
and acquired disorders that affect localized or regional areas of the
pediatric musculoskeletal system. These entities often share a common
clinical presentation and underlying pathologic process, yet they are
not confined to any one part of the skeleton nor are they caused by a
recognized systemic abnormality.
practice. Most are of no clinical importance, but occasionally these
anomalies can be deforming or life-threatening and require
interdisciplinary management. These anomalies may either occur alone,
or form the organizing features of various syndromes (1).
is used here to describe pediatric vascular disorders with primarily
dermatologic or visceral manifestations, excluding abnormalities of the
heart and great vessels. The term hemangioma
has been traditionally used to describe every type of vascular anomaly,
but Mulliken et al. have recently devised a classification system to
better clarify these lesions on the basis of their endothelial
characteristics (2,3,4).
Two main groups of disorders have been identified: the hemangiomas of
infancy, that is, vascular tumors with an early proliferative and later
involuting stage; and vascular malformations, namely, a heterogeneous
group of vascular lesions comprising dysplastic vessels, which may be
capillary, venous, arterial, lymphatic, or a combination of these.
vascular system are divided into vasculogenesis, angiogenesis, and
lymphangiogenesis. These morphogenic processes are controlled by the
interactions and ordered effects of many angiogenic and antiangiogenic
factors (5). Genes that have been implicated in
vascular tumors and malformations can be grouped according to
functional pathways, primarily the kinase receptor signaling pathways.
The main effect is on the vascular endothelial cells that play an
important role in angiogenic vessel formation.
intramuscular, or visceral. The location is most commonly in the head
and neck (60%) followed in frequency by the trunk and extremities. Most
occur singly but 20% proliferate in multiple sites.
A small cutaneous mark is present early in life, which grows rapidly to
become a raised, bosselated, vivid red color that blanches poorly.
Subsequent slow, spontaneous involution occurs with complete regression
in 70% of children by 7 years (3). Although no
definite cause has been elucidated, recent studies have characterized
abnormalities in endothelial cells from hemangiomas and have identified
a possible locus on chromosome 5q31-33 (7).
Lumbosacral hemangiomas and other lesions such as hypertrichosis and
dimpling should alert the orthopaedist to an underlying occult spinal
dysraphism.
occur in the lower dermis or muscle. The overlying skin may be only
slightly raised or bluish. On palpation, the mass is fibrofatty,
similar to the superficial hemangioma. These deep hemangiomas can be
mistaken for venous malformations (VMs) or lymphatic malformations
(LMs), which are usually soft and compressible unless thrombosed. In
the case of deep hemangiomas, spontaneous involution usually occurs.
Sonography can differentiate between a hemangioma and a vascular
malformation, but magnetic resonance imaging (MRI) results are
considered the gold standard (6,8).
scarring. Superficial ulceration and bleeding are uncommon. Up to 20%
of hemangiomas can cause significant complications including
destruction of involved tissues or obstruction of a vital structure
such as the eye or airway (6). High-output
congestive heart failure can result from large hemangiomas, and
gastrointestinal bleeding can occur because of intestinal involvement.
Kasabach-Merritt syndrome, or thrombocytopenic coagulopathy, was
recently shown to be associated with a rare vascular tumor known as kaposiform hemangioendothelioma, and not with the common hemangioma (9).
The slow-flow anomalies include capillary malformations (CMs) (port
wine stains and telangiectasias), VMs, and LMs (previously known as lymphangiomas and cystic hygromas).
Arterial malformations (AMs) and arteriovenous malformations (AVMs) are
fast-flow anomalies. Unlike the vascular tumors, vascular malformations
do not regress spontaneously and can worsen, depending on the type.
Combined, complex malformations occur. An example of this is the
Klippel-Trenaunay syndrome, a slow-flow, capillary-venous and often
lymphatic malformation (CVLM) that results in limb overgrowth. Typical
vascular malformations are outlined in Table 10.1.
These are flat, red or purple cutaneous lesions, usually on the head or
neck, which occur in less than 1% of newborns. The etiology of these
vascular malformations has not been elucidated, but linkage analysis
has identified a locus, CMCl, on 5q13-22 (11).
Most of these are cosmetic vascular birthmarks but some indicate an
underlying condition such as Sturge-Weber syndrome. This is a
nonhereditary syndrome characterized by a capillary malformation in the
trigeminal nerve distribution, and more importantly, an associated
vascular anomaly of the ipsilateral choroid and leptomeninges (1,4,6).
Children with this syndrome may develop seizures, hemiplegia,
developmental delays, and retinal damage. This disorder must be
considered when assessing a child with apparent cerebral palsy. MRI of
the brain is usually diagnostic. Facial and limb CMs may be associated
with soft tissue and bone hypertrophy, which are usually seen at birth.
These malformations may be part of a complex vascular anomaly such as
Klippel-Trenaunay syndrome, which will be discussed later. Pulsed-dye
laser treatment will improve the appearance of these lesions.
disorder seen at birth, characterized by a deep purple, serpiginous,
reticulated cutaneous pattern, usually involving the
trunk and extremities (4,12).
The cutaneous findings usually improve spontaneously but venous
dilation becomes more prominent, and atrophy of the involved limb with
leg-length discrepancy can occur (12). There are no reports in the literature about the efficacy of epiphysiodesis in this disorder.
|
TABLE 10.1 ASSOCIATIONS OF VASCULAR MALFORMATIONS IN CHILDREN
|
||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||||||||||||||||||||||||||||||||||
Rendu-Osler-Weber syndrome is an autosomal dominant disorder
characterized by multisystemic vascular malformations and hemorrhage.
Spiderlike red maculopapules are seen on the face and mucous membrane
in the first decade of life (6). The resultant
arteriovenous fistulas in the lungs and gastrointestinal tract can lead
to hemorrhage and cardiac failure. This disorder is caused by mutations
in two genes (endoglin and the activin receptorlike kinase) that have a
predominant effect on the endothelial cells. It is interesting to note
that both may be receptors for the TGF-[β] and may be important in the
switch from the activation phase of angiogenesis to the resolution
phase (13).
autosomal recessive condition that causes cerebellar ataxia followed by
progressive neuromotor degeneration. Telangiectasias develop on the
upper part of the body: the face, neck, arms, and conjunctiva.
Contractures may develop at the foot and ankle.
are typically cutaneous but can also be skeletal or visceral. Most are
solitary, but multiple lesions can occur (6). A locus for autosomal dominant multiple cutaneous and mucosal venous malformations, VMCM1, was identified on chromosome 9p21 (14). If superficial in location, VMs manifest as easily compressible masses with blue coloration (Fig. 10.1).
The swelling is worse when the limb is dependent, and improves with
elevation. These lesions cause aching discomfort and usually enlarge
gradually as the child matures. Subcutaneous lesions can cause local
sensory nerve irritation (15). Activity-related
pain, mimicking a chronic compartment syndrome, is often associated
with intramuscular lesions, and diagnosis can be difficult (15). Thrombosis is common and phleboliths can be present as early as 2 years of age (4,6). Peri-articular lesions can cause recurrent hemarthroses.
films, which show the abnormalities in most cases of deep VMs,
including phleboliths and enlargement or distortion of soft-tissue
planes (1,16).
Ultrasound and computerized tomography (CT) scans with intravenous
contrast demonstrate similar findings but do not add significant new
information to that shown by plain films. They may be used for
directing aspiration or needle biopsy if a limited specimen is useful.
MRI and magnetic resonance angiography add significant anatomic detail
and are the most informative diagnostic modalities (8).
Hemangiomas produce a very high T2 signal, presumably because of the
pooling of blood with low flow. The signal is nonuniform because of the
fibrous and fatty septae within the hemangioma. MRI can determine the
extent of deep hemangiomas and differentiate the vascular elements from
surrounding tissues. However, MRI cannot differentiate feeding arteries
from veins, and angiography is
the only way to obtain this information. Venography may not demonstrate the lesion if the flow is slow (17,18).
Angiography is helpful if an arteriovenous fistula is suspected, if
sclerotherapy is planned, or if surgical resection is being considered.
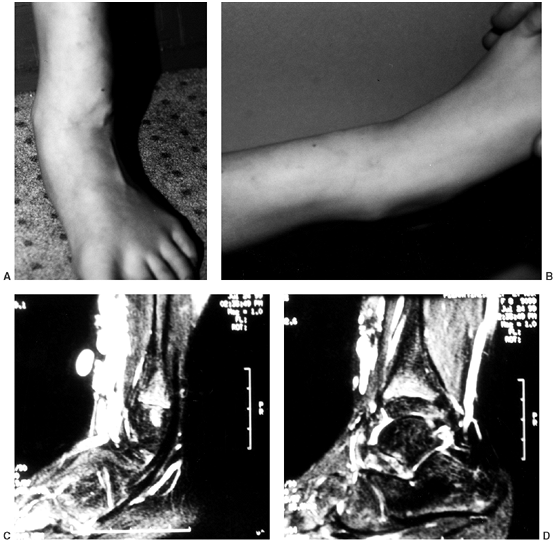 |
|
Figure 10.1 An 8-year-old girl with vascular malformation reports aching discomfort above the ankle. A:
A clinical photograph of the patient in standing posture demonstrates swelling and superficial irregularity over the anterolateral aspect of the ankle. The swelling was not tender to the touch and was easily compressible. B: With elevation of the limb, the lesion drained and was not palpable except for deep thick structures. C, D: Magnetic resonance images demonstrating high T2 signal typical of a vascular malformation. |
The platelet count is normal in contrast to the Kasabach-Merritt
phenomenon, and the prothrombin time and D-dimers are elevated. Heparin
may be required in the treatment (2).
discomfort in the extremity. Sclerotherapy, the injection of an
irritating solution, is an effective intervention, but the VMs can
recur (6). Surgical resection, often done after
sclerotherapy, is considered for large, painful lesions. Incomplete
exsanguination under tourniquet control allows dissection in a
relatively bloodless field and better visualization of the margins (15). Recurrence is common, and was noted in 48% of patients in one study (15). Pharmacologic treatment is indicated in vascular tumors with active angiogenesis but has
not been successful in the management of vascular malformations (2).
The lesions are usually present at birth and are raised soft, blue,
compressible nodules that blanch. The nodules become more numerous as
the child grows older. The main risk arises from gastrointestinal
hemorrhage, but there are significant orthopaedic problems as well. The
cutaneous lesions in the hands and feet cause pain and interfere with
function (20,22).
Hemarthrosis and stiffness can result from articular involvement.
Skeletal deformities may be secondary to pressure from adjacent
lesions, and hypertrophy may occur because of hypervascularity (20). Significant hypertrophy may require amputation.
subcutaneous tissues, along with multiple intraosseous enchondromas.
There is no consistent genetic basis (1). The
hemangiomas are present at birth in only 25% of patients; in the
remaining patients, the lesions become evident by 5 years of age. The
clinical appearance is of a blue discoloration on the skin. X-ray films
may show calcified thrombi in addition to the enchondromas. The
skeletal manifestations include short stature, limb-length inequality,
angular deformities, and scoliosis. The risk of malignant
transformation is 30%; this includes the potential for vascular lesions
to undergo transformation into spindle cell hemangioendothelioma (2).
commonly seen in the cervicofacial region, axilla, mediastinum, and
pelvis (23,24).
Although usually falling within the province of the general pediatric
surgeon, a lymphatic malformation may first be noticed as a mass of
unknown origin in a limb or as a cause of osteolysis or nerve
compression. Most are present at birth or are detected in the first 2
years of life (6). There are no known underlying molecular genetic abnormalities (5).
of anomalously formed lymphatic channels, described as microcystic,
macrocystic, or a combination thereof (3,4,6).
Superficial LMs are recognized by clear, small vesicles. Intravesicular
bleeding often occurs, causing red nodules that are seen typically in
combined lesions such as in Klippel-Trenaunay syndrome. Large, deep
malformations (also known as cystic hygromas or lymphangiomas)
present as ballottable masses with an underlying blue hue. Although
they are benign, they are locally aggressive and may compress adjacent
structures. Airway compression by cervicofacial lesions can be life
threatening. Skeletal and soft-tissue overgrowth are seen in
association with these lesions. The organs most commonly affected
include the spleen, liver, and lungs, with multiple organs involved in
75% of cases. Typically, the axial skeleton is involved (25). Chronic disseminated intravascular coagulation, as described previously in the context of VMs, can also occur in large LMs (2,6).
LMs are thought not to have the potential to develop into malignancy,
but malignancy has been seen in previously irradiated lesions (26).
nuclear imaging, CT scan, or MRI. Lymphangiomas tend to have a
characteristic appearance on MRI, with a heterogeneous low signal on
T1-weighted images (lower than that of muscle), but a very high
T2-weighted signal (higher than that of fat) (27). In pure LMs, no enhancement is noted with the use of gadolinium (8,28). Children with this disorder may also show other congenital anomalies.
spontaneously. Surgical resection of the involved tissues should be
done for large lesions or for those that compress vital structures (29,30). Recurrence rates can be as much as 40% (25).
Radical procedures, with removal of important adjacent structures, are
not indicated to alter the course of the disease. Unresectable lesions
have been treated with embolization, sclerotherapy, and interferon.
a congenital lymphedema that is autosomal dominant. Mutations have been
found in the vascular endothelial growth factor receptor 3 (VEGFR-3) gene (5). When the onset of lymphedema occurs late in the first decade or in the second decade of life, the condition is called Meige disease or lymphedema praecox. The inheritance pattern is the same. It is caused by inactivating mutations in the forkhead transcription factor FOXC2 (5).
Lymphedema is also seen as part of Turner syndrome and Noonan syndrome
(i.e., Turner phenotype with normal-appearing chromosomes and mental
retardation). Treatment in all of these cases is conservative:
elevation of the affected part when possible, and compression with a
Jobst stocking or intermittent pneumatic compression (18,31).
or the more extensive forms described later in the chapter. Solitary
intraosseous lesions are extremely rare (32).
They are lytic and well demarcated but variably circumscribed. The
appearance resembles a simple cyst, but involvement within bone is more
extensive. Curettage and bone grafting have reportedly been successful
in such cases (32).
which are a mix of capillary, venous, lymphatic, or arterial
malformations. They share a propensity for skeletal and soft tissue
overgrowth.
and Trenaunay in 1900. It has three essential features: a cutaneous
capillary-venous malformation, varicose veins, and hypertrophy of soft
tissue and bone in the involved limbs (Fig. 10.2). The vascular malformation is
usually seen early in life and typically does not cross the midline of the body (33,34,35,36).
The entire limb is not uniformly affected. The severity of the varicose
veins varies, but they tend to get larger with age and are always
present by 12 years (37). Abnormalities in
arteries and lymphatic vessels are also frequently seen. If clinically
significant arteriovenous shunting is present in addition to the
typical triad, the additional name “Parkes-Weber” has been applied (Klippel-Trenaunay-Parkes-Weber) (38).
Some authors suggest that the Parkes-Weber syndrome should be
considered as being distinct from the Klippel-Trenaunay syndrome (6).
Usually overgrowth occurs in the limb affected by the vascular
malformations, but sometimes not. The mechanism leading to overgrowth
involves increased bulk or girth and increased length and width of the
bone. Most of the size discrepancy seen is in girth, secondary to
soft-tissue hypertrophy (sparing the muscles) and lymphatic
abnormalities (37).
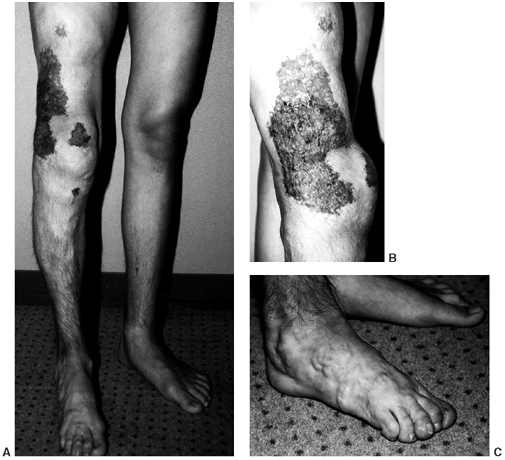 |
|
Figure 10.2 A:
A 15-year-old boy with Klippel-Trenaunay syndrome of his right lower extremities with typical findings of hypertrophy, varicosities, and superficial complex, combined vascular malformations. B, C: He had aching discomfort from the varicosities, intermittent pain from thrombophlebitis, and drainage from the superficial vascular malformations. |
mutation in a factor that is crucial for the endothelial activity
necessary for vasculogenesis and angiogenesis during embryonic
development (37,39,40).
It is presumed that this vascular malformation induces the hypertrophy
of other tissues, but there may also be primary mesodermal
abnormalities in these cases (41). Translocations (5:11, 8:14) have been reported as being associated with this syndrome (42,43).
The absence of deep venous drainage and venous hypertension is not
likely to be a causative factor, because deep veins are missing in only
14% of patients and venous flow is normal (33).
Baskerville et al. suggest that increased flow through the abnormal
capillary network and venous channels may promote overgrowth (33).
at increased risk for developing Wilms tumor, as are children with
generalized hemihypertrophy (44,45). Routine abdominal ultrasound screening is not required.
time of birth or infancy, a few cases have been reported in which
features appeared as late as 6 years of age. There is no recognized
pattern of inheritance. The lower extremities are affected at least ten
times more often
than the upper extremities. The affected limb is longer than normal in 90% of the patients (16,37,46). Usually, all bones and soft tissues in the limb are involved in the hypertrophy (Fig. 10.2).
rigorously studied over time, but there are enough reported cases of
nonuniform overgrowth to caution the physician that prediction of an
eventual discrepancy should be done only as a rough estimate.
McCullough and Kenwright described two patients in whom the discrepancy
decreased with growth (47). Severe leg-length discrepancy is uncommon. In one study only 10% of the children had a discrepancy greater than 3 cm (37).
In addition to the hypertrophy of the extremity, there is evidence of
fundamental embryologic regulatory defects, especially distally, with
25% of patients having anomalies of fingers or toes, such as
macrodactyly, syndactyly, polydactyly, and clinodactyly (46). Carpal tunnel syndrome has been reported in children (48).
Scoliosis affects at least 5% of patients, although it rarely requires
surgery. An adult with a right thoracic scoliosis and hypertrophy of
the seventh thoracic vertebra presented with severe spinal stenosis (49).
An arteriovenous fistula, medullary angioma, and extradural venous
vascular malformation contributed to the stenosis, which was treated
successfully by decompression (49). Systemic
involvement may occur. In the central nervous system, AVMs and cerebral
and cerebellar hypertrophy have been described (50,51).
The gastrointestinal and genitourinary system may be involved,
resulting in bleeding in some cases. Surface bleeding from the
hemangioma occurs in 25% of patients, and 15% have clinical pulmonary
emboli, spontaneously or after operation (33).
Congestive heart failure may occur in patients with large vascular
malformations. Despite these complications, the life expectancy is not
markedly decreased. The impact of this complex medical condition on the
child’s psychosocial development should not be underestimated.
of hypertrophied, irregular, or absent medial layers of the veins,
allows dilatation. Valves are anomalous; they are absent or obstructed.
Deep venous channels are usually present, and AVMs are uncommon (40). Lymphatic hypoplasia is common. Other tissues, such as nerve and subcutaneous tissue, may be hypertrophied.
without prominent nevi. Maffucci syndrome often includes limb-length
inequality with vascular malformations, but it is differentiated by the
presence of intraosseous enchondromas. Beckwith-Wiedemann syndrome
involves localized overgrowth but also includes neonatal hypoglycemia,
visceromegaly, macroglossia, and a predisposition for Wilms tumor.
Proteus syndrome is a more severe disorder that includes virtually all
the features of Klippel-Trenaunay, and also soft-tissue tumors,
pigmented skin lesions, and thickened palms and soles (34). Bannayan-Zonana syndrome is characterized by thoracic and abdominal lipomatosis, vascular malformations, and macrocephaly (52).
ultrasonography, MRI, MR angiography, MR lymphangiography, and
lymphoscintigraphy can provide sufficient information for diagnosis and
planning in most cases (28,37,52,53,54). Lymphatic imaging is particularly useful in the evaluation of children with massive hypertrophy (37).
When the surgeon is planning for hemipelvectomy or hip disarticulation,
arteriography may be helpful to map major vessels to anticipate
significant bleeding (16). Contrast angiography is also useful for percutaneous treatment of a vascular malformation.
Klippel-Trenaunay syndrome are usually not dramatically progressive.
Surgery has a limited role in this condition. It should be done only
for disabling problems and when the benefit is fairly predictable (54).
Initial therapy of the aching, hypertrophic limb with varicosities
should consist of compression. Intermittent pneumatic compression
should be applied at night, using a custom-fitted garment and a home
pump, inflated every 90 seconds to a pressure midway between diastolic
and systolic (55). Just before getting up in
the morning, a Jobst compression garment should be applied, and worn
throughout the day. With these measures, a marked decrease in limb
girth, resolution of cardiac overload and dependent syncope, reduced
discomfort, and marked improvement in function can be seen. A technique
of manual lymphatic drainage can also be used, if there is a
significant lymphatic component to the malformation (31).
Thrombophlebitis and pulmonary embolism are common. Consultation with a
vascular surgeon may be beneficial for most of these patients.
include: cardiac failure from shunting in children; overgrowth that is
nonresponsive to compression therapy; rapid enlargement in limb size;
bleeding from abnormal vessels in the gut, kidney, or genitalia;
coagulopathy; reconstruction in selected cases of syndactyly or
polydactyly; and amputation of severely disfiguring dysfunctional limbs
for which reconstruction is not an option and where amputation may
provide a more functional limb. The site of amputation is often
dictated by the extent and severity of the vascular malformation. A
knee disarticulation is preferred to a midthigh amputation because it
is an end-bearing stump with rotational control and better suspension;
the distal femoral physis is preserved, and overgrowth is avoided.
Midthigh amputation requires ischial weight bearing, which may be a
problem in the presence of any scarring or gluteal vascular
malformations (34,56,57).
Risks of surgery include infection, particularly in children with
abnormal lymphatic drainage, and delayed wound healing, which is
common after transverse amputations (58).
Occasionally, a proximal limb disarticulation is needed in neonates as
a life-saving procedure. For patients in this age group, hypothermia
and total circulatory arrest for up to 60 minutes have been successful
as an adjunct for minimizing blood loss (59). Pulsed-dye laser treatments can be used for treating the cutaneous vascular malformations over limited areas (37).
debulk the extremities has usually resulted in recurrence or minimal
improvement. Varicose vein ligation may provide relief to local
symptoms, but the varicosities often recur, and ligation should be
avoided if the deep venous system is not patent. Epiphysiodesis is used
in the treatment of leg-length discrepancy, but the growth patterns are
unpredictable. The procedure does not decrease the widths of the
affected extremities. Limb shortening at skeletal maturity may be the
most accurate technique. If surgery is planned, the skin involved
should be protected, and the increased risk of deep thrombosis has to
be borne in mind.
soft-tissue disorder characterized by vascular anomalies, macrodactyly,
exostoses, asymmetric hypertrophy, subcutaneous tumors, scoliosis, and
other anomalies (60,61,62,63,64). The name is derived from the Greek god, Proteus, who could change shape at will to avoid capture.
However, it has been suggested that Proteus syndrome and other similar
sporadic congenital hamartomatous syndromes such as
encephalocraniocutaneous lipomatosis and epidermal nevus syndrome
represent a phenotypic continuum that arises through somatic mosaicism.
The concept of lethal autosomal (somatic) mutations that can survive
only in a mosaic state is supported by sporadic occurrence,
distribution of lesions in a scattered or asymmetric pattern, variable
extent of involvement, and equal sex ratio (65,66). Germline loss-of-function mutations in PTEN,
a tumor suppressor gene on 10q23.3 that encodes a dual-specificity
phosphatase involved in various cell-survival pathways, have been noted
in up to 20% of patients with Proteus syndrome and 50% of patients with
Proteus-like syndrome. The PTEN syndrome spectrum includes other
disorders that are characterized by hamartomata, lipomatosis, and
neoplasia (67,68,69).
include capillary, lymphatic, capillary-venous and
capillary-lymphatic-venous types, with an overall predominance of
lymphatic vessels (70). Subcutaneous tumors,
found most commonly on the trunk, are composed of adipose and fibrous
tissue, Schwann cell structures and vascular tissue. Extraskeletal
manifestations of the disorder can include hemimegalencephaly,
opthalmologic anomalies, and splenic hyperplasia (71,72,73). Unusual neoplasms have been described, and precocious puberty may result from juvenile granulosa cell tumors (74,75).
Severe cystic emphysematous pulmonary disease and sudden death by
pulmonary thromboembolism can occur, even in young children (76,77).
MRI is helpful in delineating the nature and extent of the subcutaneous
tumors as well as in aiding in the diagnosis of the syndrome (60,71,78).
The enlarged digits may not be located on the same side as the
hemihypertrophy. The macrodactyly progresses rapidly in the first few
years of life, and slows in later childhood and adolescence. Severe
cosmetic and functional problems can result. The histology is a
hamartomatous proliferation of all mesenchymal tissues, especially the
osseous and fibrofatty components (63).
Treatment will be discussed in the section on macrodactyly later in
this chapter. A striking finding in the foot is plantar hypertrophy,
resulting in cerebriform or gyriform creasing (Fig. 10.3).
and it results in limb-length discrepancy which varies greatly.
Overgrowth and progressive atrophy have been described (61).
Angular upper- and lower-limb deformities are seen in Proteus syndrome.
Genu valgum occurs commonly, recurs after bracing, and often requires
repeated osteotomies (61,63) (Fig. 10.4). Joint contractures and angular deformities may be caused by epiphyseal exostoses (61).
reported. It can result in progressive spinal deformity and compressive neuropathy from canal stenosis (63,79,80,81). Infiltration of the spinal canal by an angiolipomatous mass can also occur, and may lead to paraplegia (79,80,82).
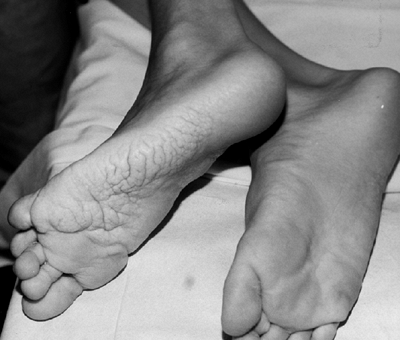 |
|
Figure 10.3 Adolescent boy with Proteus syndrome with typical gyriform creasing of the sole of his foot.
|
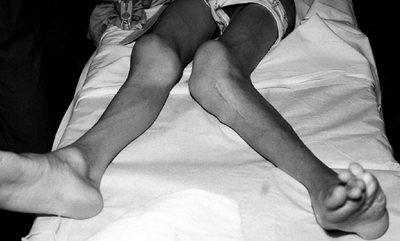 |
|
Figure 10.4
Adolescent boy with Proteus syndrome with recurrent left genu valgum after a high tibial osteotomy, just prior to repeat osteotomy. |
limb-length inequality, angular deformity, and hypertrophy. Because the
skeletal age may be delayed and the rate of progression of limb-length
inequality is not well understood, planning the treatment can be
difficult. Extensive overgrowth and joint contractures may lead to
amputation. During extensive surgical excisions, residual abnormal
tissue is often needed in the reconstruction, and postoperative lymph
drainage may be prolonged (83). Patients who
are scheduled for surgery should have a thorough assessment of their
airway and pulmonary functions because of the relatively high frequency
of pulmonary involvement and tonsillar hyperplasia (84).
Close monitoring and treatment of deep venous thrombosis is important
because of the increasingly recognized risk of fatal pulmonary embolism
(76).
This has been classified in this chapter as a complex, combined
lymphatic-venous malformation, but the pathogenesis has not been fully
elucidated.
subsequently involve adjacent bones. The joints and intervertebral
discs do not act as barriers to extension. Typically, progressive bone
resorption occurs, and the bone is replaced by fibrous tissue. The
resorption may stop spontaneously on rare occasions. The peak age at
onset is in the second and third decades of life (86).
Gorham disease is differentiated from other forms of idiopathic
osteolysis on the basis of its lack of association with neuropathy or a
genetic mode of transmission (87). It may involve any area of the skeleton but is most common in the shoulder, pelvic girdles, and spine (86,88,89,90).
It may present as a dull ache and weakness in the segments involved,
and is associated with increasing deformity and pathologic fractures.
Patients with lesions extending outside the bone have a high mortality
rate, approaching 50%, from chylothorax, chylopericardium,
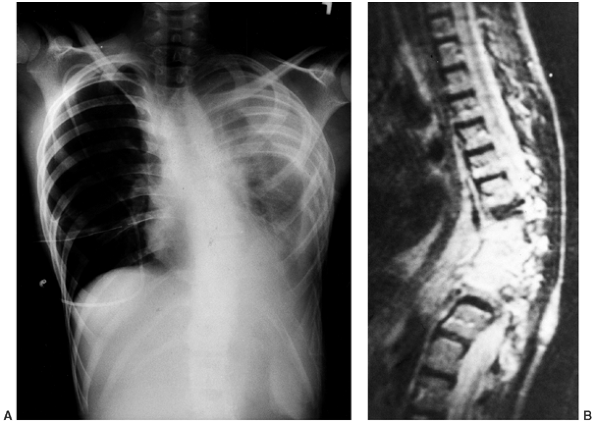
chyloperitoneum, and cachexia (23,88,91,92,93) (Fig. 10.5A).
There is subsequent disappearance of contiguous bones and tapering of
the bony remnants. No sclerosis or osteoblastic reaction is seen. If a
pathologic fracture occurs, and if the lysis is minimal, the bony
lesion may be overlooked initially. Unlike in normal fracture healing,
the bone ends eventually become tapered, and there is no evidence of
the formation of new callus. CT scan is the best method for evaluating
the extent of bone destruction. Arteriography, venography, and
lymphangiography do not reveal the intraosseous lesions (89).
Nuclear medicine scans are inconsistent in showing the lesions. MRI is
very helpful, but the signal characteristics vary depending on the
stage of the disease process (87,88,95).
The neovascular tissue is bright on T1 and T2 imaging early, but the
predominant fibrous tissue that is present late in the process is dark
on T1 and T2 imaging (Fig. 10.5B. The
differential diagnosis includes other rare causes of idiopathic
osteolysis, including syndromes that have generalized osteolysis as a
clinical feature, such as Winchester syndrome (96).
Other conditions with more focal osteolysis exist, such as idiopathic
multicentric (carpotarsal) osteolysis, which is characterized by
nephropathy and osteolysis predominantly involving the hands and feet (97,98), and
Hadju-Cheyney syndrome, in which acroosteolysis predominates (99).
 |
|
Figure 10.5 A 12-year-old boy with Gorham disease. A: Chest x-ray demonstrates a chylothorax and loss of ribs 8, 9, and 10. B:
The neovascular tissue is bright on T2, and this magnetic resonance image shows extensive vertebral destruction and paravertebral involvement. This boy subsequently died because of the disease. |
vessels, lined by endothelial cells. The vessels may be empty or may
contain proteinaceous fluid and/or blood cellular elements (91).
Fibrous connective tissue replaces the bone. There is no evidence of
malignancy or inflammation. The histologic changes are similar to
benign hemangioma or venous malformation of bone, but there is much
more extensive bone destruction (86). The cause is unclear, but perivascular cells of the lesion show the characteristics of osteoclast precursors (89). The bone shows enhanced osteoclast activity that appears to be caused by an increase in the sensitivity
of osteoclast precursors to humoral factors that promote osteoclast
formation and bone resorption, rather than to an increase in the number of circulating osteoclast precursors (100,101).
successful, but surgery, with or without radiation therapy, has been
the mainstay of treatment. Some cases, especially those without
significant involvement of chest or abdominal cavities, stabilize
spontaneously, although return of the “vanished” bone does not occur.
This complicates the evaluation of published treatment protocols. Local
resection and bone grafting, with or without internal fixation, have
not been consistently successful (86). Most
grafts are resorbed, but there is one report of the successful use of a
vascularized fibular graft in pelvic-femoral reconstruction (102). Wide resection and limb salvage, with or without prosthetic reconstruction, can be successful (86,87,103). Amputation has been required in order to achieve a satisfactory margin and improved function in some patients (86,102). Radiation has been shown to be an effective adjunct therapy, when doses of 30 Gy or more are used (86,104,105).
The management of vertebral and rib involvement with chylothorax is
difficult. Resection with adequate margins is usually not possible.
Surgical decompression with anterior and posterior fusion and
instrumentation can successfully manage neural compression and
instability at the cervicothoracic junction (106). Radiotherapy, combined with pleurodesis and pleural adhesion therapies, has proved promising in some cases (88).
asymmetries between the right and left sides of the body to a greater
degree than can be attributed to normal variation.
This may involve the length and girth of the limbs as well as of the head, trunk, and internal organs.
normal and what is not; that is, in determining whether the larger
limbs are hypertrophic or the smaller limbs are hypotrophic (107).
Differentiation of overgrowth from undergrowth is based on comparison
of the limb with its expected length, in proportion to the rest of the
body. This may be visually obvious and straightforward in typical
cases, but may be difficult to discern in milder cases. The examiner
may search for anomalies that herald the presence of an abnormal limb.
Vascular malformations and associated digital malformations or
macrodactyly usually signify overgrowth conditions; obvious muscle
hypotrophy, focal neurologic abnormality, mental retardation, or
abnormality of the joints may accompany undergrowth. If no such clues
are found, a graph of normative sitting heights can be used for
determining the patient’s trunk height percentile, and a graph of
normal lengths of the tibia and femur can be used for determining which
side of the patient corresponds to a percentile that most closely
matches the normal (47).
Nehme referred to an unpublished survey from the growth study at
Children’s Hospital, Boston (108). The maximum
mean discrepancy seen at different ages for 95.5% of the population was
approximately 1.4% (0.4 cm difference at 1 year, 0.8 cm at 10 years,
and 1.1 cm at skeletal maturity) (108). Similar data have been reported in a group of young adult army recruits (109). Pappas and Nehme have defined abnormal asymmetry as a 5% or greater difference in length and/or circumference (108). MRI and other imaging techniques can assess the tissues in the extremities and aid in establishing the diagnosis (52,107).
classified as either congenital or acquired. Acquired asymmetry can
result from injury, infection, radiation, or inflammation (110,111).
Congenital forms may be classified as total or limited. The total forms
have involvement of all organ systems, including the ipsilateral paired
organs, whereas the limited forms have only muscular, vascular,
skeletal, or neurologic involvement. Limited forms can be categorized
according to the area of involvement: classic (ipsilateral, that is,
upper and lower limbs), segmental (a single limb), facial, or crossed (110,111,112). Alternatively, these disorders can be classified as nonsyndromic (isolated) or syndromic (part of a clinical syndrome). Table 10.2 outlines the differential diagnosis of hemihypertrophy and hemihypotrophy.
hypertrophy associated with Beckwith-Wiedemann syndrome,
neurofibromatosis, Bannayan-Zonana syndrome, or vascular malformations
such as Klippel-Trenaunay and Proteus syndromes and LMs (Table 10.2).
Undergrowth may be the result of secondary nonsyndromic hypotrophy,
mosaicism for Turner syndrome, Russell-Silver syndrome, neurologic
asymmetry (cerebral palsy, polio), osteochondromatosis,
enchondromatosis, or polyostotic fibrous dysplasia (113).
There is no clear inheritance pattern. The hemihypertrophy is rarely
apparent at birth but becomes evident during the early years of growth.
The unilateral enlargement may include one ear, one half of the tongue,
the pupil of one eye, one nipple, one side of the thorax and abdomen,
the internal organs on that side of the body, and the arm and leg on
that side (111,115) (Fig. 10.6). Contrary to earlier reports, mental capacities are usually normal, unlike those of patients with idiopathic hemihypotrophy (63,114).
Cutaneous vascular lesions are not associated with nonsyndromic
hemihypertrophy. Compensatory scoliosis secondary to the leg-length
discrepancy is common, but the incidence of structural scoliosis is
also greater, even after leg-length equalization, and may be the result
of asymmetry of the vertebral body. Uncommon skeletal anomalies are
syndactyly, lobster-claw hand, developmental dysplasia of the hip, and
clubfoot (113). Genitourinary abnormalities are
also commonly associated, and these may include inguinal hernias,
cryptorchidism, and medullary sponge kidney (110,114).
malignant tumors, such as Wilms, adrenal carcinoma, hepatoblastoma, and
leiomyosarcoma (110). A recent study suggests that the incidence of malignancy in these children is approximately 5.9% (116).
It is possible that the same abnormal cellular growth control mechanism
that results in overgrowth also predisposes these children to tumor
formation (110,116). In
order to detect these tumors as early as possible, abdominal
sonographic screening has been recommended for children with
hemihypertrophy (110,117).
This is controversial, because there is little evidence that this
results in a better clinical outcome for children with Wilms or other
tumors. Also, it will not diagnose extraabdominal tumors, and the
hemihypertrophy itself is recognized in only 30% of the children prior
to the tumor diagnosis (110,118,119).
Nevertheless, current recommendations are for regular screening:
abdominal ultrasound every 3 months until age 7, then physical exam
every 6 months until skeletal maturity (72).
However, there are patients in whom resolution of the inequality takes
place in early childhood itself, and others in whom there is a
reduction in the rate of increase in adolescence. These variations make
an accurate prediction of discrepancy difficult (47,112). The tibia is overgrown as much as, or slightly more than, the femur (108). Limb girth is increased by an average of 10%, primarily because of muscle hypertrophy (108).
|
TABLE 10.2 DIFFERENTIAL DIAGNOSIS OF HEMIHYPERTROPHY AND HEMIHYPOTROPHY
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||
syndrome usually diagnosed at birth, and characterized by neonatal
hypoglycemia, macroglossia, visceromegaly, omphalocele,
hemihypertrophy, and a predisposition to form embryonal tumors, most
frequently Wilms tumor (121). The risks of
having Wilms tumor, hepatoblastoma, and neuroblastoma are higher than
in nonsyndromic hemihypertrophy; in one study, 13 of 183 children
developed a tumor by the age of 4 years (122).
include neurofibromatosis, Proteus syndrome, Klippel-Trenaunay
syndrome, and lymphangiomas (78).
These patients, however, have a higher incidence of other dysmorphic
features, including cleft palate and facial malformations, congenital
scoliosis, and genitourinary malformations. Mental retardation is
common, but Wilms tumor is not associated with the condition (127). Rarely does this syndrome require orthopaedic treatment because in most cases the discrepancy is less than 2.5 cm.
 |
|
Figure 10.6 An 8-and-a-half-year-old boy with idiopathic hemihypertrophy. A: Note the enlargement of the entire left lower extremity. The left upper extremity was enlarged as well. B: The lower extremity orthoroentgenogram demonstrates the leg-length discrepancy.
|
idiopathic hemihypotrophy, but it is characterized by overall short
stature, with most patients never exceeding a height of 152 cm (5
feet). These patients have characteristic small, triangular faces and
renal and genital malformations (127).
Scoliosis is common and may be congenital or idiopathiclike. The limb
hypotrophy is usually minimal, but as much as 5 cm has been reported (128).
have hemihypotrophy, as may patients who have multiple enchondromas or
osteochondromas, with mean discrepancies of 9 cm and 3.5 cm,
respectively, for those who have limb-length inequality. Limb
inequalities resulting from neurogenic causes vary in proportion to the
asymmetry of the neurologic involvement, rarely exceeding 2.5 cm in the
lower extremities in patients with cerebral palsy or 6 cm in patients
with polio (120). In assessing patients with
localized overgrowth or undergrowth, it should not be assumed that the
growth alteration is proportional. Shapiro described five patterns of
limb-length discrepancy and, even within a given diagnosis, multiple
patterns may be seen (120). Periodic
assessments should be done during growth, if possible, in order to
determine the pattern of overgrowth or undergrowth. Predictions can
then be made more accurately. If significant joint contractures are
present, plain radiographs or scans may not measure limb length
accurately, and CT scans may be needed. For determining whether
upper-extremity inequalities are significant enough to require
treatment, normative growth data are available (129).
increase in the size of the constituent elements of a single digit or
several adjacent digits of the hand or foot. Two types exist: the more
common static type, which enlarges from birth, the increase in size
being proportional to growth; and the progressive type, in which there
is disproportionate growth of the involved digit (130). Localized gigantism
is a term used inconsistently in the literature, and describes
macrodactyly as well as enlargement of the tissues of the hands and
feet proximal to the digits.
isolated or idiopathic condition. Macrodystrophica-lipomatosa
(fibrolipomatosis) is a nonhereditary overgrowth of all mesenchymal
elements of the digit, particularly the fibroadipose tissue (52,131). Macrodactyly may also occur in association with neurofibromatosis and vascular
malformations (capillary, venous, arterial, or lymphatic), such as Klippel-Trenaunay and Proteus syndromes (130,131,132,133).
Children with multiple enchondromatosis, Maffucci syndrome, and
tuberous sclerosis may have enlarged digits. It may be difficult to
make a diagnosis in infancy because of the delayed manifestations of
the symptoms that characterize these syndromes, such as the
cafe-au-lait spots in neurofibromatosis. MRI evaluation of the tissues
involved in the macrodactyly can be helpful in establishing a diagnosis
in the absence of specific clinical features (52).
although occasionally the dynamic type may not become apparent until
later in infancy, when relentless enlargement occurs (Fig. 10.7).
The upper extremities are more commonly affected than the lower
extremities. Unilateral involvement occurs in 95% of cases, but in
two-thirds of the patients, more than one digit on the affected side,
usually adjacent ones, are affected (130). The
second ray is the most commonly enlarged, followed in descending
frequency by the third, first, fourth, and (very rarely) fifth rays.
Syndactyly may coexist. Usually, the palmar or plantar surface is more
hypertrophied than the dorsal, resulting in hyperextension of the
metatarsal or metacarpophalangeal joints (134,135).
If two adjacent digits are affected, they grow apart from each other.
In static macrodactyly, the digits involved are approximately 1½ the
normal length and width. In the dynamic type, even more striking
enlargement may occur. The bone age may be advanced in the phalanges
involved (134,136). The metacarpals or metatarsals may be enlarged, thereby widening the hand or foot (134). The width of the forearm, leg, thigh, or arm on the affected side may also be subtly increased (136).
Not long after skeletal maturity, interphalangeal joint stiffness and
degenerative changes may supervene prematurely, even in untreated cases
(130).
macrodactyly. The most consistent feature is overgrowth of the
fibro-fatty tissue, but all tissues are enlarged in the digit involved (130,134,136).
Fibrous bands and hypertrophied adipose tissue infiltrate muscle and
nerve. A proliferation of fibroblastic tissue between the cortex and
periosteum may account for the phalangeal overgrowth (134,136).
The digital nerves are more prominent than usual, particularly in the
hand, with proliferation of epineural and perineural tissue (130,131,134). Plexiform neurofibromas are typical in neurofibromatosis (131).
In children who have met strict criteria for the diagnosis of Proteus
syndrome, macrodactyly is not associated with enlarged digital nerves
or fibroadipose tissue proliferation (133).
Occult neurofibromatosis was once thought to be the cause, but
long-term follow-up studies, including one of 26 years, have failed to
reveal the development of features of neurofibromatosis other than the
macrodactyly itself (137).
location, type, and severity of the macrodactyly. The management
considerations for the hand are very different from those for the foot.
In the hand, function is paramount, and a significant increase in the
width and length of a digit can be tolerated, whereas, in the foot,
accommodating even a small excess of width in a shoe may be difficult.
A good cosmetic result can be difficult to achieve. In the static type
of macrodactyly, debulking and shortening procedures such as phalangeal
resection and epiphysiodesis are successful. In contrast, the deformity
seen in progressive forms will usually require ray resection. Extensive
debulking can interfere with the blood supply to the skin and result in
delayed wound healing.
two-stage (3 months apart) soft tissue debulking procedure, provided
length is not a problem (130). If length is a
problem, then epiphysiodesis of the proximal phalanx, and of the
metacarpal or metatarsal, if involved, will be effective in the young
child (<6 to 8 years) (130,134,138,139). In the older child, bone shortening by phalangeal resection can be combined with staged debulking (140,141).
Ligament reconstruction or fusion with Kirschner wire fixation will
avoid the reported complications of floppy toes or secondary
deformities (137). Isolated phalangeal amputation should be avoided because of subsequent angular deformities of the adjacent toes (132,137).
effective, in the long term, for dealing with excessive forefoot
widening. Ray resection is a more definitive solution to the multiple
dimensions of enlargement that are often encountered (131,137,142,143).
One or two rays may be removed, but the first ray of the foot should
not be amputated in isolation because of its unique function in balance
and weightbearing (144). Ray resection results in good cosmetic and functional outcomes in feet with less toe involvement (144).
In order to improve correction and avoid recurrence, it has been
suggested that a wedge of adjacent tarsal bones be resected when doing
a ray resection (131). Ray resection prevents
crossover of the adjacent digits and eliminates “gap” formation and
soft-tissue enlargement. Three rays are the minimum requirement for a
foot to be functionally serviceable. More extensive involvement of the
foot would necessitate midfoot or Syme disarticulation, depending on
the extent of overgrowth.
be seen in all levels of the limb. Herring and Tolo reported a case of
macrodactyly with width increase in the absence of any overall
limb-length discrepancy (145). The family
should be counseled from the first visit that this condition is complex
and involves multiple tissues and levels. They need to know that
multiple procedures may be necessary
and
that early, appropriate, aggressive resections, involving all affected
dimensions, may decrease the total number of surgeries required. For
example, as soon as it becomes apparent that length and width may be
excessive, resection of one or several rays, with shortening or
epiphysiodesis of an adjacent ray, may be appropriate. Often, several
consultations with one or more surgeons may be necessary to help
families come to this realization.
 |
|
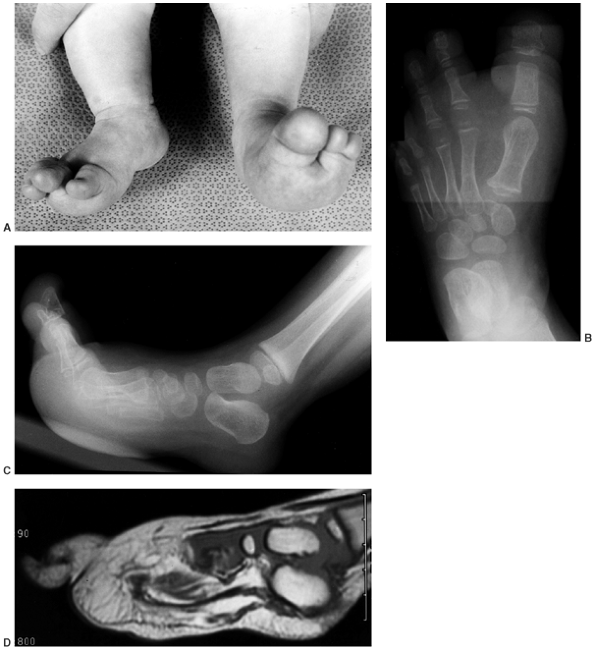
Figure 10.7 A 2-and-a-half-year-old girl with progressive macrodactyly of both feet, with macrodystrophica-lipomatosa. A:
There is significant plantar hypertrophy, resulting in hyperextension of the digits, and there is marked asymmetry in the digital enlargement. B, C: The plain radiographs demonstrate soft-tissue enlargement, as well as underlying bony enlargement. D: The magnetic resonance imaging demonstrates overgrowth of essentially all the elements of the digit, particularly the fibroadipose tissue typically seen in macrodystrophica-lipomatosa. |
The three main manifestations are acrosyndactyly, superficial or deep
constriction bands involving a digit or extremity, and intrauterine
amputation (146,148,149,150).
band syndrome is quite variable. The bands may be superficial and
incomplete or extend deeply to the underlying bone and may be
circumferential. Multiple bands may be present in a single extremity.
Swelling often results from impaired venous and lymphatic drainage
distal to the site of constriction. In the case of deep,
circumferential bands, there may be significant neurovascular
impairment (147,148,149,150,151,152). With growth, the constriction band occasionally gets more severe and may become increasingly symptomatic (147).
Upper extremity involvement is more frequent than lower extremity
involvement. Head, neck, or trunk constriction bands can occur but are
very uncommon (153). The distal aspect of the
limb, particularly the longest digits (index, long, and ring fingers,
and great, second, and third toes) are most frequently affected (146,147,148,154). Craniofacial abnormalities, including cleft lip and palate, are seen in 7% of children with constriction band syndrome (147,150,155).
of the severity of the syndrome: simple constriction rings;
constriction rings associated with deformity of the distal part, with
or without lymphedema; and constriction rings associated with
syndactyly and intrauterine amputation (156).
one-half of the children who are affected. Transverse, terminal, and
digital amputations with normal proximal skeletal development are
typical (146,147,148,154) (Fig. 10.8A). Simple syndactyly is common, whereas complex syndactyly with bony fusions is rare (148).
Acrosyndactyly or fenestrated syndactyly, in which there is an open
cleft between digits joined distally, may also occur. Hypoplastic or
absent nails are a consistent feature of the condition (147).
These feet are often rigid and more difficult to treat than idiopathic
clubfoot. Approximately 30% to 50% of the clubfeet are classified as
paralytic (149,150,152,157).
These feet show weakness of the peroneal muscles and are typically
associated with deep ipsilateral constriction bands in the lower leg.
The bands in the paralytic form of clubfoot are thought to cause a
compression neuropathy, direct muscle injury, or compartment syndrome.
Nonparalytic clubfeet associated with the disorder may or may not have
ipsilateral constriction bands; these bands are typically superficial
or incomplete and are considered to be idiopathic or resulting from
oligohydramnios (148).
can occur below the constriction bands in the upper and lower
extremities (156,158,159).
Anterolateral tibial bowing appears similar to that seen in congenital
pseudarthrosis of the tibia associated with neurofibromatosis; but, in
contrast to the latter situation, remodeling can occur and realignment
osteotomies, if required, will heal (158) (Fig. 10.8C). Rapid, spontaneous healing of osseous defects in the forearms and tibiae of infants has been described (158,159). Zionts et al. prefer the term discontinuity rather than pseudarthroses in describing these defects, because of the spontaneous healing (159).
Surgical management to equalize limb length will be required in some of
these children, depending on the predicted discrepancy at skeletal
maturity.
constriction band syndrome remain unclear. The two main conflicting
theories focus on whether the band formation is the result of factors
intrinsic or extrinsic to the embryo or fetus, but the disorder may be
more heterogeneous than had been thought.
acceptance. Torpin has proposed that entanglement of the limbs in
defects or free strands of amnion result in constriction band syndrome (160,161).
Supportive evidence includes the lack of hereditary factors, the
ultrasonographic demonstration of prenatal amniotic bands, the
involvement of the longer digits and the histologic demonstration of
amnion in constriction bands. Amniocentesis in animals produces fetal
malformations that resemble constriction band syndrome in humans. Kino
demonstrated in rats that the malformations result from subcutaneous
hemorrhages caused by excessive uterine contractions after
amniocentesis (146). A study that reviewed
children with clubfeet and constriction band syndrome showed that there
was a history of attempted first-trimester abortion in 60% of them (152).
Although an association between amniocentesis or fetal blood sampling
and the occurrence of amniotic band syndrome in humans has been
suggested (162), evidence for this remains inconclusive.
suggests that a defect of the subcutaneous germ plasm causes
soft-tissue necrosis and subsequent healing, with the formation of
constriction bands (163). There is evidence that in utero
vascular disruption from the death of a co-twin, or from placentally
derived embolic infarcts, can cause constriction band syndrome (164).
In these children, there was no ultrasound evidence of amniotic bands.
It therefore appears that this syndrome may result from factors other
than amniotic entanglement.
been traced through as many as four generations in one family. These
creases are present from birth, predominantly involve the extremities,
and disappear by 5 years of age (165). Hair/thread constriction may occur in infants, usually younger than 2 years of age, and cause circulatory compromise (166).
Strands of hair or fabric may become wrapped around fingers and toes
tightly enough to cause distal swelling and circumferential laceration
of the skin. It may be difficult, at the time, to see the hair or
thread that is causing the problem, thereby making it difficult to
distinguish the condition from a congenital constriction band. The
absence of a family history of constriction rings at birth should
suggest the diagnosis of hair constriction. Ainhum is a disorder
characterized by ulceration at the base of the fifth toe on the plantar
surface; it progresses to a circumferential constriction ring with
autoamputation. It is seen mainly in Africa (167).
 |
|
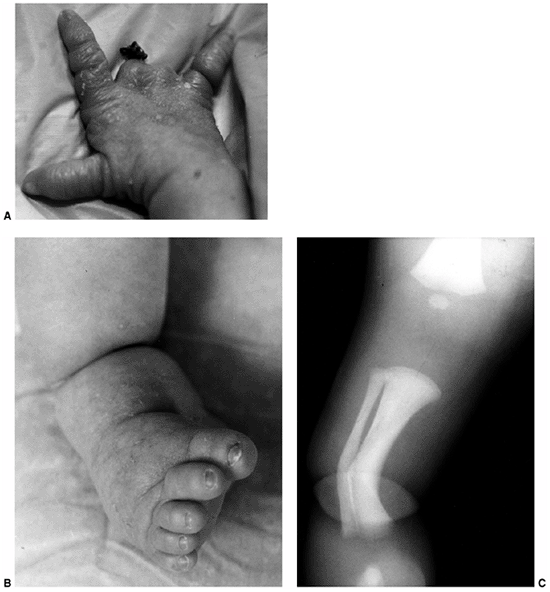
Figure 10.8 A child with congenital constriction band syndrome. A:
A clinical photograph demonstrating autoamputation of the long and ring digits through the proximal phalanges, and a circumferential constriction band in the proximal segment of the small finger. B: A clinical photograph demonstrating circumferential constriction band involving the distal lower leg, and an associated talipes equinovarus deformity. C: The radiograph of the same patient demonstrates an uncommon finding of angulation of the bone underlying the circumferential band. |
the band and closure with multiple Z-plasties is indicated: (a) in
bands that extend to the deep subcutaneous tissue or fascia; (b) if
there is edema distal to the band; (c) if there is vascular
insufficiency or neurologic deficit; and (d) if the band is increasing
in severity. In some cases, emergent surgical release in neonates may
be indicated in order to salvage the limb (Fig. 10.9). The surgical release used to be done in stages, releasing one-half of the band at a time with a 6- to 12-week interval (148,156,168).
This was because of concern that impaired venous or lymphatic flow and
skin-flap necrosis might occur if the whole band were released in one
stage, but single-stage release of constriction bands can now be done
safely (168).
band can be treated with serial manipulation and casting only if there
is no significant foot edema or evidence of neurovascular compromise.
Nonoperative management is rarely successful in these children,
especially when the clubfoot is of the paralytic variety (151,152,157).
The severity of the clubfoot appears to be more important than the
presence or severity of the constriction band in predicting the outcome
of surgical treatment (152,157). Paralytic clubfeet with deep bands often require multiple procedures and usually have poorer outcomes (169).
Resection of the constriction band should be done prior to surgical
release of the clubfoot in order to avoid neurovascular compromise.
Muscle imbalance of the foot, resulting from peroneal weakness, can be
managed by a split transfer of the tibialis anterior tendon (150,157).
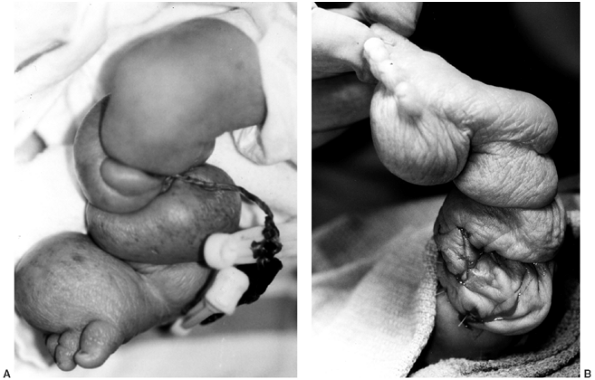 |
|
Figure 10.9
A 1-day-old girl born at 28 weeks’ gestation. An amniotic band around the umbilical cord led to fetal distress and precipitous delivery. A: A clinical photograph of the right lower extremity, with multiple deep circumferential bands. Marked distal swelling and vascular compromise with petechial formation are present. B: A clinical photograph immediately following emergent single-stage circumferential release of amniotic bands, with Z-plasty of the skin. Note decompression and reperfusion of all segments distal to the resected bands. |
generally done between the ages of 6 months and 1 year because of the
severity of the deformity and in order to allow for longitudinal growth
and function of the affected digits (154).
release of deep, symptomatic constriction bands. The constriction band
and underlying fibrous tissue extending through the skin, subcutaneous
tissue, fascia, and muscle should be completely
excised
with an adjacent 1- to 2-mm cuff of normal tissue. To avoid injury to
compressed neurovascular structures, the nerves and vessels should be
exposed proximally and distally and followed under the constriction.
Skin closure utilizing multiple Z-plasties fashioned with large
60-degree flaps helps to avoid postoperative circumferential scar
retraction. Subcutaneous fat advancement flaps (SFAFs) can also be
utilized to achieve a more normal contour to the extremity (172).
In children with ischemia of the distal limb, a fasciotomy may be
required. Surgical treatment of the constriction band is generally
performed prior to any surgical treatment of foot deformities in order
to avoid swelling and neurovascular compromise. However, while excising
a deep band in the distal aspect of a lower leg that is associated with
a clubfoot, Achilles tendon lengthening and posterior capsular release
of the tibiotalar and subtalar joints can be performed through the
incision used for band resection. If necessary, more extensive
procedures on the foot should be delayed until healing has occurred
after the excision of the constriction band, and there is resolution of
the distal edema.
syndrome, is a sclerosing bone dysplasia characterized by progressive
diaphyseal thickening and sclerosis, bone pain, and weakness (173,174).
the variability of its expression have not yet been fully elucidated,
but it appears to be associated with various missense mutations in the
gene encoding human transforming growth factor-β1 (TGFB1) on chromosome 19q13 (175,176,177).
The TGF-β family represents a class of signaling molecules that plays a
central role in morphogenesis, growth, and cell differentiation. TGFB1,
which is abundant in skeletal tissue and is stored in the bone matrix,
appears to act as a coupling factor between bone formation and
resorption. TGFB1 is secreted as a latent precursor, which consists of
a mature peptide and a latency-associated peptide (β1-LAP). TGFB1 can
exert its multiple functions only after going from this latent
precursor state to an active state in which the binding site of the
mature peptide for its receptor is no longer shielded by the
latency-associated peptide. It appears that the most common mutation in
the disorder involves an arginine-cysteine amino acid change at codon
218 (R218C) in the latency-associated peptide domain of TGFB1, which may lead to an increase in TGFB1 bioactivity (178,179,180,181).
with boys being affected more often than girls (approximately in the
ratio 3:2) (182). The age at the onset of
symptoms is highly variable, with most individuals presenting in the
first decade of life. In one study, the mean age at onset was 15 years
and 4 months, with the ages ranging from 1 year to 70 years (182).
The clinical and radiologic manifestations are also variable. The most
common symptoms are limb pain, proneness to fatigue, headache, poor
appetite, and difficulty in running. Clinical signs include muscle
weakness and atrophy, thickening of the long bones, genu valgum,
waddling gait, and exophthalmus (182). If radiographs are not performed, these children can be mistakenly diagnosed as having neuromuscular disorders (183). Basilar skull sclerosis can lead to narrowing of cranial foramina with symptomatic auditory and optic nerve compression (184).
Progression usually occurs in a slow, unpredictable fashion but
occasionally will spontaneously stop. Patients have a normal life span.
Biochemical markers of bone turnover may be useful for assessment of
disease activity, and may correlate with bone scan activity (186).
and endosteal diaphyseal sclerosis, and diaphyseal widening. The tibia
is most frequently involved, but all the long bones in the upper and
lower extremities, and the clavicle, can be affected early in the
course of the disease. The cortex becomes wide and irregular with
narrowing of the medullary cavity (Fig. 10.10).
CT scans show that the thickening and sclerosis are irregular and
nonhomogeneous, and that endosteal involvement may be more extensive
than periosteal thickening (187). Sclerosis at
the base of the skull is common but is not an early finding. The
evolution, as evidenced by radiologic studies, is usually progressive,
with increasing sclerosis of the bone, extension to the metaphysis and
epiphysis, and eventual involvement of the metacarpals and metatarsals,
pelvis, and spine (182,185,188,189).
In the spine, only the posterior elements and the posterior aspect of
the vertebral body are affected, and there is no stenosis (187,189).
Technetium-99m bone scans demonstrate uptake in the middiaphysis of the
affected long bones early in the disease, often before changes show up
on the plain radiographs. MRI shows cortical involvement with sparing
of the medullary cavities but is not helpful in diagnosis or management
(189).
abnormality unique to progressive diaphyseal dysplasia. Early changes
are typical of recent bone formation, with woven bone and lack of
haversian system development, but modeling occurs later (184). Marrow fibrosis and narrowing of the medullary canal can occur, and this may explain the occasional patient with anemia (188).
Considerable overlap exists in the classification of sclerosing bone dysplasias (190).
The diagnosis can be made by determining the inheritance pattern,
clinical and radiologic characteristics, and laboratory findings or
lack thereof. Ribbing disease has a similar radiologic appearance but
affects only the lower extremities and is not always symmetrical.
Extensive early cranial and facial involvement, an autosomal recessive
inheritance pattern, and mental retardation differentiate
craniodiaphysial dysplasia from progressive diaphyseal dysplasia.
Osteopetrosis is characterized by sclerosis throughout the skeleton,
with loss of the medullary canals. Infantile cortical hyperostosis, or
Caffey disease, is distinguished by neonatal onset, febrility,
hyperirritability, and soft-tissue swelling. Hyperphosphatasia may have
radiologic findings similar to progressive diaphyseal dysplasia, but
the alkaline phosphatase level is markedly increased. Hardcastle
syndrome is an autosomal dominant disorder with diaphyseal medullary
stenosis and sclerosis with pathologic fractures and malignant
transformation (191). Juvenile Paget disease,
infantile cortical hyperostosis, hypervitaminosis A, and fluorosis
should also be considered in the differential diagnosis.
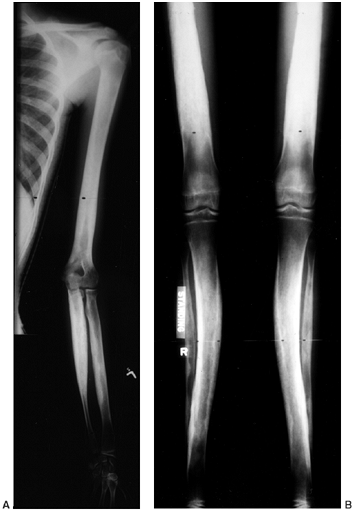 |
|
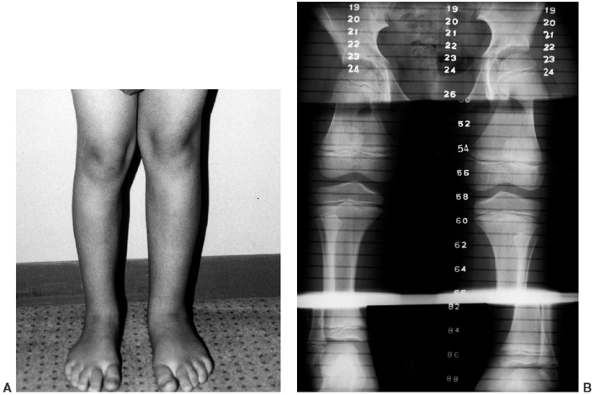
Figure 10.10 A, B:
A 17-year-old girl with diaphyseal dysplasia. Radiographs demonstrate wide and irregular cortices and marked narrowing of the medullary cavity of the long bones in the upper and lower extremities; the typical finding of genu valgum is demonstrated. |
largely supportive. Nonsteroidal antiinflammatories may help decrease
limb pain, and physical therapy can be instituted to help maximize
function. Corticosteroid administration has been shown to reduce pain
in children as well as in adults, with inconsistent reports of
decreased diaphyseal uptake on Tc-99m bone scintigraphy (192,193,194,195).
Knee flexion deformities, genu valgum, and external tibial torsion were
corrected by distal femoral and proximal tibial and fibular
osteotomies. The bone
was described as being soft and vascular but it healed uneventfully.
1922, is a rare, sclerosing skeletal dysplasia characterized by
soft-tissue contractures in childhood overlying slowly evolving linear
hyperostosis (196). The name is derived from the Greek words melos meaning “limb” and rhein meaning “to flow,” as in wax.
The typical presentation is with painless, asymmetric joint
contractures prior to 6 years of age (197,198).
The lower extremities are more frequently involved than are the upper
extremities. The underlying hyperostosis develops slowly and progresses
with age, usually more rapidly in childhood. The overlying soft tissues
may be thickened with lymphedema, and the skin is often tense, shiny,
erythematous, and sclerodermatous over the involved areas (199,200,201,202).
There is associated muscle atrophy and periarticular fibrosis with
contractures. Flexion contractures at the knee, hip, ankle and fingers,
and patellar dislocation are the most common joint deformities (198). Paraarticular ossification and synovitis can further impair joint motion (203,204).
Limb-length discrepancy and angular deformities around the knee and
ankle can result from fibrosis and physeal abnormalities. A mean of 4
cm of limb shortening was noted in one study of 11 children, although
one patient had overgrowth of the affected limb (198).
Melorheostosis does not shorten life span, but morbidity may be
considerable. Kidney anomalies, including minimal-change nephrotic
syndrome and renovascular hypertension secondary to renal artery
stenosis, have been described (205,206,207).
It has been observed that distribution of the lesions correspond to
sclerotomes (204). One hypothesis is that an
infection analogous to herpes zoster occurs, and lesions spread along
the affected nerve roots with resultant scarring and osseous changes.
The sporadic occurrence of melorheostosis and osteopoikilosis, the
so-called mixed sclerosing bone dysplasia, in families with familial osteopoikilosis (an autosomal dominant disorder) has been reported (190,208,209),
and raises the possibility that the two disorders may be related. Kim
et al. suggested that the altered expression of several adhesion
proteins may contribute to the development of the hyperostosis and
associated soft-tissue abnormalities in melorheostosis, in view of the
downregulation of human transforming growth factor-β-induced gene
product (betaig-h3) in skin fibroblasts from affected regions (210).
asymmetrical bands of sclerosis in an irregular, linear pattern often
described as molten wax flowing down the side of a candle (Fig. 10.11). In children the hyperostosis is endosteal, whereas in adults it is in an extracortical, subperiosteal location (202,203).
The hyperostosis can be located throughout the skeleton, typically on
one side of the diaphysis of long bones, the pelvis, and in the hands
and feet. The ribs, skull, and spine are affected least often. Patches
of hyperostosis, rather than a linear pattern, are present in the
carpal and tarsal bones and the epiphyses. This is similar to what is
seen in osteopoikilosis. Bone scans reveal increased uptake in the
areas involved.
Increased expression of procollagen α1(I) mRNA and secretion of
α1(I),α2(I) andα1(III) collagen has been noted in skin biopsies
overlying the bone involved (212).
osteopetrosis, osteopoikilosis, and osteopathia striata, all of which
can have similar radiographic findings. Mixed sclerosing bone dysplasia
comprises melorheostosis and osteopoikilosis and/or osteopathia striata
in the same individual (190,208,209).
The transient periosteal reaction in infantile cortical hyperostosis is
less dense and is found in different locations. Focal scleroderma may
cause soft-tissue fibrosis and contractures, but the bones are
radiologically normal (213).
Non-steroidal antiinflammatory medications can ease discomfort.
Surgical treatment including soft-tissue releases, capsulotomies, and
osteotomies are difficult to carry out, and incomplete correction or
rapid recurrences of the deformity are common, sometimes necessitating
an amputation (198,203).
Distal limb ischemia can occur when the chronically contracted and
flexed joint is extended. Osteotomies that permit shortening may avoid
this complication.
of children for lengthening, realignment of angular deformities, and
correction of joint contractures (214,215,216,217,218).
Realignment and lengthening have been done successfully, but
complications include pseudarthrosis and, in one child, ischemia that
caused pain and led to loss of function and eventual amputation of the
affected part (215,217).
is an uncommon, autosomal dominant, sclerosing bone dysplasia
characterized by numerous small foci of increased radiodensity in the
periarticular regions.
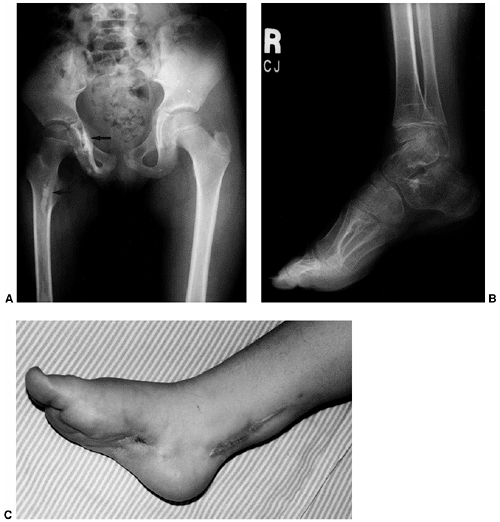 |
|
Figure 10.11 An 8-year-old girl with melorheostosis who presented with an equinovarus foot deformity. A: The classical findings of irregular linear hyperostosis are seen at the arrows. B: Patches of hyperostosis are seen in the talus and calcaneus; this is typical of melorheostotic involvement of the tarsals. C:
There was pain and swelling around the equinovarus foot. Previous surgical releases had resulted in rapid recurrence of the deformity. |
throughout life. The children have normal stature and most are
asymptomatic, but up to 20% may have mild articular discomfort
associated with joint effusions (219). The
diagnosis is often made as an incidental radiologic finding. Fractures
heal uneventfully and pathologic fractures have not been reported. The
risk of malignancy is probably not higher than in the normal
population. Osteopoikilosis is frequently seen in association with
another hereditary dermatologic condition, dermatofibrosis lenticularis
disseminata, or Buschke-Ollendorf syndrome, which is marked by the
presence of papular fibromas (220,221).
This syndrome is usually asymptomatic, but soft tissue fibrosis and
joint contractures that appear clinically similar to those seen in
melorheostosis can occur. Osteopoikilosis has also been described in
association with melorheostosis, the so-called mixed sclerosing bone
dysplasia (190,208,209).
homogeneous, bilateral, circular- to ovoid-shaped from 1 to 15 mm in
width, and are located in the metaphyses and epiphyses of long bones,
the carpus, the tarsus, the pelvis, and the scapulae (Fig. 10.12). The ribs, clavicle, and skull, are not involved (219,222). The lesions may increase or decrease in size or number (222).
This is useful in differentiating this condition from metastatic
carcinoma of breast or prostate that may have a similar radiographic
appearance. Mastocytosis should also be considered in the differential
diagnosis.
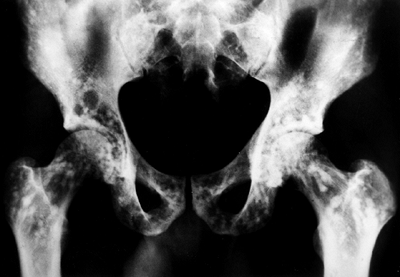 |
|
Figure 10.12 Osteopoikilosis. Extensive involvement of the pelvis is seen in this asymptomatic patient.
|
clear. The sclerotomal distribution and association with abnormalities
of mesodermal tissues suggests a relation between this condition and
other osteosclerotic disorders (220,222,224).
The concept of mosaicism, due to nondisjunction or anaphase lagging
resulting in an individual with two or more cell lines with different
karyotypes, may explain the occurrence of osteopoikilosis, an autosomal
dominant disorder, occurring along with melorheostosis, a sporadic
disorder, in so-called mixed sclerosing bone dysplasia. The
melorheostotic component may be caused by early loss of the
corresponding wild-type allele at the gene locus that gives rise to
isolated familial osteopoikilosis (190,208,209,225).
rare cases of associated fibrosis and joint contracture, the management
is the same as for melorheostosis.
the sclerosing bone dysplasias and is characterized by dense linear
striations in bone (226,227).
exhibits no physical or laboratory abnormalities. The diagnosis is
usually made on an incidental radiograph.
the tubular and flat bones with the exception of the skull and
clavicles. These lesions do not change with time. In the long bones,
the striations are parallel to the long axis and affect mainly the
metaphyses, but may extend into the epiphyses also (Fig. 10.13).
A fan-shaped pattern of linear striations can be seen in the iliac
wings, likely reflecting the growth pattern of the pelvis (227). There is no increased uptake on bone scan.
constituent of a more complex sclerosing bone dysplasia, such as mixed
sclerosing bone dysplasia, where it may be found in association with
osteopoikilosis and/or melorheostosis (224,227,228).
Osteopathia striata with cranial sclerosis (OS-CS) is a rare syndrome
characterized by longitudinal striations of the long bones and
sclerosis of the craniofacial bones in association with typical facies,
macrocephaly, conductive hearing loss, mental retardation, and other
manifestations (229,230). Severe cervical kyphosis has been noted in one patient with this disorder (231).
Sponastrime dysplasia includes osteopathia striatalike radiographic
findings, characteristic facies, and vertebral anomalies (232,233). Symptomatic osteopathia striata is also a characteristic of X-linked focal dermal hypoplasia (234).
disorder of formation of the clavicle that is recognizable at birth.
The right clavicle is most commonly affected, although bilateral
involvement has been reported in approximately 10% of cases, and rare
examples of isolated left-sided involvement have been documented. The
absence of birth trauma and the subsequent lack of exuberant early
callus formation distinguish it from acute neonatal fracture. The
appearance of the clavicular defect on roentgenographic examination,
and the lack of other characteristic bony malformations of the skull
and pelvis, distinguish this disorder from cleidocranial dysplasia.
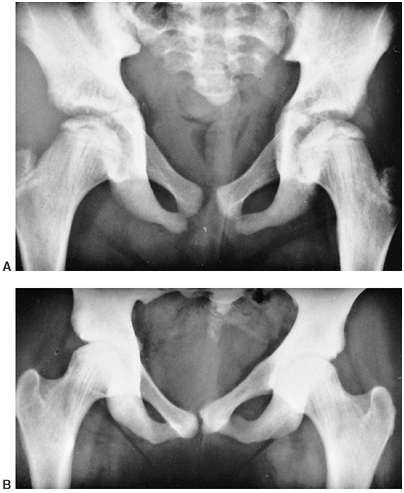 |
|
Figure 10.13 Osteopathia striata. The streaking of the proximal femurs did not change much in this patient between the ages of 12 years (A) and 25 years (B). Note the flattened femoral epiphysis.
|
clavicle is unclear. Most cases are thought to arise sporadically,
although familial cases demonstrating autosomal dominant inheritance
have been reported (235,236).
Congenital pseudarthrosis of the clavicle is thought to occur either
because of a primary intrinsic failure of clavicular development or
because of external compressive forces. The development of the clavicle
from two separate centers of intramembranous ossification was first
recognized by Mall (236) and has been confirmed by others (237,238).
It has been postulated that an intrinsic failure of coalescence of
these two centers is responsible for the development of pseudarthrosis (239). Lloyd-Roberts et al. (158)
postulated that extrinsic compression by the subclavian artery, alone
or in combination with a cervical rib, was responsible for the defect.
This interesting concept is supported by the overwhelming frequency of
right-side involvement, the normally higher location of the right
subclavian artery compared to the left, and the location of the artery
directly under the area of the defect. Rare cases of left-side
involvement have been associated with dextrocardia, and bilateral cases
have been associated with cervical ribs (240).
presents in infancy or in early childhood, with a painless prominence
and hypermobile segment over the middle third of the clavicle.
Radiographs may be necessary to distinguish the lesion from birth
fracture or cleidocranial dysplasia. Most lesions involve only the
right clavicle, although bilateral cases and rarer left-side cases have
been described. The cosmetic deformity tends to be slowly progressive,
with an increase in noticeable prominence over the pseudarthrosis and
foreshortening and dropping of the shoulder girdle over time. The
pseudarthrosis can become painful with certain overhead activities, or
with direct compression or palpation. Shoulder motion is typically
regarded as normal, and significant functional disability is unusual.
Several cases of later development of thoracic outlet compression
syndrome have been reported in association with congenital
pseudarthrosis of the clavicle (235,241,242,243,244,245,246,247,248).
are characteristic. The clavicle typically displays a definite
separation in its midportion, with the medial aspect lying superior to
the lateral aspect, owing to muscle forces and the weight of the upper
extremity. The bone ends appear bulbous, with sclerotic closure of the
marrow cavity and no evidence of callus formation or reactive bones (Fig. 10.14A). The disorder is
easily distinguishable from cleidocranial dysplasia because of the lack
of other characteristic radiographic abnormalities of the skull and
pelvis, including wide cranial sutures, coxa vara, failure of pubic
ossification, and hypoplastic ilia. Subsequent radiographs of the
clavicle will fail to demonstrate callus formation, thereby
distinguishing early cases from acute neonatal fracture.
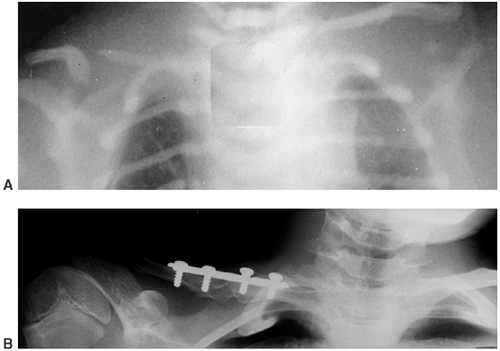 |
|
Figure 10.14 Pseudarthrosis of the right clavicle. A: Congential pseudarthrosis of the right clavicle. There are no signs of healing. B:
Radiographic appearance after solid union achieved by open reduction, internal fixation, and autogenous bone grafting in a different 8-year-old boy. |
demonstrated hyaline cartilaginous caps at both ends of the
pseudarthrosis, often joined by dense fibrous and fibrocartilaginous
tissue (249,250).
Hirata et al. demonstrated columnar distribution of chondrocytes in
different stages of maturation at the ends of the pseudarthrosis,
similar to that seen in a developing cartilaginous physis. Endochondral
ossification at these terminal ends was confirmed by tetracycline
labeling (251).
clavicle, whose cases are described in the literature, have undergone
some form of operative treatment, and therefore the true natural
progression of untreated cases is unknown. Indications for operative
management have included progressive pain and unacceptable cosmetic
deformity at the site of the pseudarthrosis, functional limitations
and, at later stages, onset of thoracic outlet compression syndrome (235,239,241,242,243,244,245,246,247,248,249,251,252,253).
Most specialists recommend delaying surgery until the patient is
approximately 3 to 6 years of age, and thereafter resecting the
pseudarthrosis followed by bone grafting and internal fixation, most
commonly with the use of a plate and screws (235,238,244,250,253) (Fig. 10.14B).
Solid union of the pseudarthrosis and acceptable cosmesis have been
regularly achieved by using these methods. Grogan et al. reported on
early operative treatment of this condition in eight patients, of whom
six were younger than 30 months (252). They
demonstrated union in all eight patients, using a technique of careful
preservation of the periosteal sleeve, resection of the pseudarthrosis,
and approximation of the bone ends without bone grafting or internal
fixation. Toledo and MacEwen pointed out the hazards of using an
intramedullary pin for fixation after operative treatment of this
condition (254), although this technique has been successfully employed by others (250,255).
pseudarthrosis using a technique of resection of the pseudarthrosis,
autogenous iliac crest grafting, and plate and screw fixation,
typically performed at approximately 3 to 6 years of age. This approach
avoids the noticeable clavicular prominence and foreshortening and
dropping of the shoulder girdle that occurs over time, and it may
eliminate the later development of thoracic outlet syndrome, although
this is unsubstantiated. Parents should be counseled that operative
treatment does replace a “bump” with a “scar,” and is not essential for
ensuring good shoulder function.
localized osteochondral overgrowth, involving single or multiple
epiphyses or ossification centers. First reported by Mouchet and Belot
as tarsomegalie, the disorder was later clarified by Trevor, who termed the disorder tarso-epiphysial aclasis (256,257). Fairbank later suggested the term dysplasia epiphysialis hemimelica,
to deemphasize tarsal involvement and to describe the predilection for
single-side involvement of the medial or lateral aspect of one or more
epiphyses (258). Although the lesion most commonly affects the medial aspect of the epiphysis, lateral
involvement, as well as involvement of entire epiphyses, has been
reported. When multiple sites are involved, unilateral distribution is
common, and the lesion can present on both sides of an affected joint.
unknown. Trevor considered it to be a congenital error of skeletal
development, in which an altered process of cell division at the
superficial zone of articular cartilage allows for persistent
proliferation and production of a large cartilaginous mass (257).
Fairbank hypothesized that the disorder was due to a localized
disturbance of the pre- or postaxial part of the apical cap of the limb
bud in early fetal development (258). To date, no clear mechanism for genetic transmission of the disorder has been described.
1 million, most of them being men. Presentation is usually in childhood
or early adolescence, although presentations in adulthood have also
been described (259). In cases of multiple
sites of involvement, the lesions are characteristically unilateral,
although bilateral cases have been reported. Azouz et al. classified
dysplasia epiphysealis hemimelica into three categories: (i) localized
(involving only one epiphysis), (ii) classic (involving more than one
bone in a single limb), and (iii) generalized (involving an entire
lower extremity from the pelvis to foot). The classic form is the most
common (260). Clinical features vary, depending
on the location of the lesion, but patients typically complain of
painless swelling or a mass on one side of a joint, limitation of
motion, and occasionally locking, angular deformity, limp, regional
muscle wasting, or limb-length discrepancy. The lower extremities are
affected most commonly, predominantly at the knee and ankle (257,258,261,262).
Other reported sites have included the capital femoral epiphysis and
acetabulum, sacroiliac joint, metacarpophalangeal joint, wrist,
shoulder, and subtalar joint (263,264,265,266,267,268,269).
hemimelica can vary depending on the age of the patient and the degree
of maturation of the lesion. The radiologic diagnosis is fairly
straightforward when the lesion is fully calcified or ossified,
appearing as an irregular, often multicentric, lobulated mass
protruding directly from one side of the affected epiphysis or tarsal
bone (Fig. 10.15). The affected ossification
center will often appear prematurely, and may be larger than the
contralateral side. Modeling abnormalities of the adjacent metaphysis
have also been reported. However, early lesions may present only with
joint space enlargement or small foci of irregular calcification, and
may resemble para-articular tumors or other disorders that involve
ectopic ossification or calcification. With maturation, the lesion
enlarges and ossifies, and becomes confluent with the underlying
epiphyseal bone (270). MRI has proven useful in
demonstrating the extent of the lesion and in identifying a potential
cleavage plane between the lesion and the underlying epiphysis (261,270,271,272).
resemble an osteochondroma, although no discrete stalk is present.
Microscopically, one may find a boundary of cartilage separating the
lesion from the underlying bony epiphysis, if the lesion has not yet
fully ossified. A thick zone of hyperplastic cartilage is
distinguishable from the rest of the cartilaginous epiphysis only by
some irregularity in the size and distribution of the chondrocytes (258). Malignant degeneration of the lesion has not been reported.
directed at addressing the patient’s complaints, while preserving the
integrity of the affected joint as much as possible. Most of the
reported cases have been treated surgically, and therefore the
long-term prognosis for untreated lesions involving the weight bearing
surface of the joint (i.e., articular lesions) is mostly unknown.
Fairbank predicted that the affected joints would go on to early
degenerative joint disease, and this has been demonstrated in some
patients after surgical intervention (261,262,267,273).
If the cartilaginous overgrowth is not on the weight bearing surface of
the joint (i.e., juxta-articular lesions), simple excision is warranted
in order to relieve pain and improve function. However, recurrence of
the lesion prior to skeletal maturity has been reported (262,263).
Fairbank described spontaneous correction of associated angular
deformities after excision, but this has not been a consistent finding (236).
The treatment of articular lesions remains somewhat controversial. Kuo
et al. did not recommend excision of articular lesions because of
unsatisfactory results in three of nine patients with lesions involving
the distal tibia and talus (262). Keret et al.
performed corrective osteotomies without excision for angular deformity
associated with articular lesions wherever an arthrogram demonstrated a
smooth joint surface (261). The authors noted
recurrence of valgus deformity at the knee after varus osteotomy in one
of three patients, requiring repeat osteotomy.
affected joint and to identify any potential cleavage plane for
surgical separation of the lesion from the underlying epiphysis.
Symptomatic juxta-articular lesions are treated by simple excision.
Intra-articular lesions are treated by excision if there is
irregularity of the articular surface. Close observation of limb length
and alignment is necessary in view of the potential for growth arrest
and angular deformity, and osteotomy and/or limb lengthening may be
required.
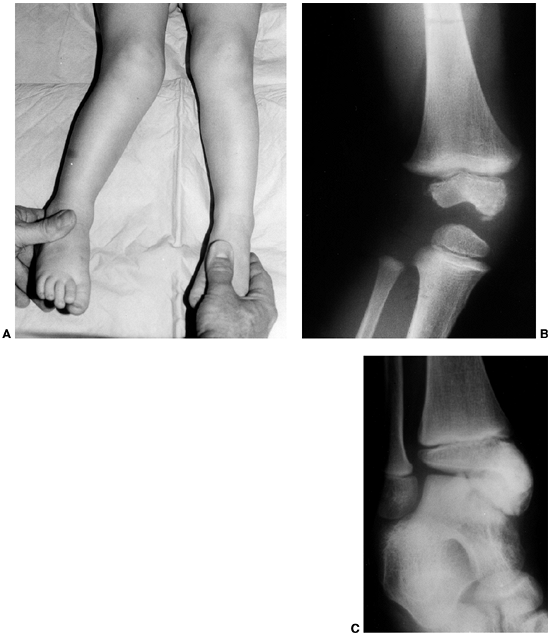 |
|
Figure 10.15 A–C:
Dysplasia epiphysealis hemimelica in a young girl. Right genu valgum has resulted from involvement of the distal medial femoral epiphysis. Associated involvement in the medial distal tibial epiphysis is demonstrated. |
disorder characterized by congenital malformations of the great toes
and by later development of a predictable pattern of heterotopic
endochondral osteogenesis of tendons, ligaments, fasciae, and striated
muscles. Although most patients present as new mutations, autosomal
dominant transmission and variable expressivity have been confirmed by
several authors (274,275,276,277). The incidence of this condition is extremely rare, affecting less than 1 in 1 million persons (278).
fibrodysplasia ossificans progressiva are unknown at this time, and
clinical, molecular, and genetic heterogeneity may exist (279,280,281).
The combination of the congenital malformation of the great toe and the
postnatal development of heterotopic bone, in characteristic patterns
of developmental gradients, suggests a possible role of the bone
morphogenetic proteins in the etiology of this disorder. Bone
morphogenetic proteins have been implicated in several embryonic
epithelial and mesenchymal interactions, such as limb formation, and
have the potential to transdifferentiate myoblasts in culture into
osteoblastlike cells. Overexpression of bone morphogenetic protein 4
(BMP 4) and its messenger ribonucleic acid (mRNA), and elevated steady
state levels of BMP 4 receptor mRNA, have been demonstrated in lesional
and nonlesional tissue from patients with this condition (282,283,284). However, a mutation in the BMP 4 gene has not been demonstrated.(285).
Additionally, high-affinity antagonists of BMP 4, such as the secreted
polypeptide noggin, are underexpressed in the cells of affected
patients (286),
thereby suggesting that an impaired BMP-antagonist response following
soft-tissue trauma could explain the explosive bone induction seen
during flare-ups.
fibrodysplasia ossificans progressiva involves aberrant morphology of
the great toe, which demonstrates shortening of the first ray,
delta-shaped proximal phalanx, interphalangeal joint fusion, and
varying degrees of hallux valgus, either alone or in combination (287).
Progressive heterotopic ossification becomes evident at a mean age of 5
years, but may be seen shortly after birth or as late as the second
decade of life. Most lesions arise spontaneously, although blunt trauma
can trigger the onset. Individual nodules can regress, although most of
them progress to form mature bone. In their review of 44 patients with
fibrodysplasia ossificans progressiva, Cohen et al. noted a predictable
pattern of involvement of heterotopic ossification, proceeding from
axial to appendicular, cranial to caudal, and proximal to distal (288).
Dorsal involvement preceded ventral involvement, with the initial
lesion characteristically occurring as a painful, erythematous,
subfascial nodule in the posterior aspect of the neck, spine, or
shoulder girdle. Kaplan et al. described the early, intermediate, and
late progression of a typical lesion in fibrodysplasia ossificans
progressiva (277). The early lesion is heralded
by pain, erythema, swelling, warmth, and tenderness, and may resemble
an infectious process. After several weeks, the swelling and pain begin
to subside, but an increase in induration occurs (the intermediate
lesion). The late lesion is present by 12 weeks, and consists of a
hard, painless mass that is evident roentgenographically (Fig. 10.16).
ankylosis of the major diarthrodial joints. Hip and knee involvement
can impair walking and sitting ability. Progressive spinal deformity is
common, and can be particularly severe when unilateral
pelvis-to-chest-wall synostosis is present (289). Humeral-to-chest-wall synostosis can lead to superior subluxation of the glenohumeral joint (290).
Involvement of the temporomandibular joint may lead to inanition with
resultant poor nutrition. Decubiti often develop because of a
combination of underlying bony prominences, poor nutrition, and lack of
mobility. Other characteristics that may be associated with this
disorder include shortened thumbs, fifth-finger clinodactyly,
conductive hearing loss, diffuse thinning of the hair on the scalp,
premature cessation of menses in female patients, and, uncommonly,
mental retardation. Exacerbating factors may include trauma to the
muscles, biopsy of lesions, surgical attempts at bone removal,
intramuscular injections, careless venipuncture, and dental therapy (287).
New lesions can occur throughout the patient’s lifetime, and most
patients become completely immobilized and confined to a wheelchair by
their third decade. The diaphragm, extraocular muscles, cardiac muscle,
and smooth muscle are characteristically spared in the disorder.
shortened survival of some patients with fibrodysplasia ossificans
progressiva. Heterotopic ossification involving the chest wall and
spine can lead to impaired chest wall dynamics and reliance on
diaphragmatic breathing (278,288).
Kussmaul et al. found a high incidence of right ventricular
abnormalities on electrocardiographic evaluation of patients with
fibrodysplasia ossificans progressiva who had severely restrictive
chest-wall disease (291). The investigators raise the possibility that cor pulmonale could eventually develop in such patients.
ossificans progressiva are initially similar in radiographic appearance
to myositis ossificans, with diffuse calcification developing
progressively to zonal osseous maturation. Sheets of mature skeletal
bone eventually form along striated muscles, fasciae, tendons, and
ligaments, causing ankylosis of the joints. Solid extraosseous bridges
can form between bones, leading to characteristic synostoses of
cervical spine to occiput, of scapula and chest wall to humerus, of
spine to pelvis, and of pelvis to femur. In addition to the
characteristic great toe deformities, abnormal cervical vertebrae with
small bodies, large posterior elements and fused lateral masses,
shortened first metacarpals, fifth-finger clinodactyly, short and broad
femoral necks, and exostoses of the proximal tibiae have been reported (Fig. 10.16 C,D,E,F) (287).
fibrodysplasia ossificans progressiva have provided insights into the
pathogenesis of the ossification. Histopathologic and
immunohistochemical studies of specimens from early preosseous lesions
reveal an abundance of primitive vascular cells and angiogenesis, with
a conspicuous absence of an acute or chronic inflammatory process. The
clinician may be misled toward a presumptive diagnosis of aggressive
fibromatosis or low-grade sarcoma at this stage if fibrodysplasia
ossificans progressiva is not considered. Intermediate lesions exhibit
scattered cartilaginous foci and evidence of endochondral ossification,
the stages proceeding in a sequence that appears entirely normal. Late
lesions demonstrate histologically normal lamellar bone, with fatty
marrow encased in fibrous connective tissue (292).
fibrodysplasia ossificans progressiva. Treatment is largely supportive,
with attempts at limiting the factors that exacerbate ectopic bone
formation, such as trauma, intramuscular injections, careless
venipuncture, and dental extractions. Padding of bony prominences and
nutritional
support may be useful in preventing decubiti. Biopsy of lesions or
surgery to excise ectopic bone must be avoided, because they always
precipitate new bone formation (287).
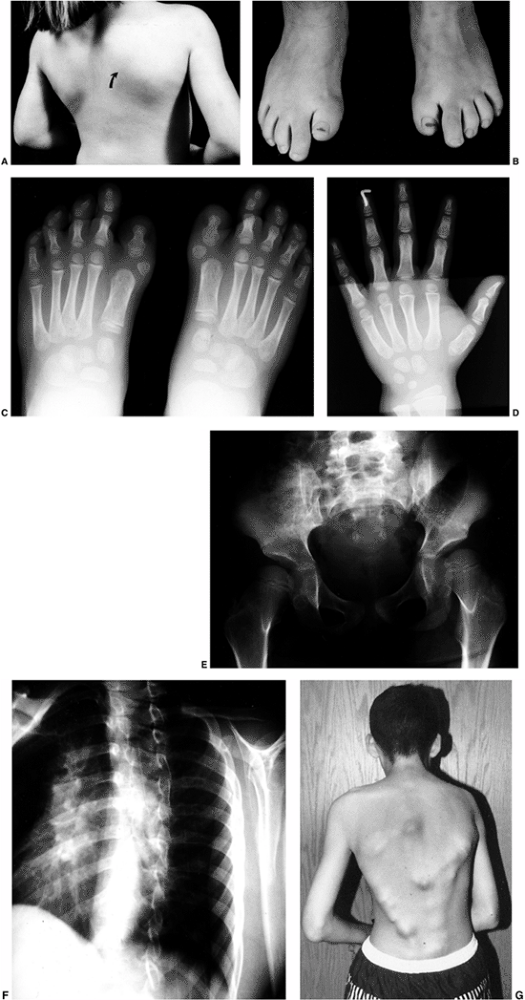 |
|
Figure 10.16
Fibrodysplasia ossificans progressiva. A 4-year-old girl at the time of presentation. She complained of right periscapular swelling, warmth, and tenderness. A: Clinical photograph demonstrating the area of right periscapular involvement (arrow). B: Characteristic great toe morphology, demonstrating shortening of the great toes bilaterally. C: Bilateral radiographs of the feet demonstrating shortened great toes, with bilateral delta phalanges and shortened first metatarsals. D: Anteroposterior radiograph of the hand, demonstrating characteristic shortening of the thumb metacarpal. E: Anteroposterior radiograph of the pelvis, demonstrating short, broad femoral necks, and exostoses. F: Early spontaneous fusion along the posterior elements and lateral masses is demonstrated. G: Clinical appearance of an older individual with advanced subcutaneous ossification and characteristic dorsal-to-ventral pattern. |
and corticosteroids has not been shown to influence the development of
the ectopic ossifications. Brantus and Meunier evaluated the effects of
intravenous administration of ethane-1-hydroxy-1-diphosphonate and oral
prednisone on early flare-ups in patients with fibrodysplasia
ossificans progressiva (293). During a mean
follow-up of 6 years in 7 patients with the disorder, the symptoms of
local swelling and pain were ameliorated in 29 out of 31 acute
flare-ups, and no ossification appeared in 21 flare-ups. However, the
percentage of acute flare-ups that normally result in ossification is
unknown; therefore it cannot be confirmed whether administering
intravenous diphosphonates during early flare-ups has any effect on
subsequent progression to ossification. Also, during the period of
study, 10 new ossifications were observed that resulted in severe
deterioration of joint mobility, and there was no change in the
radiographic appearances of preexisting ossifications.
ossificans progressiva will likely center around inhibitors of
endochondral osteogenesis. Proposed treatments have included the use of
BMP 4 inhibitors, isotretinoin (an
inhibitor of mesenchymal tissue differentiation into cartilage and
bone), and antiangiogenic and antiinflammatory factors (293,294,295,296,297). To date, such treatments should be regarded as investigational.
of nonneoplastic heterotopic ossification within soft tissues, most
often in response to localized trauma. Although the process most
commonly develops within skeletal muscle, the term itself is a
misnomer, because nonmuscular tissue may be involved, and inflammation
is rare. Adolescents and young adults, predominantly men, are affected
most frequently, although myositis ossificans has been reported in
infancy as well (298).
The development of heterotopic ossification has also been reported
after severe thermal injury, in neurologic diseases, in posttraumatic
paraplegia and brain injury, and after some orthopaedic operations,
such as total hip arthroplasty (300,301,302,303,304,305,306,307,308).
Rarer genetic and developmental forms of heterotopic ossification
exist, including fibrodysplasia ossificans progressiva, progressive
osseous heteroplasia, pseudomalignant heterotopic ossification,
Albright hereditary osteodystrophy, and parosteal fasciitis (287,294,309,310,311,312).
ossificans remains unknown. Recent investigations into the role of
extraskeletal osteogenic precursor cells, and the local factors that
induce them, may provide insights into the
origins
of ectopic bone formation in myositis ossificans, and in other
disorders characterized by the formation of heterotopic ossification (313,314).
begin approximately 1 to 3 weeks after an injury, and include localized
pain and a palpable mass. Increased warmth, swelling, and limitation of
motion may be noted in the affected area. A low-grade fever and a
mildly elevated erythrocyte sedimentation rate may be present. Limb
involvement predominates, most commonly in the quadriceps or brachialis
muscles, although virtually any region of the body can be affected. An
increase in the firmness of the lesion and a decrease in pain occur
over an 8- to 12-week period, with the development of varying degrees
of contracture of the affected part.
demonstrate a noncalcified mass in the soft tissues, typically over the
diaphyseal region of the extremity. Within 2 to 4 weeks after the
injury, floccular calcifications begin to appear within the mass; if
the cambium layer of the periosteum was involved in the initial injury,
a periosteal reaction of the underlying bone may be seen. Over a 6- to
8-week period, serial radiographs obtained at 1- to 2-week intervals
characteristically demonstrate peripheral osseous maturation of the
lesion, with a central lucent zone and a lucent line separating it from
the underlying cortex, and should be helpful in distinguishing the
lesion from an extraosseous sarcoma (Fig. 10.17).
After 5 to 6 months, mature bone is evident, and the lesion may show a
decrease in overall size. A computed tomography (CT) scan may be
particularly helpful in delineating the zonal maturation and cortical
separation in myositis ossificans when the diagnosis is unclear (Fig. 10.18) (299).
Other imaging modalities that have been utilized in the diagnosis of
this condition include bone scintigraphy, ultrasound, MRI, leukocyte
scanning, and angiography, particularly in early lesions or in
difficult cases (299,312).
In patients with a typical history of trauma and localized findings,
and with roentgenographic evidence of progressive peripheral osseous
maturation, the use of these other imaging modalities is infrequently
required.
The typical lesion consists of a central (inner) zone of
undifferentiated cells and atypical mytotic figures, which may be
impossible to distinguish from a sarcoma; an adjacent (middle) zone of
well-oriented osteoid formation in a non-neoplastic stroma; and a
peripheral (outer) zone of well-oriented lamellar bone, clearly
demarcated from the surrounding tissue. Although rarely required in
this disorder, if biopsy is performed, a specimen of sufficient size
and orientation is important for establishing the architectural pattern
of the lesion.
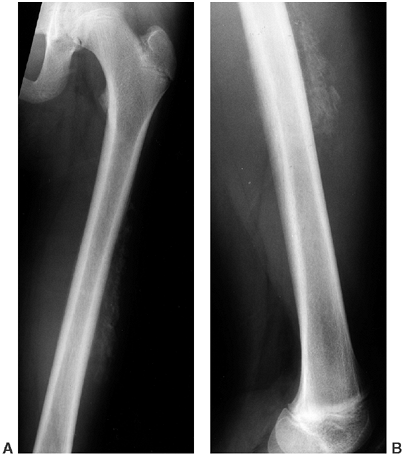 |
|
Figure 10.17
Adolescent boy with symptoms of quadriceps tightness, swelling, and discomfort, 6 weeks after sustaining an impact to his thigh during a football game. A, B: Anteroposterior and lateral radiographs of the femur demonstrate floccular calcifications within the soft tissue of the thigh, with underlying periosteal reaction in the femoral diaphysis. |
initial injury would be ideal. Nonsteroidal antiinflammatory agents can
be utilized to help diminish symptomatology in the early stages of
myositis ossificans. Hughston et al. suggested that strict rest of the
affected part, through the use of splinting, would allow for more
complete resorption of the hematoma and discourage formation of
heterotopic bone (316). Thorndike advocated a combination of rest, icing, compression bandaging, and avoidance of massage therapy (317).
Jackson and Feagin developed a treatment strategy for preventing
myositis ossificans after quadriceps contusions in young athletes (318).
They recommended a three-phase program of 24 to 48 hours of strict rest
of the extremity in extension, elevation, and icing (phase I), followed
by restoration of motion (phase II), and progressive resistance
strengthening (phase III). Ryan et al. advocated that the position of
splinting of the affected muscle should be in tension (e.g., flexion
for quadriceps contusion) (319). However, there
is presently no clear evidence that such measures prevent the
development of myositis ossificans, or modify its severity.
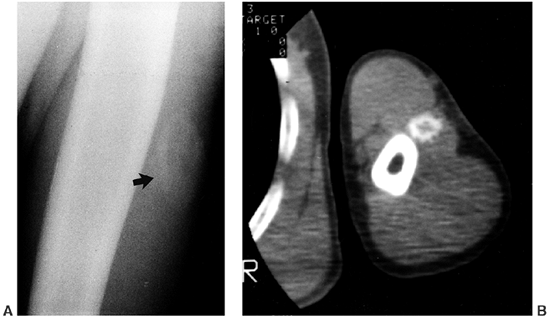 |
|
Figure 10.18 Myositis ossificans. A: Anteroposterior radiograph demonstrating myositis ossificans in the left upper arm (arrow). B:
CT scan of the same patient, delineating peripheral maturation and clear separation of the lesion from the underlying cortex of the humerus. |
can be deceptive until maturation of the lesion occurs. Many of these
lesions will become asymptomatic, and not interfere with function of
the affected part. It is therefore advisable, when considering excision
of heterotopic bone, to wait until full maturation of the lesion is
demonstrated roentgenographically or on bone scintigraphy (typically 12
to 18 months after injury), and to remove lesions only when they
interfere with function or cause persistent local symptomatology.
endochondral ossification, including both chondrogenesis and
osteogenesis, in a previously normal endochondral growth region. The
condition is considered to be idiopathic, and has been reported in
nearly every growth center of the body, including epiphyses, apophyses,
and physes. The various osteochondroses often have associated eponyms,
and their clinical features are related to their area of occurrence. As
a group, the osteochondroses have relatively well-defined natural
histories, and generally predictable outcomes (320).
The more clinically significant osteochondroses are described
throughout this text according to anatomic region, but are highlighted
here because they include features of localized disorders of bone and
cartilage (Table 10.3).
the process of disordered endochondral ossification and subsequent
necrosis remains unknown. Etiologies based upon trauma and vascular
events have been proposed. It has been suggested that the normal
process of chondrification of cartilage canals in the growth center
fails, leading to necrosis of cartilage canal vessels in the
subchondral bone and adjacent epiphyseal cartilage, or to failed
enlargement of the bony centrum and disordered proliferation of
cartilage cells within the epiphysis (320,321). In the case of lesions involving segmental epiphyseal necrosis of the subchondral bone and overlying cartilage, that is, osteochondritis dissecans,
it is not clear whether the vascular necrosis precedes or follows
necrosis of the cartilage. The osteochondroses that affect nonarticular
cartilage, so-called traction apophysitis,
may be related to mechanical disruption of the endochondral mechanism
from longitudinally directed shear forces along tendonous or
ligamentous attachments.
|
TABLE 10.3 EXAMPLES OF OSTEOCHONDROSES BY REGION
|
|
|---|---|
|
osteochondroses are dependent on the site of occurrence and the degree
of involvement, but may include local or referred pain, inflammation,
effusion, limitation of motion, gait disturbance, or, in the case of
physeal involvement such as Blount disease, visible disturbance in
growth.
characteristic for the anatomic area involved, they do share some
common radiographic features. Early involvement may appear as a
decrease in size, increase in density, or irregular architecture of the
involved centrum. Resorption of necrotic bone may lead to apparent
worsening of the radiographic appearance, with return of
radiographically evident bone as revascularization and reossification
occurs (Fig. 10.19). The resultant area may
come to appear radiographically normal, or may demonstrate permanently
altered anatomy. Delay in complete revascularization of a fragmented
necrotic centrum may result in separation of a nonunited segment, as in
osteochondritis dissecans.
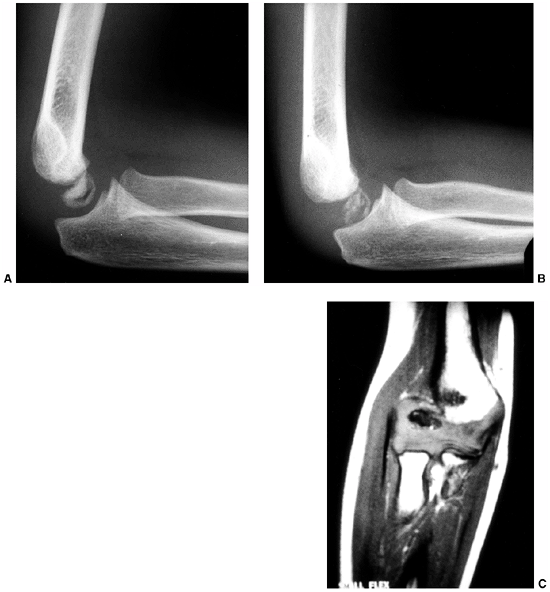 |
|
Figure 10.19 A 7-year-old boy with pain in the right elbow. A:
Lateral radiograph of the elbow when the child was initially seen, demonstrating intra-articular effusion, subchondral lucency, and sclerosis of the capitellar ossification center. B: Lateral radiograph obtained approximately 4 months later, demonstrating fragmentation of the capitellar ossification center. C: Magnetic resonance imaging demonstrating avascular changes within the capitellar ossification center. |
described, including necrosis of bone and cartilage, revascularization,
granulation tissue invasion, osteoclastic resorption of necrotic
segments, osteoid replacement along necrotic trabeculae, and,
eventually, formation of mature lamellar bone (320).
On the basis of the characteristic clinical and radiographic findings,
however, biopsy of an area of involvement is rarely indicated. It is
generally accepted that
in
osteochondritis dissecans, necrosis of the underlying subchondral bone
leads to the separation of an osteocartilaginous fragment from the
surrounding viable bone. It is interesting to note, however, that the
specimen may be purely cartilaginous and, if bone is present, it is not
always necrotic (322).
will respond to symptomatic, nonoperative treatment with complete
resolution (322,323,324,325).
A combination of rest, icing, and rehabilitation of the affected part
can help decrease the duration of symptoms, but may not influence the
final outcome (323). Surgical intervention is
typically reserved for more clinically severe forms, often to reduce
causative stress, such as bony realignment in some cases of Blount
disease, or to stimulate healing or remove intra-articular fragments as
in some cases of osteochondritis dissecans.
congenital or acquired contracture of all or part of the extensor
mechanism of the knee. Some authors call this entity congenital fibrosis; however, we prefer the term infantile quadriceps fibrosis in order to distinguish this entity from congenital dislocation of the knee, which may have quadriceps fibrosis as a feature (326,327,328).
limitation of knee flexion, thigh atrophy, genu valgum or recurvatum,
high-riding patella, and habitual patellar dislocation, which are
thought to be directly related to the extent of the quadriceps fibrosis
(326,327,329). In some patients, increases in knee flexion are possible only after obligate lateral dislocation of the patella (329).
There may be a history of repeated intramuscular injections of the
thigh and gluteal muscles in early infancy, often in relation to severe
or prolonged illness (329,330,331).
In some children, the etiology cannot be established but may be due to
some other form of direct or indirect injury to the quadriceps
mechanism. As part of the differential diagnosis, the physician should
consider mild forms of arthrogryposis, congenital dislocation of the
knee or patella, or spinal dysraphism.
implied that the disorder was caused by a muscular dysplasia of
congenital origin, superficially resembling an incomplete syndrome of
arthrogryposis (326,327,332).
Gunn established the role of intramuscular injections in infancy as the
major cause of the fibrosis, and this has since been confirmed by
others (329,330,331,333,334).
and fatty replacement of muscle are present, most commonly involving
the vastus lateralis, vastus intermedius, and iliotibial tract (326,329).
An “abnormal” attachment of the iliotibial tract to the superolateral
patella was noted by Jeffreys, which more likely represented a
mechanically hypertrophied epicondylopatellar ligament (335,336).
splinting, and stretching, has not been successful in addressing the
condition (326). Surgical intervention has
included various forms of distal muscle and fascia release, lengthening
of the quadriceps tendon, and rebalancing of the extensor mechanism,
depending upon the extent of involvement and clinical features (326,327,329,337).
In some patients, early release of the muscles through a limited
subtrochanteric exposure has been described, with good results reported
(330).
a history of repeated intramuscular injections of the thigh in infancy,
we favor early proximal release through a limited subtrochanteric
exposure as described by Sengupta (330).
by pain in an extremity and associated dysfunction of the autonomic
nervous system. This syndrome usually follows a minor trauma, but has
been associated with many medical and surgical conditions, and with
certain drugs. Although initially thought to be rare in children, it is
being recognized with increasing frequency (338,339).
The syndrome has been given a variety of names, including posttraumatic
pain syndrome, algoneurodystrophy, shoulder-hand syndrome, Sudeck
atrophy, and causalgia. The term causalgia has been used primarily to describe the burning pain following a peripheral nerve injury, whereas reflex sympathetic dystrophy has been reserved for cases that resemble causalgia but do not have a vascular or neural lesion (340). To clarify the terminology, the International Association for the Study of Pain has introduced the term complex regional pain syndrome (CRPS)
to describe the constellation of symptoms. In type I (CRPS I), which is
distinguished by the absence of a definable peripheral nerve injury,
there are two subtypes (341).
than boys, and the lower extremities are much more frequently involved
than the upper extremities (338,339,342,343).
The pain is disproportionate in duration, severity, and distribution to
that which would be expected in response to the initiating noxious
event. It may be spontaneous and aggravated by dependency or touch, or
evoked by mechanical or thermal stimuli. Hyperpathia, or pain that
persists after the stimulus has been removed, and allodynia, or pain
that is produced by a nonnoxious stimulus, such as light touch, are
common complaints. Edema is a common early finding. The temperature of
the affected extremity may be less or more than the uninvolved side,
and have a different skin color. Although chronic trophic changes are
reported as being rare in children (342), one study reported a 15% incidence of trophic changes (338).
Skin, hair, and nail growth changes, subcutaneous tissue and muscle
atrophy, and leg-length discrepancy may be seen. Motor changes include
weakness, tremor, dystonia, and reduced movement.
Psychological stressors can modify the severity of the symptoms and the
continuous pain, and accompanying disability may result in anxiety
disorders and depression.
characteristic stages: the acute stage, followed by the dystrophic
period and, finally, by atrophy (347). Recently, the staging system has been shown to be inconsistent in describing the evolution of the disorder (348).
-
The presence of an initiating noxious event or a cause of immobilization.
-
Continuing pain, allodynia, or hyperalgesia in which the pain is disproportionate to the inciting event.
-
Evidence, at some time, of edema, changes in skin blood flow, or abnormal sudomotor activity in the painful region.
-
The diagnosis is excluded by the existence of conditions that would otherwise account for the degree of pain and dysfunction.
Plain radiographs, bone scan, and thermography are rarely required for
diagnosis in children.
fracture, infection, tumor (such as osteoid osteoma), and psychological
disorders with somatic manifestations.
The patchy osteopenia typical in adults is not often present in
children. Bone scan has been advocated as a diagnostic tool in adults;
however, in children the results are highly variable, demonstrating
increased, decreased, or normal uptake in the involved extremities (343,351). The sensitivity and specificity of the bone scan are not good enough to warrant its routine use in the diagnosis of CRPS (339,343). However, bone scan is useful for ruling out occult skeletal disorders such as stress fracture or neoplasm (338). MRI may also be useful in evaluating for occult fractures, but is inconsistent in demonstrating edema in affected areas (352).
theories suggest dysfunction of the sympathetic nervous system but they
are controversial (340). Although sympathetic
influences are still viewed as the most likely underlying mechanism
leading to the development and/or propagation of CRPS, these influences
are no longer thought to cause an increase in sympathetic tone (353).
Ochoa has demonstrated that the pain and accompanying features that
resemble sympathetically mediated disturbances may be caused, instead,
by sensitization of peripheral nociceptors and mechanoreceptors, and by
the effects of vasoactive substances released at the ends of small
sensory afferent fibers (354). This “neurogenic
inflammation” may result in neuromediator accumulation or depletion at
the site of the lesion, and may play a pivotal role in the development
of CRPS.
this is achieved through a coordinated, staged, progressive approach
that uses various treatment modalities to achieve both remission and
rehabilitation (338,344,355,356).
outlines a treatment algorithm for reflex sympathetic dystrophy in
adults and children (344). It focuses on the
principles of patient motivation, mobilization, and desensitization
facilitated by the relief of pain. Physical therapy is the cornerstone
of treatment, with pharmacologic management (nonsteroidal
antiinflammatories, opioids, tricyclic antidepressants, steroids, and
drugs such as carbamazepine), regional anesthesia (for analgesia and
sympathetic blockade), neuromodulation, and psychotherapy used as
adjuncts to facilitate management. Casts and splints should not be
used in the treatment because immobilization may exacerbate the problem (342).
An interdisciplinary team approach is most likely to succeed. The
initial treatment in children, especially in early cases, should be
physical therapy. It is necessary to educate the patient and the family
about the nonprotective nature of the neuropathic pain to help them
work through the discomfort (344). More than 50% of the children will respond to physical therapy alone (342,343) or in combination with transcutaneous electrical nerve stimulation (TENS), psychotherapy, and the use of oral medication (338,356).
Treatment early in the course of the disease gives the best results.
Sympathetic nerve blocks may be useful if the above mentioned treatment
is not successful. A continuous block with a catheter technique may be
preferable to intermittent blocks (344,357).
Physical rehabilitation and behavioral therapy should be continued
along with the sympathetic nerve block. It should be noted, however,
that most studies on treatment modalities in CRPS, including the use of
spinal cord stimulation, transcranial stimulation, surgical
sympathectomy, and pharmacologic sympathetic blockade, have focused
primarily on adults with CRPS. One should be cautious when applying
these treatment modalities to children. An evidence-based treatment
algorithm for child patients is yet to be established.
one of gradual improvement, but children can have long-term persistent
pain and dysfunction (338).
JB, Glowacki J. Hemangiomas and vascular malformations in infants and
children: a classification based on endothelial characteristics. Plast Reconstr Surg 1982;69:412.
GA, Alexander MA, Harcke HT. A multiscintigraphic approach to imaging
of lymphedema and other causes of the congenitally enlarged extremity. Semin Nucl Med 1993;23:334.
PA, Ackroyd JS, Lea Thomas M, et al. The Klippel-Trenaunay syndrome.
Clinical, radiological and hemodynamic features and management. Br J Surg 1985;72:232.
RV, Frieden IJ. Presence of vascular anomalies with congenital
hemihypertrophy and Wilms tumor: an evidence-based evaluation. Pediatr Dermatol 2003;20:199.
RE, Lytle JD, VanDevanter S. The use of total circulatory arrest in the
surgery of giant hemangioma and Klippel-Trenaunay syndromes in
neonates. Clin Orthop 1983;289:237.
LG, Happle R, Mulliken JB, et al. Proteus syndrome: diagnostic
criteria, differential diagnosis, and patient evaluation. Am J Med Genet 1999;84:389.
D, Hager J, Nikolaudes N, et al. Proteus syndrome: musculoskeletal
manifestations and management: a report of two cases. J Pediatr Orthop 1992;12:106.
XP, Marsh DJ, Hampel H, et al. Germline and germline mosaic PTEN
mutations associated with a Proteus-like syndrome of hemihypertrophy,
lower limb asymmetry, arteriovenous malformation and lipomatosis. Hum Mol Genet 2000;9:765.
LG, Peters KF, Darling TN, et al. Clinical differentiation between
Proteus syndrome and hemihyperplasia: description of a distinct form of
hemihyperplasia. Am J Med Genet 1998;79:311.
M, Schwartzenruber DJ, Lukish J, et al. Principles for the surgical
management of patients with Proteus syndrome and patients with
overgrowth not meeting Proteus criteria. J Pediatr Surg 2002;37:1013.
RD, Bone HG III, Roodman GD. Interleukin-6: a potential mediator of the
massive osteolysis in patients with Gorham-Stout disease. J Clin Endocrinol Metab 1996;81:1893.
T, Sabokbar A, Itonaga I, et al. Cellular and humoral mechanisms of
osteoclast formation and bone resorption in Gorham-Stout disease. J Pathol 2001;195:624.
SF, Rosenberg A, Mankin H, et al. Gorham’s massive osteolysis. The role
of radiation therapy and a review of the literature. Int Radiol Oncol Biol Phys 1993;26:491.
KD, Fong KM, Walker QJ, et al. Gorham’s syndrome: a usually fatal cause
of pleural effusion treated successfully with radiotherapy. Thorax 1996;51:1275.
EM, Carty HM, Kalos S. Generalized enchondromatosis associated with
hemangiomas, soft tissue calcifications and hemihypertrophy. Br J Radiol 1986;59:69.
HE, Seaver LH, Jones KL, et al. Isolated hemihyperplasia
(hemihypertrophy): report of a prospective multicenter study of the
incidence of neoplasia and review. Am J Med Genet 1998;79:274.
DM, Breslow NE, Beckwith JB, et al. Screening of children with
hemihypertrophy, aniridia and Beckwith-Wiedemann syndrome in patients
with Wilms tumor: a report from the National Wilms Tumor Study. Med Pediatr Oncol 1993;21:188.
M, Bayley R, Cole T, et al. Clinical features and natural history of
Beckwith-Wiedemann syndrome: presentation of 74 new cases. Clin Genet 1994;46:168.
MR, Zucker MA. Risk of cancer during the first four years of life in
children from the Beckwith-Wiedemann Syndrome registry. J Pediatr 1998;132:398.
RM, Ainscough JF, Barton SC, et al. Distant cis-elements regulate
imprinted expression of the mouse p57K1P2 (CDKN1C) gene: implications
for the human disorder, Beckwith-Wiedemann syndrome. Hum Molec Genet 2001;10:1601.
A, Heeson S, Cooper W, et al. An association between variants in the
IGFZ gene and Beckwith-Wiedemann syndrome: interaction between genotype
and epigenotype. Hum Molec Genet 2004;13:247.
NJ, Jay Kumar S, Guille JT. Clubfeet associated with congenital
constriction bands of the ipsilateral lower extremity. J Pediatr Orthop 1995;15:599.
R. Amniochorionic mesoblastic fibrous strings and amniotic bands:
associated constricting fetal malformations or fetal death. Am J Obstet Gynecol 1965;91:65.
A, Hasbargen U, Paek B, et al. Intra-uterine fetal demise caused by
amniotic band syndrome after standard amniocentesis. Fetal Diagn Ther 2000;15:4.
P. A modified technique for the surgical treatment of congenital
constriction bands of the arms and legs of infants and children. Orthopedics (in press).
K, Gershoni-Baruch R, Van Hul E, et al. Localisation of the gene
causing diaphyseal dysplasia Camurati-Engelmann to chromosome 19q13. J Med Genet 2000;37:245.
K, Gershoni-Baruch R, Guanabens N, et al. Mutations in the gene
encoding the latency-associated peptide of TGFbeta 1 cause
Camurati-Engelmann disease. Nat Genet 2000;26:273.
K, ten Dijke P, Ralston SH, et al. Transforming growth factor–beta 1
mutations in Camurati-Engelmann disease lead to increased signaling by
altering either activation or secretion of the mutant protein. J Biol Chem 2003;278:7718.
NW, MacPherson H, Janssens K, et al. A mutation affecting the
latency-associated peptide of TGFbeta1 in Camurati-Engelmann disease
enhances osteoclast formation in vitro. J Clin Endocr Metab 2003;88:3321.
T, Kinoshita A, Yoshiura Ki, et al. Domain-specific mutations of a
transforming growth factor (TGF)-beta 1 latency-associated peptide
cause Camurati-Engelmann disease because of the formation of a
constitutively active form of TGF-beta1. J Biol Chem 2001;276:11469.
MV, Peris P, Guanabens N, et al. Biochemical markers of bone turnover
in Camurati-Engelmann disease: a report on four cases in one family. Calcif Tissue Int 1997;61:48.
KI, Wagreich JM, Granowetter L, et al. Diaphyseal medullary stenosis
(sclerosis) with bone malignancy (malignant fibrous histrocytoma):
Hardcastle syndrome. Pediatr Radiol 1996;26:675.
F, Darendeliler F, Petorak I, et al. Deflazacort treatment in
progressive diaphyseal dysplasia (Camurati-Engelmann disease). J Paediatr Child H 1999;35:401.
T, Shuke N, Sato J, et al. Scintigraphic evaluation of pamidronate and
corticosteroid therapy in a patient with progressive diaphyseal
dysplasia (Camurati-Engelmann disease). Clin Nucl Med 2001;26:680.
JM, Samilson RL, Corley CL. Melorheostosis: review of the literature
and report of an interesting case with a nineteen year follow-up. J Bone Joint Surg Am 1963;45:1191.
CJ, Papademetriou T, Bonfiglio M. Melorheostosis: a report of clinical,
roentgenographic and pathological findings in fourteen cases. J Bone Joint Surg Am 1968;50:1281.
Goede E, Fagard R, Frijns JP. Unique cause of renovascular
hypertension: melorheostosis associated with a malformation of the
renal arteries. J Hum Hypertens 1996;10:57.
D, Bonnetblanc JM, Leroux-Robert C. Melorheostosis with associated
minimal change nephrotic syndrome, mesenteric fibromatosis and
capillary haemangiomas. Dermatology 1994;188:166.
P, Pykels E, Lammens J, et al. Melorheostosis in a family with
autosomal dominant osteopoikilosis: report of a third family. Am J Med Genet 2003;119A:188.
JE, Kim EH, Han EH, et al. A TGF-beta-inducible cell adhesion molecule,
betaig-h3, is downregulated in melorheostosis and involved in
osteogenesis. J Cell Biochem 2000;77:169.
HD, Krieg P, Rutschle H, et al. Multiple malformations in a male and
maternal osteopathia strata with cranial sclerosis (OSCS). Genet Couns 2003;14:281.
G, Lacombe D, David A, et al. Osteopathia striata cranial sclerosis:
non-random X-inactivation suggestive of X-linked dominant inheritance. Am J Med Genet 2002;107:1.
K, Shah R, Shalev Y, et al. Congenital clavicular pseudarthrosis
associated with vascular thoracic outlet syndrome: case presentation
and review of the literature. Cathet Cardiovasc Diagn 1995;35:321.
H. Pseudarthrose congenitale de la clavicule et syndrome de la
traversee thoraco-brachiale. Revue de la litterature a propos d’un cas.
J Mal Vasculaires 1995;20:51.
MC, Richards RR, Hudson AR. Thoracic outlet syndrome with congenital
pseudarthrosis of the clavicle: treatment by brachial plexus
decompression, plate fixation and bone grafting. Can J Surg 1988;31:131.
D, Chevalier J, Ducasse E, et al. Arterial complications of thoracic
outlet syndrome and pseudarthrosis of the clavicle: three patients. J Mal Vasc 2003;28:79.
P, Doaz M, Riera R, et al. Venous thoracic outlet syndrome secondary to
congenital pseudarthrosis of the clavicle. Presentation in the fourth
decade of life. Eur J Vasc Endovasc Surg 2003;25:592.
SB, King JD, Marrero G. Congenital pseudarthrosis of the clavicle: a
review of the literature and surgical results of six cases. J Pediatr Orthop 1988;8:316.
Molto FJ, Bonete Lluch DJ, Garrido IM. Congenital pseudarthrosis of the
clavicle: a proposal for early surgical treatment. J Pediatr Orthop 2001;21:689.
SC, Kuester DJ, Richardson EG. Dysplasia epiphysealis hemimelica
(Trevor disease) presenting as peroneal spastic flat foot deformity: a
case report. Foot Ankle 1991;12:55.
AJ, Frawley KJ, Bellemore MC, et al. MR imaging of dysplasia
epiphysealis hemimelica: bony and soft-tissue abnormalities. Am J Radiol 1999;172:819.
G, Li M, Martin S, et al. Fibrodysplasia ossificans progressiva, a
heritable disorder of severe heterotopic ossification, maps to human
chromosome 4q27-31. Am J Hum Genet 2000;66:128.
G, Bathelier C, Mercier G, et al. FOP Consortium. Localization of the
gene for fibrodysplasia ossificans progressiva (FOP) to chromosome
17q21-22. Genet Couns 2000;11:329.
TF, Olmstead EA, Shore EM, et al. Characterization of bone
morphogenetic protein 4 receptor in fibrodysplasia ossificans
progressiva. Clin Orthop Rel Res 1998;346:38.
M, Shore EM. Mutational screening of the bone morphogenetic protein 4
gene in a family with fibrodysplasia ossificans progressiva. Clin Orthop Rel Res 1998;346:53.
J, Serrano de la Pena L, Shore EM, et al. Paresis of a bone
morphogenetic protein-antagonist response in a genetic disorder of
heterotopic skeletogenesis. J Bone Joint Surg Am 2003; 85:667.
RB, Hahn GV, Tabas JA, et al. The natural history of heterotopic
ossification in patients who have fibrodysplasia ossificans
progressiva. A study of forty-four patients. J Bone Joint Surg Am 1993;75:215.
JR, Klimkiewicz JJ, Iannotti JP, et al. Mechanism for superior
subluxation of the glenohumeral joint in fibrodysplasia ossificans
progressiva. Clin Orthop Rel Res 1998;346:130.
FS, Sawyer J, Connors S, et al. Urinary basic fibroblast growth factor.
A biochemical marker for preosseous fibroproliferative lesions in
patients with fibrodysplasia ossificans progressiva. Clin Orthop Rel Res 1998;346:59.
EA, Gannon FH, Wang Z, et al. Embryonic overexpression of c-fos
protooncogene. A murine stem cell chimera applicable to the study of
fibrodysplasia ossificans progressiva in humans. Clin Orthop Rel Res 1998;346:81.
MA, Rocke DM, Crofford LJ, et al. Treatment of patients who have
fibrodysplasia ossificans progressiva with isotretinoin. Clin Orthop Rel Res 1998;346:121.
DL, Economides AN, Wang L, et al. In vivo somatic cell gene transfer of
an engineered Noggin mutein prevents BMP4-induced heterotopic
ossification. J Bone Joint Surg Am 2003; 85:2332.
MA, Vaughn RB. Ectopic ossification after total hip arthroplasty.
Predisposing factors, frequency, and effect on results. J Bone Joint Surg Am 1977;59:345.
FS, Craver R, MacEwen GD, et al. Progressive osseous heteroplasia: a
distinct developmental disorder of heterotopic ossification. J Bone Joint Surg Am 1994;76:425.
DJ, Fornasier VL. Pseudomalignant myositis ossificans. Heterotopic new
bone formation without a history of trauma. J Bone Joint Surg Am 1980;62:1274.
T, Dubousset J, Szendroi M, et al. Characterization of bone-forming
cells in post-traumatic myositis ossificans by lectins. Pathol Res Pract 1992;188:172.
CS, Meuten DJ, Richardson DC. Ischemic necrosis of cartilage in
spontaneous and experimental lesions of osteochondrosis. J Orthop Res 1991;9:317.
DR. Contracture of the quadriceps muscle. A discussion on the etiology
and relationship to recurrent dislocation of the patella. J Bone Joint Surg Br 1964;46:492.
PHJM, Reynen HM, Arntz IE, et al. Signs and symptoms of reflex
sympathetic dystrophy: prospective study of 829 patients. Lancet 1993;342:1012.
RM, Allen RC, Malleson PN. Technetium 99m-methylene diphosphonate bone
scans in children with reflex neurovascular dystrophy. J Pediatr 1985;106:437.
CB, Sethna NF, Micheli LJ. A technique for continuous lumbar
sympathetic blockade for severe reflex sympathetic dystrophy in
children and adolescents. Anesth Analg 1988; 67:514.