (“stippled,” “popcorn,” “rings and arcs”) (see Fig. 7.3-5A)
-
May be completely lytic
-
-
Increased uptake on bone scan
-
May use computed tomography (CT) to better define bony anatomy and assess endosteal scalloping (see Fig. 7.3-5C)
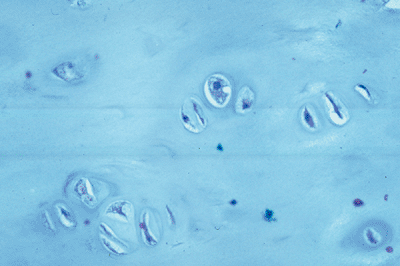
Histologic Features (see Fig. 7.3-6)
-
Hyaline cartilage
-
Relatively hypocellular with small cells with single nucleus
-
Proteoglycan matrix (pale blue staining with hematoxylin and eosin)
-
May be more aggressive-looking, especially in hands, and still be benign
-
This appearance can mimic low-grade chondrosarcoma.
-
Nuclei more pleomorphic, hyperchromatic, and anaplastic
-
Chondrosarcoma more likely in pelvis, proximal femur, proximal humerus
-
Chondrosarcoma much less likely in hand
-
More likely if symptomatic (pain) or increasing in size
-
Treatment
Nonoperative Management
Mainstay of nonoperative treatment is monitoring. X-rays
of known lesions and of pelvis should be obtained on a yearly basis,
more often if symptomatic. It has been recommended that patients with
Maffucci’s syndrome also have periodic imaging of the abdomen and brain
to screen for gastrointestinal malignancies and astrocytoma.
of known lesions and of pelvis should be obtained on a yearly basis,
more often if symptomatic. It has been recommended that patients with
Maffucci’s syndrome also have periodic imaging of the abdomen and brain
to screen for gastrointestinal malignancies and astrocytoma.
Surgical Management
Indications for Surgery
-
Biopsy is indicated for lesions that are
painful or increasing in size or that have worrisome radiologic
features (e.g., rapid growth, excessive thinning, or breakthrough of
cortex). -
If a lesion is symptomatic but not malignant, curettage and bone grafting may be successful.
-
Angular deformities may be treated with osteotomies
-
Leg-length discrepancy may be treated with epiphysiodesis or lengthening (distraction osteogenesis).
-
Lesions that transform into chondrosarcoma require wide resection.
Results and Outcome
Multiple enchondromatosis (Ollier’s disease) may be
disfiguring and impair function. Surgery may be beneficial in that
setting. Surveillance for malignant degeneration of lesions is
required. Risk of malignant degeneration (usually chondrosarcoma) is
approximately 25%.
disfiguring and impair function. Surgery may be beneficial in that
setting. Surveillance for malignant degeneration of lesions is
required. Risk of malignant degeneration (usually chondrosarcoma) is
approximately 25%.
Patients with Maffucci’s syndrome have soft tissue
hemangiomas as well as multiple enchondromas. Risk of malignancy (which
may include visceral malignancies such as brain, ovary, or soft tissue
primaries in addition to chondrosarcoma) may approach 100%. Early
detection is key to survival.
hemangiomas as well as multiple enchondromas. Risk of malignancy (which
may include visceral malignancies such as brain, ovary, or soft tissue
primaries in addition to chondrosarcoma) may approach 100%. Early
detection is key to survival.
Multiple Osteochondromas (Hereditary Multiple Exostoses)
Solitary osteochondromas (exostoses) are the most common
benign bone tumors. Hereditary multiple exostoses (HME) are far less
common. Affected individuals may have dozens or even hundreds of
osteochondromas. HME is inherited in an autosomal dominant pattern with
96% to 100% penetrance. Approximately 10% of cases have no family
history (presumed to be new, sporadic mutation).
benign bone tumors. Hereditary multiple exostoses (HME) are far less
common. Affected individuals may have dozens or even hundreds of
osteochondromas. HME is inherited in an autosomal dominant pattern with
96% to 100% penetrance. Approximately 10% of cases have no family
history (presumed to be new, sporadic mutation).
P.220
 |
|
Figure 7.3-5 A patient with enchondromatosis has ipsilateral enchondroma (A) and periosteal chondroma (B) lesions of the hand, as well as tibial (C), humeral (D), and scapular (E) lesions.
|
P.221
 |
|
Figure 7.3-6 Bland histology of an enchondroma from a patient with enchondromatosis.
|
Pathogenesis
Etiology
-
Genetic defect linked to loci EXT1 (chromosome 8p24.1), EXT2 (11p11-12), and EXT3 (19p)
-
EXT1 and EXT2 encode proteins involved in cartilage metabolism.
-
Thought to act as tumor suppressor genes
whose inactivation leads to exostosis formation and whose multiple
inactivations lead to malignancy
-
-
Autosomal dominant inheritance pattern
-
Variable penetrance in females, possibly due to an X-linked modifying gene
-
-
10% sporadic
Epidemiology
-
1 in 50,000 to 100,000
-
Slight male predominance (1.5:1)
-
Diagnosis of HME often made early in childhood (mean age 3 years) but rare before 2 years of age
Pathophysiology
-
Ectopic foci from physes that then grow independently
-
Mechanical complications and growth disturbances are more common with HME than with solitary osteochondroma.
-
Risk of malignant transformation higher
for HME, estimated at around 3%, than for solitary exostoses. When it
occurs, however, it is usually of low grade. -
Central locations (i.e., pelvis, hips, and shoulders) are more likely to undergo malignant transformation.
Diagnosis
Clinical Features
-
HME is diagnosed at an earlier age than solitary enchondromas, usually within first decade.
-
80% of those with HME will have noticeable exostoses by 10 years of age, and nearly 100% by 12 years.
-
“Knobby” prominences near joints, with the distal femur and proximal tibia being the most common sites
-
Other common sites (in decreasing order
of frequency) include proximal humerus, scapula and ribs, proximal
fibula, distal radius and/or ulna, distal fibula, distal tibia, and
foot.
-
-
Afflicted individuals within a single family can have greatly varying amounts of disease.
-
Shortened stature is seen in more severely affected individuals.
-
With severe involvement, the limbs may appear short relative to the trunk.
-
Genu valgum may be seen, as may radial
head dislocation if the radius develops a marked bow because of a
severely shortened ulna. -
Pain may develop from trauma (exostoses
may fracture), pressure on muscles or nerves, or inflammation of an
overlying bursa (common), or malignant degeneration (rare). -
Rarely, a pseudoaneurysm may develop from irritation of an adjacent blood vessel.
Radiologic Features
-
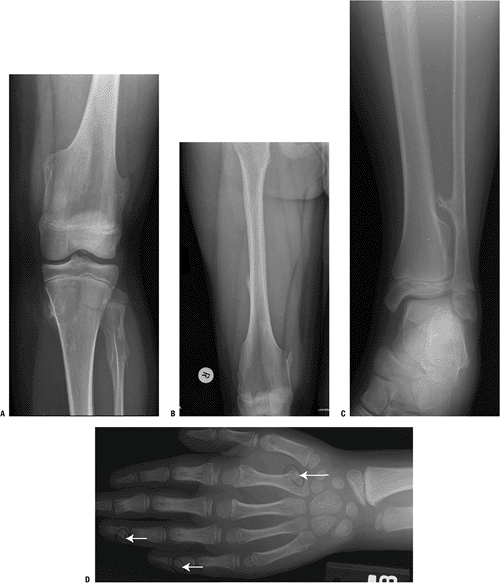
In HME, 90% of the exostoses are sessile (broad-based) and only 10% are pedunculated (narrow base with longer stalk) (Fig. 7.3-7).
-
Pedunculated lesions typically grow away from the adjacent physis.
-
-
Radiographs are diagnostic!
-
Key findings are the continuity of the
cortex of the bone with the cortex of the lesion and the appearance
that the medullary contents of the bone “flow into” and are continuous
with the inside of the lesion.
-
-
Magnetic resonance imaging (MRI) is used to evaluate the cartilaginous cap.
-
In childhood, the cap normally may be up
to 2 to 3 cm thick. In adults, the cap is usually 2 to 3 mm thick,
although up to 1.5 cm is occasionally seen.
-
Histologic Features
The lesion itself looks like normal bone, and the cartilage cap resembles the histology of a physis (Fig. 7.3-8). This histology does not differ from solitary osteochondroma.
Treatment
Nonoperative Management
Observation is the most common treatment for solitary or
multiple exostoses. Fractures of osteochondromas almost always heal
spontaneously. For patients with multiple exostoses, imaging of the
pelvis and scapulae should be obtained at the initial examination as
large lesions in these regions may go clinically undetected.
Thereafter, patients should
multiple exostoses. Fractures of osteochondromas almost always heal
spontaneously. For patients with multiple exostoses, imaging of the
pelvis and scapulae should be obtained at the initial examination as
large lesions in these regions may go clinically undetected.
Thereafter, patients should
P.222
P.223
be followed at yearly intervals. Parents and siblings should also be screened.
 |
|
Figure 7.3-7 Radiographs from a patient with hereditary multiple exostoses show numerous typical osteochondromas about the knees (A,B), ankle (C), and hand (D).
|
 |
|
Figure 7.3-8
Histology section through the cartilage cap of an osteochondroma shows the blue-staining cartilage cap with underlying trabecular bone. |
If an exostosis is painful or enlarging, an MRI should
be obtained to assess the cartilage cap. It is normal for
osteochondromas to grow in children, and malignant degeneration in
children is rare. A cartilage cap of more than 1 cm in an adult is
concerning for chondrosarcoma.
be obtained to assess the cartilage cap. It is normal for
osteochondromas to grow in children, and malignant degeneration in
children is rare. A cartilage cap of more than 1 cm in an adult is
concerning for chondrosarcoma.
Surgical Management
Indications for Surgery
-
Pain (from lesion; most often repetitive trauma, bursa)
-
Deformity (angular or leg-length discrepancy)
-
Functional limitation (e.g., decreased range of motion because of impingement, muscle irritation or tethering)
-
Spinal cord compression (uncommon)
-
Presence of pseudoaneurysm (uncommon)
-
Cosmesis (rare)
-
Rapid enlargement or other signs of malignant change
Surgical Technique
-
In children it is imperative to excise
the entire lesion, including the cap with its perichondrium and the
base and surrounding periosteum, or it will recur. -
In adults, intralesional excision may be adequate.
-
If transformation to chondrosarcoma has
occurred, wide excision is required to prevent recurrence. If there are
no metastases, prognosis is excellent.
Results and Outcome
Recurrence after resection of osteochondroma is related
to incomplete excision of the cartilage cap and is estimated to occur
2% of the time. Secondary chondrosarcomas are typically grade 1 with
good long-term survival and metastatic rates of 3% to 7%. Complication
rates as high as 13% have been reported with excision of benign
osteochondromas.
to incomplete excision of the cartilage cap and is estimated to occur
2% of the time. Secondary chondrosarcomas are typically grade 1 with
good long-term survival and metastatic rates of 3% to 7%. Complication
rates as high as 13% have been reported with excision of benign
osteochondromas.
Suggested Reading
DiCaprio MR, Enneking WF. Fibrous dysplasia: Pathophysiology, evaluation, and treatment. J Bone Joint Surg [Am] 2005;87(8):1848–1864.
Herring JA, ed. Tachdjian’s Pediatric Orthopedics, 3rd ed. Philadelphia: WB Saunders, 2002.
Lewis RJ, Ketchum AS. Maffucci’s syndrome: Functional and neoplastic significance. J Bone Joint Surg [Am] 1973;55:1465–1479.
Parekh SG, Donthineni-Rao R, Ricchetti E, et al. Fibrous dysplasia. J Am Acad Orthop Surg 2004;12:305–313.
Porter
DE, Lonie L, Fraser M, et al. Severity of disease and risk of malignant
change in hereditary multiple exostoses: A genotype-phenotype study. J Bone Joint Surg [Br] 2004;86(7):1041–1046.
DE, Lonie L, Fraser M, et al. Severity of disease and risk of malignant
change in hereditary multiple exostoses: A genotype-phenotype study. J Bone Joint Surg [Br] 2004;86(7):1041–1046.
Schmale GA, Conrad EU, Raskind WH. The natural history of hereditary multiple exostoses. J Bone Joint Surg [Am] 1994;76(7):986–992.
Shapiro F, Simon S, Glimcher MJ. Hereditary multiple exostoses: Anthropometric, roentgenographic, and clinical aspects. J Bone Joint Surg [Am] 1979;61(6):815–824.
Stieber JR, Dormans JP. Manifestations of hereditary multiple exostoses. J Am Acad Orthop Surg 2005;13:110–120.
Unni KK. Dahlin’s Bone Tumors: General Aspects and Data on 11,087 cases, 5th ed. Philadelphia: Lippincott-Raven, 1996.