
affect musculoskeletal form and function. The hematopoietic system
consists of the cellular elements in circulating blood, bone marrow,
spleen, lymph nodes, and the reticuloendothelial system. This chapter
will discuss the diseases of the hematopoietic system that have
musculoskeletal features that a pediatric orthopaedist would be
expected to diagnose and treat. Such diseases can be divided into: (a)
disorders of the bone marrow, where most of the cells of this system
are produced; (b) disorders of erythrocytes, predominantly involving
abnormalities in hemoglobin synthesis; (c) disorders of neutrophils and
lymphocytes, with accompanying immune deficiencies; (d) disorders of
monocytes and macrophages, with abnormalities of metabolism and
proliferation; (e) disorders of hemostasis, causing abnormal bleeding
or thrombosis; and (f) hematologic malignancies. The orthopaedic
evaluation and management will be discussed for each disorder, along
with recent advances in pathophysiology, molecular genetics, treatment,
and prognosis.
characterized by deficient production of one or more cell lines in the
bone marrow. Disorders characterized by bone marrow failure can cause
anemia, thrombocytopenia, leukopenia, or pancytopenia, depending on
which hematopoietic precursors are affected and at what stage of stem
cell differentiation the abnormality occurs. There are five
well-described bone marrow failure disorders with musculoskeletal
manifestations: Fanconi anemia (FA), thrombocytopenia with absent
radius (TAR) syndrome, Diamond-Blackfan anemia (DBA),
Schwachman-Diamond syndrome (SDS), and cartilage-hair hypoplasia (CHH).
The orthopaedist should be familiar with these disorders, because the
musculoskeletal manifestations that can cause significant functional
disabilities and cosmetic problems may be their first clinically
apparent signs.
bone marrow failure, skeletal anomalies, and predisposition to
malignancies. The disorder is uncommon, with an incidence of less than
1 per 100,000 live births (1). The disorder affects both sexes equally (2), and shows no clear racial dependence (3).
As skeletal anomalies are the most obvious clinical manifestations of
FA apparent at birth, the orthopaedist may be the first physician
consulted. The hand and forearm are the sites most often affected, with
a variety of radial ray differences (4, 5, 6).
Thumb hypoplasia or absence is common, although thumb duplication may
also be seen. The radius is typically either hypoplastic or absent.
Although radial ray differences are a common manifestation of FA, they
are not pathognomonic (7). Nonetheless, any
child with a radial ray deficiency should be referred to genetic
specialists and/or hematologists for evaluation of the possibility of
an underlying hematologic problem. Because 30% to 40% of patients with
FA have no clinically apparent anomalies at birth, the diagnosis may be
delayed (4,5).
Other skeletal anomalies have been reported in patients with FA, but
not often enough to be considered an integral feature of the disorder.
Patients tend to have a variety of facial abnormalities, although no
pathognomonic facies has been described. Skin pigmentation anomalies,
including café-au-lait spots, are common. As the child grows, growth retardation becomes apparent in approximately 80% of patients (4), often associated with endocrinopathies (8).
Renal anomalies are present in approximately one-third of patients with
FA, although the exact prevalence is likely underestimated because not
all patients in large series have undergone renal diagnostic imaging (9).
Cardiovascular and gastrointestinal anomalies can be found in 15% to
30% of patients. The central nervous system is often affected, with
microcephaly, hearing loss, eye anomalies, and mental retardation
apparent in up to 37% of patients (4).
Thrombocytopenia, anemia, and neutropenia can be profound. The
pancytopenia typical of FA does not typically appear until age 7 or 8
years (10), although it can occur at any age (2).
Patients with FA have a heightened susceptibility to DNA breakage,
predisposing them to malignancies. The incidence of solid tumors and
leukemia in patients with FA is estimated, respectively, at 48 and 785
times that in the general public (11). In a recent review of 1300 cases of FA reported in the literature (12),
24% of patients had at least one malignancy, and in one-fourth of these
patients the finding of malignancy preceded the diagnosis of FA. Over
the past decade, eight genes have been implicated in the pathogenesis
of FA and seven have been cloned (1). Although
FA is caused by a wide variety of mutations in any one of these eight
genes, all result in an impairment of repair of DNA breakage. The
diagnosis of FA is made, therefore, by detecting increased chromosomal
breakage in response to in vitro exposure of cells to DNA cross-linking agents (13).
covered elsewhere in this text. Hematologic treatment of FA is directed
primarily at the consequences of bone marrow failure. Medical
treatments include combinations of androgens and corticosteroids (3), as well as hematopoietic growth factors (14). However, bone marrow transplantation remains the most effective treatment for bone marrow failure in patients with FA (15),
despite the genotoxic effects of pretransplant bone marrow irradiation
and chemotherapeutic agents in these patients whose DNA repair
mechanisms are impaired. In a mouse model of FA, gene transfer has been
used to successfully reverse the susceptibility of DNA to breakage (16).
rare autosomal recessive disorder characterized by marked
thrombocytopenia and absent radii. The radial absence is complete and
almost always bilateral (17). The radial club
hands seen in TAR syndrome differ from those in FA in that the thumbs
are present in TAR syndrome, but are absent in FA if the radii are
absent (Fig. 11.1). See Table 11.1
for a comparison of FA and TAR syndrome. The thumbs are hypoplastic in
about half of patients with TAR syndrome. Thumb and finger function is
impaired to varying degrees, and wrist function is abnormal owing to
the lack of radius and abnormal carpal bones and musculature.
Typically, the ulna is hypoplastic and bowed, as in other causes of
radial club hand. In approximately 40% of the patients the humerus is
hypoplastic or absent, and the shoulder girdle is abnormal in one third
of the patients (18). Major intercalary transverse deficiencies may exist, with hands arising directly from the shoulder girdle (18,19).
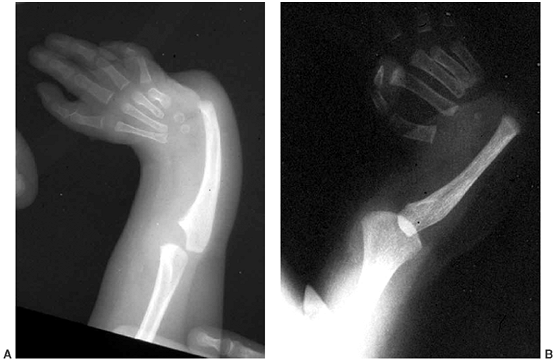 |
|
Figure 11.1 Forearm and hand radiographs in (A) Fanconi anemia and (B)
thrombocytopenia with absent radius (TAR) syndrome. Note the absence of a thumb in the child with Fanconi anemia, as is the case when the radius is completely absent. However, note the presence of the thumb in the child with TAR syndrome, despite complete absence of the radius. |
syndrome, despite the name of the disorder. In the original description
of 40 patients by Hall et al. (18), 40% had lower extremity deformities. More recent series have estimated an 80% prevalence of lower extremity deformities (20,21).
The severity of upper extremity involvement seems to correlate with the
presence and severity of lower extremity involvement (19).
As with the fingers, all five toes are typically preserved, even with
fibular hemimelia or other lower extremity deformities (19). Knee abnormalities have been studied in detail in 21 patients by Schoenecker et al. (20).
Genu varum was present in 18 patients and was apparent at birth in 12
of them. The genu varum was usually associated with varus laxity of the
knee joint rather than a fixed bony deformity. Intraarticular pathology
was found to be extensive at surgery in six patients, including
concave
medial femoral condyles. Internal rotation of the tibia was also
common. Patellar anomalies included hypoplasia, instability, and total
absence. The deformities tended to progress as the children grew,
requiring bracing. Lower extremity deformities usually recurred
following corrective osteotomies, although some have reported success
in correcting a fixed knee deformity with osteotomy (22). Anomalies of other systems, including cardiac, neurologic, and genitourinary, are reported in one third of patients (18).
|
TABLE 11.1 CLINICAL FEATURES OF FANCONI ANEMIA AND THROMBOCYTOPENIA WITH ABSENT RADIUS (TAR) SYNDROME
|
||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
profound thrombocytopenia that can cause serious bleeding within the
first few months of life (18). Viral illnesses
can exacerbate the thrombocytopenia, and patients should be kept
relatively isolated in the early months to avoid undue viral
challenges. Routine platelet transfusions are required in the first
year to prevent potentially fatal hemorrhage. Surgery should be avoided
in the first year, unless it is critical to the survival of the
patient. The radial club hands should be splinted to minimize the
progression of radial deviation that occurs as a result of the abnormal
line of pull of forearm muscles. After the first year of life, the
platelet count starts to rise spontaneously, so that for patients who
survive the first year, the musculoskeletal anomalies become the major
problems. Wrist centralization procedures, followed later by
pollicization of the index finger, if necessary for a hypoplastic or
dysfunctional thumb, can be performed once the platelet count has
improved. Bone marrow transplant is rarely indicated, as most patients
resume platelet production spontaneously (23).
only the erythropoietic cell line and associated with hand and other
skeletal anomalies. Approximately 4 in 1 million live births are
affected by DBA (24). Typically, the disorder occurs sporadically, but various forms of inheritance have been reported in 12% to 25% of cases (10,25). Approximately 30% of patients have associated skeletal anomalies (10). Among upper limb differences, triphalangeal or hypoplastic thumbs are the most common features (10). Radial hypoplasia is uncommon in DBA (26). Patients with DBA also have urogenital anomalies, and cardiac anomalies such as atrial or ventricular septal defects.
the signs of anemia do not usually develop until later in infancy.
Pallor and irritability develop slowly as red cell stores are depleted.
The anemia is normocytic but severe, with hemoglobin levels less than 4
g per dL. The exact cause of DBA is unknown, and the features of the
disorder are highly variable. A predisposition to malignancies may
exist in patients with DBA, but as no known impairment exists in DNA
repair mechanisms, the predisposition is slight compared to that of FA.
For those who do not respond to cortico-steroids, treatment options
include hematopoietic growth factors such as erythropoietin,
interleukin-3, and stem cell factor, cyclosporin A, or metaclopromide.
Transfusions of packed red blood cells (pRBCs) are sometimes required
to maintain adequate hemoglobin to sustain growth, but chelation is
also required to prevent iron overload. Bone marrow transplantation
from human leukocyte antigen (HLA)-identical donors has been used with
a 72% 2-year survival (28). Gene therapy has met with some clinical success in a small number of patients (29).
pancreatic insufficiency, bone marrow hypoplasia, metaphyseal
dyschondroplasia, and growth retardation. Patients can appear normal at
birth, but typically have low birth weights (30).
The first signs of the disorder are attributable to malabsorption,
including failure to thrive, growth retardation, and steatorrhea.
Severe respiratory infections also occur in the first year of life. Few
patients have an uneventful neonatal period.
because of skeletal abnormalities contributing to delayed growth and
deformity. Metaphyseal chondrodysplasia occurs in approximately 62% of
patients, usually in the proximal femur (30). This lesion in the proximal femur can cause coxa vara, coxa magna, pathologic femoral neck fracture, or pseudoarthrosis (30,31). Other common sites for chondrodysplasia include the knees, wrists, spine, and ribs (30,31).
Spinal deformity can include kyphosis and scoliosis. Ribs are typically
short and anteriorly flared. Long bone bowing is a common finding, and
can recur following osteotomies. Clinodactyly was reported in almost
half of the patients in one series (30).
Laboratory studies reveal that the pancreatic insufficiency gives rise
to enzymatic abnormalities, including the absence of trypsin, lipase,
and amylase in the stool. SDS can be differentiated from cystic
fibrosis by a normal sweat chloride test. Hepatic, respiratory, and
renal dysfunction also occur. Neurologic development is usually delayed.
half of the patients with SDS will live to age of 35 years, but
survival is reduced to 24 years for patients with pancytopenia and to
10 years for patients with leukemia. Early treatment includes oral
administration of pancreatic enzymes and aggressive antibiotic
treatment of infections.
The bone marrow failure may respond to growth factors and androgens, although these treatments are only temporarily effective (23). Bone marrow transplantation has been reported in seven patients, with survival in four (23,33).
by disproportionate, short-limbed dwarfism, thin, sparse hair, and
cellular immunodeficiency. The skeletal manifestations of this
condition are covered in detail in chapter 8, so only the hematologic aspects will be covered here.
immunodeficiency and anemia. The degree of immunodeficiency varies
greatly (34). Recurrent respiratory tract
infections may occur in these patients, and serious illness can result
from vaccinations with live viruses. The immunodeficiency is usually
due to diminished numbers of T lymphocytes. The anemia of CHH is an
integral feature, occurring in 73% of patients (35). Anemia may be severe in infancy, but tends to improve with growth (35).
infection and have only mild anemia, often no treatment is required.
For more severe immunodeficiency with anemia, bone marrow
transplantation may be needed. Bone marrow transplantation can correct
the immunodeficiency but not the chondrodysplasia (36).
tissues. Hemoglobin, a four-chain protein combined with iron, carries
the oxygen in erythrocytes and is vital to erythrocyte function.
Disorders of erythrocytes often involve disorders of hemoglobin
synthesis. Mutations in the genes that encode the protein chains can
cause abnormal hemoglobin molecules that affect the form and function
of erythrocytes. Iron deficiency and chronic inflammation can cause
diminished production of hemoglobin, resulting in anemia. Disorders in
number, form, or function of erythrocytes can cause significant
musculoskeletal pathology, and can complicate the treatment of other
musculoskeletal conditions.
hemoglobin synthesis. The protein component of hemoglobin is composed
of four globin chains: two α-globin chains and two β-globin chains.
Hemoglobin S refers to hemoglobin containing abnormal β-globin produced
by a single base change mutation (GAT to GTT) in the sixth codon of
exon 1 in the β-globin gene on chromosome 11 (37).
The resulting glutamic acid-to-valine substitution in the β-globin
chain predisposes the hemoglobin molecule to polymerization upon
deoxygenation, causing distortion or “sickling” of the erythrocytes
that contain the abnormal hemoglobin. Hemoglobin C contains β-globin
chains with a glutamic acid-to-lysine substitution at the same position.
genotype of the β-globin gene and the resulting proportion of
hemoglobin S. (a) SS disease results from homozygous inheritance of the
hemoglobin S mutation. All hemoglobin is hemoglobin S and the sickling
is severe. (b) SC disease results from inheritance of one hemoglobin S
allele and one hemoglobin C allele. No normal hemoglobin is produced,
but the tendency to sickle is tempered by the presence of hemoglobin C.
(c) Sβ0 disease results from inheritance of one hemoglobin S
allele and an allele with a β-thalassemia mutation that causes slightly
reduced β-globin synthesis. Some normal β-globin is produced, and
sickling is less severe. (d) Sβ0 disease results from
inheritance of one hemoglobin S allele and an allele with a
β-thalassemia mutation that causes greatly reduced β-globin synthesis.
Very little normal β-globin is produced, leading to a preponderance of
hemoglobin S and severe sickling.
malaria, explaining the persistently high prevalence of the gene in
areas of the world where malaria is endemic, such as sub-Saharan
Africa. In the United States, SCD affects 1 in 300 to 1 in 600 African
Americans (38,39). Today, all 50 states in the US screen for SCD in newborns (40). Screening allows early diagnosis and prompt treatment, which can improve the clinical course (41).
several factors cause sickle cells to occlude the microvasculature,
including abnormal cell shape, cellular dehydration, and increased
cellular adhesion to vascular endothelium. The molecular components of
abnormal erythrocyte adhesion to vascular endothelium are the targets
of new drug treatment strategies (43).
Newborns with SCD are normal in height and weight, and have no clinical
signs of the disease at birth. Growth and development are typically
delayed, however, especially in terms of weight and sexual maturation (44,45). The growth disturbance has been attributed to hematologic, hormonal, and physeal abnormalities (46). By the time growth ceases, normal height is usually reached (45).
radiography include osteopenia, biconcave vertebrae, and medullary
expansion and cortical thinning due to marrow hyperplasia (47) (Fig. 11.2). Vertebral collapse has been reported in two children (48). Osteopenia, although rarely a clinical problem (49,50), can be detected by calcaneal ultrasound and serum markers of bone turnover (51). Multifocal
pseudoaneurysmal bone cysts have also been found in one patient (52).
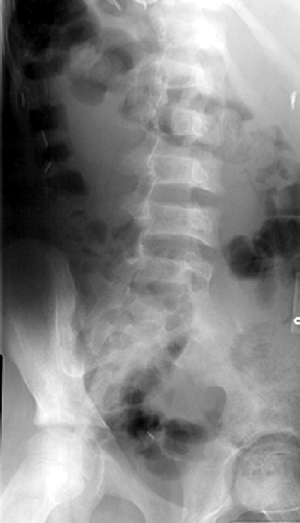 |
|
Figure 11.2 Oblique radiograph of the spine in a patient with sickle cell disease. Note biconcave vertebral bodies.
|
extremities and the back, are common manifestations of SCD. Dactylitis,
a painful swelling of fingers or toes, occurs in early childhood and is
often the first clinical manifestation of SCD. One study found a 45%
incidence of dactylitis, with 41% of affected patients experiencing
recurrent episodes until the age of 4 years (53).
No cases have been reported in children older than 6 years. The rarity
of dactylitis in older children is thought to result from a shift in
hematopoiesis from distal sites such as fingers and toes in infants to
more central sites in older children (40).
Dactylitis presents as an acutely painful and swollen digit.
Radiographs are initially normal, but may progress to demonstrate
periosteal elevation and osteolysis, mimicking osteomyelitis. Cultures
of bone aspirates can help in making a diagnosis by differentiating
between the two disorders. The pain associated with dactylitis is often
mild and is relieved by nonsteroidal antiinflammatory drugs (NSAIDs) in
infants, but can be severe in older children.
disease. Pain crises are likely underestimated by studies that use
hospital admission as a diagnostic criterion, because many painful
episodes can be treated at home. A recent long-term maintenance of pain
diaries for 39 children and adolescents showed that pain associated
with SCD occurred on 8.4% of the days covered, requiring pain
medication in 85% of the episodes (55).
In that series, 66% of the spine crises were lumbosacral, 22% were
thoracic, and 12% cervical. Patients presenting with pain crises rarely
have striking findings on physical examination. Swelling and decreased
range of motion are usually not present. Fever is variably present.
Peripheral leukocytosis and erythrocyte morphology on peripheral blood
smears have no diagnostic use in a pain crisis (60).
Analgesia is the cornerstone of treatment for a pain crisis. A stepwise
approach to analgesia is recommended, but around-the-clock opioid
analgesics are usually required ultimately. Hydration is an important
adjunct to analgesics. Oxygen supplementation has no proven benefit in
a patient who is not hypoxic (40). In a
randomized double-blind trial, it has recently been found that inhaled
nitric oxide lowers pain and reduces morphine requirements in children
with pain crises (61). Screening and treatment for nocturnal hypoxia may alleviate painful crises in children (62).
difficult to differentiate from a pain crisis. Osteomyelitis is much
less common than pain crises, with an incidence of 1.6% (63). Osteomyelitis in SCD is commonly caused by Salmonella species and Staphylococcus aureus (64, 65, 66).
As microvascular occlusion in the spleen causes repeated splenic
infarcts, patients become functionally asplenic and susceptible to
infections with encapsulated bacteria such as Streptococcus pneumonia, Salmonella, and Haemophilus (67, 68, 69). Intestinal infarcts with translocation of gut bacteria are thought to be responsible for the high rate of infection from Salmonella
and other enteric bacteria. Prompt recognition and treatment of
osteomyelitis is important. However, it is difficult to differentiate
between painful bone infarcts and osteomyelitis, and therefore the
diagnosis of osteomyelitis is often delayed (70, 71, 72, 73).
The history and physical examination findings are similar in the two
conditions, and also laboratory values such as white blood cell count,
erythrocyte sedimentation rate, and C-reactive protein. Detection of
subperiosteal fluid by ultrasonography has been reported to have 74%
sensitivity and 63% specificity for osteomyelitis in this population (74).
Others recommend supplementing ultrasound identification of a
subperiosteal fluid collection with aspiration and culture of the
collection (75). Conventional magnetic resonance
imaging (MRI) cannot differentiate between the two conditions (76).
However, in a small series, gadolinium-enhanced MRI was able to
differentiate between osteomyelitis and infarcts on the basis of the
pattern of enhancement (77). Infarcts showed as
enhancement in a ring at the periphery of the lesion, whereas
osteomyelitis showed enhancement irregularly throughout the lesion. The
specificity of this modality has yet to be tested. Technetium 99m
methylene disphosphonate bone scans can show areas of increased uptake
even in the absence of symptoms (78).
Technetium 99m sulfur colloid bone marrow scan followed by technetium
99m methylene disphosphonate bone scan has been reported to
differentiate between the two conditions (79),
but without proven consistency. Gallium scan, cultures of biopsies or
aspirated fluid, and clinical response to antibiotic treatment are the
other ways of confirming a suspected diagnosis of osteomyelitis.
tool for differentiating between painful crisis and osteomyelitis in
patients with SCD. In a painful crisis, symptoms should abate within 24
to 48 hours with hydration. Failure of symptomatic improvement, with
clinical examination and laboratory values consistent with
osteomyelitis, calls for evaluation with MRI. In the setting of a
clinical course typical for osteomyelitis, MRI evidence of
intraosseous, subperiosteal, or soft tissue fluid collection warrants
surgical drainage (Fig. 11.3). In some cases,
physical examination alone can detect a soft tissue abscess, obviating
the need for MRI, and therefore sedation, in a young child (Fig. 11.4).
In our experience, radionuclide imaging is time consuming and often
equivocal, and exposes the child to a large radiation dose. For
established or probable osteomyelitis, intravenous antibiotics are
given according to the recommendation of the infectious disease
consultants and on the basis of culture results when available. In
patients with SCD, chloramphenicol or ampicillin provides good coverage
against Salmonella, although resistant strains may require treatment with newer Sβ-lactam agents (80,81). In the absence of a fluid collection, treatment of osteomyelitis with antibiotics alone may be effective in some instances.
The cause is unknown. Patients typically present with acute pain and
swelling in one or more joints, usually the knees and elbows. Leukocyte
counts in the joint fluid are usually less than 20,000 per mm3
thereby differentiating this condition from septic arthritis. With
splinting and analgesics, symptoms generally resolve gradually over a
period of weeks to months.
Osteonecrosis of the femoral head is slightly more common than that of
the humeral head. The development of osteonecrosis is related to age
and genotype (85,86) (Table 11.2).
By age 45, nearly one third of patients have femoral head osteonecrosis
and nearly one fourth have humeral head osteonecrosis. Femoral head
osteonecrosis is bilateral in 54% of the patients, and humeral head
osteonecrosis is bilateral in 67% of the patients. Concomitant femoral
and humeral head osteonecrosis occurs in three fourths of the patients.
The genotype affects the prevalence of osteonecrosis. As with other
manifestations of the disease, patients with SS or Sβ0 disease have a higher incidence of osteonecrosis than do those with SC or Sβ+ disease (85,86).
shoulders of children. Abnormalities may show up on radiographs several
years before symptoms appear (85). The age at onset of osteonecrosis of the femoral head has been reported to correlate with outcome (87).
Osteonecrosis developing prior to 10 years of age led to a low Harris
hip score in 5 out of 14 hips studied, compared to 51 of 81 hips in
which the osteonecrosis developed between 10 and 14 years. Although
this difference is not statistically significant, it demonstrates a
trend that may point to an increased ability in younger children to
revascularize and heal osteonecrotic lesions. Plain radiographs and MRI
are used for evaluating osteonecrosis. MRI can delineate the extent and
stages of involvement (88,89). Radiographs show structural abnormalities well.
patients with SCD roughly parallels that in patients without SCD, as
covered elsewhere in this text. Containment and range of motion may be
sufficient in young children with limited involvement of the femoral
head. Core decompression has been used successfully in some early cases
(90,91). Older children or those with total involvement of the femoral head may benefit from femoral osteotomies (92). Several series have shown a high complication rate following total joint arthroplasty in patients with SCD (93, 94, 95, 96, 97).
by adequate hydration, maintenance of blood volume and oxygenation, and
prevention of hypothermia. The use of a tourniquet is allowed, as it
does not induce sickling (98). Transfusions are typically given perioperatively to keep the total hemoglobin greater than 10 g per dL (99).
found that delayed union, malunion, and joint stiffness complicate 10%
to 15% of fractures. However, fractures are not a prominent feature of
SCD.
SCD is related to erythrocyte fragility and hemolysis. The chronic
baseline anemia is generally mild and well tolerated in childhood.
However, anemia can be worsened acutely by splenic sequestration, a
sudden increase in
splenic
hemolysis that can be fatal, and by aplastic anemia, a temporary marrow
suppression triggered by parvovirus B19 infection. Acute chest syndrome
(ACS) refers to any new pulmonary infiltrate seen on a chest radiograph
in conjunction with fever and chest pain or respiratory symptoms (101). ACS can result form a wide variety of infectious or noninfectious causes, including rib infarcts (102),
and can be fatal. SCD also causes genitourinary problems, including
enuresis and priapism. Cholelithiasis is common in patients with SCD
because of ongoing hemolysis and buildup of bilirubin. Stroke is a
common and potentially devastating result of cerebral vasoocclusion or
hemorrhage. Even with appropriate, continuous treatment, 10% to 12% of
children suffer a symptomatic stroke and an additional 20% show
evidence of infarcts on MRIs of the brain (103,104).
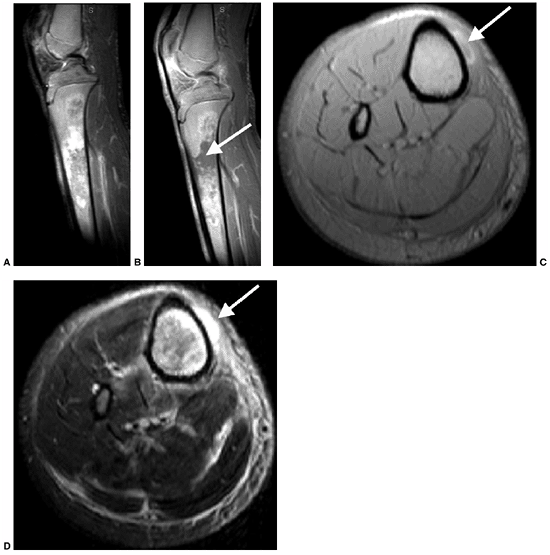 |
|
Figure 11.3
A 17-year-old boy with sickle cell disease presented with symptoms of a painful crisis in his leg. Plain radiographs of his tibia revealed no abnormalities. Failure to respond to hydration after 2 days, along with elevated peripheral white blood cell count and C-reactive protein, prompted investigation with magnetic resonance imaging. Sagittal T1-weighted images before (A) and after (B) gadolinium injection demonstrate heterogeneous enhancement throughout a large area of abnormal signal intensity in the marrow of the tibia. An intraosseous fluid collection can be seen (arrow). Axial T1-weighted (C) and T2-weighted (D) images without gadolinium demonstrate an extraosseous fluid collection (arrows) with surrounding edema. Operative corticotomy yielded purulent material. |
childhood. Sepsis used to be a major cause of mortality in this age
group. The widespread use of penicillin prophylaxis in children younger
than 5 years reduces the incidence of pneumococcal bacteremia by 84% (105).
The development of penicillin-resistant strains of pneumococcus has
prompted the use of vaccinations against pneumococcus, which can
further lower the rate of invasive infection by 80% (106).
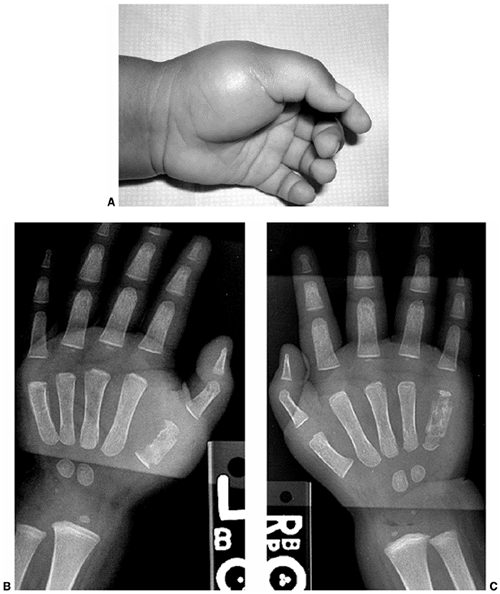 |
|
Figure 11.4 A:
A 16-month-old boy presented with a 3-week history of pain and swelling in all digits of both hands. He was treated initially as having dactylitis, and all but his left thumb swelling resolved. Physical examination revealed a fluctuant mass in the thenar eminence. Plain radiographs (B and C) revealed bone destruction in the left thumb metacarpal, as well as in both small finger metacarpals. Operative drainage of the abscess and corticotomy of the thumb metacarpal were performed. Cultures grew Escherchia coli. Four weeks of intravenous antibiotic therapy led to the resolution of symptoms and ultimate normalization of radiographs. |
recent decades. Hydroxyurea, a chemotherapeutic agent, causes increased
formation of hemoglobin F and has been found to reduce the incidence of
painful crises and ACS, as well as to reduce the requirement for
transfusion in adults (107). Several studies
have proven similar efficacy of this drug in children as young as 2
years, although the US Food and Drug Administration (FDA) has not yet
approved this drug for use in children. Many other drugs are currently
under investigation. Bone marrow transplantation has been used in
approximately 150 children with severe SCD worldwide, with 92% to 94%
survival and 75% to 84% event-free survival (108). Despite successful reversal of phenotypic SCD in transgenic mice treated with gene replacement (109), gene therapy has not yet become a reality in humans.
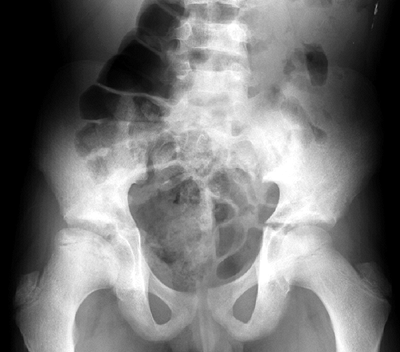 |
|
Figure 11.5
Femoral head osteonecrosis in a patient with sickle cell disease. Note the flattening of the head but congruous acetabulum. Note also the biconcave vertebrae typical of sickle cell disease. |
recessive inherited disorders of hemoglobin synthesis. Together, they
represent the most common inherited diseases worldwide (110).
The diseases and their treatments can cause an array of alterations in
skeletal dynamics that the orthopaedist should be able to recognize.
thalassemia cause deficient or nil production of either α or β-globin
chains. Alpha thalassemia results from mutations in one or more of the
four copies of the β-globin gene. One mutation results in a silent
carrier state. Mutation of two genes causes a thalassemia trait,
characterized by mild normocytic or microcytic anemia. Mutation of
three genes causes substantially diminished α-globin production and
hemoglobin H disease (named for the stable tetramer formed by the
remaining β chains) with moderate hemolytic anemia. Mutation of all
four α-globin genes causes hydrops fetalis, which is usually fatal in utero. Beta thalassemia results from mutations in the β-globin gene and is classified as (a) Sβ0 thalassemia, with reduced synthesis of β-globin or (b) β0
thalassemia, with absent β-globin synthesis. An alternate
classification of thalassemia is based entirely on clinical severity. Thalassemia major refers to severe disease, thalassemia intermedia refers to moderate disease, and thalassemia minor refers to mild disease.
most seen clinically. Children generally present with moderately severe
anemia, splenomegaly, and cholelithiasis, which may occur in response
to oxidative stress caused by infections, fever, or certain medications
(111). Patients with thalassemia major
(homozygous β thalassemia) develop severe anemia, with hemoglobin in
the 3 to 4 g per dL range within the first 6 months of life, as fetal
hemoglobin production wanes. Thalassemia major requires frequent
transfusions in order to maintain health and prolong life expectancy
beyond 5 years of age. Transfusions are generally started when the
anemia becomes clinically detrimental, and are aimed at keeping
hemoglobin levels more than 9.5 to 10.5 g per dL. Thalassemia
intermedia typically presents in the second year of life with a less
profound anemia (110).
may occur as a result of both the anemia and its treatments, include
marrow hyperplasia, short stature, skeletal dysplasia, and osteopenia.
Without transfusions to correct the severe anemia in thalassemia major,
erythropoietin secretion increases. The resulting marrow hyperplasia
causes widening of the medullary cavities and thinning of the cortices
of long bones (Fig. 11.6). This process is
initially apparent in the hands and feet, where the tubular bones
become rectangular and then convex. Premature closure of physes,
especially in the proximal humerus, can also occur (112). Marrow hyperplasia can cause dramatic expansion of calvarial bones (113).
Marrow hyperplasia in the spine is associated with back pain in adults
with thalassemia who started transfusions after 3 years of age (114).
Extramedullary hematopoiesis commonly occurs in the liver, spleen, and
chest. Extramedullary hematopoiesis in the paravertebral space can
cause spinal cord compression (115, 116, 117, 118).
MRI is helpful in detecting and evaluating this process in the spine.
Surgical decompression, radiation therapy, and transfusions are
treatment options. Marrow hyperplasia from severe anemia is not often
seen today, because of the use of maintenance transfusions.
transfusion-induced iron overload on the anterior pituitary gland and
hypothalamus. Endocrinopathies resulting from iron overload include
decreased growth hormone (GH) release or GH resistance (119), delayed puberty and hypogonadism (120), and hypoparathyroidism. In one series of transfusion-dependent patients with thalassemia major (121),
8% of boys aged 7 to 8 years had short stature (<third percentile),
as well as 12% and 15% of older boys and girls, respectively. The short
stature tends to be disproportionate, with a relatively short trunk (122). The correction of GH deficiency and induction of puberty with gonadotropins partially correct this growth disturbance (120,122).
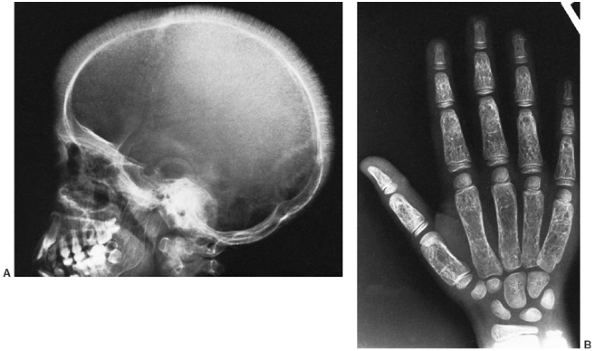 |
|
Figure 11.6 A: Lateral radiograph of the skull in a 11-year-old boy with thalassemia major. Note radial striations in the calvarium. B: Radiograph of the hand of the same patient. Note widened marrow cavities, thinned cortices, and osteoporosis.
|
chelation treatment. Iron chelation with desferrioxamine to prevent
iron overload has dramatically impacted the health status of patients
who require transfusions for thalassemia major (123,124).
Desferrioxamine, although essential in prolonging survival among
transfusion-dependent patients, causes significant skeletal dysplasia
in approximately 50% of cases (125). The
findings were a slowing of spinal growth, biconcave vertebrae that
progress to platyspondyly in some cases, and physeal widening at the
wrist and knee that, in some patients, were severe enough to resemble
rickets. Biopsies from patients with desferrioxamine-induced dysplasia
show reduced and irregular bone mineralization as well as significant
alterations in cartilage histology (126,127).
The spinal deformities are typically progressive, but metaphyseal
lesions may heal with reduction of the desferrioxamine dose or
following a switch to other iron chelators (128,129).
thalassemia major, occurring in more than 90% of patients despite
optimal transfusion and chelation (130). Bone mineral density is lower in patients who have delayed puberty or amenorrhea (131,132),
indicating a possible role for endocrinopathy in the pathogenesis of
osteopenia. Decreased bone density in patients with thalassemia is
predominantly trabecular and associated with iron deposition (133). Consistent biochemical alterations in bone turnover have not been found (134). In patients with impaired sexual maturation, bone mineral density increases in response to hormone replacement therapy (135). In GH-deficient patients, GH administration can normalize markers of bone turnover but does not increase bone density (136). Bisphosphonates were ineffective in increasing bone mineral density in two placebo-controlled trials (137,138).
although they occur less often since the widespread use of young-onset
transfusions began. The 40% to 50% incidence of fractures reported in
some early series (139, 140, 141) has decreased to 13% to 21% in recent series (142, 143, 144). However, a multicenter review (143)
found fractures to be often multiple or recurrent. The orthopaedist
treating a fracture in a child with thalassemia should consider the
problems of multiple fractures, weakened bone, high refracture risk,
and clinically significant anemia.
have led to a search for alternative medical treatments for the
thalassemias. Hydroxyurea, which stimulates hemoglobin F production,
may prove effective (145), although at the time
of writing this chapter, it has not been approved by the US FDA for
children with thalassemia. Bone marrow transplantation has been used
successfully in several centers for the treatment of severe thalassemia
(146, 147, 148), but it has not been shown to prevent osteopenia (131). Stem cell transplantation from umbilical cord blood of related donors has also been used with some success (149). Despite success in a mouse model (150), gene therapy for thalassemia is not yet a clinical reality.
aware of the health concerns relating to childhood iron-deficiency
anemia (IDA). The disease is preventable, recognizable, and treatable.
Iron deficiency, the world’s most common nutritional deficiency,
affects over 2 billion people (151). In developing countries, almost 50% of the children are anemic (152).
In the United States, approximately 5% of the children younger than 11
years meet the criteria for IDA, according the Centers for Disease
Control and Prevention (153). The highest
prevalence of IDA during childhood is in infants and adolescents, and
most cases of severe IDA occur in the first 2 years of life (154).
IDA causes typical signs of anemia, such as lethargy, failure to
thrive, and listlessness. In infancy, IDA has a significant impact on
neurologic function and development (155,156).
Iron deficiency may also predispose infants to febrile seizures, lead
poisoning, thrombocytopenia, neutrophil abnormalities, and renal
dysfunction.
abnormality in hemoglobin levels. Hemoglobin values obtained in
preprocedure screening or in the evaluation of illness should be
further evaluated if they fall to less than 11 g per dL in infants or
less than 12 g per dL in adolescents. Any concern should call for
communication with the patient’s pediatrician. A complete discussion of
current recommendations for prevention, diagnosis, and treatment of IDA
are beyond the scope of this chapter and are the responsibility of the
child’s pediatrician. However, because poor follow-up is a common
problem in IDA treatment programs (157,158),
the greater the number of physicians participating in the monitoring
and reinforcement of therapeutic strategies, the more effective the
treatment may be.
caused by either trauma or surgery. Because the total blood volumes in
children are so small, a large percentage of blood volume can be lost
very quickly even with a seemingly small amount of bleeding. Massive
blood loss is defined as the loss of 50% of blood volume within 3 hours
or the loss of 1.5 mL of blood per kg of body weight within 20 minutes (159).
Children, especially neonates and infants, are less tolerant of massive
blood loss than adults are, partly because of their limited
cardiovascular compensatory capacity (160).
Whereas a healthy adult may be able to tolerate a hematocrit of 21%, an
infant may have diminished tissue oxygen delivery at a hematocrit level
less than 24%.
transfuse following major blood loss is often made by critical care
specialists, anesthesiologists, or traumatologists. Nevertheless,
orthopaedists should be aware of the guidelines that govern transfusion
in this setting. In addition, many orthopaedic surgeries are associated
with significant blood loss, especially spinal fusion in neuromuscular
scoliosis, where over 100% of total blood volume can be lost and
replaced intraoperatively (161).
children vary considerably, depending upon the clinical scenario.
General transfusion guidelines in use at our institution have recently
been published (162), but these should not
override the actual clinical indications. Acute traumatic blood loss
with hypovolemia that does not respond to other measures warrants pRBC
transfusion. Preoperative pRBC transfusions are given to children with
clinically significant anemia who require emergent procedures. In
general, intraoperative pRBC transfusions are
begun
after a blood loss of greater than or equal to 15% of the patient’s
blood volume. Postoperative pRBC transfusions are given for a
hematocrit level of less than 24% with signs and symptoms of anemia,
including tachycardia, tachypnea, hypotension, lethargy, and poor
appetite among others. Transfusion of other blood products will be
discussed in detail later in the section on dilutional coagulopathy.
Transfusion guidelines are different for children younger than 4 months
than for older children, and different also for children with
cardiopulmonary, hematologic, or systemic illness. Other clinical
situations will exist, and transfusion decisions at our institution are
often made with the guidance of the transfusion service personnel.
surgery, and should be considered in children over 25 kg with a
hemoglobin concentration greater than 11 g per dL and no
cardiopulmonary problems (163). No more than 12% of the patient’s total volume of blood should be withdrawn at a time.
orthopaedist may treat are associated with anemia. These include
chronic infections, inflammatory diseases, and malignancies. The
pathogenesis of anemia in these conditions appears to be mediated by
cytokines that are involved in inflammation and immune responses (164). Because of the commonality of inflammatory mediators, the anemia has been termed anemia of chronic inflammation, although some experts still refer to it as anemia of chronic disease (165).
peripheral blood smear resemble those of iron-deficiency anemia. The
anemia is typically mild, with normocytic or microcytic erythrocytes.
However, elevated plasma ferritin level with low transferrin saturation
seen in anemia of chronic disease differentiates it from IDA, in which
both these markers are low. Nonetheless, IDA and systemic inflammatory
conditions can coexist.
of iron despite the low hemoglobin levels. This is thought to be the
result of cytokine inhibition of iron mobilization (164).
Hepcidin, a recently identified molecule that plays a role in the
immune response and regulates release of iron from cellular stores, may
play a central role in anemia of chronic disease (166). Another prominent feature of anemia of chronic disease is a blunted erythropoietin response (167).
For any given drop in the level of hemoglobin, the increase in
erythropoietin secretion is less than expected, and the bone marrow
response to erythropoietin is further blunted by cytokines.
treatment of the underlying pathologic process by attempting to
decrease the circulating cytokines. In addition, despite a blunted
response to endogenous erythropoietin, exogenous human recombinant
erythropoietin has been used with success in both adults and children
with this condition (168). Orthopaedists who
are involved in the treatment of children with chronic musculoskeletal
infections, inflammatory arthritides, or malignancies must be able to
recognize the often-associated anemia. Anemia that persists despite
optimal treatment of the underlying condition should prompt a
consultation with a hematologist.
include neutrophils, lymphocytes, monocytes, and macrophages.
Neutrophils serve as a first line of defense against bacterial and
fungal diseases. Neutrophils circulate in the peripheral blood and,
through a complex chemotactic process, migrate to sites of infection
where they recognize, phagocytose, and kill pathogenic microorganisms.
Lymphocytes are classified as B cells derived from bone marrow and T
cells derived from the thymus. B cells control humoral immunity and T
cells control cellular immunity. The complex interaction of the cells
of the immune system is mediated largely through cytokines, and a
discussion of this process is beyond the scope of this chapter. This
section will discuss disorders of neutrophils [chronic granulomatous
disease (CGD)], B cells [X-linked agammaglobulinemia (XLA)], and T
cells (acquired immunodeficiency syndrome) that are relevant to the
pediatric orthopedist.
infections of the musculoskeletal system, including osteomyelitis.
Therefore, despite being a rare disease, CGD should enter the
orthopaedist’s mind in the setting of atypical, unusually severe, or
difficult-to-treat infections.
neutrophils, and occurs in approximately 1 in 200,000 to 1 in 500,000
live births (169). A key component of
neutrophil function is the respiratory burst. After phagocytosis,
creation of hydrogen peroxide and hypochlorous acid in the phagosome
allow optimal killing of ingested pathogenic microorganisms. The
creation of these oxidants is dependent on nicotinamide adenine
dinucleotide phosphate (NADPH) oxidase. CGD is a group of disorders
characterized by a variety of mutations of any of the NADPH component
genes, transmitted in X-linked or autosomal patterns (170).
The mean age at diagnosis depends on the type of CGD. X-linked CGD
presents at a mean age of 3 years, whereas the autosomal recessive
types typically present at 7 to 8 years (169). The diagnosis of CGD is made by detecting in vitro dysfunction of the respiratory burst (170).
organisms commonly encountered in infections in patients with CGD include S. aureus, Gram-negative enteric bacteria, Burkholderia spp., Nocardia spp., Pseudomonas spp., and Aspergillus spp. (169). Infections with catalase-negative organisms such as S. pneumoniae and H. influenza
are uncommon because such organisms produce hydrogen peroxide that can
be used by the neutrophil for killing when NADPH oxidase is ineffective.
These infections can be superficial, such as lymphadenitis and
perirectal abscesses, or deep, such as pneumonia, liver abscesses,
osteomyelitis, and otitis media. Also, the lack of the oxidant products
of the respiratory burst, which would have acted to mediate or suppress
further neutrophil chemotaxis, allows continued recruitment of
neutrophils and the formation of granulomata.
infections of the spine are typically difficult to treat. Some experts
recommend surgical debridement with the wound left open to heal
secondarily (173), although others have reported successful medical cure of spinal Aspergillus osteomyelitis using interferon-γ and antifungals (174, 175, 176). One child with tibial Aspergillus osteomyelitis was successfully treated with interferon-γ and antifungals after failure of surgical debridement (177).
aggressive, with early surgical debridement and liberal use of
antibiotics. Accurate cultures are essential, because pathogens
uncommonly encountered in the general pediatric population are common
causes of infection in patients with CGD. The possibility of fungal
infection should always be specifically investigated with fungal smears
and cultures.
treatment of children with CGD. Routine preventative measures such as
hand-washing and good hygiene assume paramount importance in these
individuals. Prophylactic administration of interferon-γ (178), itraconazole (179), or antibiotics may help to prevent infections. Stem cell transplantation has been used with some success to “cure” CGD (180). Gene therapy has shown promise in vitro, but Phase I trials have yet to show clinical usefulness (181).
The long-term prognosis for children with CGD is variable, but fewer
than 50% of patients live beyond the second decade of life (169).
function, may present to the orthopaedist as a clinical picture of
arthritis. Arthritis occurs in XLA for unclear reasons, and may be an
initial presenting symptom in children at an average age of 2 years (182). Fifteen of 69 patients in one series had arthritis at initial presentation; only 4 of these cases were due to infection (183). The arthritis of XLA most often affects the knees, wrists, ankles, and fingers, and may be polyarticular in presentation (184). The clinical picture may closely resemble juvenile rheumatoid arthritis (185).
Synovitis is present, and the synovial tissue has a large number of
suppressor T lymphocytes, differentiating it pathologically from that
of juvenile rheumatoid arthritis (186). Septic
arthritis must be ruled out in both acute and chronic presentations
because mycoplasmal infection was the leading cause of chronic
arthritis in a series of 358 patients with XLA (187). The arthritis, if aseptic, usually responds to immune globulin treatment and antiinflammatory medication (184).
A knowledge of the clinical picture and consequences of XLA will allow
the orthopaedist to appropriately refer patients for further evaluation
and treatment.
XLA results from one of more than 750 possible mutations in the gene
for B-lymphocyte tyrosine kinase (BTK), which is necessary for B cell
maturation (189). The immunologic abnormality
of XLA therefore consists of very low numbers of mature B cells and
profoundly decreased production of all three major immunoglobulin
classes (190). The number and function of T cells are usually normal.
maternal IgG levels begin to decline in the first few months, recurrent
infections begin to appear (191). Respiratory infections are common and are typically caused by organisms such as Streptococcus spp. and Haemophilus influenzae (182). Infections are usually severe enough to require hospitalization before a diagnosis of XLA is made (192).
Therefore, the orthopaedist who is evaluating a child with unexplained
arthritis should inquire about past history of hospitalization for
respiratory infections. Infectious disease consultation should be
obtained if an infection history accompanies a clinical picture of
arthritis in young children.
hypogammaglobulinemia and very low numbers of circulating
B-lymphocytes. Treatment of XLA consists of immune globulin replacement
and aggressive treatment of infections. Immune globulin, given as a
regular prophylaxis, can lower the incidence of respiratory infections
and prolong life (193). Recurrent respiratory infections lead to chronic lung disease (CLD), and respiratory failure is a major cause of mortality (191).
immunodeficiency virus (HIV) infection both as a cause of
musculoskeletal disease and as a complicating factor in the
treatment of other musculoskeletal problems. More than 42 million people are infected with HIV worldwide (194). In the year 2000, 600,000 children were newly diagnosed with HIV infection (195), and AIDS caused 3% of infant deaths in the world (196).
Most cases of pediatric HIV infection occur in sub-Saharan Africa,
where the prevalence is increasing. Conversely, the incidence of new
infections in developed countries has decreased since the mid-1990s
with the widespread use of antiretroviral therapy (197).
transmission from an infected mother. Transmission can occur before,
during, or after delivery (198). Premature
babies are more likely to become infected than full-term babies.
Cesarean section carries only half the risk of intrapartum transmission
as compared to vaginal delivery. Postpartum transmission usually occurs
through breast-feeding. The worldwide risk of vertical transmission of
HIV to an infant from an infected mother ranges from 15% to 40% (199).
The virus infects glial cells early in the course of the infection,
presumably inciting cytokine-mediated apoptosis of neurons.
Encephalopathy occurs more rapidly in children than adults, with 10% of
infants showing signs within 1 year of infection (201). Children infected in utero
or at a very young age can develop catastrophic encephalopathy with
severe motor and cognitive dysfunction. The encephalopathy may be
either static or progressive. Initial hypotonia is replaced by severe
spasticity in a diplegic or quadriplegic pattern. Severe mental
retardation is common. Surgical intervention for spasticity in children
infected with HIV is not contraindicated as a rule. Indications and
planning for surgery should follow the principles established for
spasticity of other etiologies. Preoperative requirements are that the
child’s HIV infection is being satisfactorily treated with
antiretroviral medications and that clinically significant infections
do not pose a significant anesthesia risk.
among HIV-infected children, the orthopaedist may be faced with
infectious complications of AIDS in a child. The clinical course of HIV
infection in children differs from that in adults in several ways. The
progression from HIV seropositivity to AIDS is more rapid, with one
third of infants developing AIDS within 3 months of life if untreated (195,202). Bacterial infections are more common in children than in adults with AIDS (203),
and bacterial skin infections may be first evaluated by an
orthopaedist, even before the diagnosis of HIV infection is made.
Unusual or severe infections in an infant should prompt an
ascertainment of maternal risk factors for HIV infection. A low total
lymphocyte count, found during a routine complete blood count with
differential, can also provide a clue to the diagnosis and even
prognosis of HIV infection in young children (204).
Suspicion of HIV infection should prompt immediate consultation with an
infectious diseases specialist, given the social and medical
complexities associated with this diagnosis in a young child.
is not known, but the orthopaedist should be aware of the profound
effects of this disease on immune status. Like adults, children with
AIDS are also prone to develop neoplasias such as Kaposi sarcoma,
lymphoproliferative disorders, and smooth-muscle tumors (205), some of which may appear as masses on an extremity, leading to referral to the orthopaedist for initial evaluation.
Such children in developed countries are routinely living into
adolescence. Therefore, the pediatric orthopaedist must not look with
despair at a child with HIV infection, and should use long-term
strategies and provide lasting treatments for any musculoskeletal
problem.
derived from a common bone marrow precursor that provides important
immune functions in various parts of the body. Macrophages ingest
cellular debris, pathogens, and foreign bodies, and are particularly
abundant in the spleen, liver, lymph nodes, lungs, and bone.
Osteoclasts are a specialized form of macrophage, derived from the same
precursor. Dendritic cells are nonphagocytic antigen-presenting cells
that are thought to arise from the monocyte-macrophage stem cell. A
wide variety of diseases affect the monocyte-macrophage system. Two
diseases with musculoskeletal manifestations discussed in this chapter
are Gaucher disease, which is a lysosomal storage disease, and
Langerhans cell histiocytosis (LCH), which is a dendritic cell
proliferative disorder.
catabolic enzymes that allow toxic accumulation of metabolic pathway
products. A variety of enzyme deficiencies lead to a variety of
diseases with different manifestations (Table 11.3).
The most common lysosomal storage disease is Gaucher disease, and this
example will be discussed in detail in this chapter. Gaucher disease
has significant skeletal manifestations, and can require orthopaedic
attention for bone pain, osteomyelitis, osteopenia, pathologic
fractures, and osteonecrosis.
until 1965, when Brady et al. (207)
linked it to a deficiency of glucocerebrosidase, a membrane-bound
enzyme responsible for cleaving glucocerebroside. The lipid
glucocerebroside accumulates in macrophages, and such lipid-laden
macrophages are termed Gaucher cells. The
clinical manifestations of Gaucher disease are caused by the
accumulation of these cells in organs, resulting in organ dysfunction.
|
TABLE 11.3 STORAGE DISEASES IN CHILDREN, AND ASSOCIATED MUSCULOSKELETAL ABNORMALITIES
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
storage disease, with an autosomal recessive inheritance pattern and a
prevalence of 1 in 40,000 in the general population and 1 in 400 to 1
in 600 among Ashkenazi Jews (208, 209, 210, 211).
Three forms of Gaucher disease are recognized: type 1
(nonneuronopathic), type 2 (acute neuronopathic), and type 3 (subacute
neuronopathic). Type 1 is by far the most common form and is
characterized by hepatosplenomegaly, pancytopenia, and predominant
skeletal manifestations. Type 2 is a rare form that involves the
central nervous system and cranial nerves and usually causes death by
apnea or aspiration before the age of 2 years (212). Type 3 disease is characterized by neurologic symptoms, including seizures, that begin during adolescence (213). More than 100 disease-producing mutations of the glucocerebrosidase gene, which is located on the short arm of chromosome 1, have been identified (213), and some mutations predict the phenotype (214). The detection of glucocerebroside in blood and urine confirms the diagnosis of Gaucher disease.
the genotype and clinical type. In a series of 53 patients, Zimran et
al. (215) found that the average age at diagnosis was 25 years (range, 8 months to 70 years). Another series (214)
of 34 children and adolescents with type 1 disease found that most of
them presented before the age of 10 years. A patient with Gaucher
disease may present initially to the orthopaedist with musculoskeletal
symptoms. Bone pain or fracture is the reason for presentation in 13%
to 60% of patients (215,216).
Growth retardation is also a common musculoskeletal presenting symptom,
with 26% and 30% of patients presenting with less than the third
percentile of normal values for weight and height, respectively (214). Skeletal abnormalities are detected radiographically in 88% to 94% of patients at presentation (214,216).
which organs are affected by accumulated Gaucher cells. Splenic
involvement causes splenomegaly and can cause hypersplenism, leading to
anemia, thrombocytopenia, or pancytopenia. Liver involvement can cause
mild liver dysfunction. Impaired hepatic synthesis of clotting factors
may compound the thrombocytopenia, causing clinically significant
coagulopathy.
In a review of 602 patients with type 1 Gaucher disease from the
Gaucher registry, 21% were found to have some form of disability in
mobility related to skeletal involvement (218).
Skeletal manifestations include pain, deformity, osteopenia,
osteonecrosis, osteomyelitis, pathologic fracture, and vertebral
collapse (217). Gaucher disease is associated
with a classic abnormality that shows up on radiographs as an
“Erlenmeyer flask” deformity of the distal femur and proximal tibia,
representing impairment of remodeling (Fig. 11.7).
However, this finding is not pathognomonic for Gaucher disease, and
occurs only in 56% to 70% of patients with known Gaucher disease (215,216).
involvement. Bone crises are thought to be related to intramedullary or
subperiosteal hemorrhage (219,220)
made possible by thrombocytopenia and deficient clotting factor
synthesis. Bone crises are episodes of acute bone pain accompanied by
fever, leukocytosis, and elevated erythrocyte sedimentation rate.
Because of this clinical picture, bone crises are also known as pseudoosteomyelitis.
Blood cultures may help differentiate between bone crisis and
osteomyelitis. Further differentiation is difficult, as in SCD, and
imaging studies may not be helpful. Early in bone crises, plain
radiographs are normal, but may progress to show periosteal reaction
and areas of radiolucency (221,222). Radionuclide bone scans may show an area of decreased uptake early in the course of the process (223) and increased uptake around a photopenic area later in the course (224). MRI shows marrow edema on T2-weighted images, with or without signs of hemorrhage (218,220).
Periosteal fluid accumulation seen on MRI may indicate infection and
should be aspirated for culture under sterile conditions and
radiographic guidance.
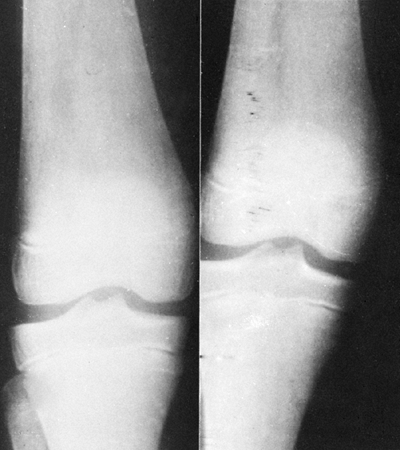 |
|
Figure 11.7
Radiographs of the knee in a child with Gaucher disease. Note the typical flaring of the distal femoral metaphysis, or Erlenmeyer flask deformity. (Photo courtesy of Henry J. Mankin, MD) |
early in the course usually requires opioid analgesics, which can be
augmented with high dose prednisolone (225).
The symptoms gradually abate over 2 to 4 weeks. Failure of the symptoms
to improve should warrant further investigation into the possibility of
osteomyelitis. Bone aspiration in an operating room setting may be
required. Osteomyelitis can follow a bone crisis, often with anaerobic
organisms, suggesting that there has been a period of ischemia (226).
Treatment of osteomyelitis in Gaucher disease parallels that in SCD as
discussed earlier, although attention should be paid to the altered
structural integrity of bone and increased bleeding risk when surgical
debridement is considered in a patient with Gaucher disease.
Osteonecrosis can follow a bone crisis, so routine radiographic
evaluation of an affected area is necessary even after the crisis
resolves.
correlate well with other signs of skeletal involvement. Back pain is
common in children with spinal involvement (227). Chronic back pain may be severe enough to require bracing.
The possibility that thrombosis plays a role in the pathogenesis of
osteonecrosis is supported by elevated D-dimer levels in patients with
Gaucher disease and osteonecrosis compared to those without
osteonecrosis (229).
Gaucher disease. Fractures occurred in 23% of 1476 patients in the
Gaucher Registry (218). The common sites of
fracture are the distal femur, proximal tibia, and femoral neck, and
65% of the fractures occur at the site of a prior bone crisis (230).
Fractures at the base of the femoral neck occur in young children and
can be complicated by coxa vara, pseudoarthrosis, and osteonecrosis.
Vertebral compression fractures occur with spinal involvement and can
lead to severe kyphosis and spinal cord compromise on rare occasions (227,231). Fracture healing is impaired in patients with untreated Gaucher disease, and delayed union and nonunion are common.
Quantitative computed tomography can also accurately measure bone
mineral density, but is not recommended in children because of the very
high radiation doses involved (234). Chemical
markers of bone turnover are also abnormal in Gaucher disease. When
compared with a control group, patients with Gaucher disease had
elevated urinary excretion of pyridinoline and deoxypyridinoline (232), as well as elevated serum levels of carboxy terminal telopeptide of type I collagen (235),
all of which are markers of bone resorption. Serum levels of
carboxyterminal propeptide of type I collagen, a marker of bone
formation, are significantly lower in patients with Gaucher disease
than in the controls (235).
quantified. Quantitative chemical shift imaging (QCSI) is an MR
spectroscopic technique that utilizes the difference in resonance
between fat and water in bone marrow to quantify the reduction in fat
fraction that occurs in Gaucher disease (236,237). Marrow infiltration in vertebral bodies measured by QCSI correlates well with disease severity (237), and the technique is reproducible (238).
A bone marrow burden score has recently been developed to allow
quantification of marrow infiltration using standard MR imaging (239),
with high inter- and intrarater reliability and sensitivity only
slightly less than that of QCSI. Several other semiquantitative
techniques using standard MRI have been developed and are currently
under investigation (234,240).
enzyme, enzyme replacement therapy (ERT) is the cornerstone of
treatment. In fact, replacement of macrophage-directed
glucocerebrosidase has become standard medical treatment for type 1
Gaucher disease (214,241, 242, 243).
Given intravenously at 2-week intervals, ERT reliably reverses anemia,
thrombocytopenia, and splenomegaly. Although marrow infiltration
responds more variably and more slowly (244, 245, 246), bone mineral density and bone pain improve with ERT (247,248). Children tend to respond more quickly and reliably than adults (248).
Enzyme replacement, if started early in life, can prevent skeletal
deformity and allow normal skeletal development and growth (249,250). A decrease in the incidence of fractures has also been observed (251).
Bone marrow transplantation has been used in patients with a variety of
lysosomal storage diseases, including Gaucher disease (252).
Also, because the pathogenesis of Gaucher disease involves an
accumulation of glucocerebroside, efforts to decrease production of
this molecule may prove effective in treating the disease (253). Gene therapy is not yet routinely available.
abnormal proliferation of a marrow-derived histiocytic cell initially
described by Langerhans in 1868 (254). The skeletal manifestations of LCH were not described in detail in the literature until a report by Fraser in 1935 (254). In 1940 Lichtenstein and Jaffe coined the term eosinophilic granuloma
of bone
One year later, Farber argued that eosinophilic granuloma of bone
belonged to the same spectrum as Hand-Schuller-Christian disease and
Letterer-Siwe disease. Later Lichtenstein grouped all three conditions
under the term histiocytosis X (254). In 1961, Birbeck used electron microscopy to detect the oblong granules in Langerhans cells (254),
but it was not until 1973 that Nezelof identified these granules in
specimens of histiocytosis X and recognized the disease as a
proliferation of Langerhans cells (255). Today, the term LCH is the preferred name of the spectrum of conditions.
The median age at diagnosis is between 1 and 3 years, but the diagnosis
can be made at any age from infancy to over 80 years (258). There is a slight male preponderance in the occurrence of the condition (259,260). Bone involvement is found in 80% to 97% of patients with LCH (257,259, 260, 261, 262, 263, 264). The skull is the most often affected bone, followed by the femur, spine, ribs, mandible, and pelvis (265, 266, 267). Bone involvement in the hands and feet is uncommon (268,269). Widespread involvement of multiple organ systems can occur, and carries a worse prognosis than isolated bone involvement (270). This chapter will discuss in detail the evaluation and management of bone lesions only, whether solitary or multiple.
It is uncommon for patients who present with solitary bone lesions to
develop secondary bone lesions. However, a series with 52 patients
found that 30% of them developed secondary bone lesions, half of which
were asymptomatic; the lesions were detected during routine skeletal
surveys (264). The prognosis in patients with
multiple bone involvement without soft tissue lesions is still
favorable, with death occurring in 1 of 22 patients in one series (257).
swelling, or limping. The pain is usually of less than 2 months
duration. Symptoms such as lethargy, cough, dyspnea, and failure to
thrive are uncommon, and may indicate widespread involvement. Because
diabetes insipidus is the most common extraskeletal abnormality that
develops in patients presenting with bone involvement (274),
specific questioning is required regarding polyuria and polydipsia.
Skin rash, jaundice, hepatosplenomegaly, tachypnea, exophthalmos,
hearing difficulties, and poor growth are important signs of widespread
involvement. Physical examination may reveal a tender mass associated
with a bone lesion in the skull, jaw, or extremities. Torticollis,
scoliosis, kyphosis, and neurologic impairment may accompany a spine
lesion (275, 276, 277, 278, 279).
The radiographic appearance of LCH of bone depends upon the phase of
the disease and the site of occurrence. In the early phase of the
disease, the lesion may appear aggressive, with a permeative pattern of
osteolysis and laminated periosteal reaction mimicking Ewing sarcoma (280, 281, 282).
Later in the course of the disease, the lesion appears less aggressive,
with well-defined margins, a narrow zone of transition, and mature or
absent periosteal reaction (269). Widening of
the medullary cavity with cortical thinning, scalloping, or penetration
are also common findings in long bone lesions (269). In the long bones, the lesion typically exists in the diaphysis or metaphysis. Epiphyseal involvement is uncommon (269,283,284).
In the skull, LCH may give rise to a lesion that appears round,
radiolucent, and “punched-out” when viewed on plain radiographs (Fig. 11.9).
The vertebral body is markedly flattened, but the posterior elements
are usually spared. In contrast to osteomyelitis, the disc spaces are
preserved in LCH. The thoracic vertebrae are affected in more than half
of the patients. Cervical spine involvement is uncommon (269), but involvement of the posterior elements in the cervical spine has been reported (285,286), as has cervical vertebral body involvement without vertebra plana (287).
the diagnostic work-up of any patient with a radiographic lesion
resembling LCH. In fact, one should consider obtaining results from
both studies, as neither is wholly sensitive: in one series of 42
patients who were studied with both modalities, bone scans missed 36 of
191 lesions (19%), and skeletal surveys missed 55 (29%) (288).
Bone scans were more effective at detecting lesions in locations that
are difficult to view clearly on plain radiographs, such as the ribs,
spine, and pelvis. Computed tomography can demonstrate the extent of
bony destruction. MRI is helpful in evaluating the extent of the
lesion, soft tissue involvement, and marrow edema. A high-intensity
signal is seen on T2-weighted images within and around the lesion (280). MRIs may be normal initially but progress to markedly abnormal within a few weeks (289).
Because many patients present with a lesion that radiographically
resembles a sarcoma of bone, MRI is essential for ruling out the soft
tissue mass that often accompanies a sarcoma.
should include a complete blood count, liver function tests, and serum
and urine osmolality if history suggests diabetes insipidus. An
oncologic consultation is essential for all patients with suspected
LCH. For patients with evidence of multiple organ involvement or for
patients younger than 3 years with multiple bone lesions,
investigations such as arterial blood gas, bone marrow aspirate,
computed tomography of the chest, abdominal ultrasonography, and
audiology, dental, and immunologic
assessments may be obtained as indicated in coordination with the oncology team (264).
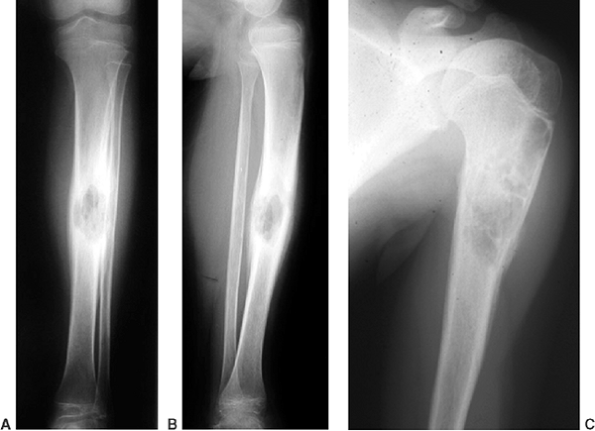 |
|
Figure 11.8 Langerhans cell histiocytosis of the tibia (A, B) and of the proximal humerus (C) in two children. In both cases, the lesions healed following biopsy, both symptomatically and by radiographic findings.
|
biopsies are easy to perform, and are recommended when skin involvement
is present. In the absence of skin involvement, bone lesions,
preferably the most easily accessible, should be biopsied. Biopsies can
be performed either open or by CT-guided needle aspiration. Some
experts have reported an accuracy of as high as 90% to 100% for needle
aspration (290,291).
Biopsies of suspicious lesions must be sent as fresh specimens, because
immunohistochemical tests for α-D-mannosidase and CD1a are not possible
with paraffin-embedded tissue.
mononuclear cell with a grooved nucleus and characteristic
racket-shaped organelles (Birbeck granules) in the cytoplasm.
Langerhans cells are considered to be antigen-presenting cells that are
members of the dendritic cell family (292,293).
Pathologic Langerhans cells (LCH cells) phenotypically represent
Langerhans cells arrested at an early stage of maturation, in that they
lack certain differentiated features of dendritic cells (294).
The exact pathogenesis of LCH is unknown, and theories of viral,
reactive, and neoplastic etiologies have been put forth with variable
supporting evidence. Detailed coverage of this debate is beyond the
scope of this chapter. Langerhans cells are not the only cells in an
LCH lesion, and are accompanied by T cells, eosinophils, and
macrophages (295) (Fig. 11.10).
in the absence of systemic involvement, is toward resolution,
irrespective of whether or not the lesions were treated (285,296). Most lesions will resolve following biopsy alone. A recent series (285)
found that of 26 patients with solitary lesions, 25 showed partial or
complete healing after a mean of 4 years, despite the fact that only 4
were treated in any way. Also, two of the four treated patients
suffered treatment-related complications. Reappearance of a trabecular
pattern is seen in bone lesions 6 to 10 weeks after diagnosis, and
complete healing is seen at 36 to 40 weeks on radiographic
investigation (297). In view of this, a failure
to heal after approximately 4 months of observation, as indicated
radiographically, may indicate the need for intervention.
that cause substantial pain or threaten pathologic fracture. There are
several treatment options for bone lesions. Some experts believe that
curettage with or without bone grafting hastens the healing process (298).
For relief of pain from bone lesions, intralesional injection of
corticosteroids has been used successfully in a small series (299) and is also recommended by others (300). Indomethacin has been used successfully in treating the symptoms of LCH in 8 of 10 children in one series (301).
A recent multicenter review of low-dose radiotherapy in 63 patients
with LCH, aged 9 to 44 years, found excellent remission rates even in
widespread disease (302). However, the risk of secondary malignancy (264,303) precludes the use of this therapy in
solitary bone involvement. For this reason, we do not use radiotherapy at our institution.
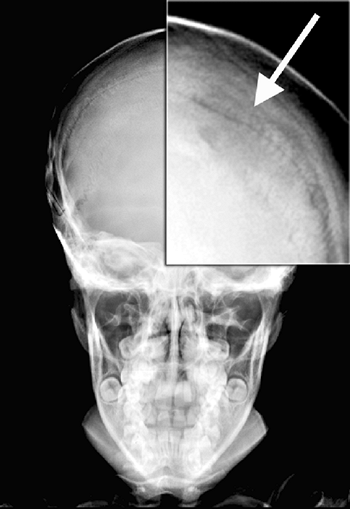 |
|
Figure 11.9 Langerhans cell histiocytosis lesion in the skull (arrow, inset).
|
with suspected LCH lesions. Skeletal surveys assist in localizing
multiple lesions, and MRI can identify a soft tissue mass that would
raise the suspicion of a sarcoma. We prefer an open biopsy with
intraoperative frozen section. Sufficient tissue is needed for
histology, immunohistochemistry, cultures, and occasionally electron
microscopy or molecular genetics studies. Intraoperative frozen section
confirms the acquisition of lesional tissue and can support the
diagnosis of LCH. If a patient presents with isolated vertebral
involvement, a skeletal survey or bone scan is performed in order to
identify a more accessible lesion for biopsy. If no such lesion is
found, CT-guided needle biopsy may be performed. After confirmation of
LCH following biopsy, most lesions are left to heal. Curettage and bone
grafting after biopsy confirmation may accelerate the natural history
of healing and may be necessary for large lesions in weight-bearing,
high-stress areas where the risk of pathologic fracture is high.
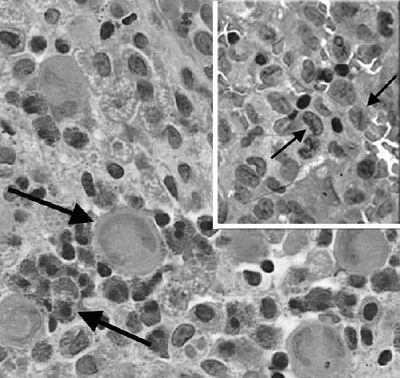 |
|
Figure 11.10
Hematoxylin and eosin stain of Langerhans cell histiocytosis. Note the eosinophils, giant cells, and Langerhans histiocytes with the coffee bean—shaped nucleus showing a central groove (arrows, inset). |
MRI is required whenever there is neurologic dysfunction, and can
differentiate between nerve root and spinal cord compression. Surgical
decompression and stabilization are indicated for spinal cord
compression, whereas bedrest with systemic or injected corticosteroids
can effectively treat nerve root compression. In the absence of nerve
root or cord compression, long-term back and neck problems from spine
LCH are uncommon (305). Widespread, multiorgan
LCH carries a poor prognosis, and its treatment is beyond the role of
the orthopaedic surgeon. Chemotherapy, interferon, and bone marrow
transplantation have been used with some success.
elements of the circulatory and hematopoietic systems. Normal
hemostasis begins with vascular injury. Injury to a blood vessel causes
vasoconstriction, helping to stem blood loss. Disruption of the
endothelium allows interaction between vessel wall components and
circulating platelets and plasma. Platelet aggregation occurs in
response to the injury in the endothelium and, with the assistance of
the von Willebrand factor (vWF), begins the process of thrombosis. In
the plasma, coagulation factors interact to form fibrin, which
completes the thrombogenic process.
The intrinsic pathway is stimulated by collagen exposure and the
extrinsic pathway by tissue thromboplastin. Both pathways result in
activation of factor X, which combines with activated factor V to
convert prothrombin into thrombin, which in turn converts fibrinogen
into
fibrin.
Of particular relevance to disorders of hemostasis are factors VIII and
IX, which combine to activate factor X in the intrinsic pathway.
Intrinsic pathway function is measured in the laboratory by the partial
thromboplastin time (PTT), and extrinsic pathway function by the
prothrombin time (PT). The functions of specific factors can be
assessed by quantifying their activity levels. Disorders of platelet
function are evaluated by the bleeding time, because platelet plug
formation is the first step in clinically apparent hemostasis.
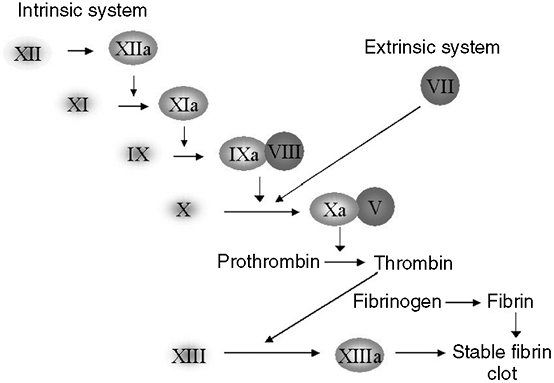 |
|
Figure 11.11 Schematic depiction of the intrinsic and extrinsic pathways of the clotting cascade. See text for details.
|
deficiency of clotting factor. Hemophilia A is a deficiency of factor
VIII, whereas hemophilia B, or Christmas disease, is a deficiency of
factor IX. In a large surveillance study in the United States, the
incidence of hemophilia was found to be 1 in 5032 live male births,
with an age-adjusted prevalence of 13.4 per 100,000 among the male sex (306).
Seventy-nine percent of these had hemophilia A. Extrapolation of the
prevalence data from the six states represented in the United States
study estimated that, in 1994, there were approximately 16,000 persons
with hemophilia nationwide (306).
sex-linked recessive pattern, largely limiting the disorder to the male
sex. Female carriers may also be symptomatic, despite the recessive
nature of the gene. Skewed random inactivation of the normal X
chromosome may result in reduced levels of clotting factor and a
consequent bleeding diathesis in these girls and women (307).
indistinguishable. The clinical manifestations of hemophilia depend on
its severity. Severe clotting factor deficiency (less than 1% factor
activity) leads to spontaneous joint and soft tissue bleeds. In
moderate hemophilia (1% to 5% factor activity), spontaneous bleeds are
uncommon, but excessive bleeding and hemarthroses can occur following
minor trauma. In mild hemophilia (5% to 20% factor activity), abnormal
bleeding is typically seen only following major trauma or surgery.
According to surveillance data, hemophilia is severe in 43% of
patients, moderate in 26%, and mild in 31% (306).
hemophilia and comprise most of the pathology that the orthopaedist
sees. The knee, elbow, and ankle are the most commonly affected joints (308, 309, 310) (Fig. 11.12).
The shoulder, hip, and wrist are less often affected. An acute
hemarthrosis causes pain, warmth, and swelling in the affected joint;
these symptoms abate over a period of a few days if properly treated.
Hemophilic arthropathy is a degeneration of joints following recurrent
hemarthroses, and can be quite destructive and debilitating. Muscle
hematomas usually occur in the pelvis, thigh, and forearm. These
hematomas cause pain, swelling, and muscle spasm, and may also induce
compartment syndrome or nerve palsies. Recurrent intramuscular bleeding
can lead to the development of pseudotumors, or “blood cysts,” in the
extremities and pelvis. Hematuria and bleeding from mucosal surfaces in
the mouth and nose also occur to a variable extent but will not be
discussed in this chapter. Other serious consequences include airway
compromise from neck hematomas and neurologic compromise from
intracranial bleeding.
bleeding from circumcision, or easy bruising from minor trauma or
immunizations. A single swollen joint from minor trauma can also be a
presenting sign of hemophilia (311). In a
recent study of children with severe hemophilia, the first clue that
pointed toward the diagnosis was hematoma in 47%, family history in
24%, and excessive bleeding after an operation in 14% (312).
Mouth, joint, and muscle bleeds prompted the diagnosis in 5% each. All
the children were diagnosed prior to 2 years of age, with 81% being
diagnosed during the first year of life and 38% in the first month of
life. However, others have found a
higher
prevalence of mild and moderate hemophilia in the 5 to 14-year-old age
group than in the 1- to 4-year-old age group, suggesting a delay in
diagnosis of milder forms of hemophilia (306).
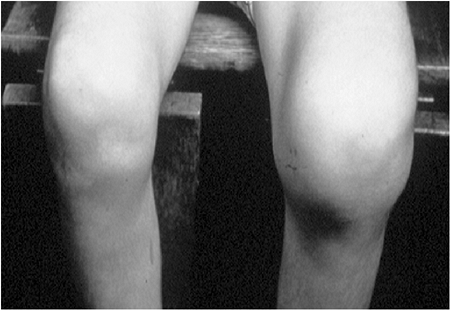 |
|
Figure 11.12
A teenager with severe factor VIII deficiency and recurrent hemarthroses of the left knee. Note massive swelling typical of hypertrophic synovitis and hemarthrosis. |
activity of the clotting factors; hemophilia exists if factor activity
is less than 20% to 50% of normal. The activated partial thromboplastin
time (aPTT) is typically prolonged because the deficient factors affect
the intrinsic coagulation pathway. The PT, platelet count, and bleeding
time remain normal. Hemophilia can be subdivided into the three grades
of severity described in preceding text on the basis of the
quantification of factor activity.
replacement of the deficient factor. Minor joint and soft tissue bleeds
can be treated with replacement of the relevant factor to a serum
factor activity level of 30% to 70% immediately, and thereafter
maintenance of 30% activity for 1 week to 10 days. Most patients under
routine care for hemophilia are able to self-administer factor
replacement, so treatment often begins at home. More severe bleeds,
including those causing nerve palsies or compartment syndrome, require
acute correction of factor activity to 100%. The administration of
factor is the first step in the acute treatment of any bleeding episode
in a patient with hemophilia, especially prior to any necessary
emergent surgical intervention. Factor replacement should be
administered prior to elective invasive procedures, even those as
simple as injections and suturing of lacerations.
available, and each has its own limitations. The discovery of
cryoprecipitate and concentrated clotting factors in the 1960s (313, 314, 315)
made the replacement of deficient factors realizable for patients with
hemophilia. However, such preparations of factor were derived from
pooled sources of human blood and therefore had the potential to
transmit disease. Exposure to hepatitis B or C was found in up to 85%
of patients with severe hemophilia in the 1980s (316, 317, 318). HIV was found to infect 70% of stored hemophilic blood samples that were collected in 1982 (319),
and by June 1991, 91% of the cases of HIV infection that were reported
to the Center for Disease Control and Prevention were in patients with
hemophilia (320). The heat-treatment of factor
concentrates, instituted in 1984, has effectively eliminated the
transmission of HIV, and newer forms of virus inactivation have limited
the transmission of hepatitis. Factors VIII and IX are mostly now
derived from recombinant DNA techniques, virtually eliminating the risk
of disease transmission. Such products are now considered the
replacement factors of choice (321), despite their higher initial cost.
exogenous factor VIII, or rarely factor IX, and if these antibodies
deactivate the exogenous factor they are termed inhibitors. Inhibitors to factor VIII have been reported in 3.6% to 52% of patients with hemophilia (322).
Once a patient develops an inhibitor, exogenous factor replacement no
longer corrects their coagulation deficit. Although bleeds do not
increase in frequency they are harder to treat. Patients with low
titers of inhibitors can often be treated simply with higher doses of
factor replacement. Patients with high titers, however, typically
cannot be treated with any exogenous human form of factor VIII. Until
recently, bleeds in these patients often became quite severe, and
elective surgery was considered contraindicated. The development of
recombinant activated factor VII (rFVIIa) has made correction of
coagulation deficits possible in patients with high-titer inhibitors
following muscle and joint bleeds and prior to elective surgery (323, 324, 325, 326).
This is possible because activated factor VII induces coagulation
downstream from factors VIII and IX in the clotting cascade. The
disadvantage of the high cost of rFVIIa is offset by improvements in
the quality of life for these patients (327), and specialists have argued against rationing its use on the basis of high cost (328).
progressive joint damage. Prompt treatment of these hemarthroses is
therefore essential. The most important aspect in treatment of an acute
hemarthrosis is factor replacement. Factor activity levels should be
increased to 50% to 100% of normal for the first 48 hours. Because of
the variable preferences for and responses to specific factor
formulations among patients with hemophilia, the choice of the factor
replacement product should be left to the patient, the patient’s
hematologist, and the hemophilia treatment center that is coordinating
the patient’s care.
procedure. Whereas some authors find it a crucial step in the
management of a painful, or “major” hemarthrosis (329),
others recommend it only in the setting of a tense hemarthrosis or when
ruling out a septic arthritis in an HIV-infected patient (330).
Regardless of the indication, arthrocentesis should be performed using
sterile technique and only after appropriate factor replacement (331).
contraindicated in these patients because of the tendency toward
bleeding complications, so pain control is usually achieved with opioid
analgesics. Cyclooxygenase 2 (COX-2) specific inhibitors may show
promise for this purpose, but have not been thoroughly studied in this
population. Ice packs may help alleviate pain and swelling.
Immobilization can be helpful for significantly painful hemarthroses,
but should be limited to 1 or 2 days, because stiffness following a
hemarthrosis can be problematic. Although rest is encouraged when the
inflammation is acute, weight bearing is generally allowed to the
extent tolerated.
pain, swelling, stiffness, and warmth should abate over a few days.
Factor replacement is continued for 10 days to 1 month, depending on
the duration of symptoms. In some cases, signs of inflammation persist
despite control of the bleeding. Oral corticosteroids may be helpful in
these cases
of subacute synovitis, although their benefits have not been clearly defined (332).
To avoid stiffness following an acute hemarthrosis, a supervised
rehabilitation program is started after bleeding has been controlled.
as guided by the patient’s hematologist. If the onset of pain was
sudden or if trauma was involved, fracture should be ruled out. Plain
radiographs detect most fractures, but MRI may be indicated in a young
child in order to detect occult epiphyseal or physeal fractures (Fig. 11.13).
A tense or very painful hemarthrosis is aspirated under sterile
technique once factor activity has been restored to 100% with
replacement. Immobilization is generally avoided unless the
hemarthrosis is very painful or threatens to cause a joint contracture.
A knee immobilizer worn while at rest may help prevent a knee flexion
contracture from developing. Ambulation training and physical therapy
for range of motion are begun as soon as symptoms permit, with weight
bearing allowed to the extent tolerated, provided the radiographs and
MRI reveal no bony abnormalities. Treatment is generally performed on
an outpatient basis unless the presence of inhibitors make frequent
infusions of activated factor VII necessary.
of joints following recurrent hemarthroses. The pathogenesis of HA is
not fully elucidated, but appears to have two components: the effect of
recurrent bleeding on the synovium and the direct effect of
intraarticular blood on cartilage (333). The
synovium in joints with hemophilic arthropathy has long been thought to
be the epicenter of joint destruction. Such synovial tissue is
hypertrophic, hypervascular, and laden with hemosiderin (334). The inflammatory nature of the synovium is attributed to the accumulation of iron deposits (335).
The synovium is typically friable and bleeds easily, setting up a
vicious cycle of recurrent hemarthroses and worsening synovitis.
Affected synovial cells grown in culture secrete collagenase,
proteinase, and interleukins that are catabolically active against
cartilage (334, 335, 336).
Hemophilic arthropathy has also been found to share similarities with
noninflammatory degenerative joint disease (osteoarthrosis) (337).
Cartilage taken from joints with HA is similar to that from
osteoarthritic joints in terms of histochemistry, proteoglycan
synthesis, glycosaminoglycan content, and DNA content (337),
thereby suggesting that a direct effect of intraarticular blood on the
articular cartilage may play an important role in the pathogenesis of
HA. This theory is supported by the finding that short-term exposure to
whole blood impairs cartilage matrix synthesis in vitro and in vivo, without synovial inflammation (338).
It is likely that these two pathways, among other possible mechanisms,
interact to produce the destruction of joints that is typically seen in
HA, and that one process alone is not wholly responsible.
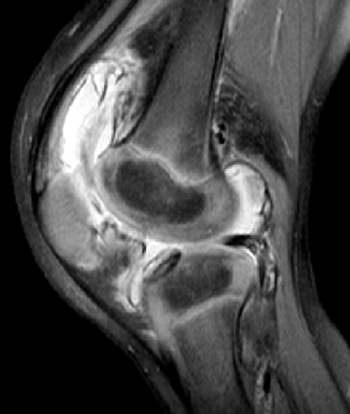 |
|
Figure 11.13
A 3-year-old boy with severe factor VIII deficiency and a high-titer inhibitor presented with traumatic knee swelling. T2-weighted MRI demonstrates massive hemarthrosis and hypertrophic synovium, while ruling out marrow edema that would indicate a fracture. |
Complete destruction of joint surfaces can take place as rapidly as
within 6 months (339), and can lead to
substantial pain and dysfunction. In Spain, a recent survey of 70
teenagers and young adults with severe hemophilia found significantly
lower quality of life than age-matched controls in six of the eight
dimensions of the SF-36 survey, especially pain and physical function (340).
Articular dysfunction was present in 85%. Similar impairments were
found in another survey of 116 patients with severe hemophilia in
France, with the lowest SF-36 scores (57.8 to 60.2) in the dimensions
of pain, vitality, and perception of general health (341).
Pain is a prominent feature of hemophilic arthropathy. A survey of 71
patients with hemophilia found that 50% of them had constant pain; 89%
had pain that interfered with their activities of daily living, and 85%
reported an impact of pain on their mood (342).
Patients had an average visual analog pain score of 4.8, with an
average of four joints each causing severe pain. Muscle strength and
anaerobic power have also been found to be inferior in boys with
hemophilia compared to age-matched controls (343). Children perceive an impact of their disease in their restricted ability to participate in sports (344), though current recommendations do allow controlled participation in physical activities (345,346).
has traditionally focused on grading the severity of joint destruction
with the aid of plain radiographs. Plain
radiographs
are easy to obtain, and clinicians are accustomed to reviewing
radiographs of arthritic joints. Early in the course of the
arthropathy, radiographs are normal. As the synovitis progresses, soft
tissue swelling can be seen. The first skeletal abnormalities
appreciable on plain radiographs include osteopenia and widening of the
epiphysis, followed later by subchondral erosions. Squaring of the
patella on a lateral view of the knee and widening of the intercondylar
notch on an anteroposterior view of the knee are typical findings at
this stage (Fig. 11.14).
Narrowing of the articular cartilage is a late finding, followed by
marked disorganization of the joint architecture with epiphyseal
enlargement and joint subluxation. Three grading systems have been
described to classify the severity of arthropathy as seen on plain
radiographs (347, 348, 349). Greene et al. (349)
found good inter- and intraobserver reliability for the three systems,
as well as good correlation between the radiographic grade of
arthropathy and the functioning of the corresponding joint.
seen on radiographs and the actual functioning of joints holds true
only for moderate or severe arthropathy. Plain radiographs do not show
early features of arthropathy. As described in the preceding text,
cartilage damage occurs early in the pathogenesis of hemophilic
arthropathy (337), but plain radiographs do not detect such damage until “joint space” narrowing is seen late in the course (347,349,350).
Ultrasonography, like plain radiographs, is noninvasive, quick, and
readily available, and has been shown to detect effusions, synovial
thickening, and cartilage damage in the knee joints of children with
hemophilia and recurrent knee bleeds (351).
However, ultrasound cannot penetrate bone, and the interpretation of
ultrasonographic images depends on the skill of the observer and on
technical factors (352).
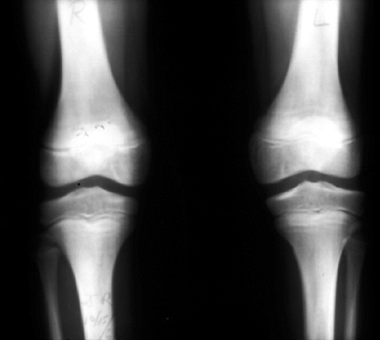 |
|
Figure 11.14 Radiographs of both knees in a child with hemophilic arthropathy. Note the widening of the intercondylar notch.
|
arthropathy and is considered by some to be the diagnostic imaging
modality of choice for evaluating the pathology of joints in hemophilia
(350). MRI can identify synovial hyperplasia
quite early in the course of hemophilic arthropathy, and the use of
intravenous gadolinium can further enhance the image of inflamed
synovium (353,354). MRI also offers easy detection and grading of subchondral bone cysts (355). Cartilage can also be imaged reliably by MRI (356).
Several prospective series, the largest of which evaluated 328 joints,
have found MRI to be superior to plain radiography for detecting
synovial and cartilage changes early in the course of arthropathy in
knees, ankles, and elbows (357, 358, 359, 360).
A comprehensive atlas of MR imaging in hemophilic arthropathy has
recently been published, giving details regarding both technique and
interpretation (361).
is prevention. Prophylactic treatment with factor replacement aims at
preventing the recurrent hemarthroses that are thought to cause
hemophilic arthropathy. As reviewed by van den Berg and Fischer (362),
many studies have shown that prophylaxis lowers the frequency of
hemarthroses and delays or even prevents hemophilic arthropathy.
Prophylaxis is more effective than on-demand treatment in minimizing
bleeding episodes (363,364),
and is recognized by the Medical and Scientific Advisory Council
(MASAC) of the National Hemophilia Foundation in the United States as
the treatment strategy of choice (365). The
specific aim of prophylaxis is to keep the factor level greater than 1%
(i.e., to convert severe hemophilia into moderate hemophilia). Several
studies have shown that, for preventing or delaying arthropathy, it is
more effective to begin prophylaxis in early childhood rather than
later (366, 367, 368), and the World Health Organization currently recommends starting prophylaxis between 1 and 2 years of age (369).
However, because of problems such as the high cost of the replacement
factors and the need for venous access devices, with their associated
complications, prophylaxis has not yet been universally accepted (362).
agent in the development of hemophilic arthropathy has led to the
development of various techniques of synovectomy to treat the chronic
synovitis seen in hemophilia. Synovectomy can be performed surgically
or chemically. Surgical synovectomy consists of open or arthroscopic
excision of the entire synovium spree of the joint; chemical
synovectomy involves injection of caustic or radioactive substances
into the joint involved.
In a subsequent follow-up study, these authors reported that none of
the 19 joints that had been treated with open synovectomy underwent
recurrent hemarthroses, and that more than half showed an improvement
in the range of motion of the joint (371).
Subsequently, several series have reported significant decreases in
bleeding, but have also reported significant complication rates (372, 373, 374, 375, 376, 377). Among the more frequent complications was loss of joint motion, occurring in up to 85% of knees (377) and 80% of elbows that underwent surgery (376).
Synovectomy of the elbow can be combined with resection of the radial
head in order to maintain range of motion when the joint is
significantly deformed (378). In younger patients without significant radial head deformity, however, radial head preservation is recommended (379). Open synovectomy of the ankle often requires multiple incisions to allow complete excision of the synovium (380).
investigation into arthroscopic synovectomy. Arthroscopic synovectomy
for hemophilic synovitis of the knee was first described by Wiedel (381) in 1985 and Klein et al. (382) in 1987. These and subsequent series reported in knees (383), ankles (384,385), and elbows (385)
replicated the decreased bleeding rates found following open
synovectomy, but also showed no change or general improvements in range
of motion. In 1992, Triantafyllou et al. (386)
retrospectively compared open versus arthroscopic synovectomy of the
knee. Those treated with open synovectomy showed a net decrease in
range of motion, whereas those treated arthroscopically showed a net
increase. Arthroscopic synovectomies required shorter hospital stays
and less factor replacement. Both groups had a decrease in bleeding
episodes. An increase in range of motion is not guaranteed following
arthroscopic synovectomy, and most experts recommend the use of
continuous passive motion (CPM) machines following knee synovectomy (382,383).
In a cost—benefit analysis, arthroscopic synovectomy of the knee has
been found to be cost effective by decreasing bleeding rates and
reducing the need for factor replacement (387).
Despite the attractiveness of arthroscopic synovectomy, it should be
noted that a thorough synovectomy is a time-consuming and difficult
procedure, given the abundant and fibrotic nature of hemophilic
synovium. Triantafyllou found that arthroscopic synovectomy of the knee
takes twice as much time to perform as open synovectomy (386). Arthroscopy of the elbow and ankle can be difficult, with the view initially hindered by hyperplastic synovium.
hemarthroses, surgical synovectomy has not been shown to prevent
arthropathy. Several long-term follow-up studies of open or
arthroscopic synovectomy have demonstrated progression of hemophilic
arthropathy despite a reduction in bleeding frequency (376,377,382,386,388,389).
It is clear from these results that, although the synovium is a
prominent source of bleeding in hemophilia, it is not the only culprit
in the development and progression of arthropathy.
initially attempted at approximately the same time as surgical
synovectomy; in 1973 Menkes et al. reported the intraarticular
injection of osmic acid for the treatment of hemophilic synovitis (390).
The lower morbidity associated with an injection allows its
administration as an outpatient procedure, with shorter factor
replacement needs and the possibility of less postprocedure stiffness.
Its minimal invasiveness makes it an option even for patients with an
inhibitor who might not be candidates for surgical intervention (391). Nonsurgical synoviorthesis can be divided into two types: chemical and radioactive.
stabilize the synovium and slow joint destruction include osmic acid,
rifampicin, oxytetracycline chlorhydrate, corticosteroids, and
hyaluronic acid. Osmic acid failed to gain popularity following the
mixed report by Menkes et al. However, rifampicin has been used in
several centers with some success (392,393). Recently, Fernandez-Palazzi et al. (394)
have begun using oxytetracycline chlorhydrate, a compound initially
used for pleurodesis. Corticosteroid injections have provided
short-term improvements in symptoms and bleeding rates in hemophilic
joints (395,396), but long-term results have been less encouraging (397).
Hyaluronic acid injection, although not purported to have a direct
effect on the inflamed synovium, may improve the functional
characteristics of the damaged hemophilic joint by improving
lubrication. Of 45 patients with hemophilia in two small series, 75%
experienced subjective improvements in symptoms lasting 1 to 24 months
following hyaluronate injection into various joints (398,399).
alternative to surgical synovectomy. Ahlberg et al. first reported the
use of radioactive gold (198Au) for synoviorthesis in hemophilia in 1969 (400), and it has been used more recently with success (401).
Disadvantages of gold include its relatively shallow depth of
penetration into the synovium, its relatively small colloidal size
(allowing leakage out of the joint following injection), and its
emission of γ-irradiation that can exert a harmful effect distant to
the joint into which it is injected. Yttrium (90Y) has advantages over (198Au)
including a greater depth of penetration, larger colloidal size, and
the fact that it is a pure β-radiation emitter. Several studies have
shown that yttrium synovectomy decreases or even eliminates bleeding in
injected joints for 1 to 12 years (402, 403, 404, 405). Chromic phosphate (32P)
is recommended by some because of its long half-life, which
theoretically minimizes the acute synovial inflammatory reaction that
is seen after exposure to agents with a short half-life. Without this
acute synovitis, the risk of immediate postsynoviorthesis bleeding is
lessened, making 32P synoviorthesis theoretically safer in
patients with inhibitors. Several series have shown that approximately
80% of the patients experience a decrease in bleeding for up to 15
years following 32P synoviorthesis (406, 407, 408).
Overall, radioactive synoviorthesis has shown encouraging results, with
over 90% of the patients experiencing a decrease in or
cessation of bleeding according to a recent metaanalysis (409).
Proponents of radiosynoviorthesis claim a potential saving of over 1
billion health care dollars in the United States if patients were
treated with radiosynovectomy rather than with surgical synovectomy (410).
dangers of radioactivity exposure in children, specifically chromosomal
damage leading to malignancy. Premalignant chromosomal changes can be
found in synovium shortly after the injection of radioactive isotopes,
but these changes disappear over time (411); besides, such changes can also be found in hemophilic joints that have not been exposed to radioactive isotopes (412). Pure β-radiation-emitting agents (90 and 186RH) do not induce chromosomal changes (412,413).
No patient has been reported to develop cancer attributable to
radiosynoviorthesis. Other potential complications of
radiosynoviorthesis include burns at the site of injection and
postinjection synovial inflammation. Clinicians who are interested in
performing these procedures should consult with radiation specialists
and other clinicians with experience in this type of procedure before
adding radiosynoviorthesis to their treatment armamentarium.
subjected to repeated hemarthroses. Knee flexion contractures are
common, as the knee is the most common site of bleeding. Contractures
usually follow the accumulation of articular damage, but can occur even
after only one hemarthrosis (414). Distension
of the joint capsule by the hemarthrosis forces the joint into a
position of flexion to increase joint volume and causes a reflex
inhibition of the quadriceps (415). Therefore,
mobilization of joints following hemarthroses is important for
preventing contractures. Immobilization for comfort following a painful
hemarthrosis should be limited to 1 or 2 days, and controlled
mobilization should be started thereafter under the guidance of a
physical therapist who has experience in treating patients with
hemophilia. Force should be avoided during range-of-motion exercises,
because it could cause trauma and stimulate further bleeding in the
joint. Gentle manual traction can improve joint mobilization following
an acute hemarthrosis (416). Pain from the
hemarthrosis or the accompanying muscle spasm can further limit the
range of motion, and such pain should be controlled adequately during
rehabilitation with analgesics, ice, and other modalities.
treatment strategies exist for improving motion or minimizing the
impact of the contracture on overall functioning. The stretching
regimens of physical therapy have not yet been proven to increase the
range of motion once a contracture has developed, but they can be
effective in minimizing the impact of a contracture by diminishing
pain, strengthening muscles, and improving posture and gait (416).
Serial casting has been recommended by many clinicians for the
treatment of established knee flexion contractures. Fernandez-Palazzi
et al. (417) achieved an average increase of 33
degrees of extension using a protocol of serial long-leg cast wedging
at a rate of 10 degrees every 2 to 3 days. One problem associated with
the passive correction of knee flexion contractures is posterior
subluxation of the tibia, especially if serial casting is used for
achieving terminal extension (417). To avoid
such subluxation during passive contracture correction, a variety of
devices have been described. A Quengel cast, with offset hinges and a
windlass mechanism, has been used with success (329), as has a modification of this device in the form of a removable orthosis (417, 418, 419).
Regardless of the device used, once the contracture has been corrected
to within 5 to 15 degrees of full extension, active exercises are
begun, with or without bracing, in order to prevent recurrent
contracture.
nonoperative treatment with casts or orthoses may require operative
procedures. Soft tissue releases were described by Hofmann et al. (420) in 1977, and included hamstring release and posterior capsulotomy. Wallny et al. (421)
retrospectively reviewed the results of this procedure in 19 patients
who were followed up for at least 5 years. Despite excellent short-term
results, improvements in extension lasted in only half of the patients.
Five patients showed deterioration after the operation. Other authors
have found disappointing results with hamstring releases (422).
An alternative to soft tissue release in the management of fixed knee
flexion contractures is an extension supracondylar femoral osteotomy.
Caviglia et al. (423) reviewed the results of
this operation in 19 patients, children and adults, with severe
arthropathy. All the patients gained enough extension to allow
ambulation. One patient developed recurvatum and another developed a
recurrent flexion deformity; both required revision osteotomies. An
Ilizarov external fixator can also be used to overcome flexion
contractures (424). Severely damaged joints
with flexion contractures can be returned to function by total knee
arthroplasty, but this procedure is rarely indicated in children and
will not be discussed in this chapter.
result from calf muscle bleeds and/or ankle arthropathy. Ankle
plantarflexion contractures impair ambulation and shoe wear, and tend
to worsen knee and hip flexion contractures. Llinas (425)
has classified ankle plantarflexion deformities according to their
primary etiology. Type 1 deformities are caused by joint distension
from recurrent hemarthroses and chronic synovitis. Stretching programs
and orthoses, combined with adequate control of hemostasis, can be
effective in overcoming this type of deformity (426).
Type 2 deformities are caused by an osteophyte on the anterior aspect
of the tibia impinging on the neck of the talus and causing a
mechanical block dorsiflexion. Treatment involves surgical cheilectomy.
Type 3 deformities result from advanced arthropathy. Typical features
of hemophilic arthropathy of the ankle include posterior capsular
contracture, Achilles tendon retraction, and collapse
of the talar dome from avascular necrosis (427,428).
Serial casting can be used in this type of deformity, although care
must be taken not to introduce secondary deformities of the midfoot if
the ankle joint itself is rigid. Surgical release can include
lengthening of the Achilles tendon, and posterior capsulotomy.
Tibiotalar fusion is reserved for cases of severe arthropathy. Type 4
ankle plantarflexion deformities result from leg pathology. Bleeds into
the calf can cause compartment syndrome, causing scarring in the
posterior compartments of the leg much like in Volkmann contracture of
the forearm. The resulting equinus contracture often requires
lengthening of the Achilles tendon, and posterior capsulotomy. If the
gastrocnemius muscle is sufficiently scarred, a knee flexion
contracture will also occur, requiring surgical treatment of the knee
at the same time. Postoperative casting and splinting is helpful in
preventing recurrent contracture (426).
significant arthropathy. Typical articular derangement that can be seen
on radiographs includes radial head enlargement, olecranon widening,
and large medial osteophytes (429).
Predominantly humeroulnar pathology is associated with decreased
flexion-extension, as well as ulnar nerve pathology from medial
osteophytes; predominantly radioulnar pathology is associated with loss
of forearm rotation, and global involvement leads to severe
contractures and significant functional limitations (429,430).
Concomitant lower limb pathology requiring the use of ambulatory aids
places a high demand on the joints of the upper extremity to bear
weight, simultaneously increasing the functional need for good joint
motion and resulting in bleeding that leads to impaired motion (431). Although recommended by some (432),
serial casting, Quengel casting, and dynamic splinting have not been
shown to be as effective in treating elbow flexion contractures as they
have in knee contractures (433). Surgical synovectomy with radial head excision has provided mixed results for range of motion (378,433,434).
The lack of proven nonsurgical or surgical means of correcting elbow
flexion contractures warrants investigation of newer procedures such as
open or arthroscopic capsulectomy.
 |
|
Figure 11.15
Computerized tomography confirmed a soft tissue hematoma in the region of the gluteus medius muscle of a 4-year-old boy with acute left buttock pain and swelling. With factor replacement, symptoms abated over 1 week. |
can cause avascular necrosis of the femoral head and significant
functional limitations (435). Hip aspiration
following hemarthrosis is recommended after adequate factor replacement
to lower the theoretical risk of intracapsular pressure on femoral head
circulation (436). Hemarthroses of the shoulder are very uncommon, especially in children (437),
and arthropathy of this joint is unlikely to be encountered by the
pediatric orthopaedist in the era of prophylaxis. Hemarthroses and
arthropathy of other joints are even less common.
The most common sites are the forearm, thigh, and calf. Muscle bleeds
typically present as stiffness, pain, swelling, and warmth. The
diagnosis can be confirmed by ultrasonography, if necessary. MRI and
computed tomography are more sensitive for detecting deep hematomas,
such as those occurring in the pelvis, but are typically unnecessary
for hematomas in the extremities (Fig. 11.15). Most bleeds resolve within a period of days if treated early with factor replacement (329).
Splinting of adjacent joints and restricted weight bearing may help to
alleviate pain in the acute phase. However, as with hemarthroses,
prolonged immobilization following muscle bleeds can lead to stiffness
and should be avoided. Rehabilitation for joint mobilization should be
begun as soon as bleeding has been controlled and symptoms have abated.
treatment of hemophilic soft tissue bleeding is the possibility of
compartment syndrome. Compartment pressures can become elevated
following untreated bleeding in the calf or forearm, and the resulting
ischemia of muscles and nerves can lead to severely disabling
consequences (439). Patients with high-titer
inhibitors are more likely to develop complications from bleeding in
muscles because of difficulty in obtaining hemostasis. If a compartment
syndrome is suspected, factor levels should be increased to 100%
acutely, and compartment pressures should be measured. If pressures are
elevated, fasciotomies should be performed
emergently
as in nonhemophilic patients. Special techniques, such as small
incisions, are not needed in patients with hemophilia; complete
compartment release is essential. Full factor replacement is instituted
preoperatively and maintained postoperatively, using activated factor
VII for patients with inhibitors. Impending compartment syndromes from
forearm bleeds can often be managed with factor replacement and close
inpatient monitoring. The resolution of symptoms within hours of factor
replacement can obviate the need for fasciotomies and potential
surgical complications.
hematomas. The most common neurologic complication from soft tissue
bleeding is femoral nerve compression from an iliopsoas hematoma (440).
Iliopsoas bleeding presents in a fashion similar to that of hip
hemarthrosis, with groin pain and hip flexion posturing. Passive
rotation of the hip is typically not painful with an iliopsoas hematoma
but is quite painful with a hip joint hemarthrosis. Ultrasound can rule
out hip effusion, and computed tomography can easily detect an
iliopsoas hematoma. The treatment of an iliopsoas hematoma involves
factor replacement, as with any other bleed, but iliopsoas bleeding
does not tend to respond as quickly as other soft tissue or joint
bleeds (438). Recurrent bleeding and prolonged
limitation of hip motion are common. Most femoral nerve palsies resolve
spontaneously following prompt and appropriate factor replacement and
limitation of weight bearing. In the upper extremity, the ulnar nerve
is the most common peripheral nerve affected by hemophilic pathology.
Large medial osteophytes associated with hemophilic elbow arthropathy
can cause ulnar nerve dysfunction, sometimes requiring surgical
transposition. The median nerve can also be acutely compressed in the
carpal tunnel with forearm bleeding (441). If control of the bleeding does not result in the return of median nerve function, carpal tunnel release is indicated.
The failure of symptoms to resolve following appropriate factor
replacement in an immunosuppressed individual raises suspicion of an
abscess. Aspiration can confirm the diagnosis, and incision and
debridement should be carried out with appropriate factor replacement.
classified pseudotumors into proximal and distal types. Proximal
pseudotumors are typically seen in adults in the proximal appendicular
skeleton or pelvis, and represent extraosseous bleeding. Computerized
tomography and MRI are helpful in evaluation (446). As conservative treatment is rarely effective (444),
treatment with surgical excision and prophylactic fixation of
substantially weakened bone is recommended, despite a high complication
rate (447). Percutaneous aspiration, curettage, and filling with fibrin glue or cancellous bone allograft have been used successfully (448), as has external beam radiation (449, 450, 451).
Distal pseudotumors typically occur intraosseously in the hands and
feet of children. These pseudotumors often resolve following long-term
factor replacement, but they may require surgical excision.
Limitation of physical activity because of physical impairment or fear
of bleeding tends to lower the incidence of fractures, whereas joint
contractures with adjacent osteopenia increase the likelihood of
periarticular fractures. Periarticular fractures can occur following
minimal trauma and should be ruled out in the setting of an acutely
painful, swollen joint following trauma, despite the tendency to
attribute the symptoms to hemarthrosis. Fractures can bleed
substantially, and even cause compartment syndrome. Therefore, factor
replacement should be begun immediately. The type and duration of
factor replacement required depends upon the disease, its type and
severity, the presence of inhibitors, and the patient’s prior responses
to factor replacement. Such decisions should be made under the guidance
of hematologic experts or the regional hemophilia treatment center.
Patients with severe hemophilia and those with fractures that may bleed
into enclosed compartments such as the forearm and leg may require
inpatient admission for close monitoring for 24 to 48 hours or longer.
Analgesia should be achieved with opioids, acetaminophen, and COX-2
inhibitors rather than with traditional NSAIDs.
hemophilia does not differ substantially from that in nonhemophilic
patients, because of the availability of factor replacement. It should
be noted, however, that any manipulation, including cast application or
change, requires preemptive correction of factor levels to at least 70%
of normal. During immobilization following casting, the patient,
family, and physician should carefully monitor for muscle or joint
bleeding that may cause tightness of the cast. The indications and
types of surgical treatment of fractures should be the same as in
nonhemophilic patients, given that hemostasis can be maintained with
factor replacement perioperatively. The fracture healing process is not
affected by hemophilia, and fracture union should be expected in
uncomplicated fractures. However, the immobilization required in the
treatment of fractures may have a significantly negative effect on
joint motion and overall function. Substantial rehabilitation may be
required in these patients, and prognostic expectations should be
adjusted accordingly (452).
prophylactic regimens has allowed hemophilic children to participate in
physical activity and organized sports. Recommended sports activities
for children with hemophilia have progressed over the past several
decades from
including only swimming to including many sports such as baseball and basketball (453). Physical exercise has been found to increase strength in hemophilic patients without increasing bleeding (454).
Regimented physical exercise has not been met with high compliance
rates in children, largely because such activities are considered
boring (455). Participation in age-appropriate
physical play and organized sports provides physical exercise as well
as a sense of normalcy in the life of the child with hemophilia. This
sense of normalcy, focusing on the health of the child rather than the
disease, is an important feature in the care of the child with any
chronic disease, including hemophilia (456).
Prophylactic factor replacement is important prior to participation in
sports, and the choice of sports should be individualized according to
the patient’s desires and functional levels. Contact sports are
generally discouraged among children with moderate or severe
hemophilia, although sports such as baseball and basketball are
possible with prophylactic factor replacement prior to each practice
and game. Measures to prevent sports injuries in children are currently
a popular topic of research, and such measures assume more importance
in children with hemophilia. As reviewed by Gilbert and Vlaskamp,
limiting the pitch counts may prevent elbow injuries during baseball,
specific training exercises may prevent knee injuries in jumping
sports, and the use of ankle orthoses may limit ankle injuries in
running and cutting sports (346). The
orthopaedist who is treating children with hemophilia must keep abreast
of developments in children’s sports safety, in order to maximize the
children’s ability to participate in physical activities with their
peers. Specific recommendations about participation in sports
activities should be made only in coordination with the child’s
treating hematologist.
children is changing. It is likely that in coming years, the
involvement of the pediatric orthopaedist in hemophilia treatment teams
will wane, because the widespread use of home treatment and
prophylaxis, begun early in life, will prevent or delay the occurrence
of profound hemophilic arthropathy until adulthood. Nonetheless,
patients with inhibitors continue to have problems with recurrent
hemarthroses and joint damage at a young age (457).
A promising future approach to treatment of hemophilia is the use of
gene therapy to create synthesis of the missing factor by the host. As
reviewed by High (458), two human clinical trials have been completed but not yet published, and three more are underway.
caused by deficient production or ineffective functioning of von
Willebrand factor (vWF), a high-molecular-weight glycoprotein that
promotes platelet aggregation in the early phases of hemostasis. vWF
also serves as a carrier protein for factor VIII in plasma, and
therefore plays an important role in intrinsic coagulation. vWD is the
most common inherited bleeding disorder, affecting 1% to 2% of children
in various populations (459,460).
The disorder can be broadly classified into three types: Type 1 vWD
refers to a partial quantitative deficiency of vWF; type 2 is a
qualitative (functional) deficiency of vWF; type 3 refers to the
virtually complete absence of vWF.
It is inherited in an autosomal dominant fashion in most cases.
Patients typically present with signs and symptoms of a mild
coagulopathy, such as easy bruising, prolonged nosebleeds, or abnormal
bleeding from surgical or dental procedures (462). In postmenarchal girls, heavy menses are a common finding (463).
Diagnosis of type 1 vWD is made by bleeding history, family history,
and low plasma levels of vWF antigen, ristocetin cofactor, and factor
VIII. Type 2 vWD comprises 15% to 30% of cases of vWD (461).
A wide variety of mutations, inherited in a variety of patterns,
accounts for several subtypes of type 2 vWD. The clinical presentation
is similar to that of type 1 vWD in most subtypes, although some
subtypes may have a presentation similar to hemophilia A because of
factor VIII deficiency caused by dysfunctional stabilization by the
abnormal vWD. Type 3 vWD is the least common major type, accounting for
approximately 5% to 10% of patients. Synthesis of vWF is impaired, and
vWF is virtually undetectable in the plasma. Factor VIII levels are
typically 1% to 5% of normal, making type 3 vWD clinically
indistinguishable from severe or moderate hemophilia A.
correcting the coagulation deficiency. The bleeding diathesis should be
corrected prior to any orthopaedic surgery or procedure likely to cause
bleeding, including injection and fracture manipulation. In most type 1
and type 2 patients, desmopressin (1-deamino-8-D-arginine vasopressin;
DDAVP), a synthetic analog of vasopressin that increases vWF and factor
VIII levels (464), is effective when given
intravenously, intranasally, or subcutaneously. Repeated infusions can
cause hyponatremia in children if given within a short period of time (465).
Amino acids such as aminocaproic acid and tranexamic acid can be used
as adjuncts to DDAVP, and can be given orally, topically, or
intravenously. For patients with Type 3 vWD or those otherwise
unresponsive to DDAVP, transfusion of blood products is the only way to
achieve and maintain hemostasis. Orthopaedic manifestations of vWD are
largely limited to type 3. Apart from the differences in factor
replacement, the treatment of muscle and joint hemorrhages and all
resulting complications in type 3 vWD parallels that in hemophilia.
refers to any condition that predisposes the individual to abnormally
increased thrombosis. Venous thromboembolic disease is a common concern
in adults, especially following prolonged immobilization or major
orthopaedic, abdominal, or pelvic surgery. Pathologic thrombosis in
children is uncommon, carrying with it a risk of venous thromboembolism
that is one tenth of the risk in adults (466).
Nonetheless, children may have several inherited conditions that cause
pathologic thrombosis, the most common of which are (a) protein C
deficiency, (b) protein S deficiency, (c) antithrombin deficiency, (d)
mutation of factor V (factor V Leiden), and (e) mutation of prothrombin
(G20210A prothrombin).
synthesized by the liver in a vitamin K–dependent fashion. Protein C
activation is mediated by thrombin. Activated protein C (APC) exerts
two antithrombotic effects: first, APC proteolytically inactivates
coagulation factors VIIIa and Va; second, it exerts a fibrinolytic
effect by inactivating plasminogen activator inhibitor-1. Deficient
production of protein C can result from one of over 150 mutations of
the protein C gene, located on chromosome 2 (467).
These mutations typically reduce APC activity to approximately 50% of
normal, and increase the risk of a venous thromboembolic event (VTE)
10-fold (468). Approximately 1 in 350 individuals (0.3%) are affected (469).
the liver in a vitamin K–dependent fashion. Protein S circulates in the
blood, mostly bound to complement factor C4b-binding protein (60%).
Free protein S (40%) inhibits thrombogenesis primarily by serving as an
APC cofactor. Over 30 mutations in the protein S gene located on
chromosome 3 have been found, any of which may cause protein S
deficiency (467). As with protein C, protein S mutations reduce activity to 50% of normal and increase thrombotic risk 10-fold (468). Protein S deficiency is estimated to be about as prevalent as protein C deficiency (468).
Inflammatory states that increase circulating levels of C4b-binding
protein can also increase the bound fraction of protein S, thereby
effectively reducing protein S activity.
inhibits factors IIa, Xa, IXa, XIa, and XIIa. The activity of
antithrombin is markedly increased by heparin. More than 50 mutations
of the antithrombin gene, located on chromosome 1, have been identified
(467). Antithrombin activity can be reduced by
either diminished production of normal protein or normal production of
a dysfunctional protein. Activity is typically reduced to 50% of
normal, thereby increasing the thrombotic risk 20-fold. Approximately 1
in 2500 individuals are affected (470).
Gln to Arg substitution at position 506 resulting from a point mutation
in the factor V gene on chromosome 1. Factor Va is normally inactivated
by APC by cleavage at one of several sites, including Arg 506. Factor
Va with the factor V Leiden mutation is resistant to cleavage by
protein C, and individuals with factor V Leiden are said to have APC
resistance. Factor V Leiden is found in approximately 4% of white
people, although its prevalence is highly variable among nationalities (468).
Heterozygosity for the factor V Leiden gene increases thrombotic risk
fourfold. Homozygosity for factor V Leiden is found in approximately 1
in 2000 whites and increases thrombotic risk 50-fold (471).
on chromosome 2 elevates prothrombin levels by 25%. This mutation
occurs in approximately 2% of white people and carries with it a
twofold to threefold increase in the risk for VTE (472).
thrombophilia consists of anticoagulation. The low rate of occurrence
of unprovoked VTE in children with known thrombophilia does not
generally warrant primary prophylaxis with anticoagulation (468).
However, the risk of thrombosis is higher than normal following
surgery, so perioperative prophylaxis with low-molecular-weight heparin
is recommended in patients with thrombophilia (473). In patients with antithrombin deficiency, antithrombin concentrates have been used effectively (474).
If a VTE occurs following surgery in a patient with thrombophilia,
anticoagulation should be administered acutely and continued for 6
months (468).
pathogenesis of Legg-Calve-Perthes disease (LCPD) has been the subject
of much recent investigation. Several series suggest that there may be
a higher rate of thrombophilic conditions in patients with LCPD
compared to the general population (475, 476, 477, 478), but this finding is not consistent (479, 480, 481, 482).
Therefore, the association between LCPD and thrombotic disorders is far
from clear and, at this point, one should neither draw etiologic
conclusions normake therapeutic decisions on the basis of this
association.
IgA-dominant immune deposits affecting small vessels; it typically
involves skin, gut, and kidney, and is also associated with arthralgias
or arthritis (483). HSP is the most common systemic vasculitis in children (484). The annual incidence ranges from 13.5 to 21.7 per 100,000, depending on the nature of the population (485,486). The occurrence of the condition in men is twice that in the women, and the disease is rare in African Americans (486). Ninety percent of the patients are younger than 10 years at presentation (486). However, the disease is uncommon in infants (487).
purpura (100%), arthritis (70%), gastrointestinal involvement (70%),
and renal involvement (37%) (488). The musculoskeletal manifestations of HSP are nonspecific.
Arthralgias are usually polyarticular, involving the large joints.
Swelling and tenderness may be present around the joint involved, but
this is not typical. The purpura are palpable and occur commonly on the
lower extremities, although they can occur anywhere (Fig. 11.16).
Gastrointestinal involvement includes abdominal pain, vomiting, melena,
and hematemesis. Renal involvement typically manifests as nephritis
with hematuria and proteinuria.
However, in approximately two-thirds of children with HSP, a
precipitating event or preceding febrile illness is not identifiable,
so such events in the history of the patient are not prerequisites for
the diagnosis (490,491).
The diagnosis is made by clinical features, because no diagnostic tests
are specific for HSP. Elevated serum IgA titers, or the presence of
IgA-rheumatoid factor in blood, or IgA deposits in skin biopsies
support the diagnosis.
Most signs and symptoms resolve after a few weeks, and few patients
have symptoms lasting longer than 1 month (490). Over 90% of the patients experience a full recovery without lasting sequelae (491).
The treatment of HSP in children is generally supportive, as the
disease course usually progresses to complete spontaneous resolution.
Severe kidney involvement may require medical treatment, but the
musculoskeletal complaints are generally self-limiting and require only
recognition.
 |
|
Figure 11.16 A skin lesion of Henoch-Schönlein purpura.
|
circulating procoagulant and anticoagulant factors. Two instances in
which this process may become apparent in the practice of pediatric
orthopaedics are disseminated intravascular coagulation (DIC) and
dilutional coagulopathy following transfusions for massive traumatic or
operative hemorrhage. The prevention, recognition, and treatment of
these states of abnormal hemostasis are essential, as they complicate
already serious illness or injury.
throughout the vascular system following serious infection or trauma.
The coagulation is typically a result of stimulation of thrombogenesis
and impairment of fibrinolysis, and is largely mediated by cytokines (492).
Cytokines that are expressed during a state of widespread inflammation
stimulate expression of tissue factor by endothelial cells, circulating
neutrophils, and monocytes. Tissue factor then activates factor VII,
setting off the intrinsic arm of the clotting cascade that ultimately
converts fibrinogen to fibrin (493). In DIC, the thrombosis progresses unchecked because the inflammatory state also impairs natural antithrombotic mechanisms (494). After thrombosis has begun in DIC, fibrinolysis to remove the coagulation is initially upregulated but ultimately impaired (495).
Widespread thrombosis causes organ damage by occluding
microcirculation, particularly in the kidneys, heart, lungs, and
central nervous system (496). Bleeding then
follows the widespread thrombosis as clotting factors are consumed.
Because mucosal and skin hemorrhages are easily detectable, bleeding
manifestations of DIC are detected more often than thrombotic
manifestations (497, 498, 499).
Most children with DIC have underlying problems such as congenital
anomalies, prematurity, malignancy, or hematologic abnormalities. DIC
can also follow severe head trauma in children (500).
A clinical coagulopathy resembling DIC can occur in children with large
venous malformations, in which intralesional thrombosis from stagnant
flow consumes clotting factors and leads to systemic coagulopathy (501).
abnormalities of hemostasis. Anemia and red blood cell fragmentation
due to hemolysis are present in 50% to 90% (499,502), and thrombocytopenia is apparent in 86% (499).
The bleeding time, PT, and PTT are prolonged because of the consumption
of platelets and clotting factors from all pathways. D-dimer, a fibrin
degradation product, and soluble fibrin monomer (sFM) can be reliably
measured and serve as indicators of fibrinolysis. Abnormalities in all
these tests can support the diagnosis of DIC, but none is pathognomonic.
inflammation and coagulation and obviate the need for treatment of the
DIC. Heparin or low-molecular-weight heparin can be used to counteract
the thrombotic tendency, but studies have shown highly variable results
(503,504).
The use of fresh frozen plasma or cryoprecipitate to counteract
bleeding or to prepare for an invasive procedure is equally risky and
controversial, as it may stimulate further thrombosis and end-organ
damage (496). Antithrombin can prevent
disseminated coagulation without increasing the bleeding risk, and has
shown promising results in clinical series in patients with DIC,
sepsis, and antithrombin deficits (505,506). Various other drugs are currently under investigation.
occurs in the setting of otherwise potentially life-threatening illness
and injury. A 42% mortality rate was found in a series of 100 patients (499). Multiorgan dysfunction causes most of the morbidity and mortality following DIC (497,498).
factors and platelets following fluid and blood resuscitation for
massive bleeding. Hemorrhage is a major cause of death in severely
injured patients who present to trauma centers (507).
Coagulopathy develops in these patients by several mechanisms:
hypothermia, acidosis, DIC from vascular injury and inflammation, and
dilution of coagulation factors and platelets during fluid and blood
resuscitation (508). Hypothermia accompanies
major trauma, often as a result of decreased motor activity, exposure
during evaluation and resuscitation, and rapid administration of
unwarmed intravenous fluids. For every degree Celsius of hypothermia,
enzymatic activity in the coagulation cascade decreases 10% (509). Coagulopathy becomes quite severe at below 34°C and virtually irreversible at below 32°C (510).
Acidosis, which also accompanies tissue trauma and hypoperfusion,
directly reduces coagulation activity and platelet function (511). Brain injury can release tissue factor, thereby causing DIC, and liver injury may impair synthesis of clotting factors.
hemorrhage begins with the shift of interstitial water into the
intravascular space because of the blood pressure dropping less than
the colloid osmotic pressure. This dilution increases with
administration of crystalloid solutions to restore blood pressure.
Administration of pRBCs further increases dilution. A unit of pRBCs
contains no functional platelets and only 35 mL of plasma. Factors V
and VIII are labile, and do not survive the processing of red blood
cells. Therefore, after transfusion of 10 U of pRBCs, it is estimated
that 70% of plasma has been lost and only 10% has been replaced,
leading to detectable abnormalities in the PT and PTT (511,512).
A recent mathematical model of blood loss and hemodilution predicted PT
to be the earliest and most sensitive indicator of coagulopathy arising
from hemodilution during hemorrhage (513).
sequestered in the spleen, are lost more slowly. Platelet counts may
remain at more than µL even after transfusion of 20 U of pRBCs (511).
essential. The most important first step is to control the hemorrhage
by surgical means. Aggressive fluid resuscitation prior to surgical
control of bleeding increases mortality (514).
If, despite surgical attempts to control hemorrhage, large-scale
transfusions are required, platelets and plasma should be
coadministered. Various recommendations have been made regarding the
ratios and timing of administration of blood products. The American
Society of Anesthesiologists recommends transfusion of blood products
in the trauma setting to maintain the PT and PTT at less than 1.5 times
normal, the platelet count at greater than 50 × 109 per L, coagulation factor activity levels at greater than 30% of normal, and fibrinogen at greater than 0.8 g per L (515). These values are unlikely to be reached until the loss and replacement of more than 150% of total blood volume (516).
Others recommend prophylactic administration of platelets and plasma
prior to detectable coagulopathy. Some physicians transfuse 1 U of
plasma for every unit of pRBCs over 10 U transfused, and 1 U of
platelets for every unit of pRBCs over 20 U transfused (511).
They recognize, however, that variability exists between patients, and
no routine algorithm will be universally successful. According to a
mathematical model, the optimal ratio of product transfusion (in units)
to prevent dilutional coagulopathy is 2:3 for plasma:pRBCs and 8:10 for
platelet:pRBCs (513). If plasma and platelet
transfusions were to be delayed, respectively, until after transfusion
of 3 and 10 U of pRBCs, coagulopathy would ensue.
clotting factors, especially when patients cannot tolerate the osmotic
load of fresh frozen plasma. Showing particular promise in small series
is recombinant factor VIIa, initially designed for use in patients with
hemophilia and high-titer inhibitors to factor VIII. The administration
of factor VIIa bypasses deficiencies in the intrinsic and extrinsic
pathways and has been shown to rapidly decrease microvascular bleeding
and save lives following dilutional coagulopathy (511).
been performed in adults, and extrapolation to children is difficult.
Nonetheless, the orthopaedist faced with severe traumatic or surgical
hemorrhage in a child should be aware of the possibility of dilutional
coagulopathy and work with the anesthesiologist and critical care
specialists to prevent, recognize, and treat this potentially fatal
event.
of platelet counts and coagulation studies as a guide to the
transfusion of platelets and plasma to keep the coagulation profile
within the parameters outlined earlier. We begin to check coagulation
profiles when estimated blood loss and replacement approaches 75% to
100% of blood volume, and then we check every 50% of blood volume
thereafter. If the profile values are within the thresholds outlined in
the preceding text but subjective surgical bleeding continues, further
blood products are given. If significant bleeding and ongoing
transfusion needs are likely to continue postoperatively, monitoring of
platelet counts and PT/PTT are required until the hemoglobin stabilizes
enough to obviate further pRBC transfusions. Strict unit cutoffs are
not practical in children because of the varying sizes of children and
consequently varying blood volumes.
children. Approximately 3250 cases are diagnosed annually in children
and adolescents, and the incidence is increasing (517, 518, 519).
Approximately 80% of childhood acute leukemia is acute lymphoblastic
leukemia (ALL), and 20% is acute nonlymphoblastic leukemia (ANLL). The
peak age at diagnosis is between 2 and 4 years. The pathogenesis of
childhood leukemia is multifactorial. A variety of genetic mutations
and chromosomal rearrangements have been shown to be associated with
acute leukemia, and these have been correlated with immunologic type
and clinical course. The Philadelphia chromosome, t(9;22)(934;q11), is
associated with an especially poor prognosis (520).
rates for children with ALL. In developed countries, current cure rates
for ALL approach or exceed 80%, depending upon the cell type, genotype,
and in some cases, the race of the patient (521, 522, 523).
important to recognize, as they may be the presenting signs and
symptoms and may be first seen by an orthopedist. Musculoskeletal pain
is common early in the course of acute leukemia, and is a presenting
complaint in 40% to 62% of patients (524,525). Pain in an extremity at presentation is seen more often in ALL than in leukocytosis (525). Almost 50% of patients in one series had gait abnormalities or refusal to walk at presentation (526).
Back pain is common at presentation and can be associated with
pathologic fractures of the vertebral bodies in the setting of severe
osteoporosis (527, 528, 529, 530, 531).
Pain and swelling in the joints, resembling juvenile rheumatoid
arthritis, is present in 11% to 13% of patients with ALL at
presentation (525, 532, 533, 534). Typically, more than one joint is involved, with knees, ankles, and elbows being most commonly affected (525).
The clinical picture of involvement of the joints can resemble septic
arthritis, and the diagnosis may be further confused by a mild
leukocytosis (535). These children may have
constitutional symptoms such as listlessness, poor appetite, and
low-grade fevers, which should raise suspicion of leukemia when they
occur in conjunction with back or joint pain.
is likely related to osteoporosis. A sixfold higher incidence of
fractures was found in a group of 61 children with ALL compared to
healthy age-matched controls (536). Most
fractures occurred in the extremities following trauma. The incidence
of fractures in ALL patients is as high as 12% to 38%, especially
during chemotherapy treatment (528,536,537).
Osteopenia is a common radiographic finding in acute leukemia. In
prospective studies, 21% to 30% of patients with ALL have bone mineral
density Z-scores below -1.645 SD (fifth percentile) as determined by
DEXA scans at the time of diagnosis (536,538). Abnormalities in bone metabolism may account for the osteoporosis induced by leukemia (538). Bone mineral density decreases further during the initial phase of intense chemotherapy (536, 537, 538).
the early stages of leukemia, even after skeletal involvement has
begun. Peripheral leukocytosis may not exist at the time of
presentation. Anemia is often present in early ALL and can provide a
clue to the diagnosis of leukemia. Platelets may be decreased in
leukemia, as opposed to the increase seen in sepsis associated with
joint infections. The erythrocyte sedimentation rate may be elevated,
but this result is also not specific.
of leukemia in a child with unexplained back or extremity pain.
Radiolucent metaphyseal bands are a common and early radiographic
finding in acute leukemia, and are thought to represent abnormal
mineralization of metaphyseal bone rather than infiltration of leukemia
cells (539). Periosteal new bone formation
along the diaphyses of long bones is also common. Radiolucent lesions
in cortical bone are also found, and most likely represent bone
destruction from leukemic infiltrates (539). These lesions can be found in the small bones of the hands and feet, metaphyses of long bones of the extremities, or skull (Fig. 11.17).
The prognostic significance of skeletal lesions in leukemia is unclear.
Abnormalities in marrow signal on MRI may also be present (540),
and MRI may be a valuable tool in evaluating a child with early
skeletal changes suggestive of leukemia in the setting of a normal
peripheral hematologic profile.
The consequences of missing the diagnosis and delaying the treatment
can be profound, so bone marrow biopsy is indicated when sufficient
clinical evidence exists to raise the suspicion of leukemia. A high
index of suspicion is needed, as no history, physical examination, or
laboratory finding is pathognomonic for acute leukemia. The diagnosis
of acute leukemia should be entertained for any
child
with unexplained musculoskeletal pain together with constitutional
symptoms, or with unusual abnormalities as seen on radiographic
examination. An oncologic consultation should be obtained in such cases.
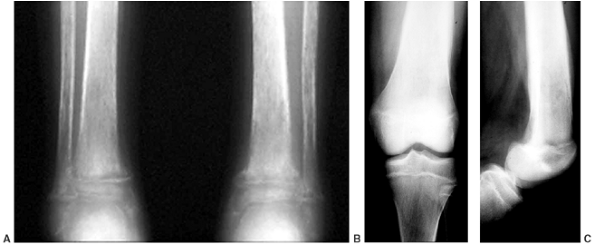 |
|
Figure 11.17 Leukemic infiltrates in the tibia and fibula (A) and distal femur (B, C)
in two patients with acute lymphocytic leukemia. The radiographic appearance of childhood leukemia can be quite variable and can mimic metabolic, infectious, or sarcomatous processes. |
leukemia. Symptomatic osteonecrosis was diagnosed in 15 of 1421
patients treated with intensive chemotherapy for childhood ALL in one
series (541). Most patients had multiple sites
of ON, with the femoral head being the most common site. The mean time
from diagnosis of ALL to diagnosis of ON was 17 months. Asymptomatic
osteonecrosis may be even more prevalent than symptomatic ON (542). Osteonecrosis can also follow bone marrow transplantation for childhood acute leukemia (543). Most cases of ON are attributed to the high doses of corticosteroids used in chemotherapy, or to bone marrow transplantation.
marrow transplantation places children with acute leukemia at high risk
for fungal infections. Cutaneous infection with fungi from the genus Aspergillus, which can be fatal (544),
may come to the attention of an orthopedist as a suspected abscess or
cellulitis. The lesion typically consists of a well-defined area of
skin necrosis surrounded by erythema and swelling, often at the prior
site of a venous access catheter. No purulent drainage is emitted.
Diagnosis is made by KOH preparations of smears from the margin of the
lesion. Treatment consists of wide excision followed by systemic
antifungal treatment under the guidance of an infectious diseases
specialist. Simple incision and drainage of a lesion is not only
ineffective, but may allow the fungal hyphae to spread into the
microvasculature, thereby contributing to widespread disease.
LG, Rosendorff J, Poole JE, et al. Comparative study of Fanconi anemia
in children of different ethnic origin in South Africa. Am J Med Genet 1994;52:279.
PF, Adler-Brecher B, Verlander PC, et al. The need for more accurate
and timely diagnosis in Fanconi anemia: a report from the International
Fanconi Anemia Registry. Pediatrics 1993;91:1116.
MP, Gertner JM, Huma Z, et al. Evaluation of growth and hormonal status
in patients referred to the International Fanconi Anemia Registry. Pediatrics 2001;107:744.
EC, Lopez KD, Huhn RD, et al. Evaluation of granulocyte-macrophage
colony-stimulating factor for treatment of pancytopenia in children
with Fanconi anemia. J Pediatr 1994;124:144.
PL, Cohn AK, Sedgwick WG, et al. Dysplasia of the knee associated with
the syndrome of thrombocytopenia and absent radius. J Bone Joint Surg Am 1984;66:421.
JA, Baraitser M, Wonke B. Autosomal dominant transmission of congenital
erythroid hypoplastic anemia with radial abnormalities. Am J Med Genet 1991;40:482.
A, Ibrahim IY, Rizk S, et al. Study of 22 Egyptian patients with
Diamond-Blackfan anemia, corticosteroids, and cyclosporin therapy
results. Pediatrics 2002;110:e44.
I, Ooka A, Brun A, et al. Gene transfer improves erythroid development
in ribosomal protein S19-deficient Diamond-Blackfan anemia. Blood 2002;100:2724.
J, Lauener R, Wick F, et al. Shwachman-Diamond syndrome: early bone
marrow transplantation in a high risk patient and new clues to
pathogenesis. Eur J Pediatr 1999;158:995.
O, Juvonen E, Dunkel L, et al. Anemia in children with cartilage-hair
hypoplasia is related to body growth and to the insulin-like growth
factor system. J Clin Endocrinol Metab 2000;85:563.
F, Siegrist CA, Ozsahin H, et al. Bone marrow transplantation in
cartilage-hair hypoplasia: correction of the immuno-deficiency but not
of the chondrodysplasia. Eur J Pediatr 1996;155:286.
J, Guilloud-Batailie M, Maier-Redelsperger M, et al. Decreased
morbidity in homozygous sickle cell disease detected at birth. Hemoglobin 2002;26:211.
MR, Thomas KA, Meyers SJ, et al. Bone mineral density of the lumbar
spine and proximal femur is decreased in children with sickle cell
anemia. Am J Orthop (Chatham, NJ) 1998; 27:43.
AT, Bererhi H, Darwish A, et al. Decreased bone mineral density in
prepubertal children with sickle cell disease: correlation with growth
parameters, degree of siderosis and secretion of growth factors. J Trop Pediatr 1998;44:194.
DJ, Bonnett C, Okolo SN, et al. Assessment of the bone status of
Nigerian children and adolescents with sickle cell disease using
calcaneal ultrasound and serum markers of bone metabolism. Calcif Tissue Int 2002;71:133.
C, Ely B, Brodecki D, et al. Characteristics of pain managed at home in
children and adolescents with sickle cell disease by using diary
self-reports. J Pain 2002;3:461.
KI, Orringer EP. Bone marrow necrosis in sickle cell disease: a
description of three cases and a review of the literature. Am J Med Sci 2000;320:342.
DL, Hibberd PL, Betit P, et al. Preliminary assessment of inhaled
nitric oxide for acute vaso-occlusive crisis in pediatric patients with
sickle cell disease. JAMA 2003;289:1136.
J, Thomas P, Serjeant GR. Septicemia caused by Salmonella infection: an
overlooked complication of sickle cell disease [Comment]. J Pediatr 1997;130:394.
FM, Sleeper LA, Weiner SJ, et al. Clinical events in the first decade
in a cohort of infants with sickle cell disease. Cooperative Study of
Sickle Cell Disease [Comment]. Blood 1995;86:776.
JB, Forsythe DA, Bertrand SL, et al. Retrospective review of
osteoarticular infections in a pediatric sickle cell age group. J Pediatr Orthop 2000;20:682.
RR, Hussein SS, Jeans WD, et al. A prospective study of soft-tissue
ultrasonography in sickle cell disease patients with suspected
osteomyelitis. Clin Radiol 2000;55:307.
MM, Hariharan V, Aradi AJ, et al. The value of ultrasound and
aspiration in differentiating vaso-occlusive crisis and osteomyelitis
in sickle cell disease patients. Clin Radiol 1999; 54:636.
DP, Heyneman LE, Ware RE, et al. MR features of soft-tissue
abnormalities due to acute marrow infarction in five children with
sickle cell disease. AJR Am J Roentgenol 1999;173:989.
H, Haramati N, Flusser G. The diagnostic role of gadolinium enhanced
MRI in distinguishing between acute medullary bone infarct and
osteomyelitis. Magn Reson Imaging 2000;18:255.
DL, Kim SK, Greene NW, et al. Differentiation between bone infarction
and acute osteomyelitis in children with sickle-cell disease with use
of sequential radionuclide bone-marrow and bone scans. J Bone Joint Surg Am 2001;83-A:1810.
JP, Rocha H, Scheld WM. Problems in salmonellosis: rationale for
clinical trials with newer beta-lactam agents and quinolones. Rev Infect Dis 1986;8:189.
P, Galacteros F, Bachir D, et al. Deformities of the hip in adults who
have sickle-cell disease and had avascular necrosis in childhood. A
natural history of fifty-two patients. J Bone Joint Surg Am 1991;73:81.
M, Kirkos J, Koussi A, et al. Avascular necrosis of the femoral head
among children and adolescents with sickle cell disease in Greece. Haematologica 2002;87:771.
EP, Neumayr LD, Haberkern C, et al. The perioperative complication rate
of orthopedic surgery in sickle cell disease: report of the National
Sickle Cell Surgery Study Group. Am J Hematol 1999;62:129.
EP, Haberkern CM, Neumayr L, et al. A comparison of conservative and
aggressive transfusion regimens in the perioperative management of
sickle cell disease. The Preoperative Transfusion in Sickle Cell
Disease Study Group [Comment]. N Engl J Med 1995;333:206.
EP, Styles LA, Colangelo LH, et al. Acute chest syndrome in sickle cell
disease: clinical presentation and course. Cooperative Study of Sickle
Cell Disease. Blood 1997;89:1787.
CH, Macklin EA, Moser FG, et al. Longitudinal changes in brain magnetic
resonance imaging findings in children with sickle cell disease. Blood 2002;99:3014.
ST, Macklin EA, Pegelow CH, et al. Silent infarction as a risk factor
for overt stroke in children with sickle cell anemia: a report from the
cooperative study of sickle cell disease. J Pediatr 2001;139:385.
C, Owusu-Ofori S. Prophylactic antibiotics for preventing pneumococcal
infection in children with sickle cell disease. Cochrane Database Syst Rev 2002;(3)CD003427.
TV, Sarnaik S, Buchanan GR, et al. Invasive pneumococcal infections in
children with sickle cell disease in the era of penicillin prophylaxis,
antibiotic resistance, and 23-valent pneumococcal polysaccharide
vaccination. J Pediatr 2003;143:438.
S, Terrin ML, Moore RD, et al. Effect of hydroxyurea on the frequency
of painful crises in sickle cell anemia. Investigators of the
Multicenter Study of Hydroxyurea in Sickle Cell Anemia [Comment]. N Engl J Med 1995;332:1317.
PJ, Almeida A, Halsey C, et al. Therapeutic challenges in childhood
sickle cell disease. Part 1: current and future treatment options. Br J Haematol 2003;120:725.
R, Westerman KA, Fabry ME, et al. Correction of sickle cell disease in
transgenic mouse models by gene therapy [Comment]. Science 2001;294:2368.
E, Keskin A, Suzer T, et al. Spinal cord compression secondary to
extramedullary hematopoiesis in thalassemia intermedia. Eur Spine J 1998;7:501.
C, Pekrun A, Bartz M, et al. Short stature and failure of pubertal
development in thalassaemia major: evidence for hypothalamic
neurosecretory dysfunction of growth hormone secretion and defective
pituitary gonadotropin secretion. Eur J Pediatr 1997;156:777.
A, Di Maio S, Baron I, et al. Final height and body disproportion in
thalassaemic boys and girls with spontaneous or induced puberty. Acta Paediatr 2000;89:1295.
NF, Nathan DG, MacMillan JH, et al. Survival in medi-cally treated
patients with homozygous beta-thalassemia [Comment]. N Engl J Med 1994;331:574.
YL, Pang LM, Chik KW, et al. Patterns of bone diseases in
transfusion-dependent homozygous thalassaemia major: predominance of
osteoporosis and desferrioxamine-induced bone dysplasia. Pediatr Radiol 2002;32:492.
Sanctis V, Savarino L, Stea S, et al. Microstructural analysis of
severe bone lesions in seven thalassemic patients treated with
deferoxamine. Calcif Tissue Int 2000;67:128.
Sanctis V, Stea S, Savarino L, et al. Osteochondrodystrophic lesions in
chelated thalassemic patients: an histological analysis. Calcif Tissue Int 2000;67:134.
A, De Sanctis V, Campisi S, et al. Treatment with deferiprone (L1) in a
thalassemic patient with bone lesions due to desferrioxamine. J Pediatr Endocrinol Metab 2000;13:677.
S, Sirikulchayanonta V, Angchaisuksiri P, et al. Abnormalities in bone
mineral density and bone histology in thalassemia. J Bone Miner Res 2003;18:1682.
E, Sioundas A, Karatzas N, et al. Bone mineral density of patients with
thalassemia major: four-year follow-up. Calcif Tissue Int 1999;64:481.
A, Conte G, Conti A, et al. Effects of 12 months rec-GH therapy on bone
and collagen turnover and bone mineral density in GH deficient children
with thalassaemia major. J Endocrinol Invest 2000;23:356.
P, Pizzarelli G, Spina M, et al. Quantitative ultrasound of bone and
clodronate effects in thalassemia-induced osteoporosis. J Bone Miner Metab 2003;21:402.
M, Abad MT, Pissard S, et al. Hydroxyurea can eliminate transfusion
requirements in children with severe beta-thalassemia. Blood 2003;102:1529.
SE, Roberts IA, Amrolia P, et al. Bone marrow transplantation for
beta-thalassaemia major: the UK experience in two paediatric centres. Br J Haematol 2003;120:289.
LM, Lankester AC, Giordano PC, et al. Paediatric allogeneic bone marrow
transplantation for homozygous beta-thalassaemia: the Dutch experience.
Bone Marrow Transpl 2003;31:1081.
F, Rocha V, Reed W, et al. Related umbilical cord blood transplantation
in patients with thalassemia and sickle cell disease. Blood 2003;101:2137.
C, Rivella S, Chadburn A, et al. Successful treatment of murine
beta-thalassemia intermedia by transfer of the human beta-globin gene. Blood 2002;99:1902.
JL, Binns HJ, Chen T, et al. Persistence and emergence of anemia in
children during participation in the Special Supplemental Nutrition
Program for Women, Infants, and Children. Arch Pediatr Adolesc Med 2002;156:1028.
S, Meert KL, Mooney JF, et al. Bleeding and coagulation changes during
spinal fusion surgery: a comparison of neuromuscular and idiopathic
scoliosis patients. Pediatr Crit Care Med 2002;3:364.
CN, Weinstein DA, Andrews NC. 2002 E. Mead Johnson Award for Research
in Pediatrics Lecture: the molecular biology of the anemia of chronic
disease: a hypothesis. Pediatr Res 2003;53:507.
JJ, Kane WJ, Laxdal SD, et al. Bone findings in chronic granulomatous
disease of childhood. A genetic abnormality of leukocyte function. J Bone Joint Surg Am 1969;51:1573.
SD, Finney T, Craver R, et al. Aspergillus osteomyelitis in patients
who have chronic granulomatous disease. Case report. J Bone Joint Surg Am 1991;73:456.
MW, Bocobo FC, Paul ME, et al. Successful medical therapy of
Aspergillus osteomyelitis of the spine in an 11-year-old boy with
chronic granulomatous disease. Pediatrics 1994;93:830.
S, Abinun M, Pistignjat B, et al. Aspergillus osteomyelitis in chronic
granulomatous disease: treatment with recombinant gamma-interferon and
itraconazole. Pediatr Infect Dis J 1996; 15:833.
N, Akasu Y, Yamane H, et al. Aspergillus osteomyelitis in a child who
has p67-phox-deficient chronic granulomatous disease. Kurume Med J 1999;46:87.
International Chronic Granulomatous Disease Cooperative Study Group. A
controlled trial of interferon gamma to prevent infection in chronic
granulomatous disease [Comment]. N Engl J Med 1991;324:509.
Giudice I, Iori AP, Mengarelli A, et al. Allogeneic stem cell
transplant from HLA-identical sibling for chronic granulomatous disease
and review of the literature. Ann Hematol 2003; 82:189.
A, Soresina A, Rondelli R, et al. Clinical, immunological, and
molecular analysis in a large cohort of patients with X-linked
agammaglobulinemia: an Italian Multicenter Study [Comment]. Clin Immunol 2002;104:221.
C, Natvig JB, Chattopadhyay H. Excessive suppressor T-cell activity of
the rheumatoid synovial tissue in X-linked hypogammaglobulinaemia. Scand J Immunol 1980; 11:455.
A, Webster AD, Furr PM, et al. Mycoplasmal arthritis in patients with
primary immunoglobulin deficiency: clinical features and outcome in 18
patients. Br J Rheumatol 1997;36:661.
ER, Vogler LB, Okos AJ, et al. B lymphocyte precursors in human bone
marrow: an analysis of normal individuals and patients with
antibody-deficiency states. J Immunol 1978; 120:1169.
P, Debre M, De Blic J, et al. Early and prolonged intravenous
immunoglobulin replacement therapy in childhood agammaglobulinemia: a
retrospective survey of 31 patients. J Pediatr 1999;134:589.
M, Le Chenadec J, Persoz A, et al. HIV-1-related encephalopathy in
infants compared with children and adults. French Pediatric HIV
Infection Study and the SEROCO Group. Neurology 2000;54:1089.
LM, Harris DR, Moye J, et al. Alternatives to HIV-1 RNA concentration
and CD4 count to predict mortality in HIV-1-infected children in
resource-poor settings. Lancet 2003;362:1625.
C, Rizzo C, Burlina AB, et al. Inborn errors of metabolism in the
Italian pediatric population: a national retrospective survey. J Pediatr 2002;140:321.
S, Abrahamov A, Hadas-Halpern I, et al. Adult-type Gaucher disease in
children: genetics, clinical features and enzyme replacement therapy. Q J Med 1993;86:565.
J, Andersson HC, Kaplan P, et al. The Gaucher registry: demographics
and disease characteristics of 1698 patients with Gaucher disease. Arch Intern Med 2000;160:2835.
G, Shapiro RS, Abdelwahab IF, et al. MR imaging in adults with Gaucher
disease type I: evaluation of marrow involvement and disease activity. Skeletal Radiol 1993;22:247.
G, Kornreich L, Hadar H, et al. Hemorrhage associated with “bone
crisis” in Gaucher’s disease identified by magnetic resonance imaging. Skeletal Radiol 1991;20:479.
G, Pastores GM, Abdelwahab IF, et al. Gaucher disease: assessment of
skeletal involvement and therapeutic responses to enzyme replacement. Skeletal Radiol 1997;26:687.
TR, Heyman S. Skeletal scintigraphy of pseudo-osteomyelitis in
Gaucher’s disease. Two case reports and a review of the literature. Clin Nucl Med 1992;17:279.
IJ, Kornreich L, Mekhmandarov S, et al. Effective treatment of painful
bone crises in type I Gaucher’s disease with high dose prednisolone. Arch Dis Child 1996;75:218.
K, Horev G, Grunebaum M, et al. The natural history of osteonecrosis of
the femoral head in children and adolescents who have Gaucher disease. J Bone Joint Surg Am 1996;78:14.
D, Rudensky B, Zimran A, et al. D-dimer assay in Gaucher disease:
correlation with severity of bone and lung involvement. Am J Hematol 2003;73:236.
L, Niggemeyer O, Kothe R, et al. Severe pathologic compression of three
consecutive vertebrae in Gaucher’s disease: a case report and review of
the literature. Eur Spine J 2003;12:97.
LA, Hoppel BE, Gerard EL, et al. Quantitative chemical shift imaging of
vertebral bone marrow in patients with Gaucher disease. Radiology 1992;182:451.
M, Akkerman EM, Venema HW, et al. Dixon quantitative chemical shift MRI
for bone marrow evaluation in the lumbar spine: a reproducibility study
in healthy volunteers. J Comput Assist Tomogr 2001;25:691.
M, van Kuijk C, Stoker J, et al. Quantification of bone involvement in
Gaucher disease: MR imaging bone marrow burden score as an alternative
to Dixon quantitative chemical shift MR imaging—initial experience. Radiology 2003;229:554.
EJ, Maas M, Akkerman EM, et al. Vertebra disc ratio as a parameter for
bone marrow involvement and its application in Gaucher disease. J Comput Assist Tomogr 2002;26:843.
NW, Brady RO, Dambrosia JM, et al. Replacement therapy for inherited
enzyme deficiency—macrophage-targeted glucocerebrosidase for Gaucher’s
disease [Comment]. N Engl J Med 1991;324:1464.
G, Schiffmann R, Parker CC, et al. Comparative efficacy of dose
regimens in enzyme replacement therapy of type I Gaucher disease
[Comment]. Blood Cells Mol Dis 2000;26:285.
Z, Okon K, Machaczka M, et al. Enzyme replacement therapy reduces
Gaucher cell burden but may accelerate osteopenia in patients with type
I disease—a histological study. Eur J Haematol 2003;70:273.
LW, Koch JA, vom Dahl S, et al. Magnetic resonance imaging of bone
marrow changes in Gaucher disease during enzyme replacement therapy:
first German long-term results. Skeletal Radiol 2001;30:496.
MR, Dardashti S, Liebman HA. Bone marrow response in treated patients
with Gaucher disease: evaluation by T1-weighted magnetic resonance
images and correlation with reduction in liver and spleen volume. Skeletal Radiol 2000;29:563.
G, Hill S, Wiggs E, et al. The efficacy of enzyme replacement therapy
in patients with chronic neuronopathic Gaucher’s disease. J Pediatr 2001;138:539.
P, Mazur A, Manor O, et al. Acceleration of retarded growth in children
with Gaucher disease after treatment with alglucerase. J Pediatr 1996;129:149.
IJ, Katz K, Kornreich L, et al. Low-dose high-frequency enzyme
replacement therapy prevents fractures without complete suppression of
painful bone crises in patients with severe juvenile onset type I
Gaucher disease [Comment]. Blood Cells Mol Dis 1998;24:296.
W, Peters C, Shapiro EG. Bone marrow transplantation as effective
treatment of central nervous system disease in globoid cell
leukodystrophy, metachromatic leukodystrophy, adrenoleukodystrophy,
mannosidosis, fucosidosis, aspartylglucosaminuria, Hurler,
Maroteaux-Lamy, and Sly syndromes, and Gaucher disease type III. Curr Opin Neurol 1999;12:167.
FM, Jeyakumar M, Andersson U, et al. Inhibition of substrate synthesis
as a strategy for glycolipid lysosomal storage disease therapy. J Inherit Metab Dis 2001;24:275.
RB, D’Angio GJ Jr. Langerhans’ cell histiocytosis (histiocytosis X):
experience at the Children’s Hospital of Philadelphia, 1970–1984. Med Pediatr Oncol 1989;17:20.
M, Girschikofsky M, Genereau T, et al. Langerhans cell histiocytosis in
adults. Report from the International Registry of the Histiocyte
Society. Eur J Cancer 2003;39:2341.
JS, Harty MP, Mahboubi S, et al. Langerhans cell histiocytosis:
presentation and evolution of radiologic findings with clinical
correlation. Radiographics 1995;15:1135.
PH, Jones CR, Steinman RM, et al. Langerhans cell (eosinophilic)
granulomatosis. A clinicopathologic study encompassing 50 years. Am J Surg Pathol 1996;20:519.
NE, Robertson WW Jr, Kilroy AW. Eosinophilic granuloma of the spine
with associated neural deficit. Report of three cases. J Bone Joint Surg Am 1980;62:1198.
E, Condom E, Canete JD, et al. Spinal cord compression by a unifocal
eosinophilic granuloma: a case report of an adult with unusual
roentgenological features. Neurosurgery 1988;23:666.
TN, Teh J, Goodman TR. Paediatric manifestations of Langerhans cell
histiocytosis: a review of the clinical and radiological findings. Clin Radiol 2003;58:269.
K, Diard F, Padovani J, et al. Unilateral mid-femoral periosteal new
bone of varying aetiology in children. Radiographic analysis of 25
cases. Pediatr Radiol 1986;16:475.
DJ, Azouz EM, Derbekyan V. Solitary lucent epiphyseal lesions in
children. Radiological case of the month. Solitary costal eosinophilic
granuloma. Skeletal Radiol 1988;17:497.
AS, Conway JJ, Miller JH, et al. Detection of bone lesions in
Langerhans cell histiocytosis: complementary roles of scintigraphy and
conventional radiography. J Pediatr Hematol Oncol 1996;18:51.
PC, Wang HS, Jaing TH, et al. From normal to abnormal MR findings
within three weeks in a solitary pelvic Langerhans histiocytosis. Skeletal Radiol 2003;32:481.
T, Silverman JF, Wakely PE Jr, et al. Fine-needle aspiration cytology
of Langerhans’ cell histiocytosis (eosinophilic granuloma) of bone in
children. Diagn Cytopathol 1991;7:261.
AW, Fanning CV, Ayala AG, et al. Percutaneous techniques for the
diagnosis and treatment of localized Langerhans-cell histiocytosis
(eosinophilic granuloma of bone). J Bone Joint Surg Am 1998;80:219.
BE, Feller AC, Pauli M, et al. Contemporary classification of
histiocytic disorders. The WHO Committee on Histiocytic/ Reticulum Cell
Proliferations. Reclassification Working Group of the Histiocyte
Society. Med Pediatr Oncol 1997;29:157.
SA, Grogan TM, Harris NL, et al. Tumours of histiocytes and accessory
dendritic cells: an immunohistochemical approach to classification from
the International Lymphoma Study Group based on 61 cases. Histopathology 2002;41:1.
RM, Thompson RC Jr, Voute PA, et al. Intralesional infiltration of
corticosteroids in localized Langerhans’ cell histiocytosis. J Pediatr Orthop 1992;12:811.
HM, Micke O, Olschewski T, et al. Radiotherapy is effective in
symptomatic Langerhans Cell Histiocytosis (LCH): long-term results of a
multicenter study in 63 patients. Int J Radiat Oncol Biol Phys 2003;57:S251.
JM, Evatt B, Jackson D. Occurrence of hemophilia in the United States.
The Hemophilia Surveillance System Project Investigators. Am J Hematol 1998;59:288.
R, Lavergne JM, Costa JM, et al. Unbalanced X-chromosome inactivation
with a novel FVIII gene mutation resulting in severe hemophilia A in a
female. Blood 2000;96:4373.
H, Richter H, Ringkamp H, et al. When are children diagnosed as having
severe haemophilia and when do they start to bleed? A 10-year
single-centre PUP study. Eur J Pediatr 1999; 158(Suppl 3):S166.
KM, Shanbrom E, Roberts HR, et al. A new high-potency
glycine-precipitated antihemophilic factor (AHF) concentrate. Treatment
of classical hemophilia and hemophilia with inhibitors. JAMA 1968;205:613.
L, Desmet VJ, Popper H, et al. Histologic patterns of liver disease in
hemophiliacs, with special reference to morphologic characteristics of
non-A, non-B hepatitis. Semin Liver Dis 1987;7:203.
ME, Gail MH, Ballard JO, et al. Natural history of human
immunodeficiency virus infections in hemophiliacs: effects of T-cell
subsets, platelet counts, and age. Ann Intern Med 1987;107:1.
Guidelines on the selection and use of therapeutic products to treat
haemophilia and other hereditary bleeding disorders. Haemophilia 2003;9:1.
AD, Gilchrist GS, Hoots WK, et al. Prospective, randomised trial of two
doses of rFVIIa (NovoSeven) in haemophilia patients with inhibitors
undergoing surgery. Thromb Haemost 1998;80:773.
JM, Roberts HR, Davignon G, et al. A randomized, double-blind
comparison of two dosage levels of recombinant factor VIIa in the
treatment of joint, muscle and mucocutaneous haemorrhages in persons
with haemophilia A and B, with and without inhibitors. rFVIIa Study
Group. Haemophilia 1998;4:790.
S, Blei F, Fetten J, et al. Human coagulation factor FVIIa
(recombinant) in the management of limb-threatening bleeds unresponsive
to alternative therapies: results from the NovoSeven emergency-use
programme in patients with severe haemophilia or with acquired
inhibitors. Blood Coagul Fibrin 2000;11:255.
H, Brewin T, Boey W, et al. Cost-utility analysis of recombinant factor
VIIa (NovoSeven) in six children with long-standing inhibitors to
factor VIII or IX. Haemophilia 2001;7:279.
M, Gracia J, Jimenez-Yuste V et al. Haematological substitutive
treatment of haemophilic haemarthroses. In: Rodriguez-Merchan EC, ed., The haemophilic joints: new perspectives. Oxford: Blackwell, 2003:20.
CT, Burke C. Double-blind studies on the use of steroids in the
treatment of acute hemarthrosis in patients with hemophilia. N Engl J Med 1970;282:639.
G, Vianen ME, Wenting MJ, et al. Iron deposits and catabolic properties
of synovial tissue from patients with haemophilia. J Bone Joint Surg Br 1998;80:540.
G, van Rinsum AC, Vianen ME, et al. Haemophilic arthropathy resembles
degenerative rather than inflammatory joint disease. Histopathology 1999;34:144.
P, Rolland N, Lebrun T, et al. Epidemiological survey of the
orthopaedic status of severe haemophilia A and B patients in France.
The French Study Group. secretariat.haemophiles@cch.ap-hop-paris.fr. Haemophilia 2000;6:23.
MA, Gulmans VA, Helders PJ, et al. Motor performance and disability in
Dutch children with haemophilia: a comparison with their healthy peers.
Haemophilia 2001;7:293.
WB, Yankaskas BC, Guilford WB. Roentgenographic classifications of
hemophilic arthropathy. Comparison of three systems and correlation
with clinical parameters. J Bone Joint Surg Am 1989;71:237.
A, Czyrny Z, Laguna P, et al. Correlation between clinical,
radiological and ultrasonographical image of knee joints in children
with haemophilia. Haemophilia 2001;7:286.
RW, Schmidt WA, Bohl-Buhler M, et al. [Technique and value of
arthrosonography in rheumatologic diagnosis. 1: Ultrasound diagnosis of
the knee joint]. Z Rheumatol 2001; 60:139.
T, Trattnig S, Male C, et al. Magnetic resonance imaging in hemophilic
children: value of gradient echo and contrast-enhanced imaging. Magn Reson Imaging 1999;17:199.
M, Kunze V, Hamann M, et al. [Hemophiliac arthropathy of the knee
joint. Gd-DTPA-enhanced MRI; clinical and roentgenological
correlation]. Rofo Fortschr Geb Rontgenstr Neuen Bildgeb Verfahr 1994;160:154.
I, Le Balc’h T, Yvart J, et al. MR imaging of hemophilic arthropathy of
the knee: classification and evolution of the subchondral cysts. Magn Reson Imaging 1992;10:67.
MB, Schmidt H, Becker S, et al. Modified magnetic resonance imaging
score compared with orthopaedic and radiological scores for the
evaluation of haemophilic arthropathy. Haemophilia 2002;8:98.
M, Lucia JF, Aguilar C, et al. Value of magnetic resonance imaging for
the diagnosis and follow-up of haemophilic arthropathy. Haemophilia 2003;9:76.
LM, Haschmeyer RH, Pettersson H. A longitudinal study of orthopaedic
outcomes for severe factor-VIII-deficient haemophiliacs. The
Orthopaedic Outcome Study Group. J Intern Med 1994;236:391.
PS, Teutsch SM, Shaffer PA, et al. Episodic versus prophylactic
infusions for hemophilia A: a cost-effectiveness analysis. J Pediatr 1996;129:424.
K, Astermark J, van der Bom JG, et al. Prophylactic treatment for
severe haemophilia: comparison of an intermediate-dose to a high-dose
regimen. Haemophilia 2002;8:753.
J, Petrini P, Tengborn L, et al. Primary prophylaxis in severe
haemophilia should be started at an early age but can be
individualized. Br J Haematol 1999;105:1109.
RM, Spencer EE, Wojtys EM. The role of arthroscopic synovectomy in the
management of hemarthrosis in hemophilia patients: financial
perspectives. Arthroscopy 2002;18:789.
F, Viso R, Bernal R, et al. Oxytetracycline clorhydrate as a new
material for chemical synoviorthesis in haemophilia. In:
Rodriguez-Merchan EC, ed. The haemophilic joints: new perspectives. Oxford: Blackwell, 2003:80.
EC, Villar A, Quintana M, et al. Intra-articular corticosteroid therapy
of haemophilic synovitis of the knee. In: Rodriguez-Merchan EC, Goddard
NJ, Lee C, eds. Musculoskeletal aspects of haemophilia. Oxford: Blackwell, 2000:57.
T, Brackmann HH, Semper H, et al. Intra-articular hyaluronic acid in
the treatment of haemophilic arthropathy of the knee. Clinical,
radiological and sonographical assessment. Haemophilia 2000;6:566.
F, Viso R, Boadas A, et al. Intra-articular hyaluronic acid in the
treatment of haemophilic chronic arthropathy. Haemophilia 2002;8:375.
A, Mikulowski P, Odelberg-Johnson O. Intra-articular injection of
radioactive gold in treatment of chronic synovial effusion in the knee.
Acta Rheumatol Scand 1969;15:81.
EC, Magallon M, Martin-Villar J, et al. Long term follow up of
haemophilic arthropathy treated by Au-198 radiation synovectomy. Int Orthop 1993;17:120.
EC, Jimenez-Yuste V, Villar A, et al. Yttrium-90 synoviorthesis for
chronic haemophilic synovitis: Madrid experience. Haemophilia 2001;7(Suppl 2):34.
GE, Girard M, Belanger R, et al. Synoviorthesis with colloidal chromic
phosphate for the treatment of hemophilic arthropathy. J Bone Joint Surg Am 1994;76:482.
E, Mikosch P, Gallowitsch HJ, et al. Clinical outcome of
radiosynoviorthesis: a meta-analysis including 2190 treated joints. Nucl Med Commun 2002;23:683.
de Vargas A, Fernandez-Palazzi F. Cytogenetic studies in patients with
hemophilic hemarthrosis treated by 198Au, 186Rh, and 90Y radioactive
synoviorthesis. J Pediatr Orthop B 2000;9:52.
T, Eickhoff HH, Raderschadt G, et al. Hamstring release and posterior
capsulotomy for fixed knee flexion contracture in haemophiliacs. Haemophilia 1999;5(Suppl 1):25.
HA, Perez-Bianco R, Galatro G, et al. Extensor supracondylar femoral
osteotomy as treatment for flexed haemophilic knee. Haemophilia 1999;5(Suppl 1):28.
PD, McMahon C, Smith OP, et al. The treatment of flexion contracture of
the knee using the Ilizarov technique in a child with haemophilia B. Haemophilia 2003;9:336.
M, Wershavski M, Martinowitz U, et al. Elbow joint, crutches and
locomotion: special reference to persons with haemophilia. Haemophilia 2000;6:556.
EC, Goddard NJ. Muscular bleeding, soft-tissue haematomas and
pseudotumors. In: Rodriguez-Merchan EC, Goddard NJ, Lee C, eds. Musculoskeletal aspects of haemophilia. Oxford: Blackwell, 2000:85.
M, Martinowitz U, Horoszowski H. The short foot syndrome—an unfortunate
consequence of neglected raised intracompartmental pressure in a severe
hemophilic child: a case report. Angiology 1986;37:128.
MS. Surgical management of the adult haemophilic blood cyst
(pseudotumor). In: Rodriguez-Merchan EC, Goddard NJ, Lee C, eds. Musculoskeletal aspects of haemophilia. Oxford: Blackwell, 2000:92.
G, Yeh HC, Gilbert MS. Computed tomography and ultrasonography of the
hemophilic pseudotumor and their use in surgical planning. Skeletal Radiol 1986;15:123.
AJ, Kessler CM, Ewenstein BM. Management of von Willebrand disease: a
survey on current clinical practice from the Haemophilia Centres of
North America. Haemophilia 2001;7:235.
RA, Economides DL, Sabin CA, et al. Assessment of menstrual blood loss
and gynaecological problems in patients with inherited bleeding
disorders. Haemophilia 1999;5:40.
JD, Gader AM, da Costa J. Proceedings: The release of plasminogen
activator and factor VIII to lysine vasopressin, arginine vasopressin,
I-desamino-8-d-arginine vasopressin, angiotensin and oxytocin in man. Br J Haematol 1974;27:363.
M, David M, Adams M, et al. Venous thromboembolic complications (VTE)
in children: first analyses of the Canadian registry of VTE. Blood 1994;83:1251.
FR, Koster T, Vandenbroucke JP, et al. High risk of thrombosis in
patients homozygous for factor V Leiden (activated protein C
resistance). Blood 1995;85:1504.
SR, Rosendaal FR, Reitsma PH, et al. A common genetic variation in the
3′-untranslated region of the prothrombin gene is associated with
elevated plasma prothrombin levels and an increase in venous
thrombosis. Blood 1996;88:3698.
D, O’Malley JP, Schorr JB, et al. Evaluation of the safety, recovery,
half-life, and clinical efficacy of antithrombin III (human) in
patients with hereditary antithrombin III deficiency. Cooperative Study
Group. Blood 1990;75:33.
CJ, Glueck HI, Greenfield D, et al. Protein C and S deficiency,
thrombophilia, and hypofibrinolysis: pathophysiologic causes of
Legg-Perthes disease. Pediatr Res 1994;35:383.
VR, Belangero WD, Ozelo MC, et al. Inherited risk factors for
thrombophilia among children with Legg-Calve-Perthes disease. J Pediatr Orthop 1999;19:84.
CJ, Crawford A, Roy D, et al. Association of antithrombotic factor
deficiencies and hypofibrinolysis with Legg-Perthes disease. J Bone Joint Surg Am 1996;78:3.
S, Reitinger T, Linhart W, et al. The role of inherited thrombotic
disorders in the etiology of Legg-Calve-Perthes disease. J Pediatr Orthop 1999;19:82.
MT, McDougall PA, Gorlin JB, et al. Prospective reevaluation of the
association between thrombotic diathesis and Legg-Perthes disease. J Bone Joint Surg Am 2002;84-A:1613.
JC, Falk RJ, Andrassy K, et al. Nomenclature of systemic vasculitides.
Proposal of an International Consensus Conference. Arthritis Rheum 1994;37:187.
R, Martinez-Taboada VM, Rodriguez-Valverde V, et al. Cutaneous
vasculitis in children and adults. Associated diseases and etiologic
factors in 303 patients. Medicine (Baltimore) 1998; 77:403.
JM, Dolezalova P, Cummins C, et al. Incidence of Henoch-Schonlein
purpura, Kawasaki disease, and rare vasculitides in children of
different ethnic origins. Lancet 2002;360:1197.
C, Calvino MC, Llorca J, et al. Henoch-Schonlein purpura in children
and adults: clinical differences in a defined population. Semin Arthritis Rheum 2002;32:149.
R, Martinez-Taboada VM, Rodriguez-Valverde V, et al. Henoch-Schonlein
purpura in adulthood and childhood: two different expressions of the
same syndrome. Arthritis Rheum 1997;40:859.
M, van der Poll T, ten Cate H, et al. The cytokine-mediated imbalance
between coagulant and anticoagulant mechanisms in sepsis and
endotoxaemia. Eur J Clin Invest 1997;27:3.
RR, Bevilacqua MP, Sawdey M, et al. Cytokine activation of vascular
endothelium. Effects on tissue-type plasminogen activator and type 1
plasminogen activator inhibitor. J Biol Chem 1988;263:5797.
S, Schneider W, Kreuz W, et al. Post-trauma coagulation and
fibrinolysis in children suffering from severe cerebro-cranial trauma. Eur J Pediatr 1999;158(Suppl 3):S197.
E, Enjolras O, Laurian C, et al. Coagulation abnormalities associated
with extensive venous malformations of the limbs: differentiation from
Kasabach-Merritt syndrome. Clin Lab Haematol 2002;24:243.
BS, Rubenberg ML, Dacie JV, et al. Microangiopathic haemolytic anaemia:
mechanisms of red-cell fragmentation: in vitro studies. Br J Haematol 1968;14:643.
N, Hasegawa H, Maki M, et al. Clinical evaluation of
low-molecular-weight heparin (FR-860) on disseminated intravascular
coagulation (DIC)—a multicenter cooperative double-blind trial in
comparison with heparin. Thromb Res 1993;72:475.
F, Chopin C, Huart JJ, et al. Double-blind, placebo-controlled trial of
antithrombin III concentrates in septic shock with disseminated
intravascular coagulation. Chest 1993;104:882.
B, Lamy M. Clinical experience with antithrombin III concentrates in
critically ill patients with sepsis and multiple organ failure. Semin Thromb Hemost 1998;24:71.
N, Moore EE, Sauaia A, et al. Predicting life-threatening coagulopathy
in the massively transfused trauma patient: hypothermia and acidoses
revisited. J Trauma 1997;42:857.
DD, Trask A, Soeken K, et al. Hypothermic coagulopathy in trauma:
effect of varying levels of hypothermia on enzyme speed, platelet
function, and fibrinolytic activity. J Trauma 1998;44:846.
WH, Wall MJ Jr, Pepe PE, et al. Immediate versus delayed fluid
resuscitation for hypotensive patients with penetrating torso injuries.
N Engl J Med 1994;331:1105.
ASo. Practice guidelines for blood component therapy: A report by the
American Society of Anesthesiologists Task Force on Blood Component
Therapy. Anesthesiology 1996; 84:732.
ST, Myllyla GJ, Vahtera EM. Hemostatic factors and replacement of major
blood loss with plasma-poor red cell concentrates. Anesth Analg 1995;81:360.
DE, Cote TR, Clegg L, et al. SEER update of incidence and trends in
pediatric malignancies: acute lymphoblastic leukemia. Med Pediatr Oncol 2002;39:554.
FM, Nachman JB, Sather HN, et al. Clinical significance of Philadelphia
chromosome positive pediatric acute lymphoblastic leukemia in the
context of contemporary intensive therapies: a report from the
Children’s Cancer Group. Cancer 1998;83:2030.
S, Pastore G, Mosso ML, et al. Population-based survival after
childhood cancer diagnosed during 1970-98: a report from the Childhood
Cancer Registry of Piedmont, Italy. Haematologica 2003;88:974.
JM, Silverman LB, Levy DE, et al. Childhood T-cell acute lymphoblastic
leukemia: the Dana-Farber Cancer Institute Acute Lymphoblastic Leukemia
Consortium Experience. J Clin Oncol 2003;21:3616.
OG, Sartain P, Ducore JM, et al. Bone pain as an initial symptom of
childhood acute lymphoblastic leukemia: association with nearly normal
hematologic indexes. J Pediatr 1990; 117:233.
JM, Atkinson SA, Fraher L, et al. Mineral homeostasis and bone mass at
diagnosis in children with acute lymphoblastic leukemia. J Pediatr 1995;126:557.
SD, Gallagher D, Warrior R, et al. The prognostic significance of the
skeletal manifestations of acute lymphoblastic leukemia of childhood. J Pediatr Orthop 1994;14:105.
G, Fama P, Lo Nigro L, et al. Marked osteoporosis and spontaneous
vertebral fractures in children: don’t forget, it could be leukemia. Med Pediatr Oncol 2003;41:450.
BE, Goldsmith DP, Athreya BH. Differentiation of systemic juvenile
rheumatoid arthritis from acute leukemia near the onset of disease. J Pediatr 1993;122:595.
RG, Buhler T, Willich E, et al. Absence of prognostic significance of
skeletal involvement in acute lymphocytic leukemia and non-Hodgkin
lymphoma in children. Pediatr Radiol 1985;15:245.
der Sluis IM, van den Heuvel-Eibrink MM, Hahlen K, et al. Altered bone
mineral density and body composition, and increased fracture risk in
childhood acute lymphoblastic leukemia. J Pediatr 2002;141:204.
SA, Halton JM, Bradley C, et al. Bone and mineral abnormalities in
childhood acute lymphoblastic leukemia: influence of disease, drugs and
nutrition. Int J Cancer Suppl 1998;11:35.
V, Wysocki M, Odrowaz-Sypniewska G, et al. Bone mass and bone mineral
metabolism at diagnosis and after intensive treatment in children with
acute lymphoblastic leukemia. Med Pediatr Oncol 2003;41:578.
M, Boccalatte MF, Silvestri D, et al. Osteonecrosis: an emerging
complication of intensive chemotherapy for childhood acute
lymphoblastic leukemia. Haematologica 2003;88:747.
AE, Paakko E, Lanning FP, et al. Osteonecrosis during the treatment of
childhood acute lymphoblastic leukemia: a prospective MRI study. Med Pediatr Oncol 1999;32:11.
P, Witvoet J, Sedel L. Avascular necrosis of the femoral head after
allogenic bone-marrow transplantation. A retrospective study of 27
consecutive THAs with a minimal two-year follow-up. J Bone Joint Surg Br 1996;78:878.