
many as 1% of all children will have pains severe enough to be
evaluated (1). Approximately 15% of healthy children reported on a health questionnaire that they had episodes of musculoskeletal pain (2).
Healthy children in day care centers have approximately one painful
episode every 3 hours, arising from play, disciplining, or interaction
with peers (3). The orthopaedic surgeon is
often the first specialist to encounter the child with joint, limb, or
back pain. In a study of subspecialty referrals of juvenile arthritis,
most children with pauciarticular juvenile rheumatoid arthritis (JRA)
(62%) were referred to orthopedic surgeons prior to referral to
pediatric rheumatology care (4). Accordingly,
it is important that the orthopaedic surgeon be able to identify the
most likely cause of the pain and either initiate treatment or refer
the patient to an appropriate medical specialist.
of childhood. It is one of the most common chronic illnesses occurring
in children. The annual incidence ranges from 7 to 20 per 100,000, with
an overall prevalence of 1 to 2 per 1,000 (5,6).
Among children who are evaluated by a physician for pain in the joints,
only 1 in 100 will eventually be diagnosed as having arthritis; but
among those who present to an orthopaedist, the frequency of arthritis
is surely higher.
orthopaedic surgeon with an in-depth understanding of the presentation,
differential diagnosis, and management of children with arthritis. With
this framework, the orthopaedic specialist should be able to identify
children with juvenile arthritis, and to differentiate arthritis from
benign pains of childhood, psychogenic pain syndromes, benign
musculoskeletal back pain, infection, malignancy, or other systemic
autoimmune diseases (lupus, dermatomyositis, and vasculitis). Infection
and malignancies, as well as congenital, mechanical, or traumatic
causes of limb or joint pain, are presented only for the purpose of
contrasting the symptoms of those conditions with those of juvenile
arthritis, because detailed presentations on those conditions can be
found elsewhere in this text.
of the great heterogeneity in the presentation and course of the
disease, there likely are multiple initiating factors, given the
setting of a susceptible host. There is no laboratory test that will
make a definitive diagnosis of arthritis. A diagnosis of juvenile
arthritis is made by taking a thorough history, performing a skilled
and comprehensive physical examination, utilizing directed laboratory
tests and imaging procedures, and following the child over time.
diagnosis and classification of juvenile arthritis. The diagnostic
criteria for juvenile chronic arthritis (JCA) were defined by the
European League Against Rheumatism (EULAR) (7) (Table 12.1).
According to the EULAR criteria, JCA is differentiated into onset
types: pauciarticular, polyarticular, juvenile rheumatoid [positive
rheumatoid factor (RF)], systemic, juvenile ankylosing spondylitis
(JAS), and juvenile psoriatic arthritis. In North America, the most
frequently used criteria for JRA have been those by the American
College of Rheumatology (ACR) (8) (Table 12.1).
These criteria define the subtypes of JRA: oligoarticular
(pauciarticular), polyarticular, and systemic. They exclude other
causes of juvenile arthritis, such as spondyloarthropathies (JAS,
inflammatory bowel disease–associated arthritis, and related diseases),
juvenile psoriatic arthritis, arthritis associated with other systemic
inflammatory diseases [systemic lupus erythematosus (SLE),
dermatomyositis, sarcoidosis, etc.] and infectious or neoplastic
disorders.
identical populations or spectra of disease; however, they have often
been used interchangeably. This has led to confusion
in
the interpretation of studies relating to the epidemiology, treatment,
and outcome of juvenile arthritis. The International League of
Associations of Rheumatologists (ILAR) has proposed (9) and revised (10) criteria for the diagnosis and classification of juvenile arthritis (Table 12.2). The term juvenile idiopathic arthritis
(JIA) has been proposed as a replacement for both JRA and JCA, and will
encompass all juvenile arthritides of unknown cause that last for more
than 6 weeks. This international compromise as regards terminology will
allow uniform interpretation of clinical and therapeutic data. Although
these criteria are not definitive (some children fit into either no
category or two or more categories), they should be thought of as a
work in progress. Recent validation of the ILAR classification criteria
has found that 80% to 88% of children could be classified, and 12% to
20% were classified as “Other” because they either did not fit into any
category or fulfilled the criteria under two categories (11, 12, 13, 14).
As techniques become available to better define the genetic risk
factors and specific triggers of juvenile arthritis, modifications to
the criteria can be made. In the remaining sections of this chapter,
the term juvenile arthritis is used to denote any type of arthritis in childhood, JIA will be used as defined earlier, and the terms JRA and JCA will be used only when referring to specific epidemiologic, therapeutic, or outcome data.
|
TABLE
12.1 Comparison Of American College Of Rheumatology (ACR) And European League Against Rheumatism (EULAR) Classifications Of Juvenile Idiopathic Arthritis |
|||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||||||||||||||||||
affecting one to four joints during the first 6 months of disease.
There are two subcategories: persistent oligoarthritis affects no more
than four joints throughout the entire course of arthritis; extended
oligoarthritis affects a cumulative total of five joints or more after
the first 6 months of disease. A child will be excluded from this
diagnostic category if any of the following five conditions are met:
psoriasis in the patient or a first-degree relative; family history of
medically confirmed HLA-B27–associated disease in at least one
first-degree relative [ankylosing spondylitis, enthesitis-related
arthritis (ERA), sacroiliitis with inflammatory bowel disease (IBD),
Reiter syndrome, or acute anterior uveitis (AAU)]; HLA-B27–positive
male with onset of arthritis after 6 years of age; a positive RF test
on at least two occasions more than 3 months apart; or systemic
arthritis. The characteristics include the age at onset of arthritis.
The patterns of occurrence of the arthritis should also be noted: large
joints only, small joints only, limb predominance (upper, lower, both),
involvement of specific joints, and symmetry of arthritis. Finally, the
occurrence of anterior uveitis, and the presence of antinuclear
antibodies (ANA) or any human leukocyte antigens (HLA) class I and II
showing predisposing or protective alleles, should be taken into
consideration. This diagnostic subgroup will certainly contain some
children with psoriatic arthritis who have not yet developed psoriasis.
It will also exclude the few children with oligoarticular disease and a
positive RF. However, these children are likely to have an early onset
of RF-positive polyarticular arthritis, or at least would be predicted
to have a more prolonged and severe course, and therefore should be
excluded from the oligoarticular group.
arthritis affecting five or more joints during the first 6 months of
disease, with negative RF and no systemic arthritis. Descriptors for
RF-negative polyarticular JIA include age at onset, symmetry of
arthritis, presence of ANA, and occurrence of uveitis. Exclusions
include: psoriasis in the patient or a first-degree relative, patient
is male and HLA-B27 positive with
onset after 6 years of age, HLA-B27–associated disease in a first-degree relative, IgM RF, or systemic JIA.
|
TABLE 12.2 Criteria For Classification Of Juvenile Idiopathic Arthritis (JIA)
|
||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||||||||||||
affecting five or more joints during the first 6 months of disease,
associated with a positive RF test on two occasions at least 3 months
apart, and the absence of systemic arthritis. Descriptors include age
at onset of arthritis, symmetry, presence of ANA, and immunogenetic
characteristics. Exclusions include: patient is male and
HLA-B27–positive with onset after 6 years of age, psoriasis in the
patient or a first-degree relative, HLA-B27-associated disease in a
first-degree relative, or systemic JIA. This disease is likely to be
the equivalent of early onset adult rheumatoid arthritis.
preceded by daily fever of at least 2 weeks’ duration, which is
documented to be quotidian for at least 3 days, and accompanied by one
or more of the following: evanescent, nonfixed, erythematous rash;
generalized lymphadenopathy; hepato- or splenomegaly; and serositis.
This type of arthritis can be described in terms of age at onset of
arthritis and the pattern of arthritis during and after the first 6
months: oligoarthritis, polyarthritis, or arthritis present only after
the first 6 months of systemic illness. Other descriptors include the
features of systemic disease after the first 6 months of disease and
the level of C-reactive protein (CRP). Exclusions include: psoriasis in
the patient or a first-degree relative, patient is male, HLA-B27
positive with onset after 6 years of age, HLA-B27–associated disease in
a first-degree relative, or IgM RF.
psoriasis or as arthritis and at least two other criteria: dactylitis,
nail abnormalities (pitting or onycholysis), or a family history of
psoriasis in at least one first-degree relative. Exclusions include the
presence of RF and systemic arthritis. Descriptors include: the age at
onset of arthritis or psoriasis, the pattern of involvement of the
joints, oligoarticular or polyarticular course, presence of ANA, and
uveitis. Exclusions include: patient is male, HLA-B27 positive with
onset after 6 years of age, HLA-B27–associated disease in a
first-degree relative, IgM rheumatoid factor, or systemic JIA.
tenderness at insertion sites of tendons, ligaments, or fascia to
bone), or either arthritis or enthesitis along with at least two of the
following characteristics: sacroiliac joint tenderness and/or
inflammatory spinal pain, presence of HLA-B27, family history of at
least one first-degree relative with a medically confirmed
HLA-B27–associated disease (e.g., ankylosing spondylitis,
enthesitis-related arthritis, sacroiliitis with IBD, Reiter syndrome,
or AAU), or onset of arthritis in a boy after the age of 6 years.
Descriptors for ERA include age at onset of arthritis or enthesitis,
patterns of arthritis, symmetry of arthritis, oligoarticular or
poly-articular disease course, and the presence of IBD. Exclusions
include: psoriasis in the patient or a first-degree relative, IgM
rheumatoid factor, or systemic JIA.
idiopathic arthritis, there will inevitably be children who do not fit
into any known category. This group of children with JIA will be
considered to have undifferentiated, or overlap, arthritis. This
category of other arthritis is defined as children with arthritis of
unknown cause that persists for at least 6 weeks, but that either does
not fulfill criteria for any other category, or fulfills criteria for
more than one of the other categories. The most common cause for the
exclusion from specific categories is the presence of psoriasis in a
first-degree relative (13).
limp, or joint pain with or without swelling requires a comprehensive
history and physical examination. The first priority is to rule out an
infection (osteomyelitis, septic arthritis, discitis), malignancy, or
orthopaedic abnormality requiring prompt intervention. The causes of
arthritis in children are extensive, and many are rare. There are no
pathognomonic presentations, and there is extensive overlap of all
types of juvenile arthritis. Laboratory tests and radiologic studies
are often uninformative, and should not be used to make or dismiss the
diagnosis of juvenile arthritis. The purpose of this section is to
provide the orthopaedist with an overview of the ways children present
with arthritis, and to generate a framework for the logical
identification of the appropriate diagnosis with a minimum of
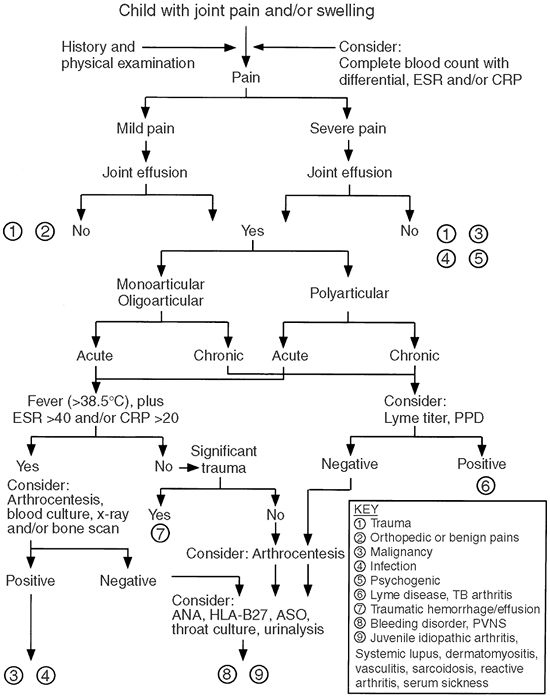
diagnostic procedures. The algorithm in Figure 12.1
provides a general guide for the evaluation of children presenting with
limb or joint pain. The first step in any evaluation of a child with
possible arthritis is to obtain a thorough history and carry out a
comprehensive review of all the systems.
Children with arthritis will frequently have mild-to-moderate pain and
stiffness (gelling) in the mornings, after a nap, after sitting in the
classroom, after a long automobile ride, or other periods of
inactivity. These complaints will generally improve within minutes or
hours of renewed activity. Although there may be some residual limp or
mild complaints of pain or stiffness, many children with JIA will be
entirely asymptomatic on presentation to the physician’s office. The
major exception to this is the child with ERA, who may have morning
pain and stiffness, but who will worsen throughout the day or with
activities, because of repeated stress on inflamed tendons and
entheses. A minority of children with JIA will have significant pain
throughout the day, and the physical exam is likely to reveal severe
arthritis in those who experience increased pain. It would be unusual
for the pain associated with JIA to keep a child from falling asleep or
to wake the child from sleep. When a child has only afternoon, evening,
and/or nighttime pains that resolve in the morning, this is typical for
benign pains of childhood (growing pains). When pain is present both
day and night and interferes with sleep, it is less likely to be due to
JIA, but, depending on the duration and location, may be associated
with malignancy, infection, or psychogenic pain syndromes.
new onset, which prompts immediate evaluation, may be due to infection,
malignancy, or trauma. Pain that is slowly worsening or unchanged over
weeks or months is typical of JIA. When pain has been present for many
years, the cause is often psychogenic or mechanical. It is not unusual
for a parent to report that a child has had pain, clumsiness, or
difficulty in walking from the time the child took his or her first
steps. If neurologic abnormalities, including cerebral
palsy, have been excluded, most of these children have only benign mechanical problems.
 |
|
Figure 12.1
Algorithm for evaluation of a child with joint pain and/or swelling. Chronic is considered less than 6 weeks. ANA, antinuclear antibody; PPD, tuberculin skin test; PVNS, pigmented villonodular synovitis; ESR, erythrocyte sedimentation rate (mm per hour); CRP, C-reactive protein (mg per dL). |
history. Children with JIA will rarely have muscular pains. True
weakness should suggest either an inflammatory or congenital myopathy.
Long-bone or periarticular pain is often seen in trauma or
malignancies. When pain is random, intermittent, or migratory,
especially if it comes and goes during the clinic visit, it is often
psychogenic in origin. In JIA, pain in the joints can be additive or
can show spontaneous improvement, but it is usually present
consistently over weeks or months.
determine in children, especially in the preverbal child. The
pain-rating scales that have been developed for children are sensitive
to the cognitive-developmental conceptualizations of children (15).
Children are asked to rate their pain on a visual analog scale (VAS),
in terms of present pain and worst pain intensity for the previous
week. Each VAS is a 10-cm horizontal line with no numbers or
descriptors. The child VAS is anchored with the developmentally
appropriate pain descriptors: happy and sad faces, corresponding to no
pain and severe pain, respectively.
Rarely does the child report severe pain. Most children with JIA will
maintain near-normal functioning. Acute arthritis, such as
postinfectious reactive arthritis including acute rheumatic fever
(ARF), is typically much more painful than JIA. Intense pain that is
nonmigratory and present night and day is more likely to represent
infection or malignancy. Severe pain that is intermittent or migratory,
disrupts school attendance, and causes sleep disturbance in a child
with normal growth and normal parameters in laboratory tests is usually
psychogenic in origin. Children with psychogenic pain syndromes are
often unable to perform normal daily activities because of the high
levels of perceived pain.
a fall or injury caused a sudden swelling and pain in a joint. However,
most children younger than 6 years fall frequently, and children of
ages 1 to 4 fall nearly every day. Although young children may have
fractures from even innocent falls, accidental trauma should not be
considered as the most likely cause of true joint swelling in this age
group.
injury or repetitive stress has caused joint or tendon pain, resting or
immobilizing the limb is often tried. Although children with arthritis
can have minimal pain when the affected joint is immobilized, they are
uniformly worse when the cast or splint is removed. If the child has
pain in the joint accompanied by effusion and stiffness, prolonged
immobilization will often result in severe restriction in the range of
motion as well as increased pain. Childhood arthritis usually improves
with activities, and every attempt should be made to keep the children
mobile. We do not limit athletic activities, but we encourage them to
engage in noncontact sports. In children with psychogenic pain
amplification syndromes, the condition will frequently be worse after
immobilization, and this may initiate or exacerbate a flare-up of
reflex sympathetic dystrophy (RSD).
the evaluation of joint pain. Fevers, whether continuous or periodic,
may be associated with infection and malignancies or with systemic
inflammatory disease. Weight loss, with laboratory evidence of
inflammation, suggests the presence of a systemic illness, such as
infection, malignancy, IBD, or systemic-onset JIA. Persistent fatigue,
sleep disturbance, and depression are often signs of a psychogenic pain
syndrome. Fatigue should not be confused with true weakness, which
could represent the onset of an inflammatory myositis.
suspected JIA. The presence of HLA-B27-associated diseases, chronic
back pains, or psoriasis could suggest the onset of an associated
disease. Other systemic autoimmune disorders, including adult
rheumatoid arthritis, often cluster in families but have no direct
pattern of inheritance. Children with psychogenic pain syndromes are
often found to have a relative, usually the mother, who has chronic
pain and acts as a role model for the pain behavior.
a complete history and screening laboratory tests, will often be
sufficient to diagnose the child with arthritis. The musculoskeletal
examination will often enable differentiation between mechanical or
psychogenic causes of pain on the one hand, and inflammatory etiologies
on the other. Children with functional pain syndromes will often
complain of pain that is out of proportion to the examination and are
often migratory or transient, even during the examination.
limitation in the range of movement of a joint, along with pain or
tenderness that is not caused by a primary mechanical disorder. Young
children will often attempt to resist a physical examination, and they
should therefore be observed closely for a limp or swollen joints
before being actually approached. It is often instructive to observe
the child playing in the waiting area and walking to or from the
examination room. All children with pain or swelling in a joint should
have a comprehensive examination of their joints. Often, children with
pain will be quite apprehensive about the examination. When a child
presents with a complaint of a single swollen or tender joint, it is
important to evaluate all joints for signs of arthritis. Frequently,
joints other than the presenting joint will be involved with arthritis.
It would therefore be wise to begin the examination at sites distant
from the point of pain, and gain the trust of the child before
approaching the painful site. Examination of a painful site will often
end the physician’s ability to further examine the child.
evaluating children with pain and swelling of joints. The selection of
specific laboratory evaluations should be guided by the history and
physical examination. For most children, a complete blood count with
differential, CRP, and erythrocyte sedimentation rate (ESR) are
indicated. This will help to identify hematologic abnormalities
suggesting malignancy, and to document the presence or absence of
systemic inflammation. The CRP is an acute-phase protein synthesized in
the liver in response to proinflammatory cytokines. The ESR is an
indirect measure of systemic inflammation and the acute-phase response.
In most children, the ESR is less than 15 mm per hour. The ESR rises in
response to the relative decline in concentration of serum albumin and
increase in acute-phase proteins, including fibrinogen and others. The
ESR may be elevated because of marked anemia or a low serum
concentration of albumin due to decreased production or loss, as in
nephrotic syndrome. Most children with arthritis will have an ESR less
than 100 mm per hour, whereas systemic arthritis, malignancies, and
infections are more likely to be associated with an ESR greater than
100 mm per hour. However, many children with oligoarticular and some
with polyarticular arthritis will have a
normal
ESR and CRP. The addition of a CRP test can be helpful in situations in
which infection is strongly suspected, because the short half-life of
this acute-phase protein results in a rapid decline in concentration
with effective antibiotic treatment, whereas the ESR may even continue
to rise.
bind to one of many potential antigens present in the nucleus of normal
human cells. ANA titers are usually considered to be elevated when they
can be identified at a dilution of 1:40, or with an absolute value of
7.5 IU per mL. The presence of an elevated ANA by itself should never
be used for diagnosing arthritis. However, the ANA does have some
utility as a screening test for JIA (18,19).
The frequency of ANA positivity is greatest in younger girls with
oligoarticular disease, and represents an increased risk for anterior
uveitis (20). When arthritis is suspected on
the basis of history and physical examination, the presence of a
positive ANA should prompt an immediate referral to an ophthalmologist
for a slit-lamp examination to evaluate for the presence of uveitis.
Even in the absence of an ANA, children with confirmed arthritis should
have a routine ophthalmologic exam with a slit lamp. However, it is
known that elevated ANA titers may be present in up to 20% of healthy
children (typically, at titers of 1:40 to 1:80), and may be induced by
recent illness or be present in first- or second-degree relatives with
SLE (21,22). Children
who have an elevated ANA, whatever its level, with no evidence of
systemic inflammation and no arthritis on examination by a pediatric
rheumatologist, are extremely unlikely to subsequently develop a
significant autoimmune disease (21,23).
recognizing IgG that has bound to antigen. RF positivity is infrequent
in children with arthritis, and rarely occurs in children younger than
7 years. When present in children with arthritis, the RF signifies a
chronic inflammatory state, and is associated with a higher frequency
of erosive synovitis and poor prognosis (24,25).
Studies in children and adults have demonstrated that a positive RF is
as likely to be present in children with diseases other than JIA as it
is in those with JIA (26,27).
Therefore there is no role for RF testing in the orthopaedic or
pediatric office evaluation of children with possible arthritis.
transient reactive arthritis, IBD, and ERA. The high familial
occurrence of ankylosing spondylitis is directly related to the
presence of HLA-B27 (28). Although HLA-B27 is
found in approximately 8% of the white population, it can be useful in
the diagnosis of ERA. It is especially important in boys above the age
of 6, where there is a family history of HLA-B27–associated illness, or
sacroiliac joint or spinal inflammatory pain.
However, there is no utility in obtaining uric acid levels as a
screening test for arthritis. The diagnosis of gout is made by
documentation of the presence of urate crystals in synovial fluid,
irrespective of serum uric acid levels.
should be performed in all children with an acute febrile
monoarthritis. The possibility of infection should also be considered
when a child with polyarticular arthritis has an acutely swollen and
tender joint, usually accompanied by fever, because this may represent
a secondary septic arthritis. The diagnosis and treatment of septic
arthritis are discussed in detail in Chapter 13.
shows evidence of inflammation. However, the total white blood cell
count in joints without septic arthritis can range from 150 to greater
than 100,000 cells per mm3 (30, 31, 32), with average counts of between 10,000 and 12,000 cells per mm3 There is often a neutrophil predominance, with a range of 18% to 88% and average of 56% (30).
A synovial biopsy should be carried out if the tuberculin test is
positive, or if the diagnosis of sarcoidosis is being considered.
of children with pain and/or swelling in the joints, predominantly for
identifying periarticular osteopenia, fractures, or other bony lesions.
In early JIA, there are no pathognomonic radiographic findings. The
diagnosis of JIA is typically made long before bony changes are
apparent. Ultrasound is often a rapid and noninvasive way to identify
an intraarticular effusion. Radionucleotide imaging with Tc 99m (bone
scan) to evaluate for osteomyelitis and malignancy will occasionally
identify other joints with subclinical inflammation, thereby suggesting
a diagnosis of JIA. Although rarely required for diagnosis, magnetic
resonance imaging (MRI) is the most sensitive technique for detecting
early articular changes in JIA (33, 34, 35).
even longer of having swelling in one or more joints, the most likely
diagnosis is juvenile arthritis. Unless there is fever, weight loss or
severe pain, it is unlikely that the child has an infection (other than
tuberculosis or Lyme disease) or a malignancy. Minor trauma does not
ordinarily result in prolonged swelling. Most children with an internal
derangement of a joint will be identified by a history of significant
trauma.
an ophthalmoscopic examination to rule out asymptomatic anterior
uveitis, and a comprehensive musculoskeletal
examination
of the affected joint(s) as well as all other joints which may be
affected though asymptomatic. We obtain a plain radiograph of the
affected joint(s), complete blood count, ESR, and ANA titer. In endemic
areas, one may consider ordering a PPD to screen for tuberculosis
and/or obtaining Lyme antibody titers. If the clinical presentation is
atypical, a bone scan is often indicated. All patients with continued
joint swelling should be started on a nonsteroidal antiinflammatory
drug, but corticosteroids should be withheld until the diagnosis of
juvenile arthritis has been confirmed. Immobilization of the affected
joint will cause loss of range of movement, and is counterproductive.
childhood is beyond the scope of this chapter. There are over 100
disorders in which arthritis may be a significant manifestation (36).
The most common classes of disorders that must be considered in the
differential diagnosis of JIA include mechanical or orthopaedic
conditions, infection, trauma, psychogenic disorders, and inflammatory
diseases. Often, the differential diagnosis will be determined by
whether the presentation is acute, subacute, or chronic, whether the
child has monoarticular or polyarticular arthritis, and whether there
are systemic signs such as fever (Table 12.3).
polyarthritis will often present with a suddenly swollen and/or painful
joint. This is in contrast to most children with JIA, who (except those
with systemic onset) often have a subacute or insidious onset. Children
with injuries can often describe exactly when and where the injury
occurred, whereas children with benign pains will frequently have a
history of pain from the time they could walk. Conversely, children
with psychogenic pain can also frequently describe an event, minor or
major injury, or illness as the initiator of their pain. However, many
children fail to fit the expected profiles, and atypical presentations
often occur. It is important to evaluate children with joint and limb
pain with a broad differential diagnosis, which can be better defined
with a thorough history and comprehensive physical exam. The presence
or absence of fever, the age and sex of the child, and associated signs
and symptoms will aid the consultant in determining the optimal
strategy for the selection of diagnostic testing.
|
TABLE 12.3 Classes Of Disorders In The Differential Diagnosis Of Juvenile Idiopathic Arthritis (JIA)
|
|||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
associated with fever, elevated neutrophil count, ESR, and CRP. This is
in contrast to monoarticular JIA, which seldom has significant systemic
inflammatory signs. However, gonococcal arthritis in sexually active
children can present with an oligoarticular, polyarticular, or
migratory pattern, with significant tenosynovitis. There are instances
in which organisms such as Staphylococcus aureus
can present with a subacute arthritis. However, this presentation is
most common for mycobacterial infections or Lyme disease. In most
cases, septic joints are extremely painful, but in JIA, swelling is
often out of proportion to the reported pain.
infection include fever and migratory arthralgia, with little or no
swelling in the joint. Early localized disease is typically manifested
by the presence of erythema migrans, the classic expanding rash that
occurs most often at the site of the
tick bite and develops within 7 days to 1 month after infection (37).
Lyme arthritis occurs months to years after the initial infection. Many
patients with untreated Lyme disease will complain of migratory
arthralgias or arthritis (38). In a recent retrospective study of 90 children with Lyme arthritis, Gerber et al. (39)
noted that the majority (63%) had monoarticular disease, but no child
had more than four joints involved. The knee was affected most often
(90%), followed by hip (14%), ankle (10%), wrist (9%), and elbow (7%),
whereas small joints were rarely involved. Most children with Lyme
arthritis do not recall a tick bite or erythema migrans (39,40).
This is in contrast to prospective studies in which 90% of children
diagnosed with Lyme disease had a history of erythema migrans. The most
likely reason for this discrepancy is that most children with erythema
migrans are identified and treated with antibiotics, and do not develop
late complications of Lyme disease. Lyme arthritis is typically a
low-grade inflammatory synovitis with a large and relatively painless
joint effusion. The ESR can be normal or elevated, and 25% of the
patients may have values greater than 60 mm per hour (39). In both children and adults, a chronic form of Lyme arthritis can persist after treatment, and is associated with HLA-DR4 and HLA-DR2 alleles (41).
Most children with Lyme arthritis can be effectively treated with a
single, 4-week course of orally administered amoxicillin or doxycycline
(39).
sterile synovitis that is an immune response to a nonarticular
infection. In most children, the reactive arthritis occurs after upper
respiratory or gastrointestinal infections, whereas in adult patients
it is more likely to occur following a genitourinary infection (42, 43, 44).
The classic presentation of reactive arthritis is the triad of
conjunctivitis, urethritis, and arthritis found in Reiter syndrome
(RS). The complete triad of RS is very uncommon in childhood. Children
account for less than 1% of all patients with complete RS, and the
ratio of boys to girls is 4:1 (44,45). A history of sexual activity could suggest infection with Chlamydia (46). Most patients with classic RS and other postinfectious reactive arthritis carry the HLA-B27 allele (44,47).
postinfectious, inflammatory arthritis. Transient synovitis of the hip
has a peak incidence, predominantly in boys (70%), at between 3 and 10
years of age. It is an idiopathic disorder often preceded by a
nonspecific upper respiratory tract infection (48).
Trauma has frequently been reported as having occurred prior to
transient synovitis of the hip, and may be a predisposing factor (49).
The onset of pain is often gradual, may be focused in the hip, thigh,
or knee, and may last for an average of 6 days. Occasionally, transient
synovitis of the hip can be bilateral (4%). The child often presents
with inability to walk or with a severe limp. There is a loss of
internal rotation of the hip, and it is usually held in flexion,
abducted, and externally rotated. There is often low-grade fever. The
ESR and white blood cell count are normal to mildly elevated (50).
Plain radiographs often show no abnormality, or may show a small
widening of the joint space. Ultrasound is a sensitive and reliable
method to confirm the presence of an effusion (50).
With rest and non-steroidal antiinflammatory drugs (NSAIDs), most
children will have complete resolution of symptoms within 2 weeks. Most
children with transient synovitis of the hip will have only a single
event, with 4% to 17% having a recurrence usually within the first 6
months after the initial onset (49).
hemolytic streptococcus. The incidence of ARF has remained relatively
constant at around 1 per 100,000 children between the ages of 5 and 17
years (51). It is very unusual for ARF to occur
in patients younger than the age of 4 years. Although ARF is rare in
developed countries, it remains the most common cause of acquired heart
disease in the developing world. In South Africa, the prevalence of ARF
has been estimated to be 690 per 100,000 (52).
poly-arthritis, usually affecting the legs first, and later the arms.
Involvement of the joints is the most common (75%) and often the first
manifestation of the disease (53). The affected
joints are often red and swollen, with pain out of proportion to the
physical findings. The arthritis of ARF is exquisitely responsive to
aspirin, and dramatic relief is often obtained within several hours
after the first dose. Residual synovitis does not usually develop.
and is the only aspect of the disease to cause significant morbidity
and mortality. The use of Doppler echocardiography has increased the
sensitivity of detection of valvar involvement in ARF, and
abnormalities have been found in as many as 90% of patients with ARF (51).
Arthralgia cannot be used as a minor criterion if arthritis is present.
A prolonged PR interval is often seen in ARF, but is not associated
with increased risk for carditis.
(<1 cm in diameter) and painless. Typically, they are present for 1
to 2 weeks. The overlying skin is not inflamed or attached to the
nodule. The most typical locations are over bony prominences. Nodules
occur in less than 10% of the patients, but are often associated with carditis.
rash, pink-to-red in color, usually affecting the trunk and
occasionally the proximal limbs, but never the face. The rash occurs
early in the disease and, if present, may persist after all other
manifestations of the disease have resolved. It occurs in less than 10%
of children with ARF, but is also associated with carditis.
movements and emotional lability. The movement disorder can often be
unilateral. It cannot be suppressed voluntarily, but is not present
during sleep. Chorea occurs in approximately 15% of children with ARF.
The interval between the streptococcal pharyngitis and the onset of
chorea can be as long as 3 months. When chorea is the only major
manifestation there may be no markers of inflammation, and
streptococcal pha ryngitis can be difficult to identify.
The diagnosis requires the presence of two major criteria, or one major
criterion and two minor criteria, and requires supportive evidence of a
preceding streptococcal infection (increased ASO/anti-DNase B, positive
rapid streptococcal antigen test, or throat culture). It is clear that
not all children who meet the Jones criteria will have ARF, and
conversely, a small number of children with ARF will not meet these
criteria.
mg/kg/d in children (8 g per day maximum), and serum salicylate
concentrations of 20 to 30 mg per dL. In the presence of carditis,
congestive heart failure, or heart block, corticosteroid therapy is
added. The typical treatment doses are 2 mg/kg/day of prednisone for 2
to 3 weeks, subsequently tapered over an additional 3 weeks. The
aspirin is usually discontinued 3 weeks after stopping the
corticosteroids. Eradication of streptococci by treatment with
penicillin is indicated in all patients with ARF, even in the absence
of a positive throat culture. Children with a history of ARF should
receive prophylactic antibiotics: intramuscular benzathine penicillin
every 3 to 4 weeks, oral penicillin V twice daily, or sulfadiazine once
per day. Patients with documented rheumatic heart disease should
continue prophylaxis indefinitely.
|
TABLE 12.4 The Modified Jones Criteria For Diagnosis Of Acute Rheumatic Fever
|
||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||||||||
PSRA typically presents as a nonmigratory oligo- or polyarthritis. It
is differentiated from ARF by the frequent presence of tenosynovitis
and the poor response to aspirin or other nonsteroidal drugs. In
addition to arthritis, other clinical manifestations include erythema
nodosum, livedo reticularis, cutaneous vasculitis, and systemic
polyarteritis nodosa (58,59).
Limited studies have suggested that further episodes of streptococcal
pharyngitis lead to an increased risk for ARF and rheumatic carditis,
and that streptococcal prophylaxis is indicated (55,57).
Among children with PSRA, there was found to be a statistically
significant increase in the frequency of occurrence of HLA-DRB1*01,
whereas those with ARF had a higher occurrence of HLA-DRB1*16, with the
occurrence of HLA-B27 being no different from the controls (60).
The association of PSRA with HLA-DRB1*01, but not with HLA-B27,
suggests that the pathogenesis of PSRA may be more similar to that of
ARF than to that of reactive arthritis. This would again support the
recommendation for prophylaxis for at least 1 year after the onset of
arthritis.
adverse immunologic response to foreign antigens mediated by the
deposition of immune complexes. Although serum sickness was first
described after injection of heterologous serum, today the most common
causes are antibiotics (penicillins and sulfonamides) and viral
infections (61, 62, 63).
Serum sickness is characterized by fever, arthralgia or arthritis,
lymphadenopathy, cutaneous eruptions (urticarial or morbilliform), and
angioedema. Both serum sickness and allergic angioedema can be mistaken
for acute-onset juvenile arthritis. However, most children with serum
sickness will spontaneously improve within a few days to weeks. For
mild disease, removal of the offending antigen and treatment with
antihistamines and non-steroidal antiinflammatory medications is
sufficient. In severe cases, a several-week course of corticosteroids
may be required.
deposition of monosodium urate crystals into the joint. The major
clinical manifestations include acute mono- or oligoarthritis,
frequently involving the first metatarsophalangeal joint, resulting in
podagra. Gout may result from either increased production or decreased
excretion of uric acid. Gout is extremely rare in children (29).
The diagnosis of gout can be confirmed only by demonstration of
negatively birefringent, monosodium urate crystals in the synovial
fluid when viewed under a polarized light microscope. Acute gout is
treated with nonsteroidal antiinflammatory medications, colchicine, and
occasionally prednisone. After the acute event has subsided,
allopurinol is utilized to decrease the level of uric acid in the serum
in order to prevent recurrences. The use of allopurinol is not
recommended during the acute phase of gout because, paradoxically, the
gout becomes worse when there is a sudden decrease in uric acid levels.
an increased incidence of musculoskeletal disorders. CF-associated
arthritis is a transient reactive arthritis often associated with
pulmonary exacerbations (64, 65, 66, 67, 68).
Teenagers and older patients with CF have a higher-than-expected
occurrence of RF-positive polyarticular JIA or adult rheumatoid
arthritis (69). Some children with CF may develop secondary hypertrophic osteoarthropathy, demonstrable on radiographs (70,71).
or chronic arthritis. There are often signs, symptoms, or laboratory
abnormalities that will aid in the diagnosis of these conditions. For a
thorough discussion of these diseases in children, several excellent
texts and reviews are available (36,72,73).
characterized by multiorgan system inflammation. Arthralgia and
arthritis affect 75% of the children with SLE. It is usually
polyarticular, and the joint pain is often out of proportion to the
physical findings. In typical cases, the arthritis responds readily to
corticosteroids, is rarely erosive (74), and does not result in deformity.
However, arthritis is frequent in childhood-onset sarcoidosis, and
typically presents as an oligoarthritis affecting the knees, ankles,
and/or elbows. It is characterized by very large effusions and boggy
synovitis with minimal pain or loss of motion. A synovial biopsy will
often be diagnostic, showing the presence of noncaseating sarcoid
granulomas.
arthritis. However, the disease most likely to be seen by the
orthopaedic surgeon is Henoch-Schonlein purpura (HSP). HSP is the most
common vasculitic syndrome in childhood, occurring in slightly more
than 1 in 10,000 children per year (76). The
classic manifestations of HSP are nonthrombocytopenic palpable purpura,
arthritis, abdominal pain, gastrointestinal hemorrhage, and
glomerulonephritis. In the complete syndrome, the diagnosis is often
clear. However, the arthritis can precede the appearance of the rash,
and the rash may be unrecognized if a comprehensive skin examination is
not done. The rash of HSP often begins on the lower extremities as an
urticarial eruption, followed by petechiae and purpura, which are most
often concentrated on the buttocks and lower extremities, especially
the ankles. The purpura will frequently recur in crops over several
weeks, resulting in multiple lesions in different stages of evolution.
The arthritis of HSP presents as a periarticular swelling and
tenderness, most commonly of large joints, with severe pain and
limitation of motion. The younger child will often refuse to use the
affected joint. The arthritis is usually transient, and resolves
without sequelae in a few days to weeks. In most children, HSP will
resolve completely within 4 weeks from onset.
Typically, the injury has been long forgotten, because many months may
pass between entry of the thorn into the skin and passage into the
joint. Often, a careful history will uncover the past trauma. Surgical
removal of the splinter and synovectomy are the only effective
treatments.
hemoglobinopathies (sickle cell disease) will present with acute pain
and swelling in the joints, resulting from hemarthrosis and localized
ischemia, respectively. A comprehensive discussion of these conditions
is found in Chapter 11.
Although effusion can occur in a joint, the pain is usually localized
to the metaphyses of the long bones. The pain in children with
malignancies is typically more severe than in JIA and will frequently
be continuous. Another frequent feature of children with malignancies
is an extreme elevation of the ESR (often >100), whereas in JIA, the
ESR is usually only moderately elevated, and may even be normal.
However, a normal ESR does not exclude malignancy. Plain radiographs
may show subperiosteal elevation, osteolytic reaction, or metaphyseal
rarefaction. In a recent study of 29 children
with
malignancy who were referred to pediatric rheumatologists, features
suggestive of malignancy included nonarticular “bone” pain (68%), back
pain as a major presenting feature (32%), bone tenderness (29%), severe
constitutional symptoms (32%), and atypical clinical features (48%) (80).
Atypical features included night sweats (14%), ecchymoses and bruising
(14%), abnormal neurologic signs (13%), and abnormal masses (7%).
Children with malignancy were more likely to have the combination of an
elevated ESR and lactate dehydrogenase (LDH) with a low platelet count
(28%).
tumor of the synovium. Although PVNS is rare in childhood, it does
frequently lead to recurrent joint swelling (81,82).
This usually results in recurrent effusions that are minimally painful,
with progressive cartilage destruction and erosion of bone. On
aspiration of the joint, a chocolate brown synovial fluid is frequently
found. The diagnosis is often confirmed by synovial biopsy showing
nodular hypertrophy, with proliferating fibroblasts and synovial cells,
and hemosiderin-laden macrophages. Surgical excision can be curative.
However, many patients have recurrences, and occasionally multifocal
disease can occur.
Although back pain may be psychogenic in origin, this symptom,
particularly in young children, has been thought to be the result of a
serious underlying organic disorder. However, there are multiple
potential pathologic causes of back pain in children and adolescents (Table 12.5).
Many of these entities are discussed elsewhere in this text and are
relatively uncommon in young children. However, benign back pain is
increasingly recognized as a common occurrence in pediatric practice.
The overall incidence of benign mechanical back pain seems to be
increasing. A large epidemiologic study found that the prevalence of
back pain in 12-year-old children was 7% and by the ages of 18 years
(females) and 20 years (males) more than 50% had experienced at least
one episode of low back pain (86).
incidence of back pain episodes. This may include the relative
deconditioning of today’s young people. Several recent studies have
implicated school backpacks as a cause of increased back pain in
children (87,88). These
authors have identified that a book bag weighing more than 15% to 20%
of the child’s weight is associated with back pain and improper use of
the backpack (one arm) can result in changes of posture and gait.
|
TABLE 12.5 Differential Diagnosis Of Back Pain In Children
|
|
|---|---|
|
diagnosed with a thorough history, comprehensive physical examination,
and selected imaging and laboratory studies. However, the orthopaedic
surgeon should be familiar with the associated signs and symptoms of
both benign and pathologic causes of back pain. Most patients with
benign mechanical back pain have intermittent symptoms that do not
interfere with sleep. These symptoms are typically improved with rest.
The pain is frequently located in the middle to lower back paraspinal
muscles with no limitation of motion. The most common causes of
significant pathologic low back pain include spondylolysis, Scheuermann
disease, and muscle or ligament injury. A history of specific trauma
may suggest a musculoskeletal origin for the pain. Continuous or severe
pain with localized point tenderness, with or without constitutional
symptoms, can be associated with infection or malignancy. Exacerbation
of the pain with a straight leg lift may suggest lumbar disc disease.
Flattening of the lumbar spine on forward flexion or tenderness over
the sacroiliac joints may represent JAS. Limitation of extension of the
cervical spine with morning stiffness and pain is frequently seen in
JIA. Osteoporosis typically causes generalized spine pain that becomes
worse with load-bearing.
treatment is conservative with rest and decreased activity. Children
with back pain who do not improve with rest can be further evaluated
with plain radiographs, a complete blood count, and ESR. If these are
normal, a continued conservative approach is indicated. Patients with
suspected
benign
mechanical pain should be counseled on proper techniques for wearing
their backpacks, or advised to use rolling backpacks or to obtain two
sets of books in order to avoid carrying them back and forth between
home and school. Many patients will improve with a physical therapy
back exercise program along with a 2 to 4 week trial of an NSAID. If
muscle spasm is suspected, a trial of rest combined with a muscle
relaxant and NSAID is frequently beneficial.
findings in their history, physical examination, or laboratory tests,
additional imaging such as computerized tomographic (CT) scan, MRI,
and/or bone scan may be utilized. CT scan is most useful for
identifying abnormalities in vertebrae or erosive changes in joints. If
disc or spinal nerve abnormalities are suspected, MRI is the superior
imaging modality. A bone scan is useful in identifying local areas of
infection, malignant infiltration, or active inflammatory arthritis.
The diagnosis of growing pains should be reserved for those children,
typically from 2 to 12 years of age, who have benign pain, precipitated
by exercise and routine physical activities. These pains usually occur
in the afternoon, the evening, or the middle of the night, but are
never present in the morning. They often respond well to massage or
analgesics. The physical examination shows no sign of synovitis, and
laboratory studies are always normal. Therapy for growing pains
includes gentle massage and stretching. Children with recurring
nighttime pains often have significant relief from a single bedtime
dose of acetaminophen, ibuprofen, or naproxen. Frequently,
acetaminophen 1 hour before and after exercise can be beneficial in
preventing pain.
that are similar to growing pains. These pains are often more frequent,
more intense, can be present at any time of the day, and are typically
increased by physical activity. These children often have distinct
physical exam findings that aid in diagnosis of this syndrome:
hypermobile joints, pes planus, and/or leg-length discrepancy.
The diagnosis of hypermobility requires three of the following:
opposition of the thumb to the flexor aspect of the forearm,
hyperextension of the fingers parallel to the extensor aspect of the
forearm, hyperextension of the elbows or knees by more than 10 degrees,
or excessive dorsiflexion of the ankle and eversion of the foot (91). Children with hypermobility will often benefit from weight training and strengthening around the hypermobile joints.
with pronation and pain in the medial side of the arch. There are often
associated mechanical strains resulting in pain in the ankles, knees,
hips, and lower back. Such children may benefit from the use of
orthotic shoe inserts.
associated with benign pains of childhood. Such children will often
have a leg-length difference of less than 2.5 cm. However, this
difference is more significant in proportion in small children. These
children are often reported as having been clumsy from the time they
began to walk. They will frequently benefit from the temporary use of
sole inserts for the shoe of the shorter leg.
associated with real or potential injury, or it is perceived in terms
of such injury. The sensation of pain is a complex process that is
dependent on multiple factors, including degree of injury, personal
experience or knowledge of others’ experience, and current emotional
and physical health. Psychogenic pain can develop without obvious
cause, as a consequence of an acute or chronic illness or following a
severe or even mild injury, but persists or worsens long after any
inciting factor has resolved. This type of pain can be localized or
diffuse. It is frequently described as more intense than other types of
pain and is often associated with changes in mood, sleep patterns, and
vocational and avocational functioning. A comprehensive discussion of
the psychogenic pain syndromes in childhood is beyond the scope of this
chapter, and the reader is referred to a number of excellent reviews (92, 93, 94, 95, 96, 97).
diagnostic dilemma for pediatricians and pediatric subspecialists.
Nearly all of these children present, with the parents and occasionally
the child believing that the pain must be due to arthritis. Much of the
difficulty in categorizing children with chronic musculoskeletal pain
is because of the variable nomenclature used by different clinicians
and researchers. Malleson et al. (98) have suggested the use of diffuse idiopathic pain syndrome, which includes primary fibromyalgia syndrome (FMS), and localized idiopathic pain syndrome, which includes RSD.
for all chronic idiopathic pain syndromes of childhood, and to
subclassify them as: (a) with autonomic dysfunction (complex regional
pain syndrome type 1 or 2, reflex neurovascular dystrophy, RSD,
algodystrophy, sympathetically mediated pain syndrome, Sudeck atrophy,
and localized idiopathic pain syndrome); (b) without autonomic
dysfunction–constant (psychogenic, psychosomatic, pseudodystrophy,
localized idiopathic pain syndrome, diffuse idiopathic pain syndrome);
(c) without
autonomic
dysfunction–intermittent (psychogenic, psychosomatic, growing pains);
(d) with multiple painful points (fibromyalgia, diffuse idiopathic pain
syndrome); and (e) hypervigilant (psychogenic, psychosomatic, growing
pains). This system has some advantages in allowing classification of
the type of chronic pain that does not meet the well-defined criteria
for fibromyalgia or RSD (93,97,99).
symptomatology that pervades all these pain syndromes is the presence
of noninflammatory pain that is disproportional to physical examination
findings and la belle indifference (an
appearance of unconcern) that most children display regarding the
severe pain and disability they are experiencing. Most patients are
female (80%), with onset typically after 6 years of age, but the
condition may be present in children as young as 3 years (94,97,100).
Another very important aspect of these pain syndromes is the ability to
move from one symptom complex to another, or to have characteristics of
multiple psychogenic syndromes simultaneously. A child may present with
localized limb pain without autonomic signs, then develop classic RSD,
which resolves only to be followed by diffuse pain with multiple
painful points or fibromyalgia.
The onset of RSD often occurs after minor trauma or after a fracture
has healed and the cast has been removed. There is an initial pain that
causes the child to stop using the affected limb. The disuse
perpetuates the pain and the extremity involved becomes painful to even
light touch (allodynia), swollen, cold, and discolored. Plain
radiographs of the affected limb may show soft tissue swelling and,
after 6 to 8 weeks, a generalized osteoporosis. Technetium 99 m bone
scans may show either a diffuse increase (early) or decrease (late) in
uptake of isotope (Fig. 12.2). The outcome for
children with RSD is thought to be generally good when intensive
physical and psychological therapy are instituted within the first year
(94,95,100).
It has also been shown that more than 50% of children with RSD who
presented after 1 year had elapsed between onset of symptoms and
diagnosis continued to have pain and prolonged dysfunction (100).
The most effective treatment for RSD is vigorous physical therapy and
careful attention to the underlying psychosocial stressors (94,95,97).
The affected limb should never be immobilized, because this will
uniformly cause a worsening of the pain during or after the period of
immobilization.
by chronic diffuse pain and localized tender points with a decreased
pain threshold. According to a US pediatric rheumatology disease
registry (102), FMS accounts for 2.1% of new
patient diagnoses by pediatric rheumatologists. Childhood-onset FMS is
similar to the adult disorder, which is characterized by diffuse pain,
tender/trigger points, irritable bowel syndrome, headaches, fatigue,
and nonrestorative sleep (99). Yunus and Masi (93)
defined the criteria for pediatric FMS as including diffuse pain and
five or more tender points. They described the prominent symptoms as:
nonrestorative sleep (100%), fatigue (91%), stiffness (79%), subjective
swelling (61%), headaches (54%), paresthesias (36%), and irritable
bowel syndrome (27%). Morning stiffness and generalized pains may
prompt the referral of a child with FMS to an orthopaedic surgeon.
Therapy for FMS consists of physical therapy with stretching and
aerobic exercise (including aqua therapy), stress reduction, and
psychological counseling.
illnesses in children. Most children with JIA have syndromes that are
unique to childhood. Even in those types of JIA that have an adult
equivalent, such as ankylosing spondylitis and psoriatic arthritis,
children often have a different pattern of onset and course of the
disease than adults. Children with arthritis are also uniquely affected
by articular inflammation which, because of their skeletal immaturity,
can result in growth disturbances. As a result of localized
inflammation, there may be an increase of ossification centers,
accelerated growth, or premature closure of epiphyses of the bones,
resulting in diminished length.
A study of the epidemiology of juvenile arthritis in Rochester, MN from
1960 to 1993 suggested that the incidence of arthritis in children may
be decreasing (6). However, the improved
recognition of Lyme disease, and the exclusion of psoriatic arthritis
and juvenile spondyloarthropathies from the definition of arthritis,
may be contributing factors in this finding.
been estimated to be between 57 and 113 per 100,000 children younger
than 16 years (105). The prevalence rates of
childhood-onset psoriatic arthritis and ERA (spondyloarthropathies)
have not been as well studied. The prevalence of JAS is reported to be
2 to 10 per 100,000, whereas juvenile psoriatic arthritis has been
reported to have a prevalence of 2 to 12 per 100,000 (106).
The incidence of juvenile spondyloarthropathies in whites of Northern
European ancestry is slightly greater than 1 per 100,000 (103,107).
However, spondyloarthropathy is the most common form of juvenile
arthritis in some Mexican and North American Indian children (108,109).
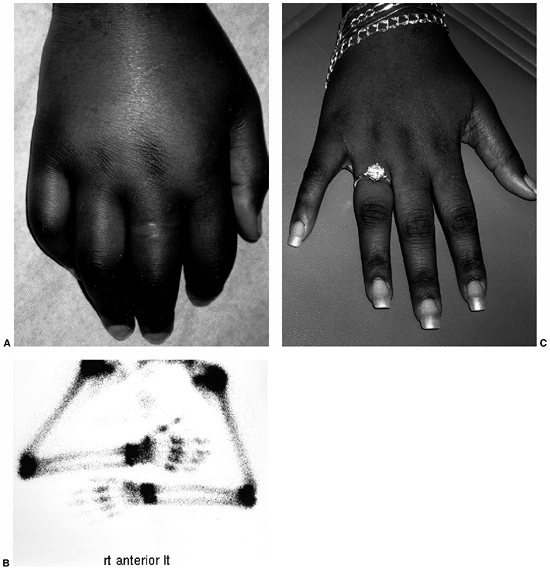 |
|
Figure 12.2 Reflex sympathetic dystrophy in a child with a 1-month history of hand swelling and pain. A: Right hand after 1 month of illness. B:
Technetium 99 m bone scan showing diffuse increase in uptake of isotope in the affected hand. In some patients, isotope uptake is diffusely decreased. C: Right hand after 3 weeks of physical therapy and psychotherapy. |
complex disorder characterized by inappropriate immunologic activation,
with failure of self-tolerance, in the setting of multiple genetic and
environmental factors in the host. JIA is a heterogeneous disorder with
multiple ages and patterns of onset, and with a highly variable course.
It is likely that multiple initiating factors are involved, including
infection, trauma, and autoimmunity, all in conjunction with genetic
predilection for arthritis.
and juvenile arthritis. Apart from HLA-B27, JIA has been associated
mostly with the HLA class II antigens, which are restricted to cells of
lymphoid origin (110). In oligoarticular
arthritis, there is an increased association with HLA-DR8, HLA-DR6, and
HLA-DR5, with relative risks of 2 to 27. This means that a child who
carries one or more of these genes has a 2- to 27-fold increased risk
of developing the disease compared to the population as a whole. The
presence of uveitis is correlated with HLA-DR5, whereas protection from uveitis is correlated with HLA-DR1 (110). Chronic uveitis has also been associated with HLA-DRB1 and HLA-DQA1 (111).
Polyarticular onset with positive RF is associated with HLA-DR4, which
parallels the association with adult rheumatoid arthritis, whereas
HLA-DR7 seems protective. RF-negative polyarticular disease is
associated with HLA-DR8, HLA-DPw3, and HLA-DQw4, with relative risk
factors of 3 to 10. Systemic-onset disease has overlapping risk
factors, showing associations with HLA-DR4, HLA-DR5, and HLA-DR8, with
relative risks ranging from approximately 2 to 7 (112).
incidence. The only immunogenic factor in common in this class of
diseases has been shown to be HLA-B27. Data from multiple immunogenetic
studies have shown that 90% of patients with JAS express the HLA-B27
antigen (113,114).
These data are supported by an animal model in which spontaneous
inflammatory disease of the gastrointestinal tract, peripheral and
vertebral joints, male genital tract, skin, nails, and heart were seen
in transgenic rats that express a functional human HLA-B27 allele (115).
antiinflammatory cytokines may be associated with chronic inflammation.
A polymorphism in the IL-1α gene was found to be associated with uveitis and pauciarticular arthritis in Norwegians (116). Children who have an IL-6 genotype, which has a relatively higher transcription rate when stimulated, may be at greater risk for systemic arthritis (117).
Of all types of JIA, systemic onset has clinical features most
consistent with an infectious process: acute onset, high fever, rash,
lymphadenopathy, and arthritis. However, there has to date been no
convincing laboratory evidence of infection in this relatively
homogeneous disease. Multiple viral and bacterial agents have been
associated with JIA (119). However, no single
or even large group of agents has been convincingly implicated in any
form of JIA. It is more probable that multiple conserved viral and
bacterial antigens, with epitopes that cross-react with human antigens,
may promote an inappropriate autoimmune response. This association is
strongest for the HLA-B27-associated diseases (120) in which arthritogenic peptides from enteric pathogens have generated specific B27-restricted CD8+ T lymphocytes; these lymphocytes have been found in arthritic joints (121).
juvenile arthritis varies widely, depending on whether the EULAR or ACR
norms are utilized. Over many years, the rates of occurrence of the
various types at onset among children with JRA have been quite
consistent, with approximately 50% of children having oligoarticular
disease, 30% to 40% presenting with polyarticular disease, and 10% to
20% having systemic onset. Only recently has psoriatic arthritis been
separated from the spondyloarthropathies and differentiated from JRA.
The subtypes of JCA show similar figures for oligoarticular (50%) and
systemic onset (11%). However, the prevalence of polyarticular disease
is only 20%, and the remainder is divided among undifferentiated
spondyloarthropathy, JAS, juvenile psoriatic arthritis, and
inflammatory bowel disease–associated arthritis (103) (Table 12.6).
At the time of writing, the current summary of the prevalence of
individual subtypes utilizing the ACR criteria is from the Pediatric
Rheumatology Data Base (102) (Table 12.6). Of 2828 children with arthritis, 11%
had systemic onset, 24% polyarticular (RF-) 1% polyarticular (RF+) 38%
pauciarticular, 24% spondyloarthritis (11% JAS), and 3% psoriatic
arthritis. Each subtype of juvenile arthritis has individual
characteristics, and each type can have widely different courses and
outcomes, thereby further emphasizing the heterogeneity of JIA.
Children with systemic arthritis frequently appear quite ill while
febrile. The fever often responds poorly to NSAIDs, but will typically
respond well to corticosteroids. In most children, the fever is
accompanied by a characteristic rash (125) (Fig. 12.3).
The rash consists of discrete, erythematous macules, which are
blanching, transient, and frequently nonpruritic. The rash is often
more pronounced on the trunk, but is often present on the extremities
and may occur on the face. Many children with systemic arthritis will
have extraarticular manifestations, including hepatosplenomegaly,
pericarditis, pleuritis, lymphadenopathy, and abdominal pain. The
extraarticular features may be present for weeks, months and,
occasionally, years prior to the onset of arthritis. Usually, the
extraarticular manifestations of systemic arthritis are self-limiting
and will resolve spontaneously or with corticosteroid therapy.
Occasionally, the pericarditis can result in tamponade. Systemic
arthritis can occur at any age, but is slightly more common before the
age of 6 years (103), and can occur rarely in adulthood, when it is referred to as adult-onset Still disease.
The condition occurs in both sexes in equal ratio, and this may support
the premise that there is an infectious trigger for systemic arthritis (126).
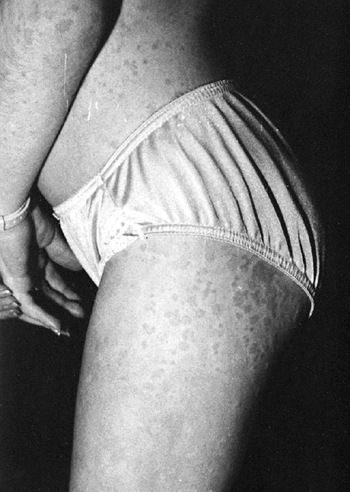 |
|
Figure 12.3 Rash associated with systemic-onset juvenile idiopathic arthritis.
|
notable for elevated acute-phase reactants. The ESR and CRP are greatly
elevated. The disease is often accompanied by anemia of chronic disease
(127, 128, 129), a leukocytosis, and a marked thrombocytosis, which may exceed 1million per mm3 Clinical experience has shown that when the platelet count remains greater than 500,000 per mm3
after 5 years, remission is unlikely. An elevation in the level of
serum ferritin has been correlated with active inflammation in some
children with systemic arthritis (130).
Patients with systemic arthritis can have coagulation abnormalities,
with generation of fibrin split products that have also been correlated
with active disease (131). Children with systemic arthritis are rarely ANA- or RF-positive.
essentially the same as for fever of unknown origin. Systemic arthritis
often presents the greatest challenge to the clinician during the phase
prior to the onset of arthritis. The diagnostic possibilities that must
be considered include infections, malignancy, IBD, SLE, and
vasculitides (polyarteritis nodosa, Kawasaki disease).
The use of glucocorticoids also may cause growth retardation as well as
Cushing syndrome in this same group of patients. When children with
systemic arthritis have active inflammatory disease, the use of human
growth hormone fails to significantly increase linear growth (134,135).
The prognosis of systemic arthritis is determined predominantly by the
course of arthritis. Approximately 50% of children with systemic
arthritis will have an oligoarticular course that is typically mild,
and in most of these children the arthritis will ultimately remit. The
remaining half of the children with systemic onset will develop a
polyarticular arthritis that can remit, but progresses in approximately
50% of the cases (25% of all systemic-onset JIA) to a severe,
unrelenting, and destructive course despite all currently available
therapeutic interventions (136). Chronic
anterior uveitis is extremely rare in systemic arthritis. Systemic
amyloidosis, usually presenting with the onset of proteinuria and
hypertension, can occur as a result of any chronic inflammatory
disease. Approximately 8% of European children with systemic arthritis
and, to a lesser degree, the other subtypes of JIA,
have been shown to develop this life-threatening complication (137).
The incidence of amyloidosis in North America is significantly lower
than that seen in Europe. The reason for this discrepancy remains
unclear.
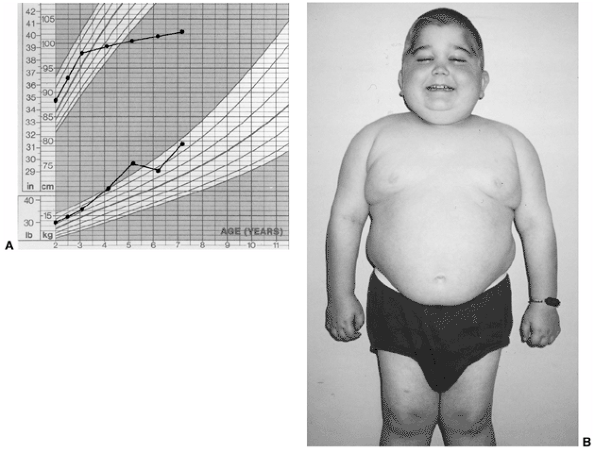 |
|
Figure 12.4 Child with systemic-onset juvenile idiopathic arthritis. A: Growth arrest due to systemic inflammation and chronic steroid use. B: The same child at age 7 with chronic polyarthritis, growth arrest, and Cushing syndrome.
|
(HLH), is a severe, potentially life-threatening complication seen
nearly exclusively in systemic arthritis. It is characterized by
macrophage activation with hemophagocytosis, and is associated with
hepatic dysfunction, disseminated intravascular coagulation with a
precipitous fall in the ESR secondary to hypofibrinogenemia, and
encephalopathy (138). It has been suggested
that antiinflammatory medications and viral infections can induce this
syndrome. High-dose corticosteroids and cyclosporine A have been shown
to improve the outcome of MAS (139,140).
characterized by arthritis in four or fewer joints during the first 6
months of disease. These children rarely have complaints of pain, do
not have associated fever, and are not systemically ill. The knee is
the most common joint affected, followed by ankles and elbows. The hips
and the small joints of the hands and feet are seldom affected.
Asymmetric oligoarticular involvement of the small joints, with or
without large-joint arthritis, is most characteristic of psoriatic
arthritis. Most children with oligoarthritis present before 6 years of
age, with girls outnumbering boys in the ratio 4:1. Oligoarthritis can
present in older children, but this late-onset type, in which there is
a male predominance and high incidence of HLA-B27, should now be
classified as ERA.
elevation of the ESR (rarely more than 80 mm per hour), but it can be
normal in some patients. The CRP is usually normal or mildly elevated.
ANAs are found in 40% to 80% of children with oligoarthritis, and are
associated with an increased risk for anterior uveitis. An RF is
generally absent in oligoarthritis. However, when an RF is present in
children with chronic oligoarthritis, it has been associated with an
aggressive and erosive disease (141).
arthritis depends on the duration of involvement of the joint. In
children with acute onset of pain and swelling in the joint, infections
(septic arthritis or osteomyelitis), trauma, hematologic causes of
hemarthrosis, and malignancy must be considered. These patients should
have a thorough evaluation, including an arthrocentesis. If the
arthritis is long standing, these causes are less likely. However, both
Lyme disease and mycobacterial infections can produce a prolonged
monoarthritis indistinguishable from JIA.
remitting course. However, in untreated children with longstanding
unilateral knee arthritis, there can be overgrowth of the affected
limb, resulting in a marked leg-length discrepancy (142,143).
There is a subgroup of children with oligoarthritis that is
indistinguishable within the first 6 months of disease, but progresses
to polyarthritis (extended oligoarticular), which is usually most
consistent with RF-negative polyarticular JIA.
the most serious complication seen in oligoarthritis, and occurs in 13%
to 34% of patients. Approximately 80% of all cases of anterior uveitis
in childhood are associated with JIA (144).
Initially, the eyes of most patients with JIA-associated uveitis appear
normal, and are asymptomatic. Of those children who will ultimately
develop uveitis, it is already present in 6% at the onset of arthritis,
but develops in most of them within 4 to 7 years after diagnosis.
Although the overall incidence and severity of uveitis seem to be
decreasing (145,146), even a low-grade chronic uveitis can result in a poor visual outcome (147).
Current guidelines for ophthalmologic examination in children with
juvenile arthritis recommend routine screening examinations, including
slit-lamp evaluation, based on age and type of onset (148) (Table 12.7).
insidious, but occasionally acute, onset of a generally symmetric
arthritis in five or more joints. It can involve both large and small
joints, and frequently affects the cervical spine and temporomandibular
joints. Typically, girls outnumber boys 3 to 1. Mild systemic features
may be present in children with polyarthritis. They may have low-grade
fevers, lymphadenopathy, and hepatosplenomegaly. The fevers are not
typically the high quotidian temperature spikes that are diagnostic of
systemic arthritis, and rash is rarely seen (8). There are at least two distinct subgroups of polyarthritis: those with and those without the presence of RF.
 |
|
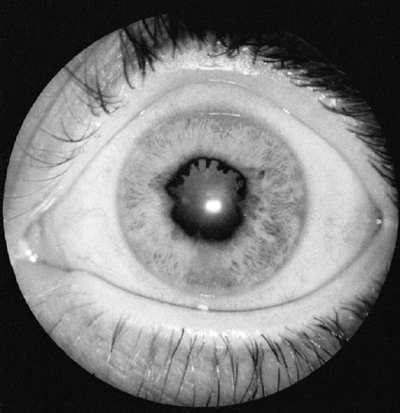
Figure 12.5
Iritis in oligoarticular juvenile idiopathic arthritis. Posterior synechiae are fingerlike adhesions between the iris and lens, and result in an irregular pupil. |
|
TABLE 12.7 Guidelines For Initial Frequency Of Screening Eye Exams In Juvenile Idiopathic Arthritis (JIA)
|
||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||||||||||||||||||||||||||||||||||||||
are positive for RF. This subtype occurs predominantly in older girls
(>8 YEARS) who are HLA-DR4 positive, and is indistinguishable from
adult rheumatoid arthritis. These children are more likely to have a
symmetric small-joint arthritis, rheumatoid nodules, and early erosive
synovitis with a chronic course. However, these children rarely develop
chronic uveitis.
elevated ESR, typically 20 to 80 mm per hour. The ESR is often a useful
measure of disease activity in children with polyarthritis (19,149).
Children with significant joint disease will often develop anemia of
chronic disease, with hemoglobin in the range of 7 to 10 g per dL,
although this is more marked in systemic arthritis (127,128).
arthritis is unusual, although an asymmetric polyarthritis and tenosynovitis can be caused by Neisseria gonorrheae.
Systemic lupus should be considered, especially in adolescent and
preadolescent girls. Reactive arthritis, inflammatory bowel-related
arthritis, juvenile psoriatic arthritis, and ERA, including JAS, should
be considered. Although juvenile dermatomyositis and scleroderma may
present with polyarthritis, the associated signs and symptoms of these
disorders usually lead to a correct diagnosis.
risk for a prolonged and destructive course. These children are
typically older girls with involvement of multiple joints (20 or more)
including the small joints of the hands and feet, early erosions, and
rheumatoid nodules. The presence of hip arthritis has been shown to be
a poor prognostic sign, and may lead to destruction of the femoral
heads (150). If polyarthritis persists longer
than 7 years, remission is unlikely. The onset of puberty seems to have
no relation to disease activity or remission (72).
If severe poly-articular (polyarticular and systemic JIA) disease with
involvement of the temporomandibular joints occurs before 5 years of
age, it can result in micrognathia (151).
published the first large report of 60 children with psoriatic
arthritis, there had been fewer than 80 cases described in the
literature in the English language. The rarity of juvenile psoriatic
arthritis was unusual, given the relatively large number of children
with psoriasis, and the fact that 7% of adults with psoriasis have
arthritis (153). Juvenile psoriatic arthritis
has historically been thought of as a juvenile spondyloarthropathy.
However, recent studies have shown that juvenile psoriatic arthritis is
a distinct entity that has been underdiagnosed, often because of the
long period from onset of arthritis to onset of psoriasis (153,154).
 |
|
Figure 12.6 Juvenile psoriatic arthritis. A: Nail pitting associated with psoriasis. B: Swelling of a single distal interphalangeal joint in a child with juvenile psoriatic arthritis.
|
There is a slight preponderance of girls (1.6 to 2.3 times as many) and
the disorder often affects young children, with the median age at onset
being 5.9 to 10.1 years. The arthritis is often an asymmetric oligo- or
polyarthritis affecting both large and small joints. At onset, patients
may have pitting of the nails (67%) (Fig. 12.6)
and a family history of psoriasis (69%) or dactylitis (39%), while less
than one-half of the children have the rash of psoriasis (13% to 43%) (103,153,154). Current criteria do not require the development of psoriasis to confirm a diagnosis of psoriatic arthritis (10) (Table 12.2).
an RF, but a positive ANA can be seen in 50% of them and is a risk
factor for uveitis. HLA-DR1 and HLA-DR6 are statistically significant
risk factors for development of juvenile psoriatic arthritis (154).
There is a mild, but not statistically significant, increase in the
presence of HLA-B27 in children with psoriatic arthritis, and these
children are more likely to have axial arthritis (152, 153, 154).
In children younger than 5 years, the presentation is often
characterized by the involvement of a small number of fingers or toes
that are relatively asymptomatic, but leading to marked overgrowth of
the digit(s).
essentially the same as for polyarthritis. However, the diagnosis of
psoriatic arthritis should be considered in a child with dactylitis,
nail pitting, asymmetric involvement of large and small joints,
arthritis of the distal interphalangeal (DIP) joints (Fig. 12.6),
or a first- or second-degree relative with psoriasis. There is rarely
the kind of fever or systemic illness that may be seen in septic
arthritis caused by Neisseria gonorrheae.
lifelong arthritis that follows a relapsing and remitting course.
Arthritis mutilans and severe DIP joint disease are unusual. However,
many of the children will have prolonged poly-arthritis that may result
in irreversible joint damage (152). Amyloidosis has been reported in the European literature as having resulted in the deaths of at least three children (152,155). Chronic anterior uveitis has been observed in up to 17% of the children (153,154),
is associated with a positive ANA titer, and is clinically
indistinguishable from the uveitis in oligo- and polyarthritis. The
uveitis associated with psoriatic arthritis may be more resistant to
treatment than the other forms of chronic uveitis associated with
childhood arthritis (72).
of arthritides that includes undifferentiated spondyloarthritis,
ankylosing spondylitis, and inflammatory bowel disease–associated
arthritis. At the onset, juvenile spondyloarthropathies are often
undifferentiated, preventing the application of adult-onset criteria
for diagnosis. The addition of other criteria (the presence of HLA-B27,
a family history of HLA-B27–associated disease, and the onset of
arthritis in a boy after 6 years of age) will increase the number of
children included in this category (10).
However, in an effort to better define the group of children who have
psoriatic arthritis, the ILAR norms now exclude even those with
ankylosing spondylitis from the diagnosis of ERA, if they have a
first-degree relative with psoriasis. This contributes a significant
number of children to the “Other Arthritis” category in that they
either fulfill no criteria or fulfill criteria for more than one
category. It is probable that in families with a genetic propensity for
psoriasis, who also carry the HLA-B27
gene, there may be two distinct mechanisms that contribute to the
development of arthritis. These disorders will begin to be categorized
more accurately as the underlying mechanisms are further elucidated by
molecular and genetic research. The current criteria will include many
of the children who have been previously diagnosed with a syndrome of
seronegativity, enthesopathy, and arthropathy (SEA syndrome) who were
shown to be at increased risk for development of classic
spondyloarthritis or JAS (156,157).
Enthesitis is identified when marked tenderness is noted at the 6, 10,
and 2 o’clock positions on the patella, at the tibial tuberosity, iliac
crest, or the attachments of the Achilles tendon or plantar fascia (72). In some children, the only manifestation of ERA may be severe enthesopathy of the heel(s) (158) (Fig. 12.7).
relatively unremarkable results. There is often systemic inflammation
with thrombocytosis and an elevated ESR. A highly elevated ESR (greater
than 100 mm per hour) is more likely to be associated with inflammatory
bowel disease in a child who meets the criteria for ERA. These patients
uniformly test negative for RF, but ANAs can be present in the same
proportion as in the general population of children (21,22,72).
 |
|
Figure 12.7 Achilles tendonitis and enthesitis in a child with enthesitis-related arthritis. (Courtesy of Dr. Ruben Burgos-Vargas.)
|
It typically presents with an acute, painful, red, photophobic eye. AAU
may resolve with no ocular residua, but some of the children will have
a persistent uveitis that is relatively resistant to therapy and can
result in blindness (161,162).
of adult criteria that require radiographic evidence of sacroilitis.
JAS most often presents in late childhood or adolescence. Children with
JAS and sacroiliac (SI) involvement are often HLA-B27-positive (82% to
95%), and boys outnumber girls in the ratio 6:1 (106).
Most children who are ultimately diagnosed with JAS will initially have
had an episodic arthritis of large joints of the lower extremities and
the tarsal bones. Regardless of axial disease, the most reliable
predictors to differentiate JAS from oligo- or polyarticular JIA are
the presence of enthesitis and tarsal disease in children who have
arthritis of the lower, but not of the upper, extremities (163).
absence of axial involvement. Only 12.8% to 24% of children with JAS
have pain, stiffness, or limitation of motion
of
the sacroiliac or lumbosacral spine at onset. A peripheral arthropathy
or enthesopathy, affecting predominantly the lower limb joints and
entheses, is seen in 79% to 89.4%. These children tend to have fewer
than five joints involved, and rarely more than ten. At presentation,
the pattern of involvement of the joints is usually asymmetric, or even
unilateral (164).
Small joints of the toes are commonly involved in JAS but are seldom
affected in other forms of JIA, with the exception of psoriatic
arthritis.
stage of onset. However, the combination of peripheral joint arthritis
with preponderance in the lower extremity, enthesitis, and SI or
lumbosacral disease, would strongly suggest the diagnosis of ERA and
possible JAS. Septic SI joint disease and osteomyelitis may present
with SI pain and limitation of motion.
to arrive at a diagnosis of JAS. Pain may be elicited over the SI
joints by direct pressure, lateral compression of the pelvis, or
distraction of the SI joints (Patrick test). Quantitation of the normal
lumbar spine flexion, by Macrae and Wright’s modification of the
Schober test (165), can identify children with
limitation of lumbar spine flexion. With the child standing upright, an
anchoring mark is made at the lumbosacral junction (dimples of Venus).
A mark is then made 5 cm below and 10 cm above the lumbosacral
junction. Then, with the child in maximal forward flexion, the distance
between the upper and lower points is measured. In general, a modified
Schober measurement of greater than 21 cm (i.e., an increase of 6 cm)
is within normal limits (72). The measurement
of fingertip to floor distance on forward flexion is not reproducible,
because it reflects both hip and back flexion and does not correlate
with the Schober index. Chest expansion is also not reliable as a test
for spine involvement in JAS (164).
and relapsing symptoms, which are frequently mild. This patient cannot
be differentiated from the child who seems to have recurrent bouts of
reactive arthritis. However, the pattern of joint disease (which often
progresses to become polyarticular) and axial disease is usually
evident after the third year of illness (164). Children with long-standing JAS have been shown to develop tarsal bone coalition that has been termed ankylosing tarsitis (166) (Fig. 12.8).
contradictory. The prognosis of JAS has been reported as being worse
according to some studies, and better according to others, than
adult-onset ankylosing spondylitis (167,168). Peripheral joint arthritis tends to be more common in child patients than in adults (72). Hip disease has been associated with a poor functional outcome (167,169) and may require total hip arthroplasty.
 |
|
Figure 12.8
Ankylosing tarsitis, a complex disorder resulting in ankylosis of the foot in a child with juvenile ankylosing spondylitis. (Courtesy of Dr. Ruben Burgos-Vargas.) |
with IBD has been reported to be 7% to 21%, and it usually occurs after
the diagnosis of the bowel disease (170, 171, 172). Two different patterns of arthritis are seen (72).
The most common type is oligo- or polyarticular arthritis of the lower
limbs. This group is less likely to meet the criteria for ERA. This
arthritis is often episodic, with exacerbation lasting 4 to 6 weeks or,
rarely, for months. The activity of the peripheral arthritis often
reflects the underlying activity of the IBD, but may also be
independent of it. The less common type of IBD-associated arthritis is
an HLA-B27–associated oligoarticular arthritis of the lower limbs, with
sacroiliitis and enthesitis, and no relationship to bowel inflammation (72).
This form is more likely to persist and progress despite control of the
bowel disease, and the course seems to be identical to that in other
children with ERA.
radiologic findings. As the disease progresses, there is often
periarticular osteopenia, localized soft tissue swelling and,
occasionally, joint space widening due to effusion or synovial
hypertrophy. Late changes seen in JIA include joint space narrowing
from cartilage loss, erosions, subluxation, and ultimately ankylosis
(Figs. 12.9 and 12.10).
Typically, erosive changes do not occur before 2 years of active
disease, and significant changes in radiographs rarely occur in less
than 6 months. Children with chronic polyarthritis will frequently
develop bony ankylosis of the carpal and tarsal joints, and in the
cervical spine.
instability can be seen, with chronic arthritis of the cervical spine.
Symptoms can range from minimal to severe neck pain and limitation of
movement of the cervical spine, to neurologic damage caused by
impingement on the spinal cord. Special precautions should be taken
prior to anesthesia in children with JIA and cervical spine arthritis,
because the rigid cervical spine and atlantoaxial instability may
predispose these patients to serious injury. Their vulnerability to
injury is further highlighted by the fact that serious traumatic
injuries have occurred in these children after spontaneous cervical
spinal fusion as a consequence of JIA (173).
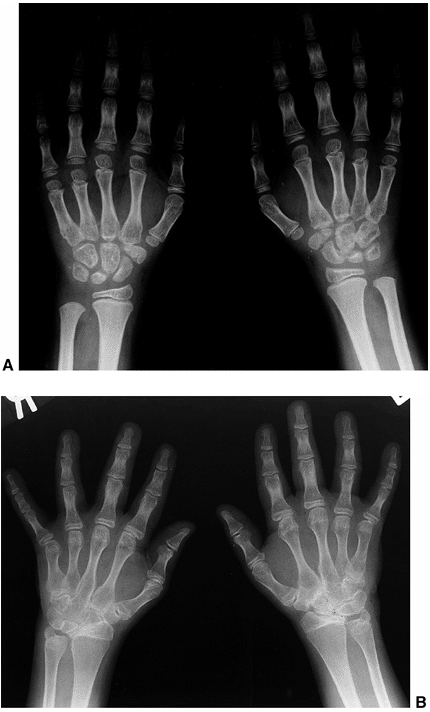 |
|
Figure 12.9 Polyarticular juvenile idiopathic arthritis with wrist and finger involvement. A: At 6 years of age, there is periarticular osteopenia and diffuse swelling of the wrist and fingers. B: At 20 years of age, there is significant carpal and carpometacarpal fusion.
|
asymmetric erosive disease (with or without regional osteoporosis), and
periosteal new bone formation. The periostitis can lead to overgrowth
of the affected bone. Although erosive changes of the DIP joints may be
seen, this is a rare occurrence (72) (Fig. 12.12).
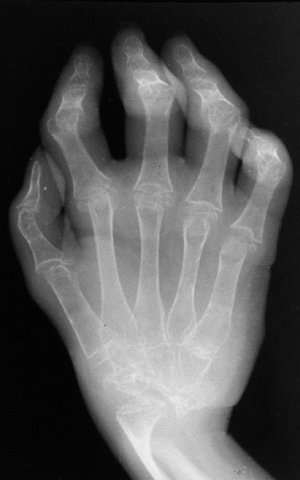 |
|
Figure 12.10
Systemic-onset polyarticular juvenile idiopathic arthritis, with prolonged arthritis resulting in severe osteopenia and destructive changes in the hand and wrist, with severe ulnar deviation. |
radiographically visible changes in the SI joints, but this may not
occur for 1 to 15 (average 6.5) years after diagnosis (159). These findings can include pseudowidening caused by erosions, sclerosis, and fusion (Fig. 12.13). Radiologic changes in the lumbosacral spine occur later in the course of JAS, and are less frequent (174).
Chronic enthesitis, particularly at the calcaneus, can result in
erosion at the insertion of the Achilles tendon or plantar fascia.
with a multidisciplinary team approach. First and foremost, the child
and family must participate in informed decision-making. The team of
health care professionals can provide comprehensive care for all facets
of this chronic disease. The team should include the primary care
physician to
coordinate
medical care and the rheumatologist for diagnosis and treatment plans.
Other valuable members of the treatment team may include the
rheumatology nurse for education and family support; social worker for
monetary and school advocacy; dietitian to minimize the effects of
corticosteroid therapy; physical and occupational therapists to
maintain and improve the strength and range of motion of the joints;
ophthalmologist for uveitis screening and treatment; and, occasionally,
the pediatric orthopaedic surgeon when surgical interventions are
indicated.
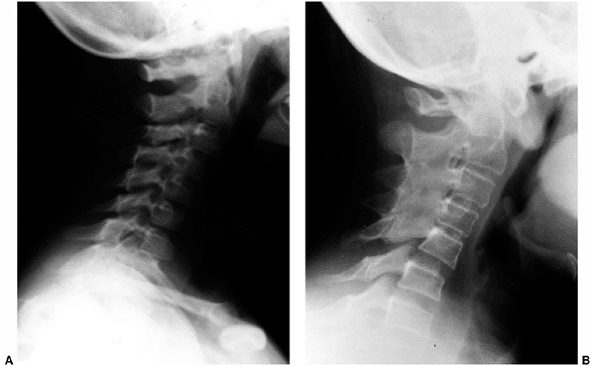 |
|
Figure 12.11 The cervical spine in a child with polyarticular JIA. A: At 6 years of age, there are no radiographic abnormalities. B: At 21 years of age, there is ankylosis of C2-C5.
|
two or more drug classes must be used simultaneously in order to
achieve disease control. Many recent advances in understanding the
mechanisms of inflammation in arthritis have led to novel therapeutic
strategies. The fundamental purpose of pharmacologic therapy is to
achieve pain control, decrease inflammation, and promote and maintain
remission. The medications used are individualized for each patient,
depending on their subtype of arthritis, degree of inflammation, and
previous response to medications.
inhibition of the biosynthesis of prostaglandins by direct action on
the enzyme cyclooxygenase (COX) (175). The
discovery of a second COX enzyme (COX-2), which is induced during the
proinflammatory cascade, and the differential inhibition of the two COX
isoforms (COX-1 and COX-2) by individual NSAIDs has provided the basis
for the development of safer NSAIDs (176). At
the time of this writing, no COX-2-specific NSAID has been approved for
use in children. In the near future, however, this exciting scientific
advance will likely change the way NSAIDs are used in the treatment of
children.
children with JIA. NSAIDs provide both analgesia and an
antiinflammatory effect. The average time-course for response to NSAIDs
is 4 to 12 weeks (177). Therefore, an NSAID is
usually tried for 4 to 8 weeks before another is substituted if there
has not been sufficient improvement. NSAIDs are generally safe and well
tolerated in most children. Abdominal pain, nausea, and vomiting are
the most common side effects, and gastrointestinal hemorrhage is rare (178). However, gastroduodenal injury is more frequent in children who are receiving high doses, or more than one NSAID at a time (179).
The use of aspirin in JIA is no longer recommended because of the risk
of Reye syndrome, increased hepatotoxicity, bleeding, and
four-times-per-day dosing.
weight, and are often proportionally greater than in adult rheumatic
diseases (Table 12.8). Preparations that come in a liquid form and have once- to twice-daily dosing are
preferred. In the United States, the most commonly used NSAID for JIA
is naproxen (10 to 20 mg/kg/day, b.i.d.). In children with fevers and
serositis associated with systemic arthritis and with JAS, indomethacin
is often the most effective NSAID (72).
Children on long-term NSAID therapy should have a complete blood count,
renal and liver function tests, and urine analysis every 6 months.
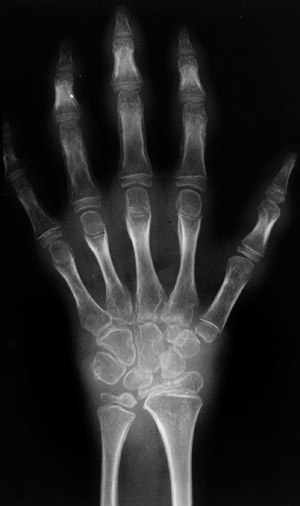 |
|
Figure 12.12
Juvenile psoriatic arthritis affecting the hand, showing metacarpophalangeal, proximal, and distal interphalangeal joint involvement, with marked fusiform swelling and periostitis. |
 |
|
Figure 12.13
CT scan of the SI joints in a child with juvenile ankylosing spondylitis, showing erosions and sclerosis of the SI joints. (Courtesy of Dr. Ruben Burgos-Vargas.) |
|
TABLE 12.8 Nonsteroidal Antiinflammatory Drugs (Nsaids) For The Treatment Of Juvenile Idiopathic Arthritis (JIA)
|
||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||||||||||||
Triamcinolone hexacetonide (1 mg per kg for large joints and 0.5 mg per
kg for medium joints) is the most commonly used agent and often
provides long-term control of inflammation. The most frequent adverse
consequence of intraarticular corticosteroids is the development of
subcutaneous atrophy at the site of injection. Systemic corticosteroids
can be used for rapid control of severe arthritis. However, long-term
use should be restricted to those children who have severe arthritis or
systemic features that do not respond to other interventions.
for treatment of juvenile arthritis. It is typically given at a dosage
of 0.5 to 1 mg per kg (with a maximum of 20 to 30 mg) once weekly,
either orally or by subcutaneous injection. It has been shown to be
superior to placebo in poly-articular and extended oligoarticular
arthritis but not in systemic arthritis (183,184), and can produce radiologically evident improvement in erosions (185).
Methotrexate has been shown to decrease the severity of uveitis in
children with JIA who were dependent on topical cortico-steroids (186).
diarrhea, and oral ulcers. Supplementation with folic acid (1 mg per
day) can usually prevent gastrointestinal complications. One of the
most significant long-term side effects of methotrexate use is the
development of liver fibrosis and cirrhosis (187, 188, 189).
Serial abnormalities of hepatic enzymes were significantly associated
with liver fibrosis in children taking methotrexate for juvenile
arthritis (190), thereby suggesting that the
current guidelines for patients with rheumatoid arthritis are also
applicable to patients with JIA (191).
increasingly in North America, for treatment of both
spondyloarthropathies and JRA/JCA (192,193).
It is typically given in the enteric-coated form at a dose of
50mg/kg/day in two divided doses. Recently, a randomized, double-blind,
placebo-controlled trial showed that sulfasalazine is both safe and
effective for the treatment of oligo- and polyarticular JCA (194).
Serious side effects have been noted in children with systemic
arthritis, and the routine use of sulfasalazine is not recommended for
this subgroup (195,196).
arthritis are still unclear, macrophage-derived cytokines, such as
tumor necrosis factor α(TNFα) appear to play a critical role in the
induction and perpetuation of the chronic inflammatory process in
juvenile arthritis. Etanercept (Enbrel) is a protein containing the
extracellular domains of 2p75 human TNF receptors attached to the Fc
portion of a type 1 human immunoglobulin. A multicenter study showed it
to be safe and effective in the treatment of juvenile arthritis (197).
The study showed that 81% of children treated with etanercept reached a
JRA 30% definition of improvement (DOI), whereas 67% had a JRA 70% DOI.
Infliximab (Remicade) is a chimeric, monoclonal anti-TNFα antibody
developed as a therapeutic agent for the treatment of diseases in which
TNFα is thought to mediate chronic inflammation. This antibody is a
recombinant IgG1κ human-murine chimeric monoclonal antibody that
specifically and potently binds to and neutralizes soluble TNFα and its
membrane-bound precursor. Infliximab has been shown to be highly
efficacious in combination with methotrexate for the treatment of
rheumatoid arthritis and, in small trials, for refractory juvenile
arthritis (198) and chronic inflammatory uveitis (199).
The major adverse events associated with the use of anti-TNFα agents
have been infections and an increased risk of contracting tuberculosis
and coccidiomycosis (200). Other
cytokine-specific monoclonal antibodies, including anti-IL6 for the
treatment of systemic juvenile arthritis, are in clinical trials at the
time of writing.
evaluated by a physical and/or occupational therapist to provide an
appropriate teaching and treatment program. Most treatment programs for
JIA will include active and passive range-of-motion exercises,
strengthening, and other modalities such as use of hot paraffin for
relief of hand stiffness. Swimming has the advantage of providing
muscle-strengthening and active range of motion without significant
weight bearing. Splinting may be used for maintaining alignment,
providing rest, and reducing flexion contractures. For children with
severe flexion contractures, a dynamic tension splint or serial casting
can be used to correct the contracture. Physical therapy for range of
motion in JAS is primarily to prevent loss of mobility and poor
functional positioning.
limited role in the management plan. With early detection and
aggressive medical management, including intraarticular corticosteroid
injections, the majority of children with juvenile arthritis have a
satisfactory outcome without significant disability. However, for those
children with persistent arthritis (treated or untreated), continued
pain, or progressive leg-length discrepancy, there is often significant
benefit from individualized orthopaedic surgical intervention. Many of
the reports of surgery for juvenile arthritis actually refer to adults
who have had arthritis since childhood. Surgical intervention in JIA
presents several problems to the management team. The small size of
children and their growth potential must be taken into consideration.
Also, in the postsurgical period, prolonged immobilization can lead to
decreased strength and range of motion, with or without active
arthritis. After a surgical procedure, intensive physical therapy will
often be required in order to mobilize the child’s joints. There is no
universal agreement about which procedures are indicated for the
treatment of complications of chronic arthritis in childhood. However,
the overall goal is to provide symptomatic relief and improved
functioning.
with JIA for relief of pain, swelling, and impaired motion of the joint
related to synovial hypertrophy. There may be short-term relief from
swelling and pain, but the range of motion may not improve or may even
worsen (201, 202, 203, 204, 205). The greatest benefit has been seen in large joints (206,207). But recurrences are common, and the ultimate outcome for children with JIA is not altered by prophylactic synovectomy (208,209). A recent study has confirmed that
elbow synovectomy in patients with JRA provided complete pain relief in
44% of patients, and 73% had a subjective good to excellent outcome (210). However, there was no significant improvement in range of motion or functional ability.
a severe contracture of the knee or hip that has been resistant to
splinting or serial casting. Initially there were encouraging reports
regarding soft tissue release (211). However, more recent reports have been less positive, showing only a modest benefit and a tendency to deteriorate (212,213). In most cases, vigorous physical therapy will avoid the need for this frequently unsuccessful operation.
joint destruction of the ankle after prolonged synovitis in oligo- or
polyarticular JIA. After puberty, a fixed and painful deformity of the
ankle is best corrected by performing a triple arthrodesis.
Occasionally, in children with isolated damage of the subtalar or
talonavicular joint, a single joint fusion may be appropriate (214). If required, these procedures may later be converted to a triple arthrodesis.
arthritis and atlantoaxial instability, there is no consensus on the
indications for prophylactic fusion. In many cases, a simple cervical
orthosis may stabilize the neck and prevent further subluxation.
However, fusion of the cervical spine (C1-C2) is indicated in children
who have progressive neurologic involvement (215,216).
arthritis. In a younger child with a fixed deformity of the knee but
good remaining joint surface and minimal active synovitis, a knee
osteotomy may result in correction of the deformity (217).
Unfortunately, the osteotomy makes a subsequent total knee arthroplasty
more difficult to perform because of the distorted anatomy.
lengthening of the ulna may be carried out to correct length
discrepancies of the forearm bones. Recently, distraction lengthening
of the ulna in six children (eight wrists), with severe destructive
changes in the wrist due to JCA, was reported (218).
This procedure was found to adequately correct the deformity and
improve functioning, and in most of the children no further splinting
was required during a follow-up of an average of 70 months (range 12 to
152 months).
there can be overgrowth of the affected limb, resulting in a marked
leg-length discrepancy (142,143).
Temporary epiphyseal stapling has been recently shown to be safe and
effective in the treatment of the leg-length discrepancy associated
with JIA (222). In this procedure, the epiphysis of the longer leg is temporarily stapled, allowing catch-up growth of the unaffected leg.
JIA who have severe destructive joint changes with functional
impairment or disabling pain. The most common joints replaced are the
hip and knee, but there may be indications for shoulder and elbow
arthroplasty.
has been severe destruction or ankylosis resulting in functional
impairment. Initial series using predominantly cemented hip
replacements showed reduction in pain and improved functional ability,
but with a significant rate of loosening and subsequent revision (223,224).
Subsequent studies have suggested an improved outcome with cementless
arthroplasty of the hip, but poor bone stock remains an indication for
cementing (225,226). In
a recent prospective study of the outcome of total hip arthroplasty in
small-proportioned patients with JRA, it was found that the miniature
anatomic medullary locking femoral component combined with an
uncemented acetabular cup resulted in a good outcome (227).
A recent study has suggested that bipolar hemiarthroplasty of the hip,
with a 79% 10-year survival, may be an alternative to conventional
joint arthroplasty (228).
is significant pain, deformity, and functional incapacity, a total knee
replacement is indicated. Initial results of total knee arthroplasty in
JIA have been encouraging, with few revisions required (229, 230, 231).
A recent long-term follow-up of total knee arthroplasty in young adults
and children with arthritis has been equally encouraging (232,233). Cementless total knee arthroplasty has been used in selected cases (234).
Recent studies have confirmed the efficacy of the procedure by
reporting an overall 99% survival for nonconstrained anatomically
graduated components prosthesis with cementless fixation (233).
with severe destruction of the elbow joint. In a recent review, Connor
and Morrey (235) evaluated the long-term
outcome for 19 children (23 elbows) who had been managed with total
elbow arthroplasty and followed up for at least 2 years. Only three
(13%) had poor results caused by late complications: aseptic loosening,
instability, and worn bushings.
when there is prolonged pain, limitation of function, and significant
joint destruction. The available data are not sufficient for evaluating
the efficacy of total shoulder arthroplasty in children. However, in
studies of adults with rheumatoid arthritis, and also in some studies
that included children, the results have been promising (236,237).
LS, Mason T, Nelson AMO, et al. Juvenile rheumatoid arthritis in
Rochester, Minnesota 1960-1993. Is the epidemiology changing? Arthritis Rheum 1996;39:1385.
RE, Southwood TR, Baum J, et al. Revision of the proposed
classification criteria for juvenile idiopathic arthritis: Durban,
1997. J Rheumatol 1998;25:1991.
I, Bidde M. Validation of the proposed ILAR classification criteria for
juvenile idiopathic arthritis. International League of Associations for
Rheumatology. J Rheumatol 2000; 27(4):1069.
MF, Mouy R, Prieur AM. Juvenile idiopathic arthritides evaluated
prospectively in a single center according to the Durban criteria. J Rheumatol 2001;28(5):1083.
JW, Thompson KL, Hanson V. The Varni/Thompson pediatric pain
questionnaire. I. Chronic musculoskeletal pain in juvenile rheumatoid
arthritis. Pain 1987;28:27.
DA, Petty RE, Fung M, et al. Persistent antinuclear antibodies in
children without identifiable inflammatory, rheumatic or autoimmune
disease. Pediatrics 1992;89:441.
RC, Dewez P, Stuart L. Antinuclear antibodies using HEp-2 cells in
normal children and in children with common infections. J Paediatr Child Health 1991;27:39.
PMG, Liard G, Siegel DM, et al. The outcome of children referred to a
pediatric rheumatology clinic with a positive antinuclear antibody test
but without an autoimmune disease. Pediatrics 1995;95:892.
AR, Chang F, Zuckner J. Markedly raised synovial fluid leukocyte counts
not associated with infectious arthritis in children. Ann Rheum Dis 1978;37:404.
LA, Shahabpour M, Van Roy P, et al. Magnetic resonance imaging of
articular destruction in juvenile rheumatoid arthritis. Arthritis Rheum 1990;33:1426.
AC, Maini RN, Pegrum GD, et al. The clinical features and HLA
associations of reactive arthritis associated with nongonococcal
urethritis. Q J Med 1979;190:323.
FM, Agarwal R, Helm J, et al. Post-streptococcal-reactive arthritis and
silent carditis: a case report and review of the literature. Pediatrics 1994;93:837.
L, et al. Human serum sickness: a prospective analysis of 35 patients
treated with equine anti-thymocyte globulin for bone marrow failure. Medicine 1988;67:40.
CG, Petty RE, Cassidy JT. The clinical progression of apparent juvenile
rheumatoid arthritis to systemic lupus erythematosus. J Rheumatol 1980;17:777.
PB, Brecker ML, Starr JL. A prospective analysis of the frequency,
course, and possible prognostic significance of the joint
manifestations of childhood leukemia. J Rheumatol 1983;10:753.
S, Carabalona R. Backpacks on! School children’s perceptions of load,
associations with back pain and factors determining the load. Spine 2002;27(2):187.
MB, Masi AT. Juvenile primary fibromyalgia syndrome. A clinical study
of thirty-three patients and matched normal controls. Arthritis Rheumatol 1985;28:138.
DD, McGuire T, Mellins E, et al. Psychosomatic musculoskeletal pain in
childhood: clinical and psychological analyses of 100 children. Pediatrics 1991;88:1093.
F, Smythe HA, Yunus MB, et al. The American College of Rheumatology
1990 criteria for the classification of fibromyalgia. Report of the
Multicenter Criteria Committee. Arthritis Rheum 1990;33:160.
RT, Berde CB, Wolohan M, et al. Reflex sympathetic dystrophy in
children. Clinical characteristics and follow-up of seventy patients. J Bone Joint Surg Am 1992;74:910.
TJ, Majd M. Reflex sympathetic dystrophy syndrome in children and
adolescents. Report of 18 cases and review of the literature. Am J Dis Child 1988;142:1325.
S, Roettcher P. Pediatric rheumatology clinic populations in the United
States: results of a 3 year survey. Pediatric Rheumatology Database
Research Group. J Rheumatol 1996; 23:1968.
DP, Jones M, Osborne J, et al. Pediatric rheumatology in the United
Kingdom: data from the British Pediatric Rheumatology Group National
Diagnostic Register. J Rheumatol 1996;23:1975.
PN, Fung MY, Rosenberg AM, et al. The incidence of paediatric rheumatic
diseases: results from the Canadian Paediatric Rheumatology Association
Disease Registry. J Rheumatol 1996; 23:1981.
C, Van Kerckhove C, Giannini EH. The iridocyclitis of early onset
pauciarticular juvenile rheumatoid arthritis: outcome in
immunogenetically characterized patients. J Rheumatol 1992;19:160.
LA, Amos CI, Wade JA, et al. Investigating the genetic basis for
ankylosing spondylitis. Linkage studies with the major
histocompatibility complex region. Arthritis Rheum 1994;37:1212.
TL, Symons JA, Ploski R, et al. A genetic association between juvenile
rheumatoid arthritis and a novel interleukin1 alpha polymorphism. Arthritis Rheum 1995;38:221.
E, Yu DTY, Meyer KH, et al. HLA-B27-restricted CD8 T cells derived from
synovial fluids of patients with reactive arthritis and ankylosing
spondylitis. Lancet 1993;342:646.
G, Baltus CAM, Van Eijk HG, et al. Anaemia of chronic disease:
diagnostic significance of erythrocyte and serological parameters in
iron deficient rheumatoid arthritis. Hum Nutr Clin Nutr 1990;40C:57.
P, Swanljung K, Siimes MA. Ferritinemia as an indicator of systemic
disease activity in children with systemic juvenile rheumatoid
arthritis. Acta Paediatr Scand 1986;75:64.
BJ, Tucker LB, Miller LC, et al. Fibrin D-dimer as a marker of disease
activity in systemic onset juvenile rheumatoid arthritis. J Rheumatol 1998;25:1620.
UM, Rooney M, Preece MA, et al. Treatment of growth retardation in
juvenile chronic arthritis with recombinant human growth hormone. J Rheumatol 1994;21:153.
UM, Jones J, Reeve J, et al. Juvenile rheumatoid arthritis. Effects of
disease activity and recombinant human growth hormone on insulin-like
growth factor 1, insulin-like growth factor binding proteins 1 and 3,
and osteocalcin. Arthritis Rheum 1997;40:332.
A, De Benedetti F, Viola S, et al. Macrophage activation syndrome in
systemic juvenile rheumatoid arthritis successfully treated with
cyclosporine. J Pediatr 1996;128:275.
R, Stephan JL, Pillet P, et al. Efficacy of cyclosporine A in the
treatment of macrophage activation syndrome in juvenile arthritis:
report of five cases. J Pediatr 1996;129:750.
EC, Goldsmith DP, Koehler MA, et al. Prevalence and outcome of uveitis
in a regional cohort of patients with juvenile rheumatoid arthritis. J Rheumatol 1997;24:2031.
Academy of Pediatrics Section on Rheumatology and Section on
Ophthalmology. Guidelines for ophthalmologic examinations in children
with juvenile rheumatoid arthritis. Pediatrics 1993;92:295.
DA, Oen KG, Petty RE. SEA syndrome revisited: a long-term followup of
children with a syndrome of seronegative enthesopathy and arthropathy. J Rheumatol 1992;19:1282.
R, Vazquez-Mellado J. The early clinical recognition of juvenile-onset
ankylosing spondylitis and its differentiation from juvenile rheumatoid
arthritis. Arthritis Rheum 1995;38:835.
O, Maldonado-Cocco JA, Suarez-Almazor ME, et al. Ankylosing spondylitis
of juvenile onset: comparison with adult onset disease. Stand J Rheumatol 1983;12:246.
D, Giannini E, Brewer E. Time course of response to nonsteroidal
anti-inflammatory drugs in patients with juvenile rheumatoid arthritis.
Arthritis Rheum 1984;27:1433.
AE, Linz C, Bern E, et al. Identification of nonsteroidal
antiinflammatory drug-induced gastroduodenal injury in children with
juvenile rheumatoid arthritis. J Pediatr 1993;122:647.
HI, Tschammler A, Horwitz AE, et al. Intraarticular corticosteroids for
chronic arthritis in children: efficacy and effects on cartilage and
growth. J Pediatr 1995;127:317.
EH, Brewer EJ, Kuzmina N, et al. Methotrexate in resistant juvenile
rheumatoid arthritis. Results of the U.S.A.-U.S.S.R. double-blind,
placebo-controlled trial. N Engl J Med 1992;326:1043.
P, Wilkes H, Southwood T, et al. Low dose methotrexate is effective in
extended oligoarticular arthritis but not in systemic arthritis of
children. Arthritis Rheum 1997;40:S47.
A, Viola S, Ramenghi B, et al. Radiologic progression in patients with
juvenile chronic arthritis treated with methotrexate. J Pediatr 1998;133:262.
PJ, Balistreri WF, Bove KE, et al. The long-term effect of methotrexate
therapy on the liver in patients with juvenile rheumatoid arthritis. Arthritis Rheum 1997;40:2226.
S, Newman AJ, Dahms BB, et al. Liver biopsy findings in patients with
juvenile rheumatoid arthritis receiving long-term, weekly methotrexate
therapy. J Pediatr 1996;128:149.
PJ, Balistreri WF, Bove KE, et al. The relationship of hepatotoxic risk
factors and liver histology in methotrexate therapy for juvenile
rheumatoid arthritis. J Pediatr 1999;131:47.
JM, Alarcon GS, Lightfoot RW, et al. Methotrexate for rheumatoid
arthritis. Suggested guidelines for monitoring liver toxicity. Arthritis Rheum 1994;37:316.
Rossum MA, Fiselier TJ, Franssen MJ, et al. Sulfasalazine in the
treatment of juvenile chronic arthritis: a randomized, double-blind,
placebo-controlled, multicenter study. Dutch Juvenile Chronic Arthritis
Study Group. Arthritis Rheum 1998;41:808.
DJ, Giannini EH, Reiff A, et al. Long-term efficacy and safety of
etanercept in children with polyarticular-course juvenile rheumatoid
arthritis: interim results from an ongoing multicenter, open-label,
extended treatment trial. Arthritis Rheum 2003;48(1):218.
V, Lurati A, Gattinara M, et al. Efficacy of infusions of an anti TNFα
antibody (infliximab) in persistently active refractory juvenile
idiopathic arthritis. Results of a two years open label prospective
study. Arthritis Rheum 2003;48(9):S92.
U, Elborgh R, Ranstam J, et al. Synovectomy of the knee in juvenile
chronic arthritis. A retrospective consecutive follow-up study. J Bone Joint Surg Br 1986;68:223.
TK, Pahle JA, Hoyeraal HM. Comparison of synovectomy and no synovectomy
in patients with juvenile rheumatoid arthritis. A 24-month controlled
study. Scand J Rheumatol 1987;16:81.
E, Savolainen A, Kautiainen H, et al. Treatment of leg length
discrepancy with temporary epiphyseal stapling in children with
juvenile idiopathic arthritis during 1957-99. J Pediatr Orthop 2003;23(3):378.
MJ, Scott RD, Thomas WH, et al. Total hip arthroplasty with cement for
juvenile rheumatoid arthritis. Results at a minimum of ten years in
patients less than thirty years old. J Bone Joint Surg Am 1997;79:44.
AG, Martin S, Zurakowski D, et al. Bipolar hemiarthroplasty in juvenile
rheumatoid arthritis: long-term survivorship and outcomes. J Arthroplasty 2002;17(8):978.
CO, Belt EA, Hamalainen MM, et al. Survivorship of AGC knee replacement
in juvenile chronic arthritis: 13-year follow-up of 77 knees. J Arthroplasty 2000;15(2):166.