
group are uncommon, and even when they do occur, they are usually
benign. The primary malignant tumors that occur predominantly in
children are two bone tumors, namely, osteosarcoma and Ewing
sarcoma/peripheral (or primitive) neuroectodermal tumor (EWS/PNET); and
one soft tissue sarcoma, rhabdomyosarcoma. In addition, there are
nonrhabdomyosarcomas, such as congenital and infantile fibrosarcoma in
young children and synovial sarcoma in adolescents. The orthopaedist
must remain alert, because the malignant tumor is an unexpected event,
and its infrequency can result in improper or delayed initial
management. The orthopaedist who sees pediatric patients but is not
prepared to manage a malignant or aggressive benign musculoskeletal
tumor needs to be comfortable with evaluating patients with these kinds
of tumors and deciding which of them should be referred to an
oncologist. This chapter reviews the common bone and soft tissue tumors
of childhood; it discusses how the patients present, what
physical
findings to expect, and what the plain radiographs may show; and it
suggests additional diagnostic and staging evaluations and treatment.
This chapter is not intended to be a definitive text on musculoskeletal
pathology, and includes only the more common tumors of childhood.
has led to dramatic improvements in the survival of children with
previously lethal sarcomas. One of the intriguing aspects of childhood
sarcomas is that, despite similar histologies, stages, and prognostic
factors, some patients respond well to treatment, whereas others seem
to be resistant to chemotherapy. To date, patients with good prognoses
cannot be distinguished from those with poor prognoses except by crude
clinical characteristics, such as the presence of metastatic disease at
diagnosis or the histologic response to preoperative chemotherapy.
Recent molecular findings in sarcomas may shed light on their biologic
behavior and their response to chemotherapy.
is to examine the chromosomes by karyotype analysis. The identification
of recurrent chromosomal abnormalities provides clues regarding sites
of potential gene mutations. Normally there are 23 pairs of chromosomes
in the nucleus of the human cell. Osteosarcomas in general have
multiple, bizarre karyotypic abnormalities: some chromosomes are
missing, some are duplicated, and some are grossly altered. To date,
all studies of high-grade osteosarcomas have shown complex karyotypes
and nonclonal chromosome aberrations superimposed on complex clonal
events (1,2). Low-grade
parosteal osteosarcoma, on the other hand, is characterized by the
presence of a ring chromosome accompanied by few other abnormalities or
none at all. Although it is usually possible to distinguish high-grade
from low-grade osteosarcoma by standard histology, the karyotype
information may be diagnostically useful in the case of other tumors.
In addition to possibly providing prognostic information, the specific
chromosomal aberrations provide clues that assist molecular biologists
who are looking for gene mutations (2).
rhabdomyosarcomas have single chromosomal translocations characteristic
of their respective histologies. In these tumors, part of one
chromosome is transposed to part of another chromosome through a
breakpoint. A novel gene and gene protein product are created that
presumably give the cell a growth advantage. The most common
translocations for these tumors are listed in Table 14.1 (3 4, 5, 6, 7, 8, 9).
the differential diagnosis of round cell tumors. Under the light
microscope, there is little to distinguish one of these tumor types
from another, and although immunohistochemistry helps to a certain
extent, it is at times difficult to be sure of the diagnosis.
Demonstration of these characteristic karyotypic findings makes
pathologists more secure in their diagnosis, and has helped with the
classification of these tumors. To perform a karyotype analysis,
short-term cultures and metaphase spreads are necessary, but these are
labor-intensive and require fresh tissue (10). More recent techniques, with fluorescent in situ
hybridization and reverse transcriptase–polymerase chain reaction,
allow rapid analysis for the presence of translocations; these
techniques can be performed on frozen tissue and sometimes even on
paraffin-embedded tissue (11,12).
Therefore, it is important to give the pathologist appropriate fresh
tissue to be snap frozen to preserve messenger ribonucleic acid (mRNA)
and allow these studies to be performed (13).
establishing the diagnosis. These rearrangements lead to novel proteins
that give the tumor cell a growth advantage. In EWS/PNET, for instance,
a fragment of the EWS gene contains DNA-binding domains of the FLY1
gene. The protein acts by disrupting pathways that regulate DNA
transcription (14). For several years, it was
difficult to make the distinction between EWS and PNET, and clinicians
were not sure whether to treat them differently. The observation that
both Ewing sarcoma, a poorly differentiated mesenchymal tumor of
uncertain cell lineage, and PNET, a tumor believed to be of neural
crest origin, shared the same chromosomal translocation led
pathologists to believe that both were related neuroectodermal tumors (15,16). As noted in Table 14.1,
further studies revealed other translocations in several of these
tumors, each such translocation specifying a different novel protein.
There is debate regarding whether one or the other of these is
associated with a better prognosis, but the treatment strategies used
today are the same for both tumors (17, 18, 19).
A recent study suggests that tumors with the type 1 transcript
(EWS-FLY1) are associated with a better prognosis than those with other
transcripts (20); however, others have disputed this (21).
Using reverse transcriptase–polymerase chain reaction technology, one
can detect small numbers of tumor cells in a bone marrow or peripheral
blood cell population (23,24).
This makes the interpretation of bone marrow aspirates more precise,
and may provide a method for the earlier detection of relapses after
treatment. It is hoped that the gene products of these translocations
can also be used in treatment strategies. Because the novel genes
formed from the translocation make a novel protein that normal cells do
not make, antibodies or targeted T cells can be generated to
specifically kill tumor cells. This is being tried in early-phase
trials of relapsed patients with rhabdomyosarcoma and EWS/PNET, and if
it works, it may be a way of treating patients who fail standard drug
therapy.
give some evidence about the pathogenesis of these tumors, and may have some prognostic and therapeutic import (25,26). Oncogenes are normal cellular genes (protooncogenes) that are necessary for the normal development and functioning of the organism (27).
When they are mutated, they may produce a protein that is capable of
inducing the neoplastic state. Oncogenes act through a variety of
mechanisms to deregulate cell growth. This is obviously a very complex
process and may involve more than one genetic event.
|
TABLE 14.1 CYTOGENETIC FINDINGS IN PEDIATRIC SOFT TISSUE NEOPLASMS
|
||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||||||||||||||||||||||||||||||
The cumulative effect alter proteins that function as growth factors
and their receptors, kinase inhibitors, signal transducers, and
transcription factors (14). The dominant
oncogenes encode proteins that are involved in signal transduction,
that is, in transmitting an external stimulus from outside the cell to
the machinery that controls replication in the cell nucleus. Mutant
cellular signal transduction genes keep the cell permanently “turned
on.” The protein products of oncogenes also function as aberrant growth
factors, growth factor receptors, or nuclear transcription factors.
These types of genes seem to have less of a role in osteosarcomas. One
recently reported exception is amplification of the HER-2/NEU/ERBB-2
protooncogene in patients with breast cancer, which confers a poorer
prognosis. Patients with this amplification are treated with a
monoclonal antibody to this protooncogene [MAb45D5, trastuzumab
(Herceptin)]. Overexpression of HER2-NEU in osteosarcoma has been reported and is associated with advanced disease and poorer prognosis (28). Although this has been disputed by some studies (29, 30, 31),
it provides the potential for treatment strategies in patients with
osteosarcoma who have amplification of HER2-NEU. A current study by the
Children’s Oncology Group is evaluating this strategy in patients at
high risk for metastatic cancer.
They act as brakes rather than as accelerators of growth. Their normal
role is to regulate the cell cycle and keep it in check.
Osteosarcomas are very frequent in patients with hereditary
retinoblastoma, both in the orbit and in the extremities, and are
unrelated to irradiation. It was subsequently learned that
osteosarcomas in these patients, as well as spontaneously occurring
osteosarcomas, carry mutations or deletions of the RB
gene. It was one of the first clues to the finding that osteosarcomas
have a genetic cause. It is estimated that approximately 60% to 75% of
sporadic osteosarcomas either have an abnormality of the RB gene or do not express a functional RB product (26).
The retinoblastoma gene is located on the long arm of chromosome 13
(13q14), and is 200 kb in length. Its product is a 105- to 110-kDa
nuclear phosphoprotein (pRB) that appears to have a cell cycle
regulatory role. The retinoblastoma protein acts as a signal protein,
or gatekeeper, to regulate the cell cycle through the transcription of
genes that mediate the cell cycle. Deactivation of the RB
gene or absence of pRB allows cells to enter the cell cycle in an
unregulated fashion, a condition that imparts a growth advantage to the
affected cell. It should be noted that one copy of the gene is
sufficient for a normal phenotype. A child born with a normal allele
and a mutant or absent allele will not manifest retinoblastoma until
some event occurs in retinoblasts to alter the normal allele. If both
copies become deranged, the normal check on the cell cycle disappears,
and the conditions for the neoplastic state are met. There are several
other mechanisms by which the function of the RB protein can be
altered; for instance, viral proteins may bind to the RB protein and
inactivate it.
Located on the short arm of chromosome 17 (17p), its product is a
nuclear phosphoprotein that has a cell cycle regulatory role similar to
that of the Rb protein. As in the case of RB,
inactivation of p53 gives the cell a growth advantage, probably because
of loss of cell cycle regulation. The p53 phosphoprotein may be
inactivated by a variety of mutations, including a single base change
(point mutation) that increases the half-life of the protein, allelic
loss, rearrangements, and deletions of the p53
gene. Each of these mechanisms can result in tumor formation by loss of
growth control. The p53 protein functions as an extremely important
cell cycle checkpoint that blocks cells with DNA damage until they can
be repaired, or directs damaged cells into apoptosis (programmed cell
death) if they cannot be repaired. Cells lacking this checkpoint can
accumulate successive genetic abnormalities and possibly become
malignant. It is estimated that approximately 25% of osteosarcomas have
detectable mutations of the p53 gene (42).
it binds to regions of other genes (DNA), and controls the expression
of genes responsible for cell cycle control (cell growth), apoptosis
(programmed cell death), and other metabolic functions, such as control
and repair of DNA damage. In concert with Rb and a variety of other
proteins, p53 acts to regulate the cell cycle through a complex cascade
of enzymes, in which Rb probably plays the central role. Apoptosis has
recently become recognized as an important mechanism by which
chemotherapy and radiotherapy kill cancer cells. p53 is involved in
this process, and appears to arrest cell division after sublethal
damage (e.g., by radiation), to give the cell time to repair DNA
defects before the next division (43, 44, 45).
If repair does not take place, the cell undergoes apoptosis and dies.
If p53 is not functional, the cell may survive and accumulate genetic
defects, leading to malignant transformation. Osteosarcomas have been
shown to have a variety of mutations of the p53 gene (46, 47, 48, 49, 50, 51, 52, 53, 54, 55).
Preliminary evidence suggests that overexpression of mutant p53 protein
(detected by immunohistochemistry) or loss of heterozygosity of the p53 gene is related to human osteosarcoma (56,57).
studied 211 adult soft tissue sarcomas by immunohistochemistry, using
monoclonal antibodies to mdm-2 and p53, and demonstrated a correlation
between overexpression of mdm-2/p53 and poor survival rates. Patients
without mutations in either gene (mdm-2/p53-) had the best survival
rates, those with one mutation (either mdm-2+/p53- or mdm-2-/p53+) had
intermediate rates of survival, and those with mutations in both genes
(mdm-2+/p53+) had the lowest survival rates. Another mechanism in which
p53 protein can be inactivated is by viral proteins that bind and
inactivate both RB and p53 protein (62).
patients with sarcomas, but mutations may also be present in all
somatic cells (germline mutations) in patients with heritable cancer (63, 64, 65, 66, 67, 68, 69, 70).
Although such defects do not appear to be common in the general
population, germline p53 mutations are present in patients who are part
of a familial cancer syndrome. These families have a variety of
cancers, often at an early age, and osteosarcomas and soft tissue
sarcomas are a fairly common occurrence in these kindreds.
Identification of patients with p53 germline mutations can be useful in
determining which patients in an affected family are at risk for
developing cancers, but much more work is needed in the area of genetic
counseling to determine how best to use this information. A recent
study showed that germline mutations were present in approximately 3%
to 4% of children with osteosarcoma, and that the detection of these
mutations was more accurate than family history in predicting the
family’s susceptibility to cancer (71).
possibility is that the p53 mutations may be potential biologic markers
of prognosis and response to treatment (chemotherapy). There is some
preliminary evidence that p53 mutations in the tumor may portend a
worse prognosis in osteosarcoma. More recently, the association of p53
with apoptosis has suggested possible strategies for chemotherapy, on
the basis of the status of the p53 pathway (44,45).
Gene therapy (replacing the missing or mutated gene by transfection
with viral carriers) is often discussed, but there are major technical
hurdles to overcome before this technology can be used for treating
cancers in humans. However, it might be possible to make tumor cells
more antigenic, or to make them more sensitive to antineoplastic drugs,
by gene transfer. Another strategy would be to alter normal cells to
make them less sensitive to damage by chemotherapeutic agents.
Currently, these techniques pose technical challenges, but they offer
realistic promise for the near future.
biology of sarcomas is multidrug resistance (MDR). MDR probably
explains why some patients respond to chemotherapy and others do not.
Drug resistance may be intrinsic (present at diagnosis) or acquired
(appearing after treatment of a tumor) (72,73).
At least four basic mechanisms of drug resistance are now recognized
under the category of the MDR phenotype. They are (a) changes in
glutathione metabolism, (b) alterations in topoisomerase II, (c)
non-P-glycoprotein (P-gp)-mediated mechanisms, and (d) P-gp-mediated
mechanisms (1,2). Recent evidence has suggested that P-gp may be of particular relevance to osteosarcoma.
MDR-1 is one member of the aneurysmal bone
cyst (ABC) superfamily of genes that encode membrane transport
proteins; these proteins function as unidirectional membrane pumps
using adenosine triphosphate hydrolysis to work against a concentration
gradient. P-gp is a 170-kDa protein that is located in the cell
membrane and functions as an energy (adenosine triphosphate)-requiring
pump that excludes certain classes (amphipathic compounds) of drugs
from the cell. This physiologic mechanism is believed to be important
in certain organ systems, such as the blood-brain barrier, placenta,
liver, kidney, and colon, for ridding the cell of unwanted toxins, but
it is also responsible for actively excluding chemotherapeutic agents,
such as Vinca alkaloids, anthracyclines,
colchicine, etoposides, and taxol (many of which are active in
osteosarcoma protocols) from the cancer cell. Another feature of the
P-gp mechanism that may have some relevance to therapeutic strategies
is that some classes of drugs can reverse the MDR phenotype by blocking
the action of the pump. These drugs include verapamil, cyclosporin A,
tamoxifen, and others.
(25% to 69%) display the MDR phenotype at diagnosis, and that relapsed
sarcomas show higher incidence and intensity of MDR expression (76, 77, 78, 79, 80, 81).
Because of the small numbers of patients in these studies, and the
variety of the methods by which MDR expression was tested, comparisons
of the studies and an accurate determination of the incidence of MDR
expression are difficult to accomplish. In addition, the age of the
patient and the type of sarcoma appear to be related to the incidence
of detectable P-gp at diagnosis. One study showed that osteosarcomas
have a higher incidence of MDR than other types of adult sarcomas (82). Serra et al. (78) demonstrated that overexpression of P-gp protein was evident in 23% of primary and 50% of metastatic osteosarcomas.
reported on 92 patients with nonmetastatic osteosarcoma of an extremity
who had been treated with chemotherapy and surgery. The study
demonstrated that an immunohistochemically determined expression of
P-gp predicted a decreased probability of the patient’s having an
event-free survival, and was more accurate in prediction than
histologic response to preoperative chemotherapy. A more recent study
failed to find a relation between MDR-1 mRNA expression and outcome in
patients treated for osteosarcoma (84).
protocols in human osteosarcoma. The drug-resistant tumor is becoming
better identified as one that has a poor histologic response to
preoperative chemotherapy and that expresses P-gp. Undoubtedly, it is
more complex than this, and other mechanisms will pertain. Several
caveats exist. One is the complexity of defining the resistant tumor.
Preoperative chemotherapy requires 10 to 12 weeks to provide an
estimate of histologic necrosis, unless ways can be found to accurately
predict percentage of necrosis by positron emission tomographic (PET)
scans, thallium scans, and/or gadolinium-enhanced magnetic resonance
imaging (MRI). Detection of P-gp at diagnosis is difficult, and no one
method has proven superior. It is probably not sufficient to
demonstrate the presence of P-gp; also important is whether the pump is
functioning to exclude cytotoxic agents from the tumor cell. Ideally,
one would like to reverse the action of the P-gp mechanism but, just as
there are no new agents to rescue patients who show poor histologic
response, the agents currently available to reverse MDR are of limited
benefit. They are potentially problematic in that they make normal
cells less tolerant of chemotherapy, and thereby increase toxicity; and
in other tumors they have not proven to be effective (73,75). The future probably lies in developing more effective reversing agents and in defining other drug-resistant mechanisms.
a bone or soft tissue mass includes neoplasia, infection, and trauma.
Infection and trauma are more common than neoplasia, and one of these
is usually the explanation of a mass or abnormality seen on a
radiograph. However, the possibility of neoplasia should not be
forgotten. The consequences of the mismanagement of a patient with a
musculoskeletal tumor can be grave (Fig. 14.1).
patient with a musculoskeletal tumor. The characteristics of the pain
can help determine the diagnosis. Ask the patient: Where is the pain?
How did it begin? Is it sharp, dull, radiating, or constant? Is it
associated with activity? Is there a particular activity that makes the
pain worse? What makes the pain better? Does it wake you at night? Is
the intensity of the pain increasing, staying the same, or diminishing?
chondroblastoma, and chondromyxofibroma) usually have a mild, dull,
slowly progressive pain that is worse at night and aggravated by
activity. Patients with malignant musculoskeletal tumors complain of a
more rapidly progressive symptom complex, not specifically related to
activity, which often awakens them at night. Occasionally, the pain
pattern is diagnostic. The pain of an osteoid osteoma is so typical
that the diagnosis should be strongly suspected from the history. This
pain is a constant, intense pain that is worse at night, and it is
almost always relieved by aspirin or nonsteroidal antiinflammatory
drugs (NSAIDs). The
pain
caused by a Brodie abscess is similar to that of an osteoid osteoma,
but the Brodie abscess pain is rarely relieved by aspirin.
 |
|
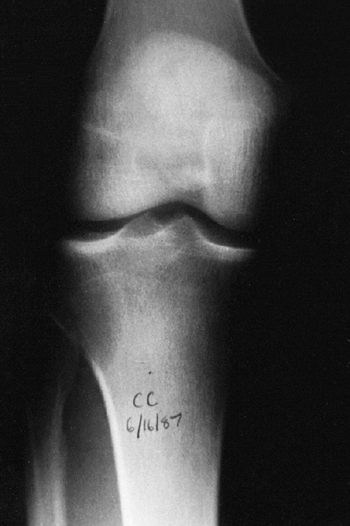
Figure 14.1
Anteroposterior radiograph of the knee of a young man who complained of it “giving way.” The orthopaedist who saw the patient suspected a derangement, and the patient eventually had arthroscopic surgery. A radiolucent lesion can easily be seen in the lateral aspect of the proximal tibial metaphysis and epiphysis. This giant cell tumor of bone was missed because the physician did not consider this diagnosis when he was examining the patient or the radiograph. By the time the tumor was recognized, it had grown so large that resection and allograft reconstruction were required. Had it been treated when this radiograph was taken, a curettage and bone graft packing, or polymethyl methacrylate packing, could probably have been done. |
a traumatic event. The specific nature of the trauma and the relation
of the trauma to the current symptoms must be evaluated thoroughly.
Trauma without a definitive fracture may be the explanation for an
abnormal radiograph, but it should not be assumed to be the
explanation, even for a periosteal reaction, unless the history is
perfectly consistent. With the increased level of organized sports for
children, there has been an increase in the incidence of fatigue
fractures, and these fractures can be confused with neoplasias. Still,
one should be cautious about ascribing a lesion to trauma.
questioned about the specifics of the injury that produced the
fracture. Most lesions that lead to a pathologic fracture are easily
recognized on a plain radiograph, but occasionally they may not be
obvious. When the traumatic event seems insignificant, a pathologic
fracture should be suspected. Patients should be asked about symptoms,
no matter how minimal, that they experienced before the fracture. Most
aggressive benign tumors and malignant tumors produce pain before the
bone is weakened enough to fracture. Inactive benign tumors such as
unicameral bone cyst (UBC) and nonossifying fibroma (NOF) are almost
always asymptomatic until the bone breaks.
but inquiries should be made. Has the child had a previous fracture?
Has the child had other illnesses? Have radiographs been taken
previously? Do not assume that the patient or the family will volunteer
significant past medical history. Ask specific questions.
decreased appetite, irritability, and decreased activity. Most patients
with musculoskeletal tumors do not have systemic symptoms, and the
presence of a systemic illness should alert the physician to the
possibility of an underlying generalized disorder or osteomyelitis.
Patients with Ewing sarcoma may have elevated temperature, weight loss,
and malaise, but this is the exception rather than the rule. Even
children with large primary malignant musculoskeletal tumors usually
appear healthy. Patients with cancer do not always present with obvious
signs of the underlying malignancy.
symptoms. If the patient is younger than 5 years of age, the mass is
usually noticed first by a parent. The parent often thinks that the
mass appeared overnight, but this is rarely the case. Teenagers may
report the presence of a mass, but often only after a few weeks or
months of waiting for it to resolve spontaneously. Painful soft tissue
masses are most often abscesses. Most soft tissue tumors, even the
malignant ones, do not produce significant symptoms until they are
large. Although most of the soft tissue masses seen in children prove
to be benign, all soft tissue masses, even those in children, should be
considered to be malignant tumors until proven otherwise. The
consequences of mistaking a malignant soft tissue tumor for a benign
tumor can be devastating, whereas the consequences of approaching a
benign tumor as if it were a malignancy are minimal.
those in the pediatric age group, should have a complete physical
examination. Not only can important information be gained about the
specific disorder being evaluated, but other significant abnormalities
may be found.
Café-au-lait
lesions of the skin are a clue that the patient has fibrous dysplasia
or neurofibromatosis. Numerous hard, nontender, fixed masses near the
ends of long bones are diagnostic of multiple osteochondroma.
Exophthalmos and otitis media indicate that the patient has
Hand-Schüller-Christian disease.
gait pattern should be recorded, muscular atrophy measured, and
abnormalities in the vascular supply and motor and sensory innervation
noted. The range of motion of the adjacent joint should be measured. If
there is a mass present it should be measured, and the presence of
erythema, tenderness, pulsations, bruit, or increased temperature
should be noted.
subcutaneous tissues, are usually benign. These can be felt best when
lubricant is applied on the overlying skin. Firm-to-hard, fixed or
tethered, tender masses, especially those deep to the superficial
fascia, are more likely to be malignant, but neurofibroma, deep lipoma,
and cyst are usually firm to the touch. Transilluminate the mass; if
light is transmitted more easily through the mass than through the
surrounding tissue, the mass is a fluid-filled cyst.
least anteroposterior and lateral plain radiographs. Good-quality plain
radiographs (at least two views, preferably at 90 degrees) are
necessary. The entire lesion must be observed. The radiograph should be
reviewed systematically. Look at the bone, all of it, and every bone on
the radiograph. Ask yourself these questions: Is there an area of
increased or decreased density? Is there endosteal or periosteal
reaction, and if there is, what are the characteristics of the
reaction? Is there cortical destruction? Is it localized or are there
multiple defects? Is the margin in the tumor well defined or poorly
defined? Is there a reactive rim of bone surrounding the lesion? Are
there densities within a radiolucent lesion? Is the bone of normal,
increased, or decreased overall density? Is the joint normal? Is there
loss of articular cartilage? Is the subchondral bone normal, thick, or
thin? Are there abnormalities in the bone on both sides of the joint?
Are there intraarticular densities? Is there a soft tissue mass? Are
there calcifications or ossifications in the soft tissue? If one looks
specifically for abnormalities, it is unlikely that an abnormality will
be missed. The pelvis and the scapula are exceptions to this rule.
Large tumors involving the pelvis or the scapula, even those with
marked destruction of bone, can be extremely difficult or impossible to
see on a plain radiograph. If there is a suggestion that the patient
has a pelvic or a scapular tumor, bone scanning and computerized axial
tomography (CT) scan or MRI are recommended.
-
Where is the tumor? This refers to the
lesion’s anatomic location: long bone or flat bone; epiphyseal,
metaphyseal, or diaphyseal; and medullary canal, intracortical, or
surface. -
What is the tumor doing to the bone? Is there erosion of the bone, and if so, what is the pattern?
-
What is the bone doing to the tumor? Is there periosteal or endosteal reaction? Is it well-developed? Is it sharply defined?
-
Are there intrinsic characteristics
within the tumor that indicate its histology? Is there bone formation
by the tumor? Is there calcification? Is the lesion completely
radiolucent?
glance, but a detailed study of all tissue present. Do not forget to
specifically examine the soft tissues visible on the radiograph.
obtaining the history, performing a physical examination, and examining
the plain radiograph. When the specific diagnosis is made from these
examinations, additional studies are requested only if they are
necessary for treatment. Often, specific treatment can be planned from
only the history, physical examination, and plain radiographs. For
example, a 16-year-old boy with a hard, fixed mass in the distal femur
that has been present for 9 years and has not increased in size for
more than 1 year complains of pain after direct trauma to this mass.
Plain anteroposterior and lateral radiographs confirm the clinically
suspected diagnosis of osteochondroma. Further evaluation to make the
diagnosis is not necessary, but if surgical resection is elected as the
treatment, CT scan or MRI may be useful in planning the operative
procedure.
possible to limit the differential to three or four diagnoses, and
appropriate additional evaluations can be requested. CT scan, MRI,
nuclear bone scanning (technetium, gallium, thallium, and indium), and
PET may reveal findings that are diagnostic, or that provide the
information required for planning a subsequent biopsy. For example, a
10-year-old boy complains of mild knee pain that has been present for 3
months, has loss of knee flexion, and on the lateral radiograph of the
distal femur there is a bone density lesion attached to the posterior
femoral cortex. From this information, the lesion is recognized as
either a parosteal osteosarcoma or an osteocartilaginous exostosis. A
technetium-99 bone scan, with increased activity in the area of the
lesion, does not distinguish between these two, but both CT scan and
MRI allow one to distinguish between a parosteal osteosarcoma and an
osteocartilaginous exostosis. A parosteal osteosarcoma is attached to
the cortex of the bone, whereas an osteocartilaginous exostosis arises
from
the
cortex and has a medullary canal continuous with the medullary canal of
the bone. CT scan or MRI is crucial in the evaluation of a patient in
this clinical setting.
values are usually normal. Only a few musculoskeletal disorders are
associated with abnormal laboratory values. The erythrocyte
sedimentation rate (ESR) is nonspecific but sensitive. Patients with
infections or malignant tumors usually have an elevated ESR, but
patients with benign disease should have a normal value. A normal ESR
value can increase the physician’s confidence that a suspected benign,
inactive lesion is just that. A markedly elevated value (>180mm per
hour) supports a diagnosis of infection and may be just what is needed
to justify an early aspiration of a bone or soft tissue lesion.
Patients with active benign or malignant musculoskeletal tumors,
particularly those with Ewing sarcoma, often have an elevated ESR, but
it is rarely greater than 80 mm per hour. C-reactive protein is another
serum value that indicates systemic inflammation. It increases and
returns to normal more quickly than ESR.
the body, but the bones and the hepatobiliary system are the
predominant sources. In the pediatric age group, conventional
high-grade osteosarcoma is associated with elevated levels of serum
alkaline phosphatase (86). Not all patients
with osteosarcoma have elevated levels of serum alkaline phosphatase,
and therefore a normal level does not exclude osteosarcoma from the
diagnosis. A minimal elevation can be observed with numerous processes,
even a healing fracture. Adults with elevated levels of serum alkaline
phosphatase secondary to bone disease are most likely to have Paget
disease of bone or diffuse metastatic carcinoma. Patients with a
primary liver disorder have elevated levels of serum alkaline
phosphatase as well, but they also have elevated levels of serum
5-nucleotidase and leucine aminopeptidase, and glutamyl transpeptidase
deficiency. The levels of 5-nucleotidase and leucine aminopeptidase are
not elevated in primary bone tumors.
measured, especially if a metabolic bone disorder is suspected. Serum
lactate dehydrogenase (LDH) level is elevated in some patients with
osteosarcoma, and patients with Ewing sarcoma with elevated levels of
the enzyme have a worse prognosis (87, 88, 89).
Elevated LDH levels may also indicate relapse in a patient who has been
treated for these tumors. Patients entering chemotherapy treatment
protocols will need to have LDH levels determined in order to stratify
them on the protocol. Other laboratory determinations are not helpful
and are not recommended.
In addition, bone scanning is the most practical method of surveying
the entire skeleton. Technetium-99 attached to a polyphosphate is
injected intravenously, and, after a delay of 2 to 4 hours, the
polyphosphate, with its attached technetium, concentrates in the
skeleton proportional to the production of new bone. A disorder that is
associated with an increase in bone production increases the local
concentration of technetium-99 and produces a “hot spot” on the scan.
The technetium bone scan can be used to evaluate the activity of a
primary lesion, to search for other bone lesions, and to indicate
extension of a lesion beyond what is seen on the plain radiograph. The
polyphosphate–technetium-99 compound also concentrates in areas of
increased blood flow, and soft tissue tumors usually have increased
activity compared with normal soft tissues. The technetium-99 bone scan
can be used to evaluate blood flow if images are obtained during the
early phases immediately after injection of the technetium-99. The
polyphosphate–technetium-99 is cleared and excreted by the kidneys, so
the kidneys and the bladder
have
more activity than other organs. The technetium-99 scan is sensitive
but nonspecific. The principal value of a radionuclide scan is as a
means of surveying the entire skeleton for clinically unsuspected
lesions. In approximately 25% of cases of Langerhans cell histiocytosis
and plasmocytoma, the bone scan is normal, or there is decreased
activity at the site of the lesion (90, 91, 92).
 |
|
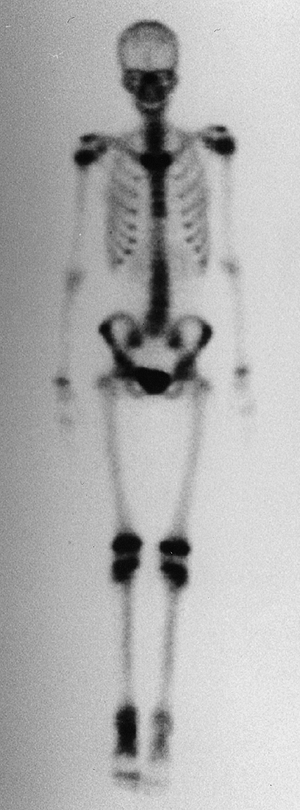
Figure 14.2
An anterior view of a whole body technetium-99 bone scan. This patient has an osteoid osteoma of her talus, and there is increased activity in the talus. There is also increased activity in the distal tibia, which is thought to be a reaction to the local increased blood flow. Technetium-99 bone scanning is an efficient means of evaluating the entire skeleton of a patient with a bone lesion. It is important to have the entire skeleton scanned, rather than limit the scan to a small part of the skeleton. |
This examination takes longer to perform (24–72 hours) than
technetium-99 scanning (2 hours), but it is believed to be useful in
the evaluation of musculoskeletal tumors (94)
because it can help differentiate a musculoskeletal infection from a
neoplasia. Gallium scans are most useful when evaluating a patient
suspected of having an occult infection. Scanning should be performed
before a surgical procedure, because the site of an operative procedure
will show increased uptake on the radionuclide scan.
Because thallium uptake correlates well with the vascular supply of a
tumor, decreased thallium uptake after therapy is a reflection of tumor
necrosis, which is the best evidence that the chemotherapy is working.
Thallium scans have not proved sufficiently specific to be clinically
useful in the evaluation of patients with tumors.
Fluoro-2-deoxy-D-glucose (FDG) PET is the type of PET used most
frequently for the musculoskeletal system. Because there is a
differential uptake of FDG between neoplastic tissue and normal tissue
(neoplastic tissue has greater uptake), it is possible to identify
neoplastic tissue on a PET scan. The role of PET in the evaluation and
monitoring of patients with musculoskeletal neoplasia is under
investigation. PET with fluorine-18-FDG has proved particularly useful
in evaluating patients with lymphoma (98,99).
improved the evaluation of bone and soft tissue tumors. The anatomic
location and extent of the tumor could be determined accurately. The
improved accuracy of anatomic localization means that less radical
surgery can be performed safely. Often a specific diagnosis can be made
or a suspected diagnosis can be confirmed after seeing the CT scan.
Smaller nodules are seen on whole-lung CT scans than are seen with
plain chest radiographs or whole-lung linear tomographs. With CT scan
the abdomen can be evaluated thoroughly without surgical exploration.
scan is not asking specific questions of the radiologist. Radiologists
do not know what specific information the orthopaedist wants; only if
specific questions are asked is the maximum value of a CT scan
realized. A specific differential diagnosis should be made from the
presentation and plain radiographs. Only then can a decision be made
regarding whether to request CT scan, MRI, both, or neither. Ask the
radiologist to determine the lesion’s location and its density and
vascularity, and to search for intralesional characteristics that may
provide a diagnostic clue. Request the radiologist to include the
contralateral normal extremity on the CT scan for comparison.
is called its “attenuation coefficient” and is measured in Hounsfield
units (HU) (Fig. 14.3). The density of water is
0 HU; tissues more dense than water have a positive value, and tissues
less dense than water have a negative value. The vascularity of a
lesion can be evaluated by measuring the increase in the attenuation
coefficient of a lesion after intravenous infusion of contrast, and
comparing this increase to that in an adjacent muscle. Normal muscle
has an attenuation coefficient of approximately 60 HU, and increases 5
to 10 HU with a bolus of intravenous contrast. Fat has an attenuation
coefficient of approximately 60 HU, and cortical bone usually has a
value of more than 1000 HU.
scanners and is less anxiety-producing, so sedation is less likely to
be needed when compared with MRI. CT scan is most useful in the
evaluation of small lesions in or immediately adjacent to the cortex
(e.g., osteoid osteoma on the surface) and lesions with fine
mineralization or
calcifications
(e.g., chondroblastoma). Percutaneous biopsies of musculoskeletal
lesions can be performed with the assistance of localization obtained
with CT scan. For all other situations, MRI has replaced CT scan.
 |
|
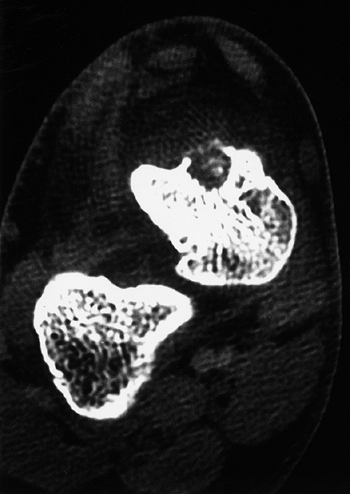
Figure 14.3
The density of a lesion can be measured on a computerized tomographic scan. This cortical bone lesion is an osteoid osteoma. The radiologist can measure the actual density of the lesion and provide information regarding the type of tissue. The measurements are made in Hounsfield units, named after a developer of computed tomography. Zero Hounsfield units is the density of water. Negative units indicate densities lower than that of water (fat measures approximately -70 Hounsfield units), and positive units indicate densities greater than that of water (cortical bone measures higher than 1000 Hounsfield units). |
proved to be the most useful tool in the evaluation of musculoskeletal
lesions. MRI produces images of the body in all three planes (axial,
sagittal, and coronal) as easily as in a single plane, and poses no
known hazards to the patient.
converts the reactions of tissue hydrogen ions in a strong magnetic
field excited by radio waves. By adjusting excitation variables, images
that are T1- and T2-weighted are obtained. A variety of techniques have
been used to produce images of improved quality compared with routine
T1- and T2-weighted images. The use of gadolinium as an intravascular
contrast agent allows one to judge the vascularity of a lesion, thereby
providing even more information about the tumor. Fat-suppression images
with gadolinium enhancement are often especially useful in
demonstrating a soft tissue neoplasia. As with CT scan, it is important
for the orthopaedist requesting MRI to discuss the case with the
radiologist. The radiologist can then determine the optimal MRI
settings required for visualizing the lesion.
physical examination and plain radiography for evaluating a
musculoskeletal lesion. The ability to view the lesion in three planes,
determine its intraosseous extent, see the soft tissue component
clearly, and have an idea of the tissue type from one diagnostic test
makes MRI a powerful tool (100). Unfortunately,
variations in technique mean that it is important that the examination
be planned carefully if the maximum information possible is to be
obtained (see Chapter 3). The radiologist must
understand what questions need to be answered from the MRI. As a rule,
it is important that the image be viewed while the scan is being done,
in order to ensure that the entire lesion is examined. T1-weighted
(with and without gadolinium), T2-weighted, and fat-suppression
techniques are the minimal images needed.
the basis of the extent of their tumor and its potential for
metastasis. These groups are called stages.
Grouping patients by their stage helps the physician predict a
patient’s risk of local recurrence and metastasis. This facilitates
making treatment decisions about individual patients and helps in the
comparison of treatment protocols. Staging systems are based on the
histologic grade of the tumor, its size and location, and the presence
of regional or distant metastases. The presence of a metastasis at the
time of presentation is a bad prognostic sign and, regardless of other
findings, puts the patient in the highest-risk stage. For patients
without metastases at presentation, the histologic grade of the tumor
is the principal prognostic predictor. Size is next in importance.
Higher histologic grade and larger tumors are associated with the worse
prognoses.
musculoskeletal tumors. The task force on malignant bone tumors of the
American Joint Commission on Cancer Staging and End Result Studies
published a staging system for soft tissue tumors in 1977 (101), which was revised in 1987 (102,103).
This staging system is based on the histologic grade (G), local extent
or size (T), whether the nodes are involved (N), and metastases (M).
The tumors are separated into three histologic grades (G1, low grade;
G2, medium grade; G3, high grade) and two sizes (T1 for less than 5 cm,
T2 for equal to or greater than 5 cm). Patients with nodal involvement
are designated N1, and those without nodal involvement are designated
N0. Patients with metastatic disease are designated M1, and those
without metastatic disease are designated M0. There are four stages,
with subclasses in each stage. Tumors at stage I are associated with
the best prognosis, and tumors at stage IV with the worst prognosis (Table 14.2).
system. This system is used more often by orthopaedists involved in the
management of patients with musculoskeletal tumors. It was designed to
be simple, straightforward, and clinically practical. The tumors are
separated into only two histologic grades (I, low grade; II, high
grade) and two anatomic extents (A, intracompartmental; B,
extracompartmental). Patients with metastatic disease in either a
regional lymph node or a distant site are grouped together as stage
III. Each bone is defined as its own separate anatomic compartment. The
soft tissue anatomic compartments are defined as muscle groups
separated by fascial boundaries (Table 14.3). There are five stages in this system (see Table 14.4).
introduced four terms to indicate the surgical margin of a tumor
resection. These terms are in common use, and provide a means of
describing the relation between the histologic extent of the tumor and
the resection margin. The surgical margins are defined as intralesional, marginal, wide, and radical.
An intralesional margin is the surgical margin achieved when a tumor’s
pseudocapsule is violated and gross tumor is removed from within the
pseudocapsule. An incisional biopsy and a curettage are two common
examples of an intralesional margin. A marginal surgical margin is
achieved when a tumor is removed by dissecting between the normal
tissue and the tumor’s pseudocapsule. This is a surgical margin
obtained when a tumor is “shelled out.” A wide surgical margin is
achieved when the tumor is removed with a surrounding cuff of normal,
uninvolved tissue. This is often referred to as en bloc resection. A radical surgical margin is achieved when the tumor and the entire compartment
(or compartments) are removed together. This usually is accomplished
only with an amputation proximal to the joint that is just proximal to
the lesion (e.g., an above-knee amputation for a tibial tumor). As a
rule, benign lesions can be managed with an intralesional or marginal
surgical margin, but malignant tumors require a wide surgical margin.
Radical surgical margins are reserved for recurrent tumors and the most
infiltrative malignancies.
|
TABLE 14.2 REVISED AMERICAN JOINT COMMISSION STAGING SYSTEM FOR SOFT TISSUE SARCOMA
|
|||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||||||||||||||||||||||
patient with a bone or soft tissue tumor, and it should be performed
only after careful planning (105, 106, 107).
Often biopsy proves unnecessary after the patient has been thoroughly
evaluated, the diagnosis having been made by the clinical setting and
radiographic findings. When a biopsy is required, the prebiopsy
evaluation improves the chance that adequate and representative tissue
will be obtained, the least amount of normal tissue will be
contaminated, and the pathologist will make an accurate diagnosis.
Biopsies performed without an adequate prebiopsy evaluation are more
likely to produce unsatisfactory results.
|
TABLE 14.3 ANATOMIC COMPARTMENTS IDENTIFIED FOR STAGING OF MUSCULOSKELETAL TUMORS
|
||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||||||||||||||||||||||||||||
suspected by the physician after the evaluation, or to determine which
diagnosis, from among a limited differential diagnosis, is correct. In
addition to providing confirmation for a specific diagnosis, the tissue
obtained must be sufficient for histologic grading. It must be
representative of the tumor and, because many musculoskeletal tumors
are heterogeneous, the specific site from which the tissue is taken is
important. The surgeon who is willing to assume the surgical management
of the patient, regardless of the diagnosis, should perform a biopsy on
the patient’s tumor. The biopsy incision and the tissue exposed during
the biopsy must be excised with the tumor, if a wide surgical
margin
resection proves to be necessary. If the surgeon who performs the
resection has planned and performed the biopsy, the patient has a
better chance of limb salvage and less risk of local recurrence (106).
The surgeon should consult with the radiologist and the pathologist
before performing the biopsy to get their suggestions for the best
tissue to obtain. Discussing the case with the pathologist before the
biopsy also allows the pathologist to be better prepared when he or she
is expected to make a diagnosis from a frozen section.
|
TABLE 14.4 STAGING OF MUSCULOSKELETAL TUMORS
|
|||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||||||||||||
Usually, they can be performed without general anesthesia or hospital
admission, thereby saving the patient expense as well as the need for
general anesthesia. The needle track may be seeded with tumor, and
should be excised at the time of the definitive resection. Needle
biopsy and fine-needle aspirate biopsy must be planned just as open
biopsy is planned, and the responsible surgeon should decide how the
biopsy is to be performed. Needle biopsy and fine-needle aspirate
biopsy are most useful for lesions whose clinical presentations are
diagnostic, and when treatment is either nonsurgical or requires
presurgical therapy. Although an experienced pathologist can usually
make the correct diagnosis from a well-done needle biopsy or a
fine-needle aspirate biopsy, more mistakes are made with these
techniques than with open biopsy, and histologic grading can be
difficult or impossible without open biopsy (108,109,111).
treatment, especially limb-salvage resection. The skin incision and
deep dissection should be made so that they can be resected with the
tumor at the time of definitive limb-salvage operation. Longitudinal
skin incisions are better than transverse skin incisions. The
dissection should be as limited as possible, flaps should not be
raised, and neurovascular bundles should not be exposed. The dissection
should be through a muscle, not between muscles. The tumor’s
pseudocapsule and a portion of the tumor should be excised as a block
and sent to the pathologist. A frozen section analysis should be done,
even when there are no plans for immediate additional surgery. The
pathologist should be certain that adequate tissue is available for
diagnosis. Only when a biopsy is performed on dense bone is it
impossible to obtain a frozen section analysis. The pathologist should
set aside tissue for subsequent examination with an electron
microscope. Some tissue should be kept frozen in the event that
immunohistochemistry is required.
should be deflated before closing the wound, and complete hemostasis
must be ensured. The tumor should be manipulated as little as possible.
Do not use a compressive bandage to exsanguinate the extremity, but
rather elevate the extremity for 3 to 5 minutes before inflating the
tourniquet.
closing the wound. The hematoma from the biopsy may contain tumor
cells, and will require resection if surgery is the treatment. The
wound can be drained, but the exit site of the drain must be in line
with the incision and close to it. The drain track will need to be
resected with the tumor and the biopsy incision if a wide surgical
resection is required.
incisional biopsy, is indicated. An excisional biopsy is appropriate
when the lesion is small, and can be excised with a cuff of normal
tissue. An excisional biopsy may be appropriate even when a major
resection is required. If the preoperative evaluation strongly supports
the diagnosis of a malignancy, particularly one for which a frozen
section analysis will be difficult to do, an excisional biopsy should
be considered. The choice between an incisional biopsy and an
excisional biopsy is usually easy to make. A clinically obvious
exostosis on the proximal tibia should have an excisional biopsy, if it
is to be performed at all. A surface osteosarcoma, diagnosed on the
basis of the results of plain radiography and CT scan or MRI, can
undergo an excisional biopsy with a resection. A large aggressive
lesion within the distal femur and invading the adjacent soft tissues
should undergo incisional biopsy. This decision is more difficult when
the evaluation reveals a small, active, possibly low-grade malignancy
on the proximal humerus or the distal radius. An incisional biopsy
exposes uncontaminated tissues to the tumor, and if the tumor proves to
be a malignancy, the definitive resection is more complicated. If the
lesion can be treated with curettage or a marginal excision, the
incisional biopsy leads to the least functional loss. The final
decision is made for each patient on the basis of not only the tumor’s
characteristics but also the patient’s preference. Some patients want
to take the fewest chances, and are willing to accept the possibility
of slight overtreatment, whereas others choose to take one step at a
time. It is the surgeon’s responsibility to explain the situation to
the patient so that an informed decision can be made.
pathologist is able to examine the entire lesion, thereby improving the
accuracy of the pathologic examination. Musculoskeletal tumors are
often heterogeneous, and the amount of tissue obtained with an
incisional biopsy is always limited. It can be particularly difficult
to distinguish active benign cartilage tumors from low-grade
chondrosarcomas. When the entire lesion, especially its connection with
the adjacent bone and soft tissue, is seen, the distinction is made
more easily.
biopsy: osteomyelitis is more common than bone tumors, especially in
children, and osteomyelitis often mimics neoplasia. The reverse is also
true; therefore, when performing a biopsy, even when the diagnosis
seems obvious, culture the tumor and biopsy the infection.
musculoskeletal pathology text, and only those tumors that are common
are discussed. The authors have tried to confine
the discussion to pertinent information regarding the tumors, their evaluation, and their treatment.
and from Garre osteomyelitis. It is a benign tumor and accounts for 11%
of the benign bone tumors in Dahlin’s series from the Mayo Clinic (113).
The patient is usually a young boy (boys are affected more commonly
than girls, at a ratio of 3:1; 80% of the patients are between 5 and 24
years of age at the time of their initial symptoms) with intense pain
at the site of the lesion.
causing the pain, which is sharp, piercing, worse at night and, almost
without exception, completely relieved by aspirin or NSAIDs. The pain
is not related to activity. The relief obtained with aspirin and NSAIDs
is most likely the result of their ability to block the action of
prostaglandins. If a patient has the typical pain of an osteoid
osteoma, but it is not relieved by aspirin, the diagnosis of an osteoid
osteoma should be doubted. The patient may have pain before any
abnormality appears on the plain radiograph, and often the patient has
had an electromyogram, a myelogram, or an arthrogram in an attempt to
find the cause of the pain, before the typical plain radiographic
changes are seen. Some patients are suspected of having a psychosomatic
disorder before the osteoid osteoma is found. A technetium-99 bone scan
will show increased uptake before changes appear on the plain
radiographs.
 |
|
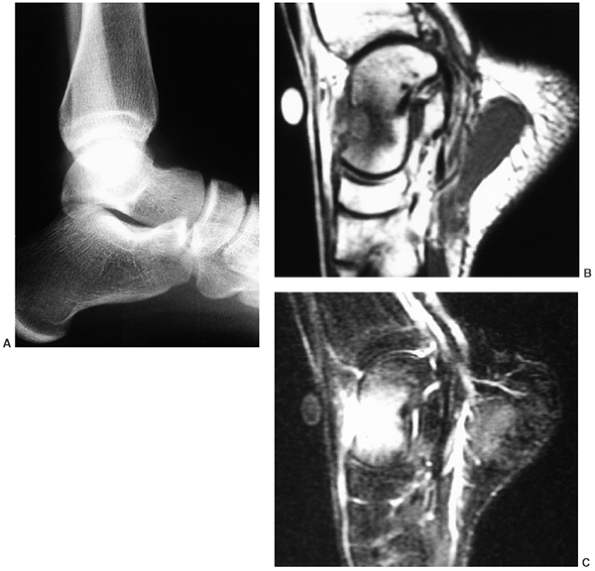
Figure 14.4 A:
Plain radiograph, lateral view, of an 18-year-old woman’s ankle. She complained of severe pain for 6 months, which was totally relieved by aspirin. There is a small erosion in the anterior neck of her talus. (Her computed tomography scan is seen in Fig. 14.3.) B: The sagittal view of a T1-weighted magnetic resonance image shows the lesion in her anterior talus. C: The T2-weighted magnetic resonance image reveals the extensive edema that is characteristic of osteoid osteoma. |
them are found in the femur or the tibia. The other half are
distributed throughout the rest of the skeleton (Fig. 14.4). The proximal femur is a common site. It is also
a site at which it may be difficult to find the lesion. Young patients
with persistent pain in the groin, the middle thigh, or the knee should
be suspected of having an osteoid osteoma. The other common location of
an occult osteoid osteoma is the spine. When osteoid osteoma arises in
the spine, it is usually located in the posterior elements (Fig. 14.5).
Osteoid osteoma in the spine does not elicit a significant bony
reaction, and is very difficult to see on plain radiographs. The
patient presents most commonly with a painful scoliosis (114,115).
When a patient with scoliosis complains of back pain, osteoid osteoma
should be considered. A technetium-99 bone scan is particularly useful
when the clinical presentation suggests an osteoid osteoma but the
lesion cannot be found on the plain radiograph.
physical examination, with the exception of scoliosis in patients with
osteoid osteoma of the spine. The child may walk with a limp and have
atrophy of the extremity involved. If the bone with the osteoid osteoma
can be palpated directly, it will be tender. Local erythema and
increased temperature are not seen, and joint motion is normal. Serum
and urine laboratory values are normal.
radiograph is of dense reactive bone, and is usually diagnostic. The
lesion itself (the nidus, less than 15 mm in diameter) is radiolucent,
but is often not seen on the plain radiograph because of the density of
the intense bone reaction that surrounds it. The nidus may be on the
surface of the bone, within the cortex, or on the endosteal surface.
Lesions on the endosteal surface have less reaction than lesions within
or on the cortex. The lesion and the reaction are associated with
increased uptake on the radionuclide study (technetium-99 bone scan) (116). The nidus is best demonstrated by a CT scan (117).
The distance between the CT scan sections should be small (1 to 2 mm),
so that the nidus is not missed. The window settings of the CT scanner
should be adjusted so that the dense reaction around the lesion does
not obscure the small, low-density nidus. When the nidus is found, it
helps to have the distance from a bony landmark to the nidus measured
on the scan, so that the nidus can be found at the time of surgical
removal. MRI has been used to examine osteoid osteoma, and though the
diagnosis may be suspected, the associated edema and reaction make the
diagnosis less specific than with CT scan.
 |
|
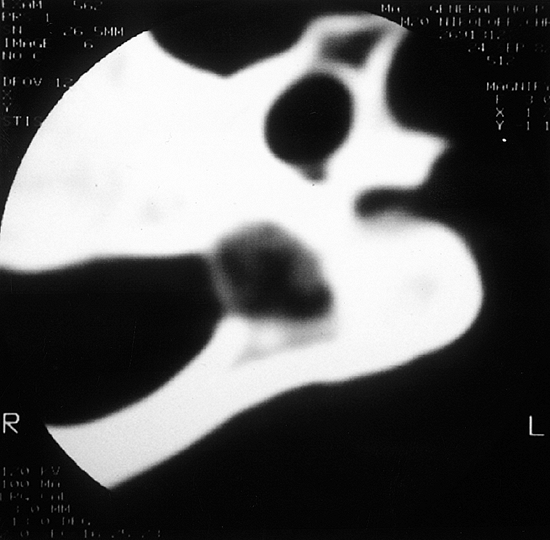
Figure 14.5
Computerized tomography scan cut through a lesion in the pedicle of a teenager with neck pain. The lesion has all of the characteristics of osteoid osteoma. It could not be seen clearly on the plain radiograph, but there was a discrete focus of increased uptake on a technetium-99 bone scan. |
red and surrounded by dense white bone. The nidus is small, usually not
more than 5 to 10 mm in diameter. A lesion that is identical
histologically to the nidus of an osteoid osteoma, but larger than 2
cm, is called an osteoblastoma. The nidus is composed of numerous vascular channels, osteoblasts, and thin, lacelike osteoid seams (Fig. 14.6). Multinucleated giant cells may be seen, but are not common.
healing lesion that eventually involutes over a period of years, and
the nidus completely ossifies. When this occurs the pain of an osteoid
osteoma can resolve spontaneously (118).
Occasionally, a patient uses aspirin or NSAIDs to control the symptoms
until the pain disappears, but most often the intensity of pain, the
time it takes for the lesion to heal spontaneously, and the amount of
medication required are not tolerable, and the patient elects to have
surgery. Kneisl and Simon (119) treated 24
patients with osteoid osteoma. Thirteen were operated on immediately,
and all had complete relief of pain. Nine others were treated with
NSAIDs. Of these, three subsequently elected to have surgery, but the
six others also eventually became free of pain (an average of 33
months). Complete removal of the nidus relieves the patient’s pain.
Partial removal may provide temporary relief, but the pain usually
returns (120). Only the nidus needs to be excised. The reactive bone around the nidus does not have to be removed.
The procedure is performed under general anesthesia, but usually can be
done without hospitalization. By using CT scan to control placement, a
needle biopsy is performed to confirm the diagnosis. Then, through the
same needle track a radiofrequency electrode with an internal
thermistor is placed in the nidus. After the procedure, the patient’s
activities were not limited after the heat ablation. There have been no
complications. Other closed methods of treatment have been reported (124).
The conventional method is a block resection of the nidus and most of
the surrounding reactive bone. The other
method
is a curettage of the nidus. The advantage of the block resection is
the assurance that all of the nidus is removed, but this technique
requires removal of a segment of the cortex and produces a marked
reduction in the strength of the bone. The defect created by the
excision may need to be corrected by bone grafting, and the patient’s
extremity may need to be protected for an extended period. The
advantage of the curettage technique is that the bone is not weakened
significantly, and bone grafting is not required. However, with
curettage it is more difficult to be certain that all of the nidus is
removed.
 |
|
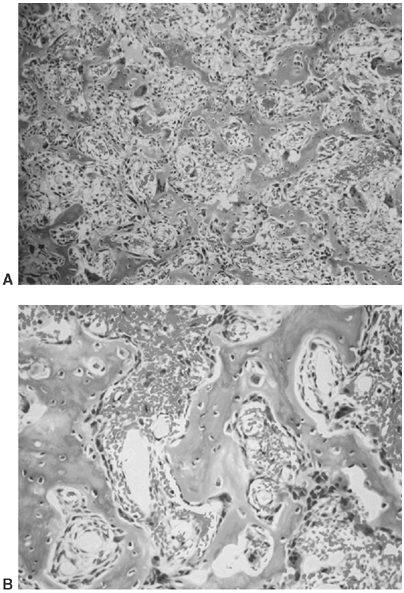
Figure 14.6 A:
Typical histologic appearance of an osteoid osteoma. There is immature (woven) bone lined with osteoblast. Between the woven bone is a vessel-rich fibrous stroma. There is no atypia, and the few mitotic figures are normal (10 × magnification). B: Higher magnification (40 ×) of the histology of the osteoid osteoma shown inA. The woven bone lined with osteoblast is easily seen. The red blood cells indicate the intense vascularity that is typical of this lesion. |
must be accurately localized preoperatively, and seen intraoperatively;
when this is not possible, block excision is preferred. Intraoperative
radionuclide scanning and intraoperative tetracycline-fluorescence
demonstration have been reported as methods of finding the nidus in the
operating room and assuring the surgeon of its complete removal (125, 126, 127, 128).
We have not found these techniques to be necessary. Preoperative
planning and careful localization of the nidus is the most important
means of ensuring that the nidus can be found during the operation.
because it is histologically identical to osteoid osteoma, but larger.
Unlike osteoid osteoma, osteoblastoma is not surrounded by dense
reactive bone. Cementoblastoma of the jaw is histologically identical
to osteoblastoma. Osteoblastoma is less common than osteoid osteoma,
accounting for less than 1% of the primary bone tumors in Dahlin’s
series (113).
life (50% of the patients are between 10 and 20 years of age, although
the age range is from 5 to 35 years) with back pain (approximately 50%
of the lesions are in the spine). The pain of an osteoblastoma is not
as severe as the pain of an osteoid osteoma, and aspirin or NSAIDs do
not have such a dramatic effect. There are no physical findings that
are characteristic of osteoblastoma. When the tumor is in the spine,
the patient has decreased motion of the spine in the area involved.
Osteoblastomas are tender, and direct palpation often localizes a
lesion even when it cannot be seen on a plain radiograph.
patient often has a limp and mild atrophy, and complains of pain
directly over the lesion. Laboratory examinations of blood and urine
show normal results. The appearance of osteoblastoma on a radiograph is
variable. It is usually a mixed radiolucent, radiodense lesion, more
lucent than dense. There is minimal reaction in the surrounding bone.
Lesions in the spine may be difficult or impossible to see when
initially examining the plain radiograph, but when located by other
studies, the subtle abnormality on the plain radiograph can usually be
appreciated.
the location of an osteoblastoma are an irregular cortex, loss of
pedicle definition, and enlargement of the spinous process. As with
osteoid osteoma, a technetium-99 bone scan is the best method of
localization. On a radionuclide scan an osteoblastoma shows increased
uptake, and technetium-99 bone scanning is an excellent method of
initially screening a patient suspected of having an osteoblastoma. In
lesions of the spine, although the bone scan localizes the lesion, it
is not specific enough to help in the planning of a surgical resection.
diagnosis and extent of the lesion. On the CT scan, the lesion usually
“expands the bone” and has intralesional stippled ossifications and a
high attenuation coefficient (100 HU or more).
bone and adjacent structures. A wide surgical resection is preferred
when practical, but an extended curettage is sufficient for most cases.
The surrounding bone should be removed to the extent possible. Most
osteoblastomas are controlled by the extended curettage, but recurrence
is not uncommon, and some can be locally aggressive. It is difficult to
give a percentage risk of local recurrence, but in our experience, it
is less than 10% of cases. Although irradiation has been tried in the
management of these patients, there is little evidence that it is of
benefit.
nidus of an osteoid osteoma. There should not be abnormal mitoses,
although mitotic activity may be observed. There are osteoblasts,
multinucleated giant cells, seams of osteoid, and a rich vascular bed.
Schajowicz and Lemos (129) suggested that a subset of osteoblastoma be termed malignant osteoblastoma.
They believe that this subset has histologic features that are worse
than those of the usual osteoblastoma, is more aggressive locally, and
is more likely to recur after limited surgery. Rarely, an osteoblastoma
metastasizes but still meets the histologic definitions of a benign
tumor, although it should probably be classified as low-grade
osteosarcoma.
spindle cells produce bone. There are two major variants that have
significantly different clinical presentations and prognoses. The more
common osteosarcoma is called classic high grade, or conventional, and
the other is juxtacortical. Some authors separate juxtacortical
osteosarcomas into parosteal and periosteal. Less common variants of
osteosarcoma (e.g., intracortical, soft tissue, radiation-induced,
Paget disease) are not discussed in this text.
the patients present during the second decade of life; more than 75%
are between 8 and 25 years of age) with symptoms of pain and a mass
around the knee. In approximately half of the patients, the lesions are
located in the distal femur or the proximal tibia. The proximal
humerus, proximal femur, and pelvis are the next most common sites. The
pain precedes the appreciation of the mass by a few weeks to 2 or 3
months. Boys and girls are affected with equal frequency. The patient
does not have systemic symptoms, and the patient usually feels well.
The mass is slightly tender, firm-to-hard, and fixed to the bone but
not inflamed. The adjacent joint may have mild restriction of motion
except in the rare (less than 1%) patient who presents with metastases
or multiple focal osteosarcoma. One-half of all patients have elevated
serum alkaline phosphatase (extremely high serum alkaline phosphatase
values indicate a worse prognosis), and approximately one-fourth of all
patients have elevated serum LDH level (an elevated LDH level also is
associated with a worse prognosis). The rest of the laboratory values
for blood and urine are normal.
diagnostic. The typical lesion is located in the metaphysis, involves
the medullary canal, is both lytic (radiolucent) and blastic
(radiodense), and has an extraosseous component and a periosteal
reaction suggestive of a rapid growth (Codman triangle or sunburst
pattern) (Figs. 14.7 and 14.8).
Many osteosarcomas have a soft tissue component, of a fluffy density
suggestive of neoplastic bone, adjacent to the more obvious bone
lesion. Those osteosarcomas that consist primarily of cartilage or
fibrous tissue are almost purely radiolucent. Telangiectatic
osteosarcoma, a histologic variant of classic high-grade osteosarcoma,
may be mistaken on a radiograph for an ABC or a giant cell tumor. This
will not be a clinical problem for the pathologist if adequate clinical
information is provided by the surgeon.
osteosarcoma. The extent of the lesion, especially the intraosseous
component, is more clearly defined by MRI. The lesion can be seen in
all three planes, and its soft tissue extension is easily appreciated.
It is critical that the entire bone be included on at least one plane
(usually the coronal view). The tumor should be viewed with at least a
T1-weighted (with and without gadolinium) image, a T2-weighted image,
and a fat-suppressed image.
 |
|
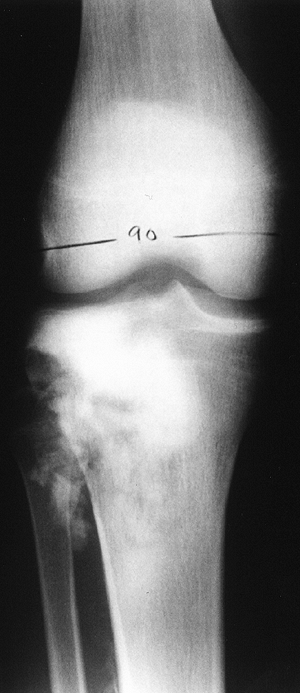
Figure 14.7
Anteroposterior plain radiograph of an 18-year-old man with an osteosarcoma of his proximal tibia. There is increased density in the proximal tibia associated with cortical destruction and extraosseous bone formation. Biopsy was confirmatory. |
 |
|
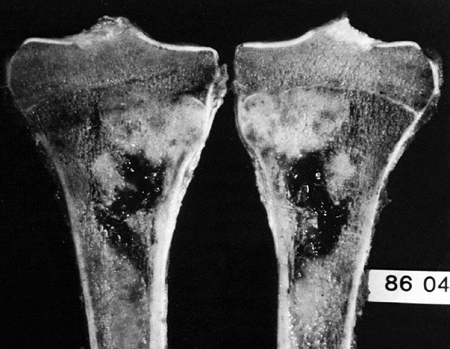
Figure 14.8
Classic high-grade osteosarcoma of the proximal tibia. The tibia was bisected for examination. The tumor is composed of an osteoblastic component in the metaphysis, which is up to, and just through, the epiphyseal plate; there is also a more distal cystic component. The tumor has penetrated the cortex, and has a small extracortical component. The patient had not received preoperative chemotherapy, but was treated successfully with limb salvage resection and knee arthrodesis. The patient received postoperative adjuvant chemotherapy, and has been continuously free of disease for 4 years. |
surgical margin, and the anatomic extent of the tumor is the principal
determinant of what operation will be required. MRI is the best method
of determining the anatomic extent of an osteosarcoma. The relation of
osteosarcoma to the major neurovascular bundle should be determined.
The muscles that have been invaded by the soft tissue component should
be identified. Involvement of the adjacent joint must be looked for,
the intraosseous extent measured, and the presence of metastasis noted.
Talking to the radiologist before MRI is performed helps to ensure that
all this information is obtained.
because a relatively high percentage (approximately 10%) of patients
present with pulmonary metastasis.
the area of the tumor. Occasionally it is useful in determining the
intraosseous extent, although MRI is more accurate. More importantly,
technetium-99 bone scanning is an excellent screen of the entire
skeleton for occult bone lesions. This screening process is the most
important reason for obtaining a bone scan. On rare occasions a lung
metastasis is seen on the bone scan, but usually a hot spot in the
chest on the bone scan is secondary to involvement of a rib.
osteosarcoma, and each is graded for the degree of malignancy. The
histologic type is determined by the predominant cell type of the
tumor. Although initially it was thought that the different types had
distinct prognoses, it is now recognized that if matched for size and
histologic grade, all types have the same prognosis. Even
telangiectatic osteosarcoma, which was originally described as having a
particularly poor prognosis, is thought to have the same prognosis as
the other classic high-grade osteosarcomas.
fibroblastic, mixed, and telangiectatic osteosarcomas. These tumors are
graded on a scale of either 1 to 3 or 1 to 4. The higher the histologic
grade, the worse the prognosis. Most osteosarcomas are grade 3 or 4,
and of the mixed type. The tumor is composed of a mixture of neoplastic
cells, but must contain malignant spindle cells making osteoid.
Atypical mitoses are common, and small areas of necrosis are usually
seen (Fig. 14.9).
adjuvant chemotherapy and surgical resection. The standard protocol
consists of chemotherapy (neoadjuvant; usually three or four courses of
a multidrug regimen), then surgical resection, and finally additional
chemotherapy (Fig. 14.10). The entire treatment
takes almost 1 year. The surgical resection can almost always be done
without an amputation of the extremity, and less radical surgery is
being performed now compared with only a few years ago. Picci et al.
demonstrated that patients whose tumors have more than 90% necrosis
after preoperative chemotherapy do not require as wide a surgical
margin as patients whose tumors show less necrosis (130,131).
The use of neoadjuvant chemotherapy has not produced increased survival
compared with postoperative adjuvant chemotherapy, but it does make
surgery easier, and gives the pediatric oncologist a predictor of the
patient’s chance of survival.
osteosarcoma are doxorubicin (Adriamycin), high-dose methotrexate, and
cisplatin. Most chemotherapy protocols include these three drugs in
various dosage schedules, in addition to one or more other drugs. The
development of granulocyte-stimulating factor (GSF) to counteract bone
marrow suppression has allowed intensification of the treatment with
fewer complications; GSF is now used routinely. Overall survival has
increased to more than 60%,
with even better survival rates being reported for patients with greater than 90% necrosis of the tumor after chemotherapy (132, 133, 134, 135, 136, 137, 138, 139, 140, 141, 142).
 |
|
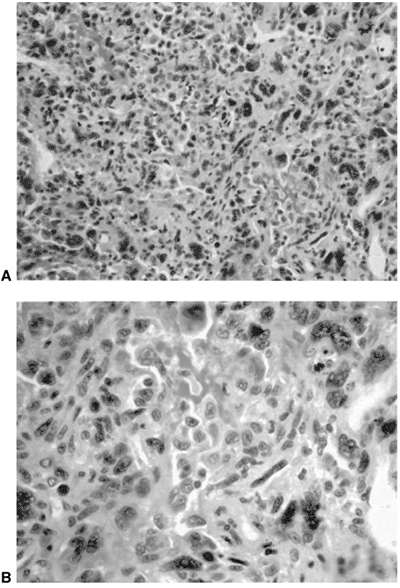
Figure 14.9 A:
Typical histologic appearance of an osteoblastic osteosarcoma. There is immature bone being formed from cells that vary in size, shape, and amount of nuclear material. These findings are typical of malignant cells (10 × magnification) B: Higher magnification (40 ×) of the osteosarcoma in (A). The nuclear detail is more clearly seen, and the bone seemingly coming directly from these bizarre cells makes the diagnosis of an osteosarcoma. |
largest of osteosarcomas. Amputation is done in fewer than 20% of all
cases (142, 143, 144, 145, 146). The accepted incidence of local recurrence is between 5% and 10%, which does not seem to increase the incidence of death (145). This is an area of concern, however, because it appears that most patients with local recurrence die of osteosarcomas (143,145).
One explanation is that local recurrence is a sign of a more aggressive
tumor, not solely the consequence of poor surgery. That being said,
however, the insistence on wide margins is paramount.
method of treatment for patients with pathologic fractures. There is an
increased incidence of local recurrence if limb-salvage resection is
performed, but this increased incidence of local recurrence does not
seem to increase the risk of death (143, 147, 148, 149, 150).
The usual treatment of a patient with a pathologic fracture and
osteosarcoma is to treat the fracture closed, give neoadjuvant
chemotherapy, and perform limb salvage if negative surgical margins can
be obtained.
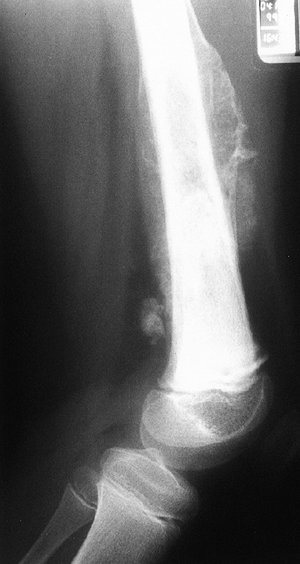 |
|
Figure 14.10
This plain radiograph is a lateral view of the distal femur of a patient who has had standard preoperative chemotherapy. The original lesion had a large extraosseous component that has been reduced in size, and there has been “maturing” of the periosteal reaction. The patient’s pain diminished, and the range of motion in her knee returned to normal. |
external surface of the bone behave differently from those that arise
from within the medullary canal. They are less aggressive locally, have
less potential for distant metastasis, and occur less commonly than
conventional osteosarcoma. There seem to be two distinct juxtacortical
osteosarcomas, parosteal and periosteal; neither is common, and how
distinct they are from each other remains a topic of debate. Parosteal
osteosarcoma is most commonly located in the posterior aspect of the
distal femur, and is composed of bone and low-grade malignant fibrous
tissue. Periosteal osteosarcoma is more often located in the diaphysis,
and is composed of bone and cartilage with malignant spindle cells
were the first to describe parosteal osteosarcoma. They postulated two
distinct types of lesions, a benign parosteal bone-forming tumor, and a
malignant bone-forming tumor. However, all
osteosarcomas
are defined as malignant, and have the potential to metastasize. The
patient’s age at presentation varies over a greater range (10 to 45
years) than in classic high-grade osteosarcoma, and the median age at
presentation tends to be slightly higher (152, 153, 154, 155, 156).
The patient usually reports a painless mass that blocks motion in the
adjacent joint. This is most often knee flexion, because the posterior
distal femur is the most common site of a juxtacortical osteosarcoma (113).
Occasionally the patient has a mild, dull ache in the area of the
tumor, but the symptoms are minimal. The mass is fixed, hard, and
nontender. The adjacent joint may have limited passive and active
motion because of the mechanical block from the tumor. Inflammation is
not observed. The laboratory values of the patient’s blood and urine
are normal.
The lesion arises from the cortex, which may be normal or thickened.
The juxtacortical osteosarcoma often wraps around the bone, with the
periosteum between the tumor and the underlying cortex. This growth
pattern (wrapping around the bone) produces the “string sign” on the
plain radiograph, with a thin radiolucent line between the lesion and
the cortex of the bone. The lesion itself is dense, and has the pattern
of bone. There is increased uptake on a technetium-99 bone scan. The
appearance of the lesion on a CT scan is characteristic, and
distinguishes a juxtacortical osteosarcoma from an exostosis (157).
Juxtacortical osteosarcoma is attached to the cortex growing out into
the soft tissue and may invade the cortex, but the normal cortex is
intact (152,153,156).
An exostosis arises from the cortex, and the cortex of the normal bone
becomes the cortex of the exostosis, with the medullary canal of the
bone communicating with the medullary canal of the exostosis. This
relation, and also the intraosseous extension of the tumor, are seen
more easily with MRI than with CT scan.
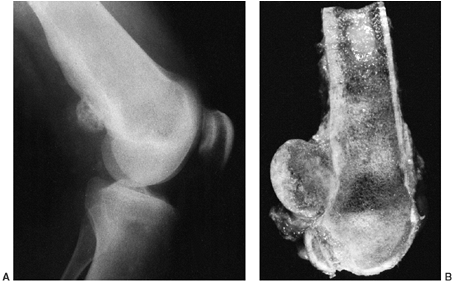 |
|
Figure 14.11 A:
Lateral radiograph of the distal femur and knee of a patient with a parosteal osteosarcoma. The posterior distal femoral cortex is thickened and slightly irregular. The radiodensity adjacent to the posterior cortex is the central portion of the parosteal osteosarcoma. Surrounding this bony mass is a nonossified component of the tumor, composed primarily of fibrous tissue, but with some cartilage. This patient was treated with limb-salvage wide resection of the distal femur, and underwent reconstruction with an osteoarticular allograft. No chemotherapy was used, and the patient has remained free of disease for 5 years. B: The parosteal osteosarcoma is larger than it appears on the plain radiograph. The cap of fibrous tissue and cartilage can be seen covering the bony center. The tumor is attached to the cortex, but does not extend through it. This gross relation is similar to that of an exostosis, and may lead to a mistaken histologic diagnosis. The gross difference between an exostosis and a parosteal osteosarcoma is that the stalk of an exostosis is cortical bone that blends with the cortex of the host bone, and the medullary canal of the stalk and host bone are connected (see Fig. 14.13B). The parosteal osteosarcoma, conversely, is attached to the cortex, but the cortex of host bone is intact, and the medullary canal does not communicate with the parosteal osteosarcoma. |
be difficult to interpret and, on the basis of histology alone, the
lesion may be mistaken for an exostosis. This is particularly true when
juxtacortical osteosarcoma is not suspected by the clinician, or when
the pathologist does not examine the radiograph. This lesion, more than
most other lesions, is diagnosed by its clinical and radiographic
presentation,
and is confirmed by histology. An excisional biopsy is recommended in
most cases. The lesion is composed of regularly arranged bone, with a
background of usually bland spindle cells and fibrous tissue. A
cartilage cap is often present. Parosteal osteosarcomas are graded
histologically on a scale of 1 to 3 (155).
Higher-grade lesions, especially those with medullary involvement, have
a greater risk of metastasizing (usually to the lung) than those of
lower grade without medullary extension (154, 158, 159, 160, 161, 162).
preoperative studies, a wide excisional biopsy is recommended. The
cortical margin should be generous and the tumor pseudocapsule should
not be disturbed. When a lesion from the posterior distal femur is
resected, the neurovascular bundle can usually be freed from the lesion
without dissecting the pseudocapsule, but the posterior capsule of the
knee and the posterior aspect of the femoral condyle must usually be
resected with the tumor. Those lesions that wrap around the bone and
show gross invasion of the medullary canal may require a resection that
includes the entire end of the bone.
the lesion without an amputation. Most patients do not need adjuvant
chemotherapy because the cure with surgery alone is approximately 80% (152,153,155,156,163,164).
Patients with histologic grade 3 lesions probably should receive
adjuvant chemotherapy, especially if the medullary canal has been
invaded. However, the relevant data are limited (155).
It is most common in the anterior proximal tibia, and it tends to be
diaphyseal in location. It has a sunburst appearance on the plain
radiograph, and is composed of malignant cartilage and bone (167). A wide surgical resection is recommended (154,165,166,168).
The role of adjuvant chemotherapy is controversial; as with parosteal
osteosarcoma, probably only patients with grade 3 lesions should
receive it.
merely observed. Others are successfully treated with a curettage. In
general, when a curettage is indicated it is best to visualize the
cavity thoroughly and carefully remove the entire tumor under direct
visualization. The use of a high-speed burr can help assure complete
removal of all tumor cells and is especially advised for giant cell
tumor of bone. The use of a local adjuvant such as phenol or a laser is
controversial but common. In children, malignant bone tumors, with the
exception of lymphoma, should be surgically resected. The surgical
resection of Ewing sarcoma is controversial, but if resection is opted
for the principles discussed apply. The goal of surgical treatment of
these malignant bone tumors is to resect all of the tumor. To
accomplish this some of the adjacent normal tissue must be removed
because the tumor infiltrates these tissues. The extent of involvement
of the normal tissue is impossible to determine preoperatively,
although MRI is a reasonably accurate method of determining the extent
of the tumor. The greater the amount of adjacent tissue removed, the
less likely the patient is to have a local recurrence; therefore as
much adjacent tissue as is practical should be removed. Although the
goal of treatment is to eliminate local recurrence, it is not practical
to try to guarantee that local recurrences never happen, because such
an approach would lead to excessive surgery for most patients without a
proven benefit in survival.
should be removed, it is useful to first determine what is the minimal
amount of tissue that must be removed in order to completely remove the
tumor as shown on an MRI. Then add as much additional adjacent tissue
to the resection as possible without changing the functional impact of
the surgery. For example, if 15 cm of a distal femur must be removed,
it is just as functional to replace 25 cm (i.e., a 10 cm bone margin)
as it is to replace 17 cm (which would be only a 2 cm margin). If
adjacent muscle has been invaded to the extent that what remains is not
functional, all of the muscle should be removed. If no additional
adjacent tissue can be removed without impacting on the patient’s
functioning, either an adjuvant is needed, or the patient should
undergo the more functionally impacting resection.
extremities, but the decision is often difficult because the surgical
margin achieved with an amputation would almost always be much better
than the one obtained with a limb-salvage resection. In a limb-salvage
operation, local recurrence is more common, the time to recovery is
longer, the complexity of the surgery is greater, there are more local
complications, the chance of needing additional surgery is increased,
and the safe level of physical activity is lower as compared to an
amputation, but the patient retains his or her own foot or hand. Since
the introduction of adjuvant chemotherapy, especially preoperative
(neoadjuvant) chemotherapy, and the availability of CT scanning and
MRI, limb-salvage surgery has become more common. The materials used in
reconstruction, and the surgeons’ experience with these materials and
techniques, have improved to such an extent that limb-salvage
resections have become commonplace in most medical centers. Nowadays, a
patient rarely needs to undergo an amputation for a sarcoma of the
extremity.
surgical resection will remove so much tissue that the remaining limb
will be less useful than a prosthesis. To make this decision the
patients have to indicate how they want to use the extremity. The more
sedentary the patient,
the
greater the amount of tissue that can be removed in limb-salvage
surgery without amputation becoming the better option, and vice versa.
In general, bone, joints, arteries, veins, and muscles can be removed
and still leave the extremity functional. Even the need to resect a
major nerve is not in itself an indication for an amputation. It is
when a combination of these tissues, including a major nerve, has to be
resected that amputation should be seriously considered. Each patient’s
situation and preferences should be considered individually.
adults. For children, however, the surgeon encounters problems
including growth, small size, and (it is hoped) greater longevity, all
of which make reconstruction more challenging. The options for limb
salvage include osteoarticular and intercalary allografts, metallic
prostheses, and vascularized and nonvascularized autograft transplants.
All of these are used at various times by the tumor surgeon.
Rotationplasty is another option somewhere between limb salvage and
amputation; it is occasionally useful in very young patients (100,169,170).
bone reconstruction is necessary. Very young children with bone tumors
of the foot and ankle are usually best treated by amputation of the
ray, or Syme amputation, or below-knee amputation. Soft tissue sarcomas
at these sites may be resectable without amputation, if it is possible
to achieve wide margins with or without adjuvant irradiation. The use
of a metatarsal head in the tibial stump (to prevent stump overgrowth)
has been described, and seems to be useful (171,172).
growth of the opposite extremity and the limb-length inequality that
follows (173). For the child who is within 2 or
3 years of completion of growth, this is not a significant issue.
Fortunately, most malignant bone tumors occur within this age group or
in older children. In a patient who is between 10 and 13 years of age
there is sufficient growth potential (particularly in the distal femur,
proximal tibia, and proximal humerus, the most common sites for
malignant bone tumors) that if limb-salvage surgery is done, special
attention is needed to achieve near-equal limb lengths at maturity.
Equal or near-equal limb lengths can be achieved with traditional
methods initially making the operated leg longer and, if needed, by
epiphysiodesis. Usually, for patients whose limb-length inequality will
be 2 cm or less, no surgical adjustment is necessary and the patient
can use a lift if desired. For limb-length inequality between
approximately 2 and 5 cm, an epiphysiodesis is usally the best means of
achieving equal limb lengths. For limb-length differences greater than
5 cm, some type of limb lengthening or rotationplasty is advised.
resection will almost certainly include an epiphyseal center. One
option is to reconstruct with an osteoarticular allograft, and treat
the limb-length discrepancy using standard methods of epiphysiodesis,
closed femoral shortening, or limb lengthening. The allograft has the
advantage of not disturbing the adjacent growth plate. Growth of the
adjacent growth plate may be arrested by placing a prosthetic stem
across it. One difficulty with the allograft approach is in obtaining a
graft of the appropriate size. For children older than 10 years, it is
usually possible to use a small adult bone. The use of grafts with open
growth plates is not advised because they will fracture through the
allograft physis. It is advantageous to use an osteoarticular allograft
for the proximal tibia because it provides a site of attachment for the
patellar ligament. The results of allografts for limb salvage in
osteosarcoma are reasonably good, but the patient should not expect
normal limb function (173, 174, 175, 176, 177, 178, 179, 180, 181). A good or excellent result, based on a functional evaluation system devised by Mankin et al. (182),
can be expected in approximately 65% to 70% of patients. It should be
noted that there are no ideal measures of function after limb salvage
(although several have been developed), and this remains an area of
investigation. In general, if patients can return to normal walking
activities without supports or braces, it is considered to be a good
result. Seldom can they return to contact sports or activities
involving running (183). Complications include infection, nonunion, fracture, and joint instability (182,184,185).
If the patient survives, he or she may need joint arthroplasty at some
time in the future, but by then the patient should be old enough that
growth is no longer a consideration. Growth equalization can be
achieved by contralateral epiphysi odesis, limb shortening, or
ipsilateral lengthening (186,187). The experience with limb lengthening in these patients is limited because limb length is seldom a major issue.
Modular prostheses are now available that allow the surgeon to
construct an implant of suitable length in the operating room (188,189). Custom implants are seldom necessary. Some prostheses have the ability to be expanded as the child grows (174, 188, 190, 191, 192, 193, 194, 195, 196, 197, 198).
There are many methods that can be used to lengthen endoprosthetic
devices. Some require a major operative procedure, some can be done
with limited surgery, and a few endoprostheses can be lengthened
without surgery. The efficacy and longevity of these “growing”
prostheses have yet to be established. Usually it is possible to
achieve at least 2 cm of length per procedure. In very young patients,
this must be repeated every 6 months until maturity, at which point
revision to an adult prosthesis may be necessary. There are few data on
these prostheses, but at least one report shows that it is possible to
gain 2 to 18 cm in length, and to have equal limb lengths at skeletal
maturity (195). The issues of prosthetic
failure, loosening, wear of polyethylene, and infection remain
unresolved. The choice between an implant or
a
reconstructive procedure has to be made by the surgeon and the
patient’s family. Prostheses are more functional initially, but their
longevity is unknown. Expandable prostheses have a high failure rate (188).
More than 80% require revision by 5 years, and the revision rate
appears to be higher in uncemented prostheses. Many of these patients
will have knee stiffness, and the infection rate, especially in the
tibia, may be as high as 38%, although this can be improved with the
liberal use of gastrocnemius flaps (188). The
modular prosthesis has been reported to have a 5-year revision-free and
amputation-free survival rate of 75%, presumably because it is
mechanically stronger and less complex (188).
Functioning appears to be better in children who are older than 8 years
at the time of the reconstruction. Rehabilitation is more difficult
with allografts, but they hold the promise of superior longevity. It is
very important to tell the patient in either case that the function
will not be normal, and that neither method of reconstruction will
return the patient to sports activities. For lower-extremity sarcomas
near the knee or hip, it is our opinion that patients older than 8 to
10 years with open growth plates are best treated with allografts to
preserve the adjacent growth center, but that skeletally mature
patients may be better served by prostheses because of the easier
recovery and rehabilitation. In either case, subsequent revisions or
eventual amputation may be necessary.
equal limb length that can be done for patients who have had resections
of malignant bone tumors. This probably should be done only after the
patient has completed chemotherapy.
performed, sparing the adjacent joints and occasionally the epiphyseal
plates. In these patients, reconstruction can be carried out with
intercalary allografts and/or vascularized fibulae (178,179,199).
A method of using an allograft to provide initial stability, augmented
by a vascularized fibular graft to achieve quicker union and long-term
healing potential, has recently been described (200, 201, 202). This technique is especially helpful when only a small segment of the epiphysis remains after the resection.
If the rotator cuff and part of the deltoid can be preserved,
reconstruction with an osteoarticular allograft yields good results (207,208).
Some surgeons prefer to use an endoprosthesis with the allograft in
order to prevent late collapse of the joint and fracture (175,209).
In either case, the allograft position allows for attachment of the
rotator cuff, which is preferable to a prosthesis. For extraarticular
resections in which both the deltoid and the rotator cuff are
sacrificed, arthrodesis, using allograft, vascularized fibula, or both,
is indicated (210). It is difficult to achieve
an arthrodesis, but when it works, it is very functional. When a formal
Tikhoff-Linberg resection including the scapula is necessary, the
reconstruction is more difficult; leaving a flail upper extremity may
be the only option (203,206,211,212).
or older patients who want to be athletically active, rotationplasty is
an option (213, 214, 215, 216, 217, 218, 219, 220, 221, 222, 223, 224, 225, 226, 227). In these patients, above-knee amputation would lead to a very short stump with a poor lever arm (228).
Rotationplasty, by taking advantage of the tibia and foot, provides a
longer lever arm and an active “knee” joint. It also avoids resection
of the major nerves, so that phantom pain is not an issue. The physical
appearance without the prosthesis is disturbing to some patients, but
with a prosthesis they look similar to other amputees and function much
better than above-knee amputees do (216, 219, 229, 230, 231, 232). The technical details are well described elsewhere (218), and the technique has been described for lesions of the proximal tibia and the proximal femur (223, 224, 225, 226, 227, 228, 229, 230, 231, 232, 233, 234, 235); more recently, similar procedures have been described for the upper extremity (236).
It is very important to have frank discussions with the patient and his
or her family about the appearance and expected outcome of this
reconstruction. It can be very helpful to the patient to be able to
meet another patient who has undergone rotationplasty, or at least view
a video and meet with an experienced physical therapist and
prosthetist. Interestingly, young patients do not view this as an
amputation, because the foot is still present, and the long-term
psychological outcomes in these patients have been very good.
more likely it is that amputation will be the better treatment choice.
A hip disarticulation has so much more functional consequence compared
to an ankle disarticulation that limb-salvage surgery for a tumor in
the proximal femur is always more valuable compared to limb-salvage
surgery for a tumor in the calcaneus.
amputation. These patients’ tumors have proved that they are more
aggressive or more extensive than had been appreciated at the time of
their initial treatment and therefore need more aggressive surgery than
usual. In these circumstances limb salvage is less likely to be
effective and should be done with caution.
believed to be effective in the treatment of malignant tumors of the
musculoskeletal system. The extremely high incidence of metastatic
disease in patients with osteosarcoma (more than 80%) and Ewing sarcoma
(more than 85%), and some promising results in patients with metastatic
sarcoma, prompted the use of adjuvant chemotherapy in patients who did
not have documented disease but in whom the risk of having subclinical
metastases was high (237). The
early results were exciting, and even the use of what was considered
minimal amounts of less-than-optimal drugs improved survival. These
early studies led to the acceptance of adjuvant chemotherapy for Ewing
sarcoma, osteosarcoma, and rhabdomyosarcoma (238, 239, 240, 241, 242, 243, 244, 245, 246, 247, 248, 249, 250). There are no chemotherapeutic agents that are believed to be effective for chondrosarcoma (251), and the use of chemotherapy for soft tissue sarcomas (except rhabdomyosarcoma) remains controversial (252, 253, 254)
and it is now standard for the initial chemotherapy for patients with
Ewing sarcoma, osteosarcoma, and rhabdomyosarcoma (42, 135, 136, 139, 237, 255, 256, 257, 258, 259, 260, 261, 262, 263, 264, 265, 266, 267, 268). Neoadjuvant chemotherapy
is a term used to indicate that the patient receives chemotherapy
before the definitive treatment of the primary lesion. This was
initially used as a means of treating patients with osteosarcoma who
were waiting for the production of a custom prosthesis. The effect of
chemotherapy on the tumor was considerable and of prognostic
significance, and this has led to the routine use of preoperative
chemotherapy.
three skeletal malignancies (Ewing sarcoma, osteosarcoma, and
rhabdomyosarcoma) for which chemotherapy is used. All these protocols
use more than one drug, usually three to five. Most protocols are
between 9 and 12 months in duration. Approximately one-third of the
chemotherapy is given preoperatively, and the remainder is given after
surgery.
-
Doxorubicin (Adriamycin), a cytotoxic
anthracycline antibiotic that passively enters the cell to diffuse into
the nucleus, where it binds nucleic acids and disrupts DNA synthesis (237).
It is cardiotoxic, myelosuppressive, and produces alopecia. It is given
intravenously in divided doses over 6 months, with 450 mg per m2 recommended as the maximum dose. -
Methotrexate is an antimetabolite that
inhibits dihydrofolic acid reductase. This interferes with DNA
synthesis and repair, and alters cellular replication. When
admini-stered in high doses (12 mg per m2 intravenously),
leucovorin or citrovorum factor is given to the patient to rescue the
normal cells. Leucovorin is a chemically reduced derivative of folic
acid and is used by the cells to complete normal cell functions without
the need for dihydrofolic acid reductase. Tumor cells seem less able to
use leucovorin than normal cells, and this difference allows
methotrexate to be effective against malignant tumors. The primary side
effects of methotrexate are gastrointestinal, including nausea,
vomiting, and loss of appetite. -
Cisplatin is a heavy metal that is
thought to cause intrastrand cross-links in DNA, and thereby interfere
with the DNA. It is given intravenously in doses of 75 to 100 mg per m2 repeatedly over the course of the treatment. The principal side effect of cisplatin is nephrotoxicity. -
Cyclophosphamide (Cytoxan) is a synthetic
drug chemically related to nitrogen mustard. It cross-links DNA and
interferes with DNA functions. It is given intravenously at a dose of
40 to 50 mg per kg in divided doses over 4 to 5 days. The major side
effects of cyclophosphamide are gastrointestinal disorders and
myelosuppression. -
Ifosfamide is a synthetic analog of cyclophosphamide, with similar actions. It is given intravenously at 1.2g/m2/day for 5 days.
-
Vincristine is an alkaloid from the
periwinkle plant. It is thought to arrest dividing cells in the
metaphase state by inhibiting microtubule formation in the mitotic
spindle. It is given intravenously at weekly intervals at doses of 1.4
mg per m2 in adults and 2.0 mg per m2 in children. The major side effect of vincristine is peripheral neuropathy. -
Bleomycin is a cytotoxic glycopeptide antibiotic from a strain of Streptomyces verticillus
that inhibits DNA synthesis. It also probably inhibits ribonucleic acid
(RNA) and protein synthesis. It is given intravenously at 0.25 to 0.50
U per kg once or twice per week. The most serious side effect of
bleomycin is a 10% incidence of severe pulmonary fibrosis. -
Actinomycin D (Dactinomycin) is one of a number of actinomycin antibiotics from Streptomyces.
It binds to DNA by intercalation with the phenoxazone ring. This
inhibits the DNA from being a template for RNA and synthesizing itself.
It is given intravenously at 0.5 mg per day for 5 days. Dactinomycin
produces nausea and vomiting and is myelosuppressive.
depending on the specific diagnosis, the protocol, the response of the
patient, and the aggressiveness of approach of the medical oncologist.
(osteocartilaginous exostosis, osteochondroma), chondrormyxofibroma,
periosteal chondroma, chondroblastoma, and chondrosarcoma. The benign
tumors are common, especially enchondroma and exostosis, whereas
chondrosarcoma is extremely rare in the pediatric age group (113,269,270).
probably not a true neoplasia. It may be the result of epiphyseal
growth cartilage that does not remodel and persists in the metaphysis,
or it may result from persistence of the original cartilaginous anlage
of the bone (271). Both possibilities have been suggested as the cause of this common benign tumor. Most patients with a solitary enchondroma
present with either a pathologic fracture through a lesion in the phalanx, which is the most common location (271,272)
(the proximal humerus and the distal femur are the other common
locations for enchondroma), or a history of the lesion having been an
incidental finding on a radiograph taken for another reason (Fig. 14.12). Enchondromas are common lesions that account for 11% of benign bone tumors (113,273),
and they do not need to be removed. However, they may be difficult to
diagnose. Usually, the diagnosis can be made from the clinical setting
and the plain radiograph. Forty percent of enchondromas are found in
the bones of the hands or feet, usually a phalanx. An enchondroma
should not produce symptoms unless there is a pathologic fracture.
There are no associated abnormalities of blood or urine. The femur and
proximal humerus are the next most common sites. Enchondromas are
located in the metaphysis and are central lesions in the medullary
canal. The bone may be wider than normal, but this is caused by the
lack of remodeling in the metaphysis rather than by expansion of the
bone by the tumor. The cortex may be either thin or normal; the lesion
is radiolucent in the pediatric age group, but at later stages it shows
intralesional calcifications. There is usually no periosteal reaction.
In the pediatric patient, UBCs have a similar radiographic appearance,
but they are most common in the proximal femur and the proximal
humerus. The appearance of an enchondroma on MRI is typical. The
cartilage matrix has an intermediate signal intensity on the
T1-weighted image and a high signal intensity on the T2-weighted image (100, 274, 275, 276, 277, 278). It has a sharp margin with the adjacent bone, without peripheral edema.
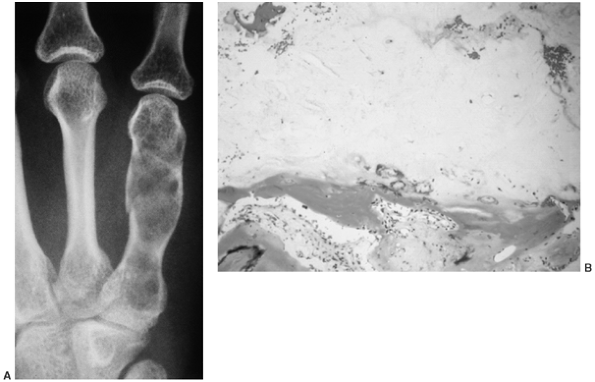 |
|
Figure 14.12 A:
This enchondroma of the fifth metacarpal is typical. The shaft is enlarged, and the lesion is radiolucent. This patient had been aware of this lesion since she was 10 years of age. She had sustained numerous pathologic fractures, and decided to have it curetted. The curettage was done after the fracture had healed. B: Enchondroma can have varied histologic appearances with varying cellularity, but generally the cartilage, the amorphous material in the center of the image, has few chondrocytes. Typically, the cartilage is lined by a thin band of bone, and the adjacent marrow is normal. Often there is considerable calcification within the cartilage component of the lesion (10 × magnification). |
biopsy is necessary. Repeat plain radiography and physical examination
should be performed in approximately 6 weeks, then every 3 to 6 months
for 2 years. Although there are reports of solitary enchondromas
differentiating into chondrosarcomas, usually late in adult life, this
does not occur frequently enough to justify the removal of all
enchondromas. The patient should be advised that after age 30 years, if
the lesion becomes painful or enlarges, it should be considered a
low-grade chondrosarcoma and be surgically resected.
Pathologists have difficulty distinguishing between active enchondroma
(most pediatric patients have active lesions) and low-grade
chondrosarcoma. The clinical course is the best measure of the lesion’s
significance, and an incisional biopsy alters the status of the lesion
and makes subsequent evaluation difficult. If the patient or the
patient’s parents insist on biopsy, it is best that the entire lesion
be removed.
far fewer than those with solitary enchondromas. Multiple enchondroma
was originally described in the late
1800s by Ollier (279).
Most patients with Ollier disease have bilateral involvement but with
unilateral predominance. These patients have growth deformities, both
angular and in length. The deformities of the extremities should be
managed surgically in order to maintain the function of the limbs,
without specific regard to the enchondroma. Patients with Ollier
disease have an increased risk of developing secondary chondrosarcoma
later in life and should be so advised (280, 281, 282).
The incidence of secondary chondrosarcoma in patients with Ollier
disease is not known, but may be as high as 25%. The pelvis and
shoulder girdle are the most common locations of secondary
chondrosarcoma.
Patients with this disorder have an even greater risk of developing
malignant cartilage tumors than do patients with Ollier disease; more
importantly, they have a great risk of developing carcinoma of an
internal organ (284).
are used interchangeably. This lesion was first described in the early
1800s and occurs commonly. Although the pathogenesis of this lesion is
not known, an abnormality or injury to the periphery of the growth
plate has been suggested as the cause (271). It
has been shown in an experimental animal study that the periphery of
the growth plate can be traumatized and a typical exostosis can be
produced.
in by a parent who has just noticed a mass adjacent to a joint. The
patient usually has no symptoms. An occasional patient has loss of
motion in the adjacent joint, and this is attributable to the size of
the mass. Often, the patient may have been aware of the mass for months
or even years, and says that it has been slowly enlarging. Some
patients have pain resulting from irritation of an overlying muscle,
repeated trauma, pressure on an adjacent neurovascular bundle, or
inflammation in an overlying bursa. On physical examination, the mass
is nontender, hard, and fixed to the bone. The rest of the physical
examination may show no abnormality.
The mass is a combination of a radiolucent cartilaginous cap with
varying amounts of ossification and calcification. The amount of
calcification and bone formation increases with age. The base may be
broad (sessile exostosis) or narrow (pedunculated exostosis). In both
types, the cortex of the underlying bone opens to join the cortex of
the exostosis, so that the medullary canal of the bone is in
continuity. This can usually be appreciated on the plain radiograph
itself, but if not, CT scan or MRI establishes this finding and
confirms the diagnosis.
to grow. They may continue to grow well into the third decade of life.
This is not a sign of malignancy. After 30 years of age, growth of an
exostosis is an indication for removal (285).
The growth rate is not steady, and occasionally a lesion grows more
rapidly than expected. Removal of the lesion in a child is indicated
only for those patients who have symptoms attributable to pressure on a
neurovascular bundle or irritation of the overlying muscle. Removal of
the lesion in a young child may result in damage to the growth plate
and recurrence of the lesion. Degeneration of the lesion into a
malignancy is extremely rare in children and uncommon in adults. The
definition of malignant degeneration of a solitary exostosis is
confusing. By convention, an exostosis is considered to be malignant in
a patient as old as 30 years or older if there is an enlarging
cartilage cap and when the cap is more than approximately 2 cm thick.
This so-called malignant degeneration is more common in lesions of the
scapula, the pelvis, and the proximal femur. Although it is often
stated that the risk of malignant degeneration is as high as 20%, the
real incidence is not known. It is probably less than 5%. The risk of
metastasis from this lesion is less than 1% (273, 277, 286, 287, 288, 289, 290).
looks like a cauliflower. It has an irregular surface covered with
cartilage. The cartilage is usually less than 1 cm thick, except in the
young child, in which it may be 2 or 3 cm thick. Deep in the
cartilaginous cap there is a variable amount of calcification,
enchondral ossification, and normal bone with a cortex and cancellous
marrow cavity. Typically, the microscopic appearance of the
cartilaginous cap is that of benign hyaline cartilage.
A patient may have three or four lesions, but more often there are 10
to 15. Usually, the patient has exostoses of all shapes and sizes. They
are concentrated in the metaphysis of the long bones, but may be in the
spine, the ribs, the pelvis, and the scapula. On physical examination,
they are hard, fixed masses adjacent to joints. Patients with multiple
exostoses are usually shorter than average but not shorter than the
normal range. The affected joints show loss of range of motion,
especially forearm rotation, elbow extension, hip abduction and
adduction, and ankle inversion and eversion.
autosomal dominant gene with a variable penetrance, and there is an
approximately 50% chance that a child of a parent with the heritable
gene will show clinical manifestations of this condition (291,294,295). The disease may manifest with extensive involvement in the parent, but with minimal involvement in the child, or vice versa.
In most patients with multiple heritable exostoses, the radiographic
appearance of the proximal femur is diagnostic. The femoral neck is
short and broad with multiple bony excrescences.
chondrosarcoma (287, 288, 294, 296, 297, 298, 299, 300, 301, 302). Secondary chondrosarcoma is extremely rare in the pediatric age group (303,304).
Occasionally, one or more of the exostoses need to be removed in order
to relieve the pain related to repeated local trauma, or to improve the
motion of the adjacent joint. Lesions in the pelvis and the spine
should be observed closely because they have the greatest risk of
undergoing malignant degeneration. We do not recommend that these
lesions be removed simply because they are present.
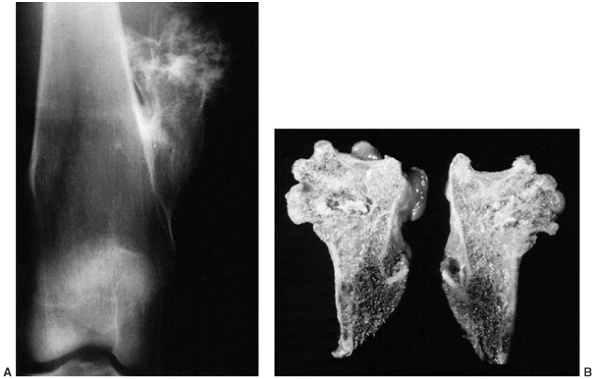 |
|
Figure 14.13 A:
Anteroposterior radiograph of the distal femur with a typical pedunculated exostosis. The cortex of the lesion blends into the cortex of the femur. The exostosis has an irregular proximal end that is covered by a cartilaginous cap. The pathologic material is the cartilage, but what is seen on the radiograph is the bone formed by enchondral ossification of the cartilage. The mass was repeatedly hit when this patient played football. It was marginally excised, and a small rim of the cortex of the femur was removed along with the exostosis, because occasionally any residual cartilage that lies at the base of the stalk can lead to a recurrence. B: Gross bisected specimen from the same patient. The femoral attachment is inferior, and the cortex of the femur can be seen blending with the cortex of the exostosis. The medullary canal of the femur is filled with hematopoietic marrow, and appears dark. The hematopoietic marrow extends into the base of the exostosis, but most of the marrow of the exostosis is fatty. There is a thin, cartilaginous cap. In a child, a cartilaginous cap more than 1 cm thick is of no concern, but in an adult a cartilaginous cap of more than 1 cm is considered to indicate early malignant degeneration to secondary chondrosarcoma. |
multiple, to be examined and to undergo radiography at least yearly.
Patients are told to report symptoms or increasing size immediately.
usually of the male sex (men are more frequently affected than women,
at a ratio of 2:1) in the second or third decade of life (113,305).
The patient complains of a dull, steady pain that is usually worse at
night. The only positive physical finding is tenderness over the
involved area, and occasionally a deep mass can be detected.
Approximately one-third of chondromyxoid fibromas occur in the tibia,
usually proximally. It is a radiolucent lesion that involves the
medullary canal but is eccentric and erodes the cortex (273,306) (Fig. 14.14). It may be covered by only periosteum, and is often mistaken for the more common ABC. The solid nature of chondromyxofibroma versus
the cystic nature of an ABC, as seen on MRI, is a means of
differentiating between these two lesions. The natural history is not
known because the condition itself is infrequent and surgical treatment
is nearly universal. Thorough curettage and bone grafting are
recommended. Recurrence is a risk, and patients and parents should be
so advised.
to be a variant of giant cell tumor of bone. Codman’s detailed
description in 1931 was of an “epiphyseal chondromatous
giant cell tumor” (307). Jaffe and Lichtenstein in 1942 (308) suggested that it be called benign chondroblastoma,
and distinguished it from giant cell tumor of bone. Codman was
particularly interested in the shoulder because he thought this lesion
was found principally in the proximal humerus (Fig. 14.15).
It has since become clear that chondroblastoma is found in many bones,
but the proximal humerus is the most common site (approximately 20%) (113, 309, 310, 311).
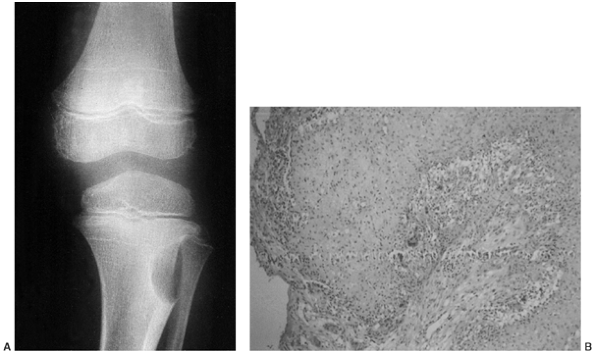 |
|
Figure 14.14 A:
Anteroposterior radiograph of a chondromyxofibroma of the proximal lateral tibia. The lesion is typically an eccentric, radiolucent abnormality that usually destroys the cortex but is contained by the periosteum. As in this case, the radiographic appearance of chondromyxofibroma is often similar to that of an aneurysmal bone cyst. B: Chondromyxofibroma does not have typical hyaline cartilage. It is a cellular lesion with areas of chondroid matrix, myxoid matrix, and abundant fibrous tissue. It can be mistaken for a low-grade malignancy if the diagnosis of chondromyxofibroma is not considered (4 × magnification). |
The patient with a chondroblastoma is usually in the second decade of
life, with an open growth plate, but the condition may occur in older
patients as well. The initial symptom is pain in the joint adjacent to
the lesion, and the patient usually presents with joint complaints. The
findings on physical examination also may suggest an intraarticular
disorder because most patients have an effusion and diminished motion
in the adjacent joint. Frequently the patient is believed to have
chronic synovitis; he or she does not have other symptoms or abnormal
physical findings. The patient’s laboratory data are normal.
In children, it is the most common neoplastic lesion of the secondary
ossification center (312); in adults, only giant
cell tumor of bone involves the secondary ossification center more
often. In children, osteomyelitis is the most common condition that can
produce a lesion in the secondary ossification center. On the plain
radiograph, the lesion is radiolucent, usually with small foci of
calcification (313).
The calcification is best seen on a CT scan. There is usually a
reactive rim of bone surrounding the lesion and, sometimes, metaphyseal
periosteal reaction. The edema associated with chondroblastoma can be
appreciated on MRI (Fig. 14.16). There is
increased uptake on a technetium-99 bone scan. Chest radiography or CT
scan should be performed, because chondroblastoma is one of the benign
bone tumors that can have lung implants and still be considered benign (314).
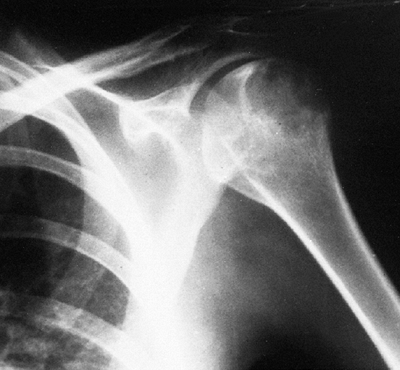 |
|
Figure 14.15
Radiograph of a chondroblastoma in the proximal humeral epiphysis. This is a typical Codman tumor. The lesion is both epiphyseal and metaphyseal, has an irregular reactive border, and has intralesional calcifications, although these are difficult to see on plain radiographs. Giant cell tumor of bone is similar to chondroblastoma except that there are no intralesional calcifications. This patient was treated with curettage and bone grafting. |
similar appearances on plain radiographs, but they should not be
confused with each other. Osteochondritis dissecans produces an
abnormality in the subchondral bone; in chondroblastoma, on the other
hand, the subchondral bone is almost always normal. Patients with
chondroblastoma have more of an effusion than patients with
osteochondritis dissecans, and their pain is constant and not related
to activity as it is in patients with osteochondritis dissecans.
typical, and is rarely confused with other diagnoses. It consists of
small cuboidal cells (chondroblasts) closely packed together to give
the appearance of a cobblestone street (308, 309, 310, 311).
In addition, there are areas with varying amounts of amorphous matrix
that often contains streaks of calcification, and usually there are
numerous multinucleated giant cells. Chondroblastoma is not as vascular
as osteoblastoma; there are few, if any, mitoses, and no abnormal ones (Fig. 14.17).
should be treated when found. Curettage is the treatment of choice, but
it should be a thorough curettage and should extend beyond the reactive
rim (311). The lesion should be seen adequately
at the time of the curettage, which usually means that the joint should
be opened. Iatrogenic seeding of a joint is not a significant risk, and
intraarticular surgical exposure is recommended if this facilitates
visualization. Most recurrences are cured with a second curettage, but
a rare lesion can be locally aggressive and requires a wide resection (310).
Chondroblastoma of the pelvis frequently behaves more aggressively than
that in long bones, and an initial wide excision is recommended if it
can be done with limited functional loss and morbidity.
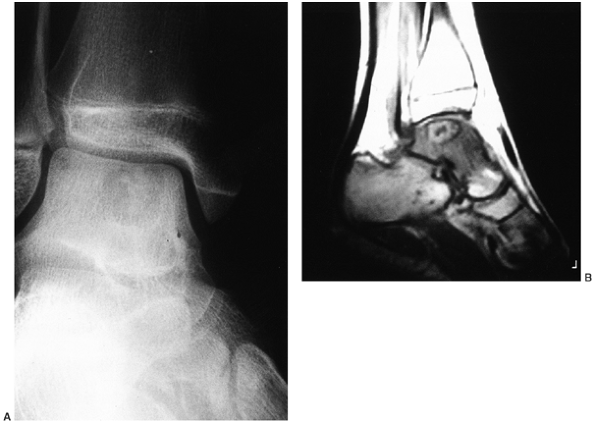 |
|
Figure 14.16 A:
This is an anterior view of a 14-year-old girl’s ankle. She complained of pain and swelling and had limited range of motion in the joint. There is a radiolucent lesion in the talus, close to the subchondral bone. There are central calcifications. B: The sagittal T1-weighted magnetic resonance image reveals the lesion and an associated edema. This lesion is a chondroblastoma, and was treated with curettage. |
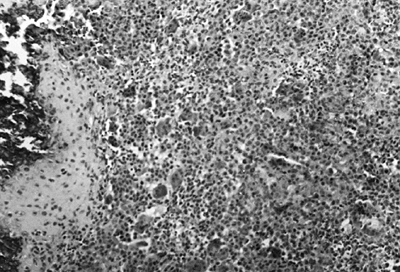 |
|
Figure 14.17
Histologic appearance of a chondroblastoma. The tumor consists of cuboidal cells (i.e., chondroblasts), varying amounts of amorphous matrix (some of which is calcified), and multinucleated giant cells. Calcification is seen (left). The cuboidal cells fit together in such a manner that they have the appearance of cobblestones (original magnification × 10). |
diagnosis is made, and the risk of growth disturbance from the tumor or
its treatment is minimal. When the patient is younger than 10 years
old, care should be taken not to damage the growth plate.
The patient usually has pain at the site of the lesion. More than half
of such lesions are found in the proximal humerus, and the others are
evenly dispersed through the long bones. The lesion can often be
palpated. It is a nontender, hard mass that is fixed to the bone. The
plain radiograph is typical (Fig. 14.18).
Periosteal chondroma is a scalloped defect on the outer surface of the
cortex, occasionally with intralesional calcifications and minimal
periosteal reaction. On microscopic examination, periosteal chondroma
is benign cartilage, but it appears more active than enchondroma. It
has been mistaken for chondrosarcoma. Because local recurrence is a
risk, a wide excision, including the underlying cortex, is the
treatment of choice.
Fibrous cortical defects are subperiosteal and erode into the outer
surface of the cortex, whereas NOFs are medullary lesions that thin the
cortex from within. Up to 40% of children have this lesion, which is
found most often between the ages of 4 and 8 years (323).
Ninety percent of the lesions found are in the distal femur. These are
asymptomatic lesions that are found only if a radiograph is taken for
another reason or when the patient has a pathologic fracture. The
patient shows no abnormal physical findings, and the serum and urine
chemistries are normal.
the basis of the clinical presentation and plain radiographic findings (318). Biopsy is rarely necessary for
diagnosis. Two radiographic appearances are possible. The one that most authors refer to as fibrous cortical defect is a small (<0.5 cm) radiolucent lesion within the cortex, with a sharply defined border (113). There is little or no increased uptake on the technetium-99 bone scan.
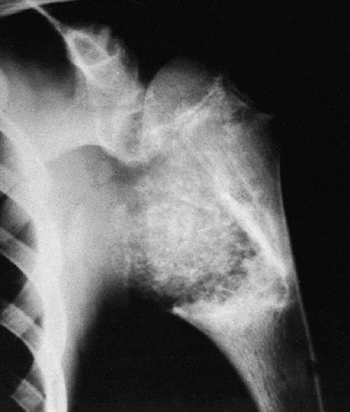 |
|
Figure 14.18
Radiograph of the shoulder of a 12-year-old boy. The large periosteal chondroma involves the medial aspect of the metaphysis. Most such tumors are smaller. This patient had no symptoms; the lesion was found by the boy’s pediatrician on routine physical examination, and an incisional biopsy confirmed the diagnosis. The lesion was only partially removed. The tumor did not change appreciably during 2 years of follow-up. |
This lesion appears to have arisen from within the medullary canal. It
is surrounded by a well-defined thin rim of reactive bone. There should
be no acute periosteal reaction unless there has been a fracture. There
may be slightly increased uptake on the technetium-99 bone scan.
Multiple NOFs occur in approximately 20% of the patients.
Multinucleated giant cells are common, and areas of large, lipid-laden
macrophages can often be seen. Hemosiderin is often present within the
fibroblastic stromal cells and multinucleated giant cells. There is no
bone formation within the lesion, and mitoses are not seen.
needs no treatment, but should be observed. Radiographs at 3- to
6-month intervals for 1 to 2 years are suggested. These lesions heal
spontaneously. NOF may need surgery. The indication for surgery is to
reduce the risk of a pathologic fracture. It is difficult to predict
who is at risk of sustaining a pathologic fracture. The less active and
the smaller the child, the less risk there is of the child sustaining a
pathologic fracture. NOFs that are less than 50% of the diameter of the
bone can be merely observed, because they have little risk of fracture,
but curettage and packing with bone graft should be considered if they
enlarge (324, 325, 326).
Patients who present with NOFs that are more than 50% of the diameter
of the bone have a risk of fracture with even a minimal injury, and
should at least be considered for surgery. According to Arata et al. (326),
such patients have an increased risk of developing pathologic
fractures. Many patients with these large NOFs elect not to have
surgery, and reduce their activity instead. This is an alternative
treatment.
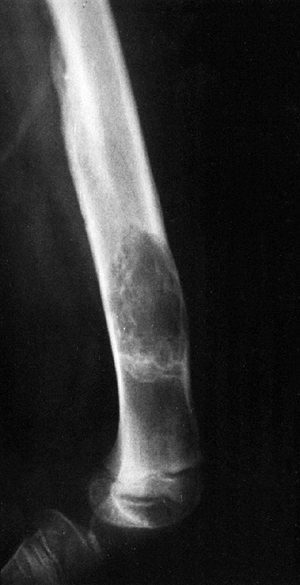 |
|
Figure 14.19
Lateral radiograph of the distal femur with a large nonossifying fibroma (NOF). The patient had sustained a pathologic fracture that had healed, but the lesion persisted. NOF is usually a metaphyseal, radiolucent, irregular lesion surrounded by a reactive rim of bone. As is often the case, the cortex surrounding a large NOF is thin and appears to be expanded. Although this lesion eventually heals spontaneously, its large size and persistence after pathologic fracture indicated that curettage and bone grafting were necessary. NOFs that replace more than half of the bone should be curetted and grafted. |
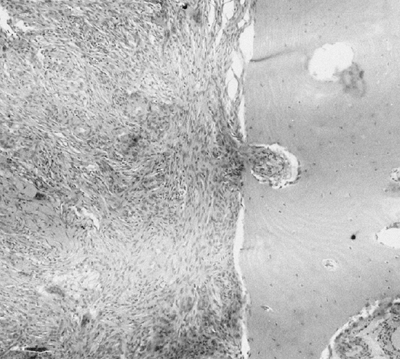 |
|
Figure 14.20
Low-power histologic view of a typical nonossifying fibroma (NOF). The fibroma is composed of benign fibrous tissue and multinucleated giant cells. Hemosiderin is often present. The NOF is invading cortical bone (right) (original magnification 10 ×) |
have the fractures treated nonoperatively if possible. The fracture
should heal without difficulty in a normal length of time. There is no
evidence that the healing of the fracture increases the chances of
spontaneous healing of an NOF, or of any other benign lesion. NOF
usually heals spontaneously, and this may happen after the fracture;
but usually the fracture callus obscures the radiolucent lesion, and
the physician is fooled into thinking that the lesion is healing even
when it is not. When the callus has remodeled and the cortices become
distinct on the radiograph, the lesion can be seen again. Patients with
pathologic fractures must be followed until the callus has remodeled
sufficiently so that a final determination can be made about
the
status of the underlying NOF. If it persists after the fracture has
healed, curettage and bone grafting should be considered.
developmental abnormality caused by a somatic mutation leading to a
defect in the formation of bone (see Chapter 7).
It is a common disorder that produces a variety of symptoms and
physical findings. Most patients (approximately 85%) have a single
skeletal lesion (monostotic fibrous dysplasia), whereas the remainder
have numerous lesions (polyostotic fibrous dysplasia). The patients
with polyostotic fibrous dysplasia may have only two or three small
areas of involvement, or may have extensive skeletal abnormalities with
grossly deformed bones.
presents without symptoms, and the lesion is found when a radiograph is
taken for unrelated reasons (327, 328, 329, 330).
Occasionally the child presents with a pathologic fracture or angular
deformity. A rib is the most common location of monostotic fibrous
dysplasia, but any bone can be involved. There are no physical findings
that are specifically associated with monostotic fibrous dysplasia, and
the café-au-lait lesions and endocrine abnormalities sometimes found in
patients with polyostotic fibrous dysplasia do not occur in patients
with the monostotic variant. Serum and urine chemistries are normal in
patients with fibrous dysplasia.
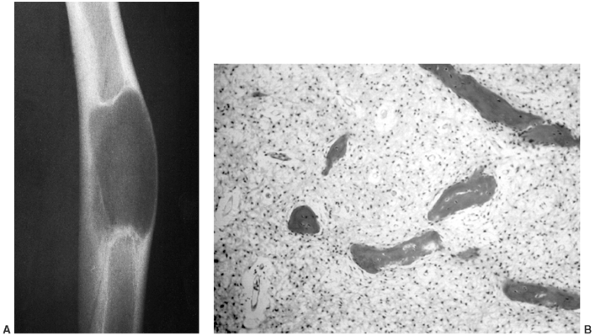 |
|
Figure 14.21 A:
Radiograph of a fibrous dysplasia in the diaphysis of a long bone. The ground-glass appearance, the thin cortex, and the angular deformity of the bone are all typical features of fibrous dysplasia. Because this lesion was large and the patient had an angular deformity, a cortical bone graft was placed within the cortex to increase the strength of the bone. Curettage of the lesion probably does not increase local control, but should be performed if it can be carried out easily. Fibrous dysplastic bone is structurally weak, and cortical grafts are more likely to improve the strength of the bone and not be resorbed by the host. B: Histologic appearance of fibrous dysplasia. The tumor is mostly fibrous tissue composed of collagen and fibroblast. Small bits of bone and osteoid, often having a “C” or an “O” shape, seem to have been sprinkled on the fibrous tissue. Osteoblasts are not seen, and the bone seems to be produced by the fibroblastic cells (40 × magnification). |
It is a medullary process that typically produces a ground-glass
appearance on the radiograph. The lesion is usually diaphyseal. The
diaphysis is larger than normal, and the ground-glass appearance of the
medullary canal blends into the thinned cortex so that it is difficult
to define the border between the medullary canal and the cortex. When
typical-appearing lesions are seen in a single bone or in a single
limb, the diagnosis is almost certain. There may be an angular
deformity in the bone, especially when the lesion is large. The lesions
may mature with age and become radiodense or cystic. Fibrous dysplasia
shows
excessive
uptake on a technetium-99 bone scan, out of proportion to what one
might predict from the plain radiographic appearance.
presents at about the age of 10 years with an angular deformity of a
bone (327, 331, 332, 333, 334).
The most common deformity is varus of the proximal femur, or shepherd’s
crook deformity. The light brown skin lesions with irregular borders
are called coast of Maine café-au-lait spots. The lesions that have smooth borders and are associated with neurofibromatosis are called coast of California café-au-lait spots.
as associated endocrinopathies, and vascular tumors have been seen in
association with fibrous dysplasia (333, 335, 336, 337, 338). McCune-Albright syndrome is a triad of fibrous dysplasia, café-au-lait spots, and precocious puberty (335).
The lesions in polyostotic fibrous dysplasia tend to be unilateral
rather than bilateral. The radiographic appearance of the lesion is the
same as in patients with monostotic disease. The structural strength of
bones with fibrous dysplasia is reduced because of the poorly organized
trabecular pattern and the thinned cortex. The weakness of the bones
leads to the deformities that are usually present.
monostotic and polyostotic forms, is composed of fibrous tissue with
normal-appearing nuclei and irregularly shaped strands of osteoid and
bone (Fig. 14.21B). There are few, if any,
osteoblasts present, and the osteoid and bone seem to arise directly
from the background fibrous stoma. The bone is irregularly organized,
and often has a “C” or an “O” shape. Multinucleated giant cells are
rare and there are few mitoses, none of which is abnormal. Nodules of
cartilage may be present in typical fibrous dysplasia.
surgical treatment. Occasionally, a solitary lesion will be painful and
curettage with grafting is required. Small lesions can be packed with
cortical cancellous bone graft (autogenous or allogenic), whereas large
lesions are probably better treated with cortical bone grafts. A
special circumstance is a lesion in the femoral neck. These lesions
seem to be associated with a risk of fatigue fractures, and cortical
bone grafting is recommended (339). Resorption
of the bone graft with recurrence of fibrous dysplasia can occur, and
the patient should be seen in follow-up for up to 5 years. Occasionally
it is necessary to augment the bone with long-term or permanent
internal fixation to prevent repeated fractures and relieve pain.
Progressive bone deformity is unusual in patients with monostotic
fibrous dysplasia. Patients with polyostotic fibrous dysplasia are more
often in need of surgical therapy. Bone deformity is the most common
indication. The proximal femur is the most challenging bone to manage.
Once a varus deformity develops, it is important not only to do a bone
graft (preferably cortical bone), but also to correct the angular
deformity with a valgus osteotomy. Rigid internal fixation is
recommended (327, 340, 341, 342, 343, 344, 345, 346, 347, 348).
osteofibrous dysplasia lesion, which is found in the mandible and the
anterior cortex of the tibia in children. It is benign, but may be
locally aggressive. It is not a healing
NOF. The patients usually do not have symptoms, and are brought to the
physician’s attention by a parent who has noticed an anterior bowing or
mass in the tibia. The lesion is almost always located within the
anterior cortex of the tibia, and is best seen on the lateral
radiograph (Fig. 14.22). There are often
numerous radiolucent lesions with a rim of reactive bone. On the
technetium-99 bone scan, there is increased uptake in the area of the
lesion.
Some authors believe that osteofibrous dysplasia is a type of fibrous
dysplasia, but this is probably not the case as most histochemical and
genetic data suggest otherwise (355,356).
Osteofibrous dysplasia arises from the cortex and involves the
medullary canal late in the disease process. It is usually associated
with a bowed tibia, and quickly recurs if curetted. There is not much
data on patients with osteofibrous dysplasia who have been
adequately followed up. Usually, many of these lesions progress slowly, and are eventually resected.
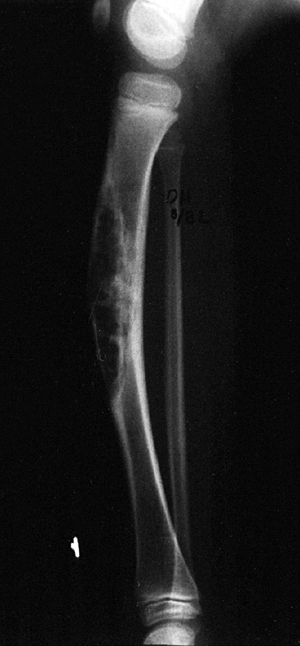 |
|
Figure 14.22
Lateral radiograph of the leg of a patient with osteofibrous dysplasia. Found almost exclusively in the tibia, this lesion involves the anterior cortex, and can extend into the medullary canal. Usually the tibia is bowed anteriorly. |
in a patient younger than 10 years of age. Incisional biopsy is not
necessary in most cases, because the clinical presentation is
diagnostic. Also, the biopsy reveals only a small portion of the
lesion, and does not change the initial management. Bracing probably
does not prevent progressive bowing, but can be tried if there is an
angular deformity. If the lesion progresses before closure of the
growth plate, biopsy and resection are suggested. If the patient
presents after closure of the growth plate, especially if the lesion is
large (more than 3 or 4 cm in diameter) or has aggressive features on
plain radiographs, a biopsy is suggested. If an adamantinoma is found,
a wide resection is recommended. If the biopsy reveals osteofibrous
dysplasia, it is best to excise the entire lesion for a complete
histologic examination to rule out the possibility of there being a
focus of adamantinoma. If the lesion is small (less than approximately
3 cm) and the patient has no symptoms, continued observation is
suggested.
osteofibrous dysplasia. In adamantinoma, however, the patient is
usually older (third decade of life), and the lesion appears more
aggressive on the radiographs (e.g., soft tissue extension, acute
periosteal reaction, large size, involvement of the medullary canal),
although this is not always so. It has been suggested that there is
another type of adamantinoma that looks very similar to osteofibrous
dysplasia, even on histologic examination. One must be suspicious of
the diagnosis of osteofibrous dysplasia, especially in a progressive
lesion in a patient older than 10 years of age (357,358).
is going to be observed, the patient should undergo radiography at
least every 6 months until the lesion heals or is resected. Typical
adamantinoma has a risk of metastasizing, but it is not known whether
the adamantinoma that looks like osteofibrous dysplasia can metastasize
(353).
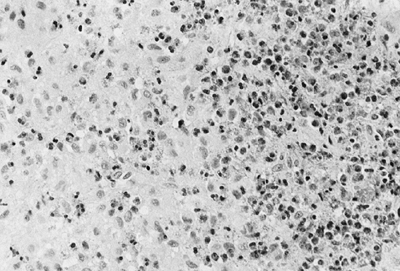 |
|
Figure 14.23
Low-power view of an eosinophilic granuloma (Langerhans granuloma). The eosinophils are numerous, but it is the presence of histiocytes that defines this tumor. The histiocytes are large cells with a clear, folded nucleus and a prominent nucleolus (original magnification × 10) |
In 1941 Farber suggested that eosinophilic granuloma,
Hand-Schüller-Christian disease, and Letterer-Siwe disease were related
(361). Later, Lichtenstein published an article agreeing with Farber, and suggested the term histiocytosis X (362).
This is a disorder of the Langerhans histiocytes, and although
eosinophils are a common component of the lesion, they are not
necessary for the diagnosis (Fig. 14.23).
between the ages of 5 and 15 years. The skull is the most commonly
involved site (363) (Fig. 14.24).
Many of the skull lesions are probably not diagnosed because the only
abnormality is a painless, small, spontaneously resolving lump in the
scalp. The vertebral bodies and the ilium are the next most common
sites of involvement (364, 365, 366).
When the lesions occur in the long bones, they may weaken the bone to
such an extent that the patient presents with activity-related pain
suggestive of a fatigue fracture, or with a pathologic fracture. The
lesion is a radiolucent abnormality with sharp borders of transition
and often no reaction by the host bone. An apparent central sequestrum
of bone may be seen. Eosinophilic granuloma usually results in
increased uptake on a technetium bone scan, but as many as 25% of the
lesions will not be associated with abnormal bone scans (90, 91,367, 368, 369, 370, 371, 372) (Fig. 14.25).
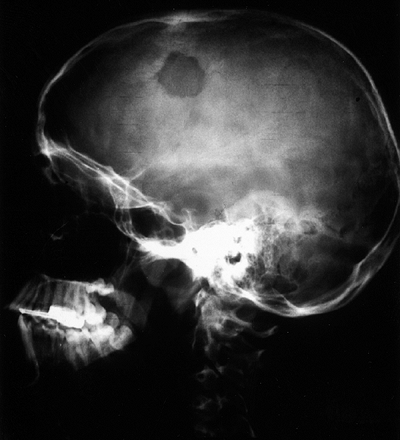 |
|
Figure 14.24
This is a lateral plain radiograph of the skull of a patient with a solitary Langerhans cell histiocytosis (eosinophilic granuloma). The skull is the single most common site of involvement for eosinophilic granuloma. Plain radiographs of the skull are recommended when eosinophilic granuloma is suspected. These lesions rarely require treatment. |
Hand-Schüller-Christian disease, but should be evaluated on
presentation to exclude the presence of that syndrome. The easiest way
to evaluate the patient for diabetes insipidus is to obtain a lateral
skull film in order to observe the size of the sella turcica, and test
a first voided urine specimen after overnight fluid restriction to
determine whether the patient can concentrate his or her urine. Liver
enzymes should be determined. A skeletal survey should also be done to
look for other lesions. Multiple lesions can be seen in eosinophilic
granuloma. If any of these parameters is abnormal, the patient should
be referred to a pediatric hematologist. We routinely seek a
consultation from a pediatric hematologist for all patients with
Langerhans cell histiocytosis.
treated, and many children underwent irradiation, curettage, or
excision. At present, it is believed that most of these lesions are
self-healing, and that no specific treatment is necessary (360). Intralesional injection of corticosteroids has been recommended by some physicians (373, 374, 375).
lesion, exophthalmos, and diabetes insipidus. Patients with this
disorder should be treated with systemic cortico-steroids or
chemotherapy (376, 377, 378, 379).
cell histiocytosis. Most patients present before 3 years of age with
skin, visceral, and brain lesions, and they may or may not have bone
lesions. They require aggressive chemotherapy (363, 380, 381, 382).
but they may not be simple to treat. These common lesions are usually
found when the patient sustains a pathologic fracture. Their
radiographic appearance is so typical that most can be diagnosed
without a biopsy (Fig. 14.26). The proximal humerus and the proximal femur are the sites that account for 90% of UBCs (383, 384, 385, 386, 387). The cysts seem to arise from the epiphyseal plate, are immediately adjacent to the plate, and extend into the metaphysis (388, 389, 390, 391).
The metaphyseal bone does not remodel and the metaphysis is broader
than normal, but not broader than the width of the epiphyseal plate. A
thin rim of bone borders the UBC. The surrounding bone is not reactive,
and there is no acute periosteal reaction. When the cyst becomes mature
(latent), usually after the patient reaches the age of 10 years, the
epiphysis grows away from the lesion. The UBC eventually heals
spontaneously and fills in with bone. No evidence of its previous
existence is seen.
fracture. Some lesions remain small and do not present a significant
risk to the patient. Other lesions are large (e.g., proximal humeral
lesions), are situated in high-stress anatomic sites (e.g., the femoral
neck), or persist after the patient has become a young adult, and in
these cases treatment is indicated. Only those patients with UBCs who
are at risk for pathologic fracture should be treated (392, 393, 394, 395, 396, 397).
Injection is recommended rather than an operative procedure because the
results of injection are equal to those of curettage and bone grafting,
whereas the risk, recovery time, and cost are less. Intracystic
injection of cortico-steroids is the treatment of choice as an initial
means of stimulating the cyst to heal (398,399).
Before the use of corticosteroid injections became common, curettage
and autogenous bone grafting had been the most common treatment.
Operative treatment with curettage and autogenous bone grafting is now
reserved for those lesions that do not respond to repeated injections
of corticosteroid (383, 398, 400, 401, 402).
The number of injections that should be tried before deciding to
operate is still a matter of debate. The authors give at least three
injections approximately 1 month apart before determining whether it is
necessary to resort to an open operative procedure. Injecting
autogenous bone marrow is a technique advocated by some (403).
In a study of ten patients whose UBCs were treated with autogenous
marrow, all were free of pain within 2 weeks of the injection, and all
healed completely within 1 year. Other materials have also been
injected into UBCs with good results (404).
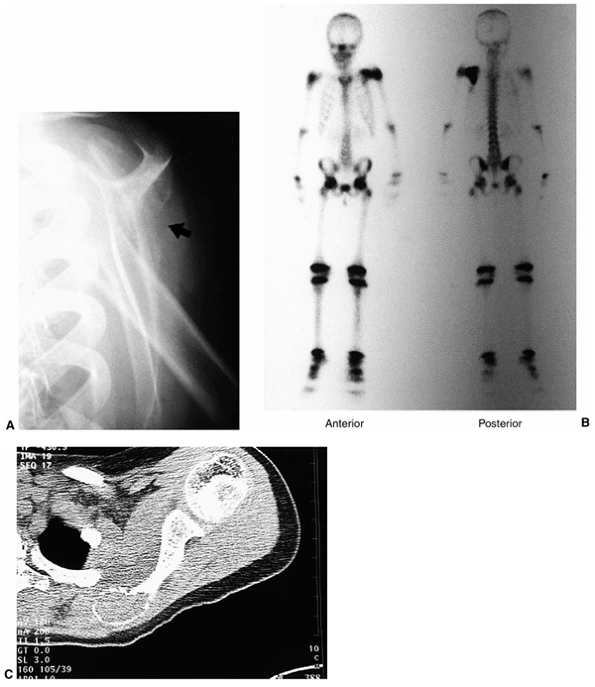 |
|
Figure 14.25 A: This plain radiograph is an oblique view of an 8-year-old girl’s scapula. The arrow points to a subtle area of cortical erosion. The patient complained of pain, and there was a tender mass over the scapula. B:
On the anterior view of the same patient’s technetium-99 bone scan, the increased uptake in the scapula is easily seen. This patient has a solitary Langerhans cell histiocytosis (eosinophilic granuloma), but her lesion is associated with increased uptake. This is true in most cases. C: On the computed tomography (CT) scan, a lesion is seen in the medial border of the scapula. After biopsy, curettage without graft was curative. |
It has been used extensively and is an established method of treatment
of a UBC. It should be performed under anesthesia (usually general
anesthesia), and with the aid of fluoroscopic visualization. An 18- or
20-gauge spinal needle is passed percutaneously into the cyst. The wall
of the cyst is penetrated easily by an 18-gauge needle with the stylet
in place. Rotating the needle as it is pushed through the bone often
helps it penetrate the cortex. Clear yellow or slightly bloody fluid
should be obtained. If no fluid is aspirated, the diagnosis of UBC
should be questioned and a biopsy of the lesion must be performed.
introduced into the cyst as far from the first as possible. A
radiopaque dye (usually Renografin 60) is injected into the cyst to
confirm that it is unicameral, and that all parts fill with dye.
Frequently the draining veins are seen shortly after the cyst is
injected. If the cyst has more than one cavity, each one should be
injected.
is important, but methylprednisolone acetate (Depo-Medrol) is probably
the most commonly used steroid. There is no standard amount of
corticosteroid as such, but usually, 80 mg is sufficient for a small
cyst, and up to 160 mg may be used for large cysts. There are two
techniques
for
corticosteroid injection. One is to inject the cortico-steroid under
pressure, with the second needle occluded, in order to rupture the cyst
wall. The other is to inject the corticosteroid without pressure, using
the second needle as a vent. It is unclear which method is better. A
repeat radiograph is taken after 1 month, and if there is no evidence
of early healing (e.g., increased thickness of the reactive wall), a
repeat injection is done. Repeated injections are often needed, but
more than three to five will probably not be beneficial.
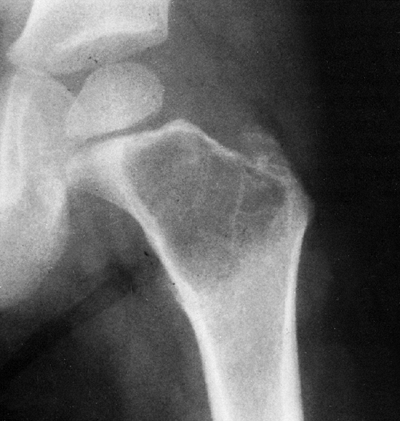 |
|
Figure 14.26
Anteroposterior radiograph of a proximal femoral unicameral bone cyst (UBC). UBCs are radiolucent lesions immediately adjacent to the growth plate, and extend into the metaphysis. This UBC is considered active because it is immediately adjacent to the growth plate and the patient is younger than 10 years. Large lesions in the proximal femur should be treated because of the risk of pathologic fracture. Initial treatment should be a corticosteroid injection. |
curetted and packed with bone graft. When the cyst is adjacent to the
growth plate, care should be taken not to damage the epiphyseal
cartilage during curettage (406). Autogenous bone or allograft cortical cancellous bone can be used for packing the cavity (383,400,407,408).
Freeze-dried cortical cancellous allograft is particularly advantageous
because it is associated with an excellent healing rate and little, if
any, incidence of complications; also, a secondary incision is not
required for obtaining the autogenous bone graft. Calcium sulfate
tablets are an alternative material that can be used for filling the
cavity (409,410).
Some investigators believe that this lesion occurs only in association
with another bone tumor, whereas others recognize ABC as a primary
diagnosis (412,415,416).
ABCs often occur in association with a number of benign tumors (e.g.,
giant cell tumor, chondroblastoma, osteoblastoma), or with osteosarcoma
(412). When it is a secondary lesion, the
primary lesion is usually obvious, and the ABC component is limited to
only a small portion of the tumor. Secondary ABCs are classified by
their underlying diagnosis. The presence of a secondary ABC does not
change the therapy or prognosis of the underlying primary tumor.
More than 50% of these cysts arise in large tubular bones, and almost
30% occur in the spine. The patient usually complains of a mild, dull
pain, and only rarely is there a clinically apparent pathologic
fracture. The physical examination usually shows normal results, and
there are no abnormal laboratory findings associated with ABC.
It resorbs the cortex and elevates the periosteum, generally making the
bone wider than the epiphyseal plate. Usually, there is a thin shell of
reactive periosteal bone, but occasionally this bone cannot be seen.
When
ABC arises in a long bone, it is metaphyseal. When it arises in the
spine, it originates in the posterior elements but it may extend into
the body and, not uncommonly, will extend also to an adjacent vertebral
body or rib. The radiograph of an ABC may appear identical to those of
giant cell tumor of the bone and telangiectatic osteosarcoma (401,412). The periosteal reaction appears to be aggressive, and the lesion may be mistaken for an aggressive or malignant tumor (385). ABCs may arise in the cortex and elevate the periosteum with or without involving the medullary canal.
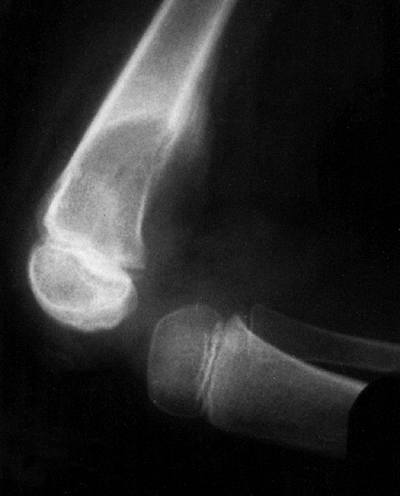 |
|
Figure 14.27
Radiograph of a distal femur with an aneurysmal bone cyst involving the distal metaphysis and extending through the posterior cortex. As in this patient, an aneurysmal bone cyst may have the appearance of an aggressive tumor. When the cyst erodes through the cortex it is usually contained by the periosteum, which reacts and produces bone. The differential diagnosis should include aneurysmal bone cyst, osteosarcoma (telangiectatic variant), Ewing sarcoma, and osteomyelitis. This patient was successfully treated with curettage and bone grafting. |
lesion should have a density of approximately 20 HU, and this does not
increase with intravenous contrast injection. When the patient lies
still for 20 to 30 minutes, the cells in the fluid within the cyst
cavity settle, and a fluid/fluid level can be seen (417).
Similar findings can be seen on MRI. Fluid/fluid level was originally
described in ABC but has subsequently been seen in a number of other
lesions, so it cannot be considered to be diagnostic of ABC. An ABC has
an increased uptake of technetium on the bone scan, but often the scan
has a central area of decreased uptake (418).
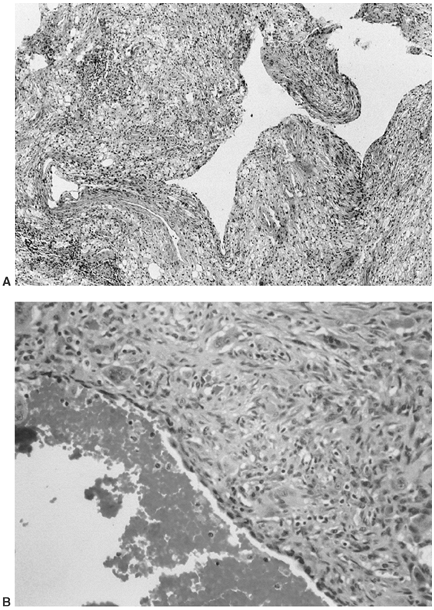 |
|
Figure 14.28 A:
Low-power view of the tissue lining an aneurysmal bone cyst. The lining is composed of fibrous tissue with multinucleated giant cells, foamy histiocytes, hemosiderin and, often, spicules of immature bone (not seen). The fronds and spaces are typical (original magnification × 10). B: Higher-power view (40 ×) of aneurysmal bone cyst. Benign spindle cells, vessels, hemosiderin, and multi-nucleated giant cells make up the solid component. There is a cystic component filled with blood. |
then they should be curetted and packed with bone graft. The
pathologist should be advised in advance, and the possibility of a
telangiectatic osteosarcoma should be discussed. It is uncommon for the
histologic appearance of an ABC to be mistaken for that of a
telangiectatic osteosarcoma, although the radiographic and gross
appearances can be identical. On gross inspection, an ABC is a cavitary
lesion with a villous lining. Microscopic examination reveals the
lining to be composed of hemosiderin-laden macrophages, multinucleated
giant cells, a fibrous stroma, and usually small amounts of woven bone (Fig. 14.28).
The microscopic appearance of the lining of the ABC is similar to that of a giant cell tumor of bone.
packing of bone graft. The first recurrence can be recuretted and
grafted. Embolization may be used in order to decrease blood loss
during surgery, and has been associated with fewer recurrences (419,420). Whether embolization is necessary or helpful is debatable. Cryosurgery has also been reportedly tried (383,411,421).
Cryosurgery may produce complications, and it is not considered
necessary in most cases. It may play a role in the treatment of
recurrent lesions. Definitive resection can be performed when the
consequences of the resection are minimal, but it is absolutely
necessary only when the lesion has a particularly aggressive clinical
growth pattern.
present a particularly challenging problem. The lesion always involves
the posterior elements, but can also involve the vertebral body. The
patients initially complain of pain at the site of the lesion, but the
ABC is often not found until the patient has nerve root or cord
compression. Radiotherapy has been used in the management of patients
with ABC of the spine, but surgery is recommended for all patients as
the initial approach. Most cases are controlled with simple curettage.
Usually the posterior elements are resected, and any involvement of the
pedicles or the body is curetted. If complete laminectomy is performed,
a short posterior fusion is advised. Radiotherapy can be used in the
postoperative period, but usually this is reserved for the rare case of
rapid recurrence with soft tissue infiltration. Postoperative MRI or CT
scan is recommended as a baseline study against which to compare any
later scans.
Both have the same chromosomal translocation between chromosomes 11 and
22, similar presentations, identical treatments, and almost identical
histologic characteristics. PNET is also called Askin tumor, and was
originally identified from tumors classified as Ewing sarcoma.
EWS/PNETs are thought to arise from the neural crest. At least 90% of
them have a characteristic chromosomal translocation
[t(11:22)(q24:q12)]. This translocation leads to a novel fusion protein
called EWS-FLI1 (423,424).
approximately 15%, used to be the most lethal of all primary bone
tumors before the use of adjuvant chemotherapy became routine (425).
Before adjuvant chemotherapy came into use, most patients were treated
with irradiation alone. With improved survival associated with adjuvant
chemotherapy the role of surgery has been reevaluated, and there is
evidence, albeit only from retrospective studies, that surgical
resection combined with chemotherapy produces improved survival rates,
compared with survival after irradiation and chemotherapy (259, 425, 426, 427, 428, 429, 430, 431, 432, 433, 434, 435, 436).
have generalized symptoms of fever, weight loss, and malaise, but this
is not the usual presentation. Male patients outnumber female patients
by a ratio of 3:2, and most patients are between the ages of 5 and 30
years. Any bone may be affected. The femur is the most common site of
origin (20%); the pelvis and the humerus are also common sites. There
is usually a soft tissue mass associated with the bone lesion, and this
mass can often be palpated during a physical examination. The mass is
warm, firm, and tender, and it may be pulsatile. There are no specific
abnormal laboratory values that are diagnostic of EWS/PNET, but the
sedimentation rate is often increased. Elevated LDH level indicates a
poor prognosis (267,422).
diffuse destruction of the bone, extension of the tumor through the
cortex, a soft tissue component, and a periosteal reaction (437) (Fig. 14.29).
The periosteal reaction may produce a Codman triangle, an “onionskin”
appearance, or a sunburst appearance. These suggest an aggressive
lesion that has rapidly penetrated the cortex and elevated the
periosteum. The extraosseous soft tissue mass and medullary canal
involvement can be seen on CT scans and MRI scans, and are usually more
extensive than what might have been expected from the appearance of the
plain radiograph (100,438,439).
determining the intramedullary extent of EWS. The inflammation around
the tumor is seen more easily with MRI than with other studies, and the
extent of inflammation is often more than would be suggested from the
findings of other tests. The technetium bone scan is most useful in
finding occult bone metastasis. Approximately 5% of these patients
present with pulmonary metastasis.
small, round, cell tumor. The EWS/PNET cell has a distinct nucleus with
minimal cytoplasm and an indistinct cytoplasmic border (15, 440, 441, 442).
The cells are similar and mitoses are uncommon. Necrotic areas are
usually seen. There are glycogen granules in the cytoplasm, and these
produce the positive periodic acid Schiff (PAS) stain on routine
histologic examination. The intracellular glycogen granules are
diastase positive (i.e., exposure to diastase will break the glycogen
down, eliminating PAS staining). Under the electron microscope, the
glycogen can be seen as dense cytoplasmic granules. Increasingly,
genetic analysis is being done in EWS/PNET in order to identify the
11:22 translocation as a means of establish the diagnosis.
and etoposide. Actinomycin D, a drug used earlier, is currently used
less often. Most protocols begin with two to four courses of
chemotherapy before a decision is made on how to manage the primary
tumor. This usually results in a significant reduction in the size of
the primary tumor. Surgical resection is recommended if the
consequences of the resection (limitation or loss of function) are
acceptable to the patient (429, 430, 431, 456, 457, 458).
If the margins are close and viable tumor is present in the resected
specimen, postoperative irradiation is recommended. If the primary
tumor cannot be resected without undue morbidity, irradiation alone can
be used (243, 244, 446, 459, 460, 461, 462, 463, 464, 465, 466, 467, 468).
The total dosage should be kept as low as possible, usually around 50
Gy, and certainly less than 60 Gy, because dosages of more than 60 Gy
are associated with an unacceptable incidence of irradiation-associated
sarcomas at a later time (469, 470, 471, 472, 473, 474, 475, 476).
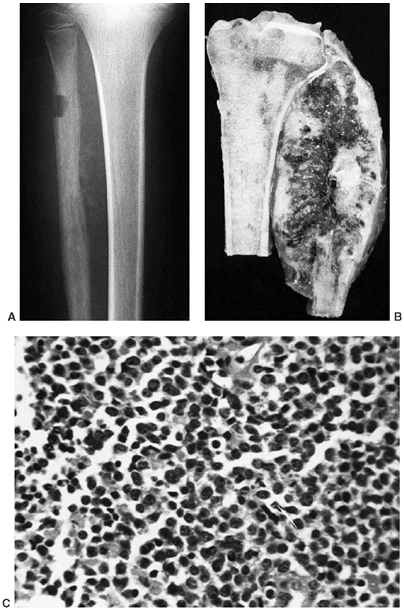 |
|
Figure 14.29 A:
Anteroposterior radiograph of the proximal tibia and fibula of a patient with Ewing sarcoma involving the proximal fibula. The fibular cortical detail is lost, and erosion of the medial surface, soft tissue mass, and periosteal reaction—all typical findings of Ewing sarcoma—are present. The combination of these findings is indicative of an aggressive process. Acute osteomyelitis may have this appearance, but the patient would usually have other signs of infection. The defect in the lateral aspect of the fibula is attributable to an incisional biopsy of the bone. A biopsy of the bone should not be performed if there is sufficient soft tissue extension. This will lower the risk of pathologic fracture. In addition, the extraosseous tumor is usually more easy to cut, and the histologic appearance is better. B: Gross specimen of Ewing sarcoma of the proximal fibula, similar to the case in A. The tumor has replaced the proximal fibula, and there is a large soft tissue mass, with invasion of surrounding muscles and no involvement of the tibia. This patient chose to have an immediate amputation, although this is not standard treatment. C: Histologic appearance of Ewing sarcoma. The nuclei are easily seen, and there are nucleoli within each nucleus. The cells are small and round, with very little variation in appearance of the nuclei. Mitoses are rare. The cytoplasm is faint and difficult to see, and the cytoplasmic borders are poorly defined (original magnification × 10). |
without metastasis reveal a 5-year disease-free survival of greater
than 65% (467). Patients who present with
metastasis have less chance of being cured, but should be treated
aggressively because some will survive.
frequency, and still they are both rare tumors (478).
Hemangioma, the fibromatoses, neurilemoma, and neurofibroma are more
common. The physician must be aware of the possibility of malignant
soft tissue tumor in the child and evaluate any lump carefully (479).
arteriovenous malformation. Its origin is controversial. It is
important that the abnormality be recognized as a benign lesion that in
certain circumstances regresses spontaneously, and in others
infiltrates the muscle and occasionally the bone. Hemangiomas are the
most common tumors in infancy and childhood, and account for 7% of
benign soft tissue tumors in all age groups. They are most common in
the head and neck regions, but may also be found in internal organs,
especially the liver. Often an intrahepatic hemangioma can be seen on a
CT scan of the abdomen taken for another reason, and its existence is
of little concern.
a classification of vascular tumors of soft tissue. The borderline
malignant and malignant vascular tumors are not pertinent to this
discussion; therefore, only the benign tumors are included:
-
localized hemangioma
-
capillary hemangioma (including juvenile type)
-
cavernous hemangioma
-
venous hemangioma
-
arteriovenous hemangioma (racemose hemangioma)
-
epithelioid hemangioma (angiolymphoid hyperplasia, Kimura disease)
-
hemangioma of granulation tissue type (pyogenic granuloma)
-
miscellaneous hemangiomas of deep soft tissue (synovial, intramuscular, neural)
-
angiomatosis (diffuse hemangioma)
benign vascular tumors. The juvenile hemangioma variant of capillary
hemangioma occurs in 1 out of every 200 live births. They may be
cutaneous or deep, and are usually seen within the first few weeks of
life, often enlarging for the first 6 months but then regressing and
becoming 75% to 95% involute by the age of 7 years. Capillary
hemangiomas do not require treatment.
type, but do not spontaneously regress and may require treatment. They
most commonly arise within muscle, and invade tissue planes
extensively. The patient often presents with complaints of swelling,
tenderness, and inflammation secondary to thrombophlebitis within the
hemangioma. This inflammation resolves within a few days, and can be
treated with local heat and oral aspirin. The noninflamed hemangioma is
soft and ill defined. The patient may have either no symptoms at all,
or the sensation of heaviness or a tight feeling in the extremity. On
the plain radiograph there are often small, smooth, round
calcifications called phleboliths. The
appearance of hemangiomas on MRI is almost completely diagnostic,
because they are composed of smooth, regular blood vessels and normal
fat.
with the major vascular tree, and do not easily fill with contrast for
angiography or venography; they are better visualized with MRI.
Occasionally a tourniquet proximal to the hemangioma permits filling of
the tumor veins at the time of venography or angiography (Fig. 14.30).
If an intravenous injection does not demonstrate the hemangioma, the
dye can be injected directly into the hemangioma. CT scan, particularly
if performed with intravenous contrast, is almost always diagnostic.
The hemangioma has varying densities with multiple dye-filled areas.
Biopsy may be performed to confirm the diagnosis, but often the
clinical presentation is sufficiently characteristic to render biopsy
unnecessary. Resection is not necessary unless the patient has repeated
bouts of inflammation or complaints of discomfort (usually a full or
tight feeling), or the parents are anxious about the mass.
entire muscle (or muscles) involved is resected. These lesions are
probably best considered as congenital abnormalities that involve most
of the veins in the extremity. When the grossly involved veins are
resected, the surrounding vessels dilate, resulting in clinical
recurrence. Hemangiomas do not undergo malignant degeneration, and
although they can produce significant abnormalities in the extremity,
surgical resection is rarely curative. However, resection may reduce
the symptoms. Irradiation has been used with varying benefit.
Embolization has also been used in patients who have severe pain.
Direct injections have been used, but there are few data regarding the
results, and no published reports of this management have been found.
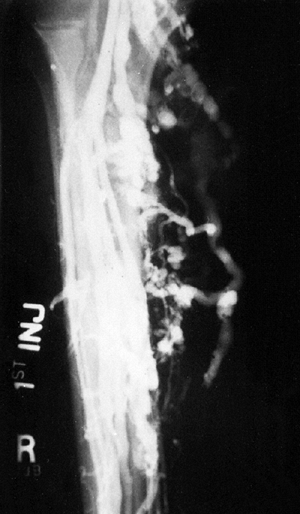 |
|
Figure 14.30
Venogram of a patient with hemangioma of the calf. The hemangioma communicated with the deep venous system and was easily filled when normal veins were injected with contrast; this is not always the case. This patient had two pulmonary emboli, and the hemangioma was confined to the gastrocnemius muscle. Therefore, the entire gastrocnemius muscle was resected. |
hamartoma, and not a true neoplasia. The solitary lesions are more
frequent, especially in the vertebral bodies where they are most often
found (Fig. 14.31). Solitary lesions may occur
in any bone, but the skull is the second most common site. These
lesions do not produce symptoms, and are usually found when a
radiograph is taken for another reason. The radiograph and CT scan are
diagnostic. The bone has a honeycomb appearance, with increased
trabecular markings around radiolucencies.
present during the first or second decade of life, with either mild
discomfort or pathologic fracture. The viscera and skin of these
patients may be involved. When multiple sites are involved, they are
usually the long bones of the extremities and the short bones of the
hands and feet. Treatment should be symptomatic, with curettage and
bone grafting for lesions that weaken the bone. Lesions that do not
produce symptoms or that are not associated with a risk for fracture
should be merely observed. They usually resolve with time.
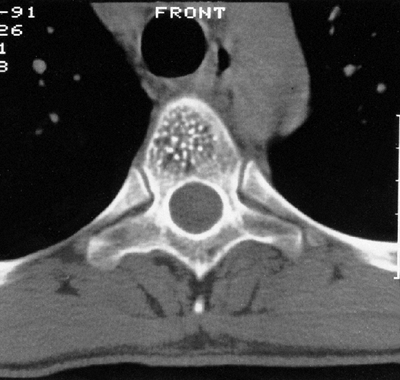 |
|
Figure 14.31
Computerized tomography scan of a typical hemangioma of the vertebral body. The small foci of increased density are thickened trabeculae of bone, and the low-density areas are filled with the hemangiomatous tissue. |
and rarely malignant. Extraabdominal desmoid, or aggressive
fibromatosis, is the most common benign fibrous lesion seen in children
(481,482). The less common lesions are not discussed in this text. Enzinger and Weiss (480) have discussed them elsewhere.
enlarging mass. The mass is deep, firm, and slightly tender but is not
inflamed. The adjacent joint is normal. A soft tissue mass can be seen
on a plain radiograph, but there are no distinguishing features.
Calcifications are not expected to be present within the mass.
in the lesion, but some large masses will not display increased uptake.
Often, even when the lesion is immediately adjacent to the bone, there
is no increased uptake of technetium. On CT scan, the mass has a
density similar to that of muscle, but it is usually more vascular and
can be distinguished best from the surrounding tissue by performing the
CT scan with an intravenous contrast. On MRI, the classic collagen
bundles produce a relative signal void (dark on T1- and T2-weighted
images) but, because the cellularity varies, fibromatoses may have an
appearance similar to any soft tissue neoplasia (483,484).
The cell of origin is believed to be the myofibroblast. The histologic
appearance and cell of origin of fibromatosis are identical to those of
plantar fibromatosis and Dupuytren contracture, but those lesions are
not as clinically aggressive as fibromatosis. Although they too recur,
they do not extend proximally from the feet or hands, as they do in
aggressive fibromatosis.
local excision rarely removes the entire tumor. Often, even after
resection, the pathologist may find a positive margin during
examination under a microscope. Fortunately, the presence of a positive
margin at the initial resection does not always lead to a local
recurrence, and it is recommended that the patient be observed for a
local recurrence. Approximately half of the patients will develop
recurrent disease regardless of the histologic margin. When lesions
recur they must be widely excised if local control is to be achieved.
Patients younger than 10 years have a greater risk of developing a
local recurrence than older patients. When a wide surgical margin is
ensured during the resection of the recurring lesion, local control is
usually achieved.
When the second surgical margin is also positive on microscopic examination, irradiation is recommended (482). Most lesions will be controlled with this combination. Low-dose methotrexate and vinblastine (377,486)
have been used for treating some patients who have an aggressive form
of fibromatosis, and the initial reports have been encouraging;
however, the effect of this treatment is unpredictable. Fibromatosis
has a variable clinical course, and the treatment needs to be
individualized for each patient.
 |
|
Figure 14.32
Histologic appearance of fibromatosis. The lesion infiltrates a muscle, and is more cellular than the typical fibromatosis. A wide resection was attempted, but microscopy revealed a positive margin. One year later, the patient had not had a recurrence (original magnification × 10). |
Neurilemomas, or schwannomas, arise from the nerve sheath. They occur
most often in early adulthood, and are usually solitary and
slow-growing. The patient usually presents with a painless mass, and
has a Tinel sign when the mass is tapped. The mass may be from any
nerve, but it is often in the superficial tissue arising from a small
sensory nerve. When arising from a spinal nerve root, the foramen may
be enlarged because of the pressure of the tumor on the bone. Nerve
dysfunction is uncommon, and is seen only when the nerve is compressed
between the tumor and an adjacent rigid structure. Patients with
superficial nerve lesions usually present early with small tumors, but
deep-seated lesions may be large before they are discovered (Fig. 14.33).
Neurilemoma is rarely seen in patients with von Recklinghausen disease
because neurofibroma is the type that is common in these patients.
Neurilemomas are nodular masses with a distinct capsule and are easily
separated from the nerve of origin. Under the microscope, they appear
as a combination of a cellular area (Antoni A) and a myxoid area
(Antoni B). The Antoni A area is composed of benign spindle cells that
tend to have their nuclei stacked with intervening cytoplasm (Fig. 14.34). This nuclear stacking produces a palisaded appearance, and the arrangement of alternating nuclei and cytoplasm is called a Verocay body.
The Antoni B area is composed of myxomatous tissue which has less
cellularity than does the Antoni A area. Neurilemomas are treated by
marginal excision without sacrificing the affected nerve. Neurilemomas
do not usually recur.
multiple lesions. Approximately 90% are solitary and are not
characteristic of von Recklinghausen disease. They may arise in the
skin or be associated with a recognizable peripheral
nerve.
Like neurilemomas, they usually present as a painless mass with a Tinel
sign. Unlike neurilemomas, however, they tend to be intimately
associated with the nerve fibers. Fortunately, most arise from small
cutaneous nerves and can be removed without loss of nerve function.
Histologically, neurofibromas are not encapsulated, and they invade the
nerve fibers and, rarely, the adjacent soft tissue. The cells are
elongated and wavy, and have dark-staining nuclei. There is a collagen
matrix composed of stringy-looking fibers. Neurites are usually seen
within the lesion. Surgical resection is recommended for those lesions
that are solitary and not associated with a major nerve. Lesions
arising from a major nerve can be resected, but the nerve fascicles
should be split and the neurofibroma should be removed from between
them. Neither solitary neurilemoma nor neurofibroma is associated with
a significant incidence of malignant degeneration, but patients with
neurofibromatosis have a small risk of developing neurofibrosarcoma.
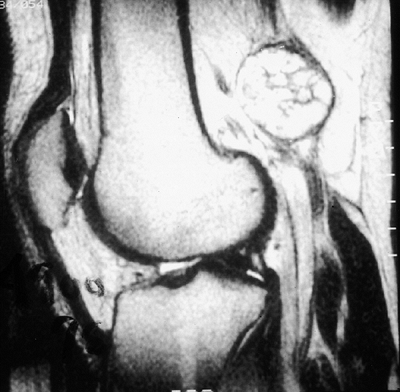 |
|
Figure 14.33
This is a sagittal view of a T1-weighted magnetic resonance image. The round, well-circumscribed mass posterior to the femur is within the peroneal nerve. It proved to be a schwannoma. Schwannomas have a typical appearance on magnetic resonance images. If they arise from a major nerve, as is the case in this patient, the nerve can usually be traced into the lesion. The schwannoma is smooth, slightly oblong, and has both bright and intermediate signals. |
 |
|
Figure 14.34
Histologic appearance of a neurilemoma (Antoni A area). The nuclei are stacked, giving the lesion a palisaded appearance (original magnification × 10). |
It was once thought to occur in adults and children with almost equal
frequency but, since the middle 1970s, most of the adult tumors that
were once called rhabdomyosarcoma have been reclassified as malignant
fibrous histiocytoma. Rhabdomyosarcoma is believed to be extremely rare
in adults, but it is the most common malignant soft tissue tumor in
patients younger than 15 years. It accounts for approximately 3.5% of
childhood malignancies, and there are approximately 350 new cases per
year in the United States (266,491). There are four histologic patterns: embryonal, botryoid-type, alveolar, and pleomorphic (492, 493, 494).
identical to the embryonal pattern, but is considered as a separate
entity because of its appearance on gross examination. A botryoid
rhabdomyosarcoma is an embryonal cell type that involves a hollow
viscus. Pleomorphic rhabdomyosarcoma is a histologic type seen in
adults, and is the least common. Embryonal rhabdomyosarcoma is the most
common type, and usually arises in the head, the neck, the
genitourinary tract, and the retroperitoneum. It is rare in the
extremities. Botryoid rhabdomyosarcoma tends to occur in the first
decade of life. The current treatment is a combination of chemotherapy,
surgery, and, if the malignancy is not totally excised, irradiation.
When chemotherapy is given preoperatively, the surgery required is less
radical, and adequate surgical margins are more easily achieved.
extremities than in the trunk, and is seen in older children and young
adults, usually between 10 and 25 years of age (247, 266, 495, 496, 497, 498, 499, 500).
The patient presents with a rapidly growing, painless mass deep within
the muscle. This occurs with equal frequency in the upper and lower
extremities. There are no clinical or laboratory findings that
distinguish rhabdomyosarcoma from other soft tissue tumors.
Approximately 70% of the tumors will have a translocation between
chromosome 13 and chromosome 2, whereas another 30% will have the
translocation between chromosome 13 and chromosome 1. Prognostic
variables include histologic subtype, size of the tumor, site of the
tumor, and age of the patient (504, 505, 506, 507).
Alveolar subtype, larger tumors, patients older than 10 years, and
location in an extremity are associated with a poor prognosis.
Therefore, the patients that the orthopaedist treats tend to do worse
than those treated by the urologist and the otolaryngologist. Alveolar
rhabdomyosarcoma, like the other subtypes, is treated with a
combination of chemotherapy and surgery (246, 247, 266, 508, 509, 510, 511, 512). Irradiation can be used if total surgical resection cannot be achieved without excessive
morbidity. If the lesion is small it should be totally resected
initially. If a rhabdomyosarcoma lesion occurs in an extremity,
preoperative chemotherapy should be considered. Chemotherapy often
makes total resection of such a lesion feasible. A wide surgical margin
is recommended (250, 511, 513, 514, 515).
Regional lymph nodes should be sampled and irradiation of the regional
nodes should be considered. Preoperative irradiation is reserved for
lesions that would require an amputation in order to obtain a wide
margin. Postoperative irradiation is used wherever preoperative
irradiation has not been given, and the surgical margins are positive
for tumor (510, 516, 517, 518).
Survival is related to the stage of the disease, but the overall
survival for a patient with rhabdomyosarcoma in an extremity is
approximately 60% (487, 488, 505, 509, 510, 519, 520).
This lesion consists of poorly differentiated rhabdomyoblasts with
limited collagen matrix. The rhabdomyoblasts are small, round-to-oval
cells with dark-staining nuclei and limited amounts of eosinophilic
cytoplasm (Fig. 14.35). Cross-striations are
not seen regularly. Alveolar rhabdomyosarcoma is composed of small,
round-to-oval tumor cells loosely arranged together in groups by dense
collagen bundles. This arrangement of cells in groups produces an
alveolar appearance; hence the name.
representation from both the Pediatric Oncology Group and the
Children’s Cancer Study Group, has been the dominant group treating
rhabdomyosarcoma in the United States. Their coordinated efforts have
resulted in major advances in the management of this malignancy (250). Their staging system for patients with rhabdomyosarcoma is currently in use (498,500,507,521) (see Table 14.5).
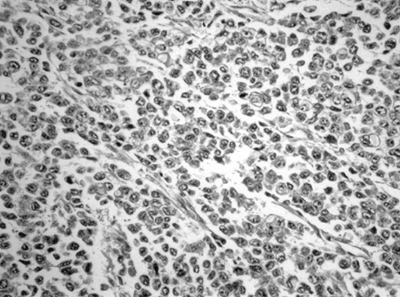 |
|
Figure 14.35
Rhabdomyosarcoma is a small blue cell malignancy. It has a histologic appearance similar to Ewing sarcoma and peripheral (or primitive) neuroectodermal tumor (EWS/PNET). There may be areas (not on this image) of sufficient differentiation such that muscle fibers with cross-striation are seen, but often the tumor is not sufficiently differentiated to produce even minimal amounts of muscle. |
tissue whose cellular characteristics suggest that the tumor arises
from primitive synovial cells, but it rarely occurs within a joint (253,480).
It has a typical chromosomal translocation, t(x;18)(p11.2;q11.2).
Unlike other soft tissue sarcomas, synovial cell sarcomas occur
frequently in the hand and foot (522). They are usually in the deep soft tissues near a joint. These tumors account for 10% of all soft tissue sarcomas (480).
Most patients are between 15 and 35 years of age, with male patients
being slightly predominant in number. Patients with synovial cell
sarcoma often experience pain before they have palpable masses, and
many patients give a history of having pain for 2 to 4 years before the
lesion was found. Synovial cell sarcoma, although rare, may be the
explanation for persistent pain in a young patient (477, 523, 524, 525, 526).
mass. Up to 25% of these patients have metastasis to regional lymph
nodes, and the lymph nodes should therefore be examined carefully (527). The patient’s blood and urine laboratory values are usually normal.
neck, and trunk account for approximately 15% of the lesions, whereas
the upper and lower extremities account for more than 50%. Almost 10%
of the lesions occur in the hands or feet.
ossifications within the tumor, and these are often seen on plain
radiographs. The radiodensities are usually very low. Small, irregular
calcific foci, or irregular ossification within a soft tissue tumor,
should suggest the diagnosis of synovial cell sarcoma. The CT scan
demonstrates a soft tissue mass, with calcified densities deep within
the tumor. Although the small foci of calcification or mineralization
are not seen as
well with MRI as with CT scan, MRI is preferred to CT scan as the staging test (528).
This is true for all soft tissue masses. Neurofibrosarcoma and
fibrosarcoma also may have intralesional calcification, but synovial
cell sarcoma is the most common tumor with intralesional densities.
|
TABLE 14.5 STAGING SYSTEM OF THE INTERGROUP RHABDOMYOSARCOMA COMMITTEE
|
||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
tumor with areas of epithelioid or glandular appearance interspersed
with areas having spindle cell appearance (480). Usually the spindle cell component predominates (Fig. 14.36). Some synovial cell sarcomas have only the spindle cell component and are called monophasic synovial cell sarcomas.
There seem to be no clinically significant differences between the two
types. Mitoses are usually present, but the tumor is more difficult to
grade histologically than other soft tissue sarcomas. Synovial cell
sarcoma is almost always a high-grade soft tissue sarcoma.
In adults and older children with synovial cell sarcoma, as in those
with other soft tissue sarcomas, preoperative radiotherapy is used in
conjunction with nonradical surgery in an attempt to salvage the
extremities. Approximately 15% of synovial sarcomas occur in the feet (522).
It used to be thought that the scarring from irradiation precluded its
use in the feet and hands but, with modern techniques, adjuvant
irradiation and marginal resection can be performed in most sarcomas of
the feet or hands, with preservation of the functioning of the
extremity. Preoperative irradiation and surgery are recommended for
most soft tissue sarcomas of the feet. This approach has been
successful in saving extremities and controlling the disease locally,
but the incidence of metastatic disease remains high, at slightly more
than 50% (253, 478, 524, 525, 529, 532, 536, 537).
synovial lining of a joint. There have been reported cases of synovial
cell sarcoma having arisen from within a joint, but this is decidedly
rare. Most synovial sarcomas arise within periarticular soft tissues
and do not invade joints. Synovial chondromatosis and pigmented
villonodular synovitis (PVNS) arise from synovial tissue and are found
in joints, bursa, and tendon sheaths. They are the only two neoplasias
that commonly occur in joints.
It occurs most often in the knee, but can arise in any joint, tendon
sheath, or bursa. Its cause is unknown, and it has no recognized
familial pattern of occurrence. The subliminal lining of the joint
produces small nodules of hyaline-like cartilage that are extruded from
the synovial lining to become loose bodies within the joint (Fig. 14.37).
If they become large, the cartilage may become necrotic; if they have
blood supply they may undergo enchondral ossification. In both cases,
they can be seen on plain radiographs. Without the calcification or the
ossification, the cartilage is radiolucent and not visible on routine
radiographs
commonly between the ages of 20 and 50 years. The most common joint
involved is the knee, with the elbow next in frequency, and the hip
third. The patient usually presents with mild discomfort, minimal loss
of motion, and an effusion in a joint. There may be a history of
locking. The knee may appear normal on examination, but usually there
is a moderate-to-large effusion, limited motion, and a boggy synovium.
The plain radiographs show nothing abnormal, or only small
intraarticular calcified bodies. The arthrogram is diagnostic, showing
an irregular synovial
surface and normal-to-thinned synovial fluid. The MRI scan is usually diagnostic.
 |
|
Figure 14.36 A:
Low-power view of the spindle component of a synovial cell sarcoma. This lesion is composed of malignant spindle cells with a minimal amount of matrix. At a higher power, mitotic figures are seen. Other areas of this tumor have a glandular appearance, which is why synovial cell sarcoma is a biphasic tumor (original magnification × 10) B: Synovial cell sarcoma does not arise from within joints. It may be monophasic to biphasic. This is a monophasic synovial sarcoma. It is composed of fibrous stroma cells with minimal matrix formation. As can be seen in this image, the direction of the fibers is often at right angles, so that there are areas where the fibers run horizontally (center) and other areas where the fibers run vertically (lower right) (40 ×) |
 |
|
Figure 14.37
Low-power view of synovial chondromatosis. The nodules of cartilage are formed within the synovial lining and extruded into the joint to produce loose bodies. The nodules can undergo enchondral ossification if they have a blood supply (top right). |
 |
|
Figure 14.38
Pigmented villonodular synovitis is often diagnosed on the basis of the grossly thickened and frondlike synovium. The histologic findings consist of multinucleated giant cells, mononuclear cells, a fibrous stroma and, usually, hemosiderin deposits. There is no atypia, and mitotic figures are rare. |
removal of the loose bodies. Usually synovectomy is performed, but
recurrence is common as the synovial lining is regenerated (538). The process seems to have a limited natural course, and the production of new loose bodies ceases after 1 or 2 years.
be a true neoplasia, although there have been suggestions that it may
be caused by an infectious process (480,539). The synovial lining becomes proliferative and hypertrophic (Fig. 14.38).
It can involve a joint (most commonly the knee) or a tendon sheath.
When tendon sheaths are involved, PVNS usually occurs in the hand or
the foot. The patient presents with a swollen joint that is usually
painless. The synovial tissue is boggy on examination. The fluid in the
joint has old, dark blood in it, and it is common for the diagnosis to
be suspected first when the joint is aspirated just before the
injection of contrast material for arthrography. The arthrogram or MRI
scan is diagnostic, with a thickened shaggy lining and demonstration of
dark pigment signal on MRI (540)
The plain radiograph is usually normal except for the soft tissue
swelling, but occasionally the proliferative synovial tissues invade
the bones adjacent to the joint. This happens most frequently when the
hip joint is involved. MRI is the best radiographic method to evaluate
the extent of the lesion. Bone invasion can be appreciated, as can the
extent of enlargement of the synovial cavity. Synovectomy is the
treatment of choice, but there is a high incidence of recurrence
(approximately 50%) (539). Intraarticular
injection of radioactive materials (dysprosium or yttrium) has been
used successfully as a means of controlling recurrent disease. In some
patients recurrence produces only minimal symptoms, and the patients
may accept the chronic swelling. As long as the bones remain
uninvolved, there is no absolute indication for surgical removal.
Patients are followed with clinical examination and plain radiography.
For patients with diffuse involvement, recurrence is common (more than
75%), but most of the patients have little or no progression and
symptoms, and therefore do not need treatment.
JA, Gebhardt MC, Kozakewich HP. Cytogenetic aberrations in
osteosarcomas: nonrandom deletions, rings, and double-minute
chromosomes. Cancer Genet Cytogenet 1994;77:81.
CR, Tschernyavsky SJ, Woodruff JM, et al. Molecular diagnosis of clear
cell sarcoma: detection of EWS-ATF1 and MITF-M transcripts and
histopathological and ultrastructural analysis of 12 cases. J Mol Diagn 2002;4(1):44–52.
M, et al. The der(17)t(X;17)(p11;q25) of human alveolar soft part
sarcoma fuses the TFE3 transcription factor gene to ASPL, a novel gene
at 17q25. Oncogene 2001;20(1):48–57.
VS, Kuhne T, Jundt G, et al. Molecular diagnosis of Ewing tumors:
improved detection of EWS-FLI-1 and EWS-ERG chimeric transcripts and
rapid determination of exon combinations. Diagn Mol Pathol 1998;7(1):29–35.
JA, Kozakewich HP, Hoffer FA, et al. Diagnostic relevance of clonal
cytogenetic aberrations in malignant soft-tissue tumors. N Engl J Med 1991;324:436.
FG, Chatten J, D’Cruz CM, et al. Molecular assays for chromosomal
translocations in the diagnosis of pediatric soft tissue sarcomas. JAMA 1995;273:553.
S, Pack S, Kumar D, et al. Detection of EWS-FLI-1 fusion in Ewing’s
sarcoma/peripheral primitive neuroectodermal tumor by fluorescence in
situ hybridization using formalin-fixed paraffin-embedded tissue. Hum Pathol 1999;30:324.
S, Cavazzana AO, Llombart-Bosch A, et al. Comparison of Ewing’s sarcoma
of bone and peripheral neuroepithelioma: an immunocytochemical and
ultrastructural analysis of two primitive neuroectodermal neoplasms. Arch Pathol Lab Med 1994;118:608.
D, Herrmann C, Jurgens H, et al. Malignant peripheral neuroectodermal
tumor and its necessary distinction from Ewing’s sarcoma: a report from
the Kiel Pediatric Tumor Registry. Cancer 1991;68:2251.
P, Henry-Amar M, Triche TJ, et al. Is neuro-ectodermal differentiation
of Ewing’s sarcoma of bone associated with an unfavourable prognosis? Eur J Cancer 1995;31A:307.
KR, Triche TJ, Kinsella TJ, et al. Prognostic value of histopathology
in Ewing’s sarcoma: long-term follow-up of distal extremity primary
tumors. Cancer 1991;67:163.
DN, Sommergruber W, Muehlbacher K, et al. Variability in gene
expression patterns of Ewing tumor cell lines differing in EWS-FLI1
fusion type. Lab Invest 2000;80(12):1833–1844.
DC, Grier HE, Swallow MM, et al. Detection of circulating tumor cells
in patients with Ewing’s sarcoma and peripheral primitive
neuroectodermal tumor. J Clin Oncol 1997;15:583.
Alava E, Lozano MD, Patino A, et al. Ewing family tumors: potential
prognostic value of reverse-transcriptase polymerase chain reaction
detection of minimal residual disease in peripheral blood samples. Diagn Mol Pathol 1998;7(3):152–157.
JK, van de Vijver MJ, Cleton-Jansen AM, et al. Overexpression of the
HER-2 oncogene does not play a role in high-grade osteosarcomas. Eur J Cancer 2004;40(7):963–970.
JY, Aviv H, Benevenia J, et al. HER-2/neu and p53 in osteosarcoma: an
immunohistochemical and fluorescence in situ hybridization analysis. Cancer Invest 2004;22(1):16–24.
SH, Bernards R, Rogelj S, et al. A human DNA segment with properties of
the gene that predisposes to retinoblastoma and osteosarcoma. Nature 1986;323:643.
Y, Dockhorn-Dworniczak B, Blasius S, et al. Analysis of mutant p53
protein in osteosarcomas and other malignant and benign lesions of
bone. J Cancer Res Clin Oncol 1993;119:172.
F, Ueda T, Huvos AG, et al. p53 and MDM2 alterations in osteosarcomas:
correlation with clinicopathologic features and proliferative rate. Cancer 1997;79:1541.
B, Gebhardt MC, Kloen P, et al. Screening for TP53 mutations in
osteosarcomas using constant denaturant gel electrophoresis (CDGE). Hum Mutat 1993;2:274.
A, Mangham DC, Reynolds GM, et al. Overexpression of p53 protein in
primary Ewing’s sarcoma of bone: relationship to tumour stage, response
and prognosis. Br J Cancer 1999; 79:1185.
T, Toguchida J, Yamamuro T, et al. Allelotype analysis in
osteosarcomas: frequent allele loss on 3q, 13q, 17p, and 18q. Cancer Res 1992;52:2419.
DE, Holden ST, Steel CM, et al. A significant proportion of patients
with osteosarcoma may belong to Li-Fraumeni cancer families. J Bone Joint Surg Br 1992;74:883.
J, Yamaguchi T, Dayton SH, et al. Prevalence and spectrum of germline
mutations of the p53 gene among patients with sarcoma. N Engl J Med 1992;326:1301.
L, Sexsmith E, Gottlieb A, et al. Germline p53 mutations are frequently
detected in young children with rhabdomyosarcoma. J Clin Invest 1995;95:1606.
D, Li FP, Strong LC, et al. Germ line p53 mutations in a familial
syndrome of breast cancer, sarcomas, and other neoplasms. Science 1990;250:1233.
JF, Smith-Sorensen B, Friend SH, et al. Germline mutations of the p53
tumor suppressor gene in children with osteosarcoma. J Clin Oncol 1994;12:925.
HS, Grogan TM, Haddad G, et al. P-glycoprotein expression: critical
determinant in the response to osteosarcoma chemotherapy. J Natl Cancer Inst 1997;89:1706.
K, Hirata M, Takeshita H, et al. Relationship between P-glycoprotein
positivity, doxorubicin binding ability and histologic response to
chemotherapy in osteosarcomas. Cancer Lett 1999;138:203.
N, Scotlandi K, Barbanti-Brodano G, et al. Expression of P-glycoprotein
in high-grade osteosarcomas in relation to clinical outcome. N Engl J Med 1995;333:1380.
R, Sampietro G, Collini P, et al. Prognostic value of clinicopathologic
characteristics including neuroectodermal differentiation in osseous
Ewing’s sarcoma family of tumors in children. Tumori 1999;85:101.
BR, Pinckney L, Etcubanas E. Relative efficacy of radiographic and
radionuclide bone surveys in the detection of the skeletal lesions of
histiocytosis X. Radiology 1980;134:377.
AR, Tashjian JH, Lazarus K, et al. Nuclear medicine studies in
evaluation of skeletal lesions in children with histiocytosis X. Radiology 1981;140:787.
MA, Kirchner PT. Scintigraphic evaluation of primary bone tumors:
comparison of technetium-99m phosphonate and gallium citrate imaging. J Bone Joint Surg Am 1980;62:758.
M, Yeh SD, Yeung H, et al. Thallium-201 scintigraphy for the evaluation
of tumor response to preoperative chemotherapy in patients with
osteosarcoma. Cancer 1997;80:1507.
L, Waxman A, Binney G, et al. Thallium-201 scintigraphy in bone
sarcoma: comparison with gallium-67 and technetium-MDP in the
evaluation of chemotherapeutic response. J Nucl Med 1990;31:567.
M, Rydholm A, Persson BM. Aspiration cytology of soft-tissue tumors:
the 10-year experience at an orthopedic oncology center. Acta Orthop Scand 1985;56:407.
CL, Rosenberg AE, Rosenthal DI. Controlled thermal injury of bone:
report of a percutaneous technique using radiofrequency electrode and
generator. Invest Radiol 1989;24:888.
DI, Alexander A, Rosenberg AE, et al. Ablation of osteoid osteomas with
a percutaneously placed electrode: a new procedure. Radiology 1992;183:29.
DI, Hornicek FJ, Wolfe MW, et al. Percutaneous radiofrequency
coagulation of osteoid osteoma compared with operative treatment. J Bone Joint Surg Am 1998;80:815.
SJ, Cook AJ, Weiner DS, et al. Treatment of osteoid osteoma by computed
tomography guided excision in the pediatric patient. J Pediatr Orthop 1990;10:510.
LA, Goris M, Bleck EE, et al. Intraoperative skeletal scintigraphy for
localization of osteoid-osteoma in the spine: case report. J Bone Joint Surg Am 1980;62:143.
P, Sangiorgi L, Rougraff BT, et al. Relationship of
chemotherapy-induced necrosis and surgical margins to local recurrence
in osteosarcoma. J Clin Oncol 1994;12:2699.
P, Bohling T, Bacci G, et al. Chemotherapy-induced tumor necrosis as a
prognostic factor in localized Ewing’s sarcoma of the extremities. J Clin Oncol 1997;15:1553.
RL, Craft AW, Van der Eijken JW, et al. Randomized trial of two
regimens of chemotherapy in operable osteosarcoma: a study of the
European Osteosarcoma Intergroup. Lancet 1997;350:911.
AJ, Ettinger LJ, Nachman JB, et al. Treatment of nonmetastatic
osteosarcoma of the extremity with preoperative and postoperative
chemotherapy: a report from the Children’s Cancer Group. J Clin Oncol 1997;15:76.
G, Ferrari S, Mercuri M, et al. Neoadjuvant chemotherapy for extremity
osteosarcoma: preliminary results of the Rizzoli’s 4th study. Acta Oncol 1998;37:41.
G, Ferrari S, Donati D, et al. Neoadjuvant chemotherapy for
osteosarcoma of the extremity in patients in the fourth and fifth
decade of life. Oncol Rep 1998;5:1259.
MP, Goorin AM, Horowitz M, et al. Adjuvant chemotherapy of high-grade
osteosarcoma of the extremity: updated results of the
Multi-institutional Osteosarcoma Study. Clin Orthop 1991;270:8.
MP, Goorin AM, Miser AW, et al. The effect of adjuvant chemotherapy on
relapse-free survival in patients with osteosarcoma of the extremity. N Engl J Med 1986;314:1600.
G, Ferrari S, Mercuri M, et al. Predictive factors for local recurrence
in osteosarcoma: 540 patients with extremity tumors followed for
minimum 2.5 years after neoadjuvant chemotherapy. Acta Orthop Scand 1998;69:230.
F, Picci P, Bacci G, et al. Limb sparing versus amputation in
osteosarcoma: correlation between local control, surgical margins and
tumor necrosis. Istituto Rizzoli experience. Ann Oncol 1992;3(Suppl 2):S23.
BT, Simon MA, Kneisl JS, et al. Limb salvage compared with amputation
for osteosarcoma of the distal end of the femur: a long-term
ontological, functional, and quality-of-life study. J Bone Joint Surg Am 1994;76:649.
SP, Temple HT, O’Keefe RJ, et al. The surgical treatment of patients
with osteosarcoma who sustain a pathologic fracture. Clin Orthop 1996;324:227.
A, Sferopoulos NK, Tillman RM, et al. The surgical treatment and
outcome of pathological fractures in localised osteosarcoma. J Bone Joint Surg Br 1996;78:694.
H, Friedman L. Dedifferentiated parosteal osteosarcoma with high-grade
osteoclast-rich osteogenic sarcoma at presentation. Skeletal Radiol 1998;27:574.
DS, Yasko AW, Raymond AK, et al. Conventional and dedifferentiated
parosteal osteosarcoma: diagnosis, treatment, and outcome. Cancer 1996;78:2136.
HM, Malinin TI, Mnaymneh WA. Massive allograft implantation following
radical resection of high-grade tumors requiring adjuvant chemotherapy
treatment. Clin Orthop 1985;197:88.
W, Malinin TI, Makley JT, et al. Massive osteoarticular allografts in
the reconstruction of extremities following resection of tumors not
requiring chemotherapy and radiation. Clin Orthop 1985;197:76.
MC, Flugstad DI, Springfield DS, et al. The use of bone allografts for
limb salvage in high-grade extremity osteosarcoma. Clin Orthop 1991;270:181.
BA, De Bari A, Krajbich JI. Massive allografts in the treatment of
osteosarcoma and Ewing sarcoma in children, and adolescents. J Bone Joint Surg Am 1995;77:54.
FJ Jr, Mnaymneh W, Lackman RD, et al. Limb salvage with osteoarticular
allografts after resection of proximal tibia bone tumors. Clin Orthop 1998;352:179.
WF, Dunham W, Gebhardt MC, et al. A system for the functional
evaluation of reconstructive procedures after surgical treatment of
tumors of the musculoskeletal system. Clin Orthop 1993;286:241.
P, Burgos-Flores J, Ocete-Guzman JG, et al. The management of
limb-length discrepancies in children after treatment of osteosarcoma
and Ewing’s sarcoma. J Pediatr Orthop 1995;15:561.
WG, Yang RS, Eckardt JJ. Endoprosthetic bone reconstruction following
malignant tumor resection in skeletally immature patients. Orthop Clin North Am 1996;27:493.
WP, Grimer RJ, Carter SR, et al. Longitudinal growth following a
growing endoprosthesis replacement of the distal femur in the
skeletally immature. In: Eighth International Symposium on Limb Salvage (ISOLS). Italy: Florence, 1995.
OS, Cannon SR, Briggs TW, et al. Stanmore custom-made extendible distal
femoral replacements: clinical experience in children with primary
malignant bone tumours. J Bone Joint Surg Br 1997;79:927 (erratum 1998;80:562).
GJ, Schraffordt Koops H, Verb RP, et al. First clinical experience with
a noninvasively extendable endoprosthesis: a limb-saving procedure in
children suffering from a malignant bone tumor. Artif Organs 1997;21:413.
JJ, Safran MR, Eilber FR, et al. Expandable endoprosthetic
reconstruction of the skeletally immature after malignant bone tumor
resection. Clin Orthop 1993;297:188.
E, Gebhardt MC, Jennings LC, et al. The results of transplantation of
intercalary allografts after resection of tumors: a long-term follow-up
study. J Bone Joint Surg Am 1997;79:97.
M, Gasbarrini A, Malaguti C, et al. Intraepiphyseal resection of the
proximal tibia and its impact on lower limb growth. Clin Orthop 1999;358:111.
R, Manfrini M, Ceruso M, et al. A new reconstruction for metadiaphyseal
resections: a combined graft (allograft shell plus vascularized
fibula). Preliminary results. In: Brown KLB, ed. Complications of limb salvage: prevention, management and outcome. Montreal: International Symposium of Limb Salvage, 1991:319.
R, van Horn JR, Biagini R, et al. The Tikhoff-Linberg procedure for
bone tumors of the proximal humerus: the classical extensive technique
versus a modified transglenoid resection. Arch Orthop Trauma Surg 1990;109:63.
G, Assenmacher S, Schmit-Neuerburg KP. Tikhoff-Linberg procedure for
bone and soft tissue tumors of the shoulder girdle. Arch Surg 1999;134:252.
MI, Sim FH, Chao EY. Limb salvage for neoplasms of the shoulder girdle:
intermediate reconstructive and functional results. J Bone Joint Surg Am 1996;78:1872.
MC, Roth YF, Mankin HJ. Osteoarticular allografts for reconstruction in
the proximal part of the humerus after excision of a musculoskeletal
tumor. J Bone Joint Surg Am 1990; 72:334.
VP, Satku SK, Mitra AK, et al. Function following limb salvage for
primary tumors of the shoulder girdle: 10 patients followed 4 (1-11)
years. Acta Orthop Scand 1994;65:55.
MM, Sugarbaker PH, Lampert M, et al. The Tikhoff-Linberg procedure:
report of ten patients and presentation of a modified technique for
tumors of the proximal humerus. Surgery 1985;97:518.
F, Kotz R, Knahr K, et al. Rotationplasty for limb salvage in the
treatment of malignant tumors at the knee: a follow-up study of seventy
patients. J Bone Joint Surg Am 1991;73:1365.
FP Jr, Glasser DB, Otis JC, et al. The Van Nes tibial rotationplasty: a
functionally viable reconstructive procedure in children who have a
tumor of the distal end of the femur. J Bone Joint Surg Am 1990;72:1541.
der Windt DA, Pieterson I, van der Eijken JW, et al. Energy expenditure
during walking in subjects with tibial rotationplasty, above-knee
amputation, or hip disarticulation. Arch Phys Med Rehabil 1992;73:1174.
R, Millesi H, Kotz R. Resection-replantation for primary malignant
tumours of the arm: an alternative to forequarter amputation. J Bone Joint Surg Br 1995;77:176.
WW, Sullivan MP, Fernbach DJ, et al. Adjuvant chemotherapy in primary
treatment of osteogenic sarcoma: a Southwestern Oncology Group Study. Cancer 1975;36:1598.
SA, Cabner BA, Young RC, et al. Treatment of osteogenic sarcoma. I.
Effect of adjuvant high-dose methotrexate after amputation. Cancer Treat Rep 1979;63:739.
G, Murphy ML, Huvos AG, et al. Chemotherapy, en bloc resection, and
prosthetic bone replacement in the treatment of osteogenic sarcoma. Cancer 1976;37:1.
J, Glaubiger D, Lichter A, et al. Local control of Ewing’s sarcoma of
bone with radiotherapy and combination chemotherapy. Cancer 1980;46:1969.
M, Razek A, Perez C, et al. Local control and survival related to
radiation dose and volume and to chemotherapy in non-metastatic Ewing’s
sarcoma of pelvic bones. Int J Radiat Oncol Biol Phys 1978;4:367.
N, Paed D, Traggis D, et al. Improved outlook for Ewing’s sarcoma with
combination chemotherapy (vincristine, actinomycin D and
cyclophosphamide) and radiation therapy. Cancer 1976;38:1925.
DM, Soule EH, Lawrence W Jr, et al. Extremity lesions in the Intergroup
Rhabdomyosarcoma Study (IRS-I): a preliminary report. Cancer 1982;49:1.
W Jr, Hays DM, Heyn R, et al. Surgical lessons from the Intergroup
Rhabdomyosarcoma Study (IRS) pertaining to extremity tumors. World J Surg 1988;12:676.
K, Beron G, Delling G, et al. Neoadjuvant chemotherapy of osteosarcoma:
results of a randomized cooperative trial (COSS-82) with salvage
chemotherapy based on histological tumor response. J Clin Oncol 1988;6:329.
G, Bacci G, Picci P, et al. Telangiectatic osteogenic sarcoma of the
extremities: results in 17 patients treated with neoadjuvant
chemotherapy. Clin Orthop Relat Res 1991;270:99.
K, Bieling P, Bielack S, et al. Local control and survival from the
Cooperative Osteosarcoma Study Group studies of the German Society of
Pediatric Oncology and the Vienna Bone Tumor Registry. Clin Orthop Relat Res 1991;270:79.
G, Picci P, Ruggieri P, et al. Neoadjuvant chemotherapy for the
treatment of osteosarcoma of the limbs: preliminary results in 100
patients treated preoperatively with high doses of methotrexate i.v.
followed by cisplatin (i.a.) and adriamycin. Chir Organi Mov 1991;76:1.
J, Munarriz B, Pastor M, et al. Long-term follow-up and prognostic
factors in Ewing’s sarcoma: a multivariate analysis of 116 patients
from a single institution. Oncology 1998;55:20.
S, Mercuri M, Rosito P, et al. Ifosfamide and actinomycin-D, added in
the induction phase to vincristine, cyclophosphamide and doxorubicin,
improve histologic response and prognosis in patients with
non-metastatic Ewing’s sarcoma of the extremity. J Chemother 1998;10:484.
G, Picci P, Ferrari S, et al. Primary chemotherapy and delayed surgery
for nonmetastatic osteosarcoma of the extremities: results in 164
patients preoperatively treated with high doses of methotrexate
followed by cisplatin and doxorubicin. Cancer 1993;72:3227.
H, Kosloff C, Nirenberg A, et al. Prognostic factors in the response of
primary osteogenic sarcoma to preoperative chemotherapy (high-dose
methotrexate with citrovorum factor). Natl Cancer Inst Monogr 1981;56:221.
G, Picci P, Ferrari S, et al. Neoadjuvant chemotherapy for Ewing’s
sarcoma of bone: no benefit observed after adding ifosfamide and
etoposide to vincristine, actinomycin, cyclophosphamide, and
doxorubicin in the maintenance phase. Results of two sequential
studies. Cancer 1998;82:1174.
M, Malawer M, Woo S, et al. Ewing’s sarcoma family of tumors: Ewing’s
sarcoma of bone and soft tissue and the peripheral primitive
neuroectodermal tumors. In: Pizzo P, Poplack D, eds. Principles and practice of pediatric oncology. Philadelphia: Lippincott-Raven, 1997:831.
K, Beron G, Kotz R, et al. Neoadjuvant chemotherapy for osteogenic
sarcoma: results of a cooperative German/Austrian study. J Clin Oncol 1984;2:617.
MJ, Bloem JL, Eulderink F, et al. Cartilaginous tumors: correlation of
gadolinium-enhanced MR imaging and histopathologic findings. Radiology 1993;186:813.
R, Baddeley H, Pringle JS, et al. Primary bone tumors: MR morphologic
appearance correlated with pathologic examinations. Acta Radiol 1991;32:290.
SE, Pike EJ, Ward WG, et al. Dedifferentiated chondrosarcoma in
patients with multiple osteochondromatosis: report of a case and review
of the literature. Skeletal Radiol 1997;26:370.
H, Lichtenstein L. Benign chondroblastoma of bone: a reinterpretation
of the so-called calcifying or chondromatous giant cell tumor. Am J Pathol 1942;18:969.
AC, Culver JE Jr, Keats TE. Histological nature of the cortical
irregularity of the medial posterior distal femoral metaphysis in
children. Radiology 1971;99:389.
W, Dudley H, Barry R. The natural history of fibrous dysplasia: an
orthopaedic, pathological, and roentgenographic study. J Bone Joint Surg Am 1962;44:207.
F, Butler A, Hampton A, et al. Syndrome characterized by osteitis
fibrosa disseminata, area of pigmentation and endocrine dysfunction,
with precocious puberty in females. N Engl J Med 1937;216:727.
M, Robboy SJ, Kim S, et al. Cushing syndrome, sexual precocity, and
polyostotic fibrous dysplasia (Albright syndrome) in infancy. J Pediatr 1975;87:917.
BH, Bray EWD, Meyer LC. Multiple osteotomies with Zickel nail fixation
for polyostotic fibrous dysplasia involving the proximal part of the
femur. J Bone Joint Surg Am 1987;69:691.
JT, Kumar SJ, MacEwen GD. Fibrous dysplasia of the proximal part of the
femur: long-term results of curettage and bone-grafting and mechanical
realignment. J Bone Joint Surg Am 1998;80:648.
JW, Shih CH, Chen WJ. Osteofibrous dysplasia (ossifying fibroma of long
bones): a report of four cases and review of the literature. Clin Orthop 1992;278:235.
M. Salton K, Horiwuchi H, et al. Comparative study of fibrous dysplasia
and osteofibrosis dysplasia: histopathological, immunohistochemical,
argyophillic nucleolar organizer and DNA ploidy analysis. Pathol Int 2001;51:603–611.
O, Ibarazi K, Shimabukura H, et al. Packing with high-porosity
hydroxyapatite cubes alone for the treatment of simple bone cyst. Clin Orthop Res 1993;293:287–292.
HM, Taminiau AH, Fleuren GJ, et al. Adamantinoma of the long bones: a
clinicopathological study of thirty-two patients with emphasis on
histological subtype, precursor lesion, and biological behavior. J Bone Joint Surg Am 1994;76:1482.
S, Sommelet D, Lascombes P, et al. Treatment of Langerhans-cell
histiocytosis in children: experience at the Children’s Hospital of
Nancy. J Bone Joint Surg Am 1994;76:1513.
L. Histiocytosis X: integration of eosinophilic granuloma of bone,
“Letterer-Siwe disease,” and “Schuller-Christian disease” as related
manifestations of a single nosologic entity. Arch Pathol 1953;56:84.
SE, Wenger DE, Gilchrist GS, et al. Langerhans’ cell histiocytosis
(histiocytosis X) of bone: a clinicopathologic analysis of 263
pediatric and adult cases. Cancer 1995;76:2471.
J, Brown L, Herkowitz H. Vertebra plana-like lesions in children: case
report with special emphasis on the differential diagnosis and
indications for biopsy. J Spinal Disord 1991;4:480.
AS, Conway JJ, Miller JH, et al. Detection of bone lesions in
Langerhans cell histiocytosis: complementary roles of scintigraphy and
conventional radiography. J Pediatr Hematol Oncol 1996;18:51.
DM, Mullan BP, Wiseman GA, et al. Bone scintigraphy evaluated in
diagnosing and staging Langerhans’ cell histiocytosis and related
disorders. J Nucl Med 1996;37:1456.
Nieuwenhuyse JP, Clapuyt P, Malghem J, et al. Radiographic skeletal
survey and radionuclide bone scan in Langerhans cell histiocytosis of
bone. Pediatr Radial 1996;26:734.
RM, Thompson RC Jr, Voute PA, et al. Intralesional infiltration of
corticosteroids in localized Langerhans’ cell histiocytosis. J Pediatr Orthop 1992;12:811.
HW, Pruszczynski M, Lemmens JA, et al. Eosinophilic granuloma of bone:
results of treatment with curettage, cryosurgery, and bone grafting. J Pediatr Orthop B 1998;7:253.
RB Jr, D’Angio GJ. Langerhans’ cell histiocytosis (histiocytosis X):
experience at the Children’s Hospital of Philadelphia, 1970–1984. Med Pediatr Oncol 1989;17:20.
O, Marchetti PG, Bartolozzi P. Final results obtained in the treatment
of bone cysts with methylprednisolone acetate (Depo-Medrol) and a
discussion of results achieved in other bone lesions. Clin Orthop 1982;165:33.
KF Jr, Bright RW, Fitzgerald SP, et al. Solitary unicameral bone cyst:
treatment with freeze-dried crushed cortical-bone allograft. A review
of one hundred and forty-four cases. J Bone Joint Surg Am 1976;58:636.
O, Marchetti PG, Bartolozzi P. The effects of methylprednisolone
acetate in the treatment of bone cysts: results of three years
follow-up. J Bone Joint Surg Br 1979;61:200.
KF, Sell KW, Brown RH. Solitary bone cyst: treatment with freeze-dried
cancellous bone allograft. A study of one hundred seventy-seven cases. J Bone Joint Surg Am 1969;51:87.
V, Sissons HA. Aneurysmal bone cyst: a review of 123 cases including
primary lesions and those secondary to other bone pathology. Cancer 1988;61:2291.
M, Arato G, Ezzati A, et al. Aneurysmal bone cyst: its pathogenesis
based on angiographic, immunohistochemical and electron microscopic
studies. Pathol Oncol Res 1998;4:277.
O, Zucman J, Melot T, et al. The Ewing family of tumors: a subgroup of
small-round-cell tumors defined by specific chimeric transcripts. N Engl J Med 1994;331:294.
VS, Kuhne T, Jundt G, et al. Molecular diagnosis of Ewing tumors:
improved detection of EWS-FLI-1 and EWS-ERG chimeric transcripts and
rapid determination of exon combinations. Diagn Mol Pathol 1998;7:29.
DJ, Dahlin DC, Dauphine RT, et al. Ewing’s sarcoma: a
clinicopathological and statistical analysis of patients surviving five
years or longer. J Bone Joint Surg Am 1975;57:10.
TM, Hamlin DJ, Enneking WF, et al. Magnetic resonance imaging of bone
and soft tissue tumors: early experience in 31 patients compared with
computed tomography. Skeletal Radiol 1985;13:134.
P, Llombart-Bosch A, Contesso G. Small round blue cell tumors in bone:
prognostic factors correlated to Ewing’s sarcoma and neuroectodermal
tumors. Semin Diagn Pathol 1996;13:250.
M, Wijnaendts LC, van der Linden JC, et al. Peripheral primitive
neuroectodermal tumour and extra-osseous Ewing’s sarcoma: a
histological, immunohistochemical and DNA flow cytometric study. Virchows Arch 1995;425:611.
H, Newton WA Jr, Soule EH, et al. Pathologic features of extraosseous
Ewing’s sarcoma: a report from the Intergroup Rhabdomyosarcoma Study. Hum Pathol 1988;19:442.
S, Marina NM, Luo X, et al. Treatment of children with peripheral
primitive neuroectodermal tumor or extraosseous Ewing’s tumor with
Ewing’s-directed therapy. J Pediatr Hematol Oncol 1998;20:55.
RB, Asmar L, Newton WA Jr, et al. Ewing’s sarcoma of soft tissues in
childhood: a report from the Intergroup Rhabdomyo-sarcoma Study, 1972
to 1991. J Clin Oncol 1997;15:574.
G, Ferrari S, Avella M, et al. Non-metastatic Ewing’s sarcoma: results
in 98 patients treated with neoadjuvant chemotherapy. Ital J Orthop Traumatol 1991;17:449.
ME Jr, Perez CA, Tefft M, et al. Multimodal therapy for the management
of primary, nonmetastatic Ewing’s sarcoma of bone: an Intergroup study.
Natl Cancer Inst Monogr 1981;56:255.
A, Cotterill S, Malcolm A, et al. Ifosfamide-containing chemotherapy in
Ewing’s sarcoma. The second United Kingdom Children’s Cancer Study
Group and the Medical Research Council Ewing’s Tumor Study. J Clin Oncol 1998;16:3628.
AW, Cotterill SJ, Bullimore JA, et al. Long-term results from the first
UKCCSG Ewing’s Tumour Study (ET-1). United Kingdom Children’s Cancer
Study Group (UKCCSG) and the Medical Research Council Bone Sarcoma
Working Party. Eur J Cancer 1997;33:1061.
LH, DeLaney TF, Tsokos M, et al. Ifosfamide and etoposide plus
vincristine, doxorubicin, and cyclophosphamide for newly diagnosed
Ewing’s sarcoma family of tumors. Cancer 1996;78:901 (erratum 1997;79:867).
K, Crowley J, Balcerzak SP, et al. A Southwest Oncology Group and
Cancer and Leukemia Group B phase II study of doxorubicin, dacarbazine,
ifosfamide, and mesna in adults with advanced osteosarcoma, Ewing’s
sarcoma, and rhabdomyosarcoma. Cancer 1998;82:1288.
S, Hoffmann C, Jabar S, et al. Evaluation of prognostic factors in a
tumor volume-adapted treatment strategy for localized Ewing sarcoma of
bone: the CESS 86 experience. Cooperative Ewing Sarcoma Study. Med Pediatr Oncol 1999;32:186.
R, Jurgens H, Burgers JM, et al. Prognostic factors in the treatment of
Ewing’s sarcoma. The Ewing’s Sarcoma Study Group of the German Society
of Paediatric Oncology, CESS 81. Radiother Oncol 1987;10:101.
SS, Woo SY, Huang LY, et al. Non-metastatic Ewing’s sarcoma: twenty
years of experience suggests that surgery is a prime factor for
successful multimodality therapy. Int J Oncol 1999; 14:1039.
Y, Kun LE, Brooks MT, et al. Ewing’s sarcoma: local tumor control and
patterns of failure following limited-volume radiation therapy. Int J Radiat Oncol Biol Phys 1991;21:1501.
CA, Tefft M, Nesbit ME Jr, et al. Radiation therapy in the multimodal
management of Ewing’s sarcoma of bone: report of the Intergroup Ewing’s
Sarcoma Study. Natl Cancer Just Monogr 1981;56:263.
A, Perez CA, Tefft M, et al. Intergroup Ewing’s Sarcoma Study: local
control related to radiation dose, volume, and site of primary lesion
in Ewing’s sarcoma. Cancer 1980;46:516.
J, Sauer R, Burgers JM, et al. Radiation therapy as local treatment in
Ewing’s sarcoma: results of the Cooperative Ewing’s Sarcoma Studies
CESS 81 and CESS 86. Cancer 1991;67:2818.
CA, Tefft M, Nesbit M, et al. The role of radiation therapy in the
management of non-metastatic Ewing’s sarcoma of bone: report of the
Intergroup Ewing’s Sarcoma Study. Int J Radiat Oncol Biol Phys 1981;7:141.
SS, Torrey M, Link MP, et al. A multidisciplinary study investigating
radiotherapy in Ewing’s sarcoma: end results of POG #8346. Pediatric
Oncology Group. Int J Radiat Oncol Biol Phys 1998;42:125.
CR, Gledhill R, Chevalier LM, et al. Osteogenic sarcoma following
treatment with megavoltage radiation and chemotherapy for bone tumors
in children. Med Pediatr Oncol 1980;8:375.
JF Jr, Wexler LH, Marcus RB, et al. Second malignancies after Ewing’s
sarcoma: radiation dose-dependency of secondary sarcomas. J Clin Oncol 1996;14:2818.
Vu B, de Vathaire F, Shamsaldin A, et al. Radiation dose, chemotherapy
and risk of osteosarcoma after solid tumours during childhood. Int J Cancer 1998;77:370.
M, McGuire MH, Herbold DR. Magnetic resonance imaging of soft tissue
masses: an evaluation of fifty-three histologically proven tumors. Magn Reson Imaging 1988;6:237.
WA Jr, Soule EH, Hamoudi AB, et al. Histopathology of childhood
sarcomas: Intergroup Rhabdomyosarcoma Studies I and II.
Clinicopathologic correlation. J Clin Oncol 1988;6:67.
RJ, Corpron CA, Hays D, et al. Extremity sarcomas: an analysis of
prognostic factors from the Intergroup Rhabdomyosarcoma Study III. J Pediatr Surg 1996;31:191.
WM, Garnsey L, Beltangady MS, et al. Prognosis in children with
rhabdomyosarcoma: a report of the Intergroup Rhabdomyosarcoma Studies I
and II. Intergroup Rhabdomyo-sarcoma Committee. J Clin Oncol 1990;8:443.
RB Jr, Crist WM, Maurer HM, et al. Prognosis of children with soft
tissue sarcoma who relapse after achieving a complete response: a
report from the Intergroup Rhabdomyosarcoma Study I. Cancer 1983;52:44.
K, Crowley J, Balcerzak SP, et al. An Intergroup phase III randomized
study of doxorubicin and dacarbazine with or without ifosfamide and
mesna in advanced soft tissue and bone sarcomas. J Clin Oncol 1993;11:1276.
DM, Raney RB, Grist WM, et al. Secondary surgical procedures to
evaluate primary tumor status in patients with chemotherapy-responsive
stage III and IV sarcomas: a report from the Intergroup
Rhabdomyosarcoma Study. J Pediatr Surg 1990;25:1100.
MD, Hanfelt JJ, Tefft MC, et al. Radiation therapy for
rhabdomyosarcoma: local failure risk for clinical group III patients on
Intergroup Rhabdomyosarcoma Study II. Int J Radiat Oncol Biol Phys 1997;38:797.
RB Jr, Gehan EA, Hays DM, et al. Primary chemotherapy with or without
radiation therapy and/or surgery for children with localized sarcoma of
the bladder, prostate, vagina, uterus, and cervix: a comparison of the
results in Intergroup Rhabdomyosarcoma Studies I and II. Cancer 1990;66:2072.
W Jr, Gehan EA, Hays DM, et al. Prognostic significance of staging
factors of the UICC staging system in childhood rhabdomyosarcoma: a
report from the Intergroup Rhabdo-myosarcoma Study (IRS-II). J Clin Oncol 1987;5:46.
E, Rao BN, Bowman L, et al. Outcome of treatment for pediatric sarcoma
of the foot: a retrospective review over a 20-year period. J Pediatr Surg 1997;32:1181.
AS, Fontanesi J, Luo X, et al. Synovial sarcoma in children and
adolescents: the St. Jude Children’s Research Hospital experience. J Clin Oncol 1994;12:2360.
D, Thum P, Harms D, et al. Synovial sarcoma in children and
adolescents: a report from the Kiel Pediatric Tumor Registry. Cancer 1991;67:1667.
HA, Albus-Lutter CE, Gortzak E, et al. Regional lymph node metastases
in patients with soft tissue sarcomas of the extremities: what are the
therapeutic consequences? Eur J Surg Oncol 1988;14:151.
ME, Pratt CB, Webber BL, et al. Therapy for childhood soft-tissue
sarcomas other than rhabdomyosarcoma: a review of 62 cases treated at a
single institution. J Clin Oncol 1986;4:559.
R, Treuner J, Koscielniak E, et al. Synovial sarcoma of childhood and
adolescence: report of the German CWS-81 study. Cancer 1993;71:3647.
A, Casanova M, Massimino M, et al. Synovial sarcoma: report of a series
of 25 consecutive children from a single institution. Med Pediatr Oncol 1999;32:32.
ML, Spurbeck WW, Pappo AS, et al. The impact of margin of resection on
outcome in pediatric nonrhabdomyosarcoma soft tissue sarcoma. J Pediatr Surg 1999;34:672.
J, Pappo AS, Parham DM, et al. Role of irradiation in management of
synovial sarcoma: St. Jude Children’s Research Hospital experience. Med Pediatr Oncol 1996;26:264.