
Editors: Frassica, Frank J.; Sponseller, Paul D.; Wilckens, John H.
Title: 5-Minute Orthopaedic Consult, 2nd Edition
Copyright ©2007 Lippincott Williams & Wilkins
> Table of Contents > Ehlers-Danlos Syndrome
Ehlers-Danlos Syndrome
Paul D. Sponseller MD
Description
-
Ehlers-Danlos syndrome is a family of
disorders involving abnormal collagen that produces connective tissue
laxity, with many resultant abnormalities in the skeleton, vasculature,
eyes, and other systems.-
At least 11 subtypes of Ehlers-Danlos syndrome have been identified, with varying patterns of inheritance and genetic causes (1,2).
-
The age at diagnosis varies from infancy to adulthood.
-
-
Classification (1–3):
-
Type I: Gravis (classic):
-
Aneurysms
-
Rupture of hollow viscus
-
Skin hyperextensibility
-
Bruising
-
Pigmented areas
-
Hernias
-
-
Type II: Mitis:
-
Similar manifestations but milder
-
-
Type III: Benign hypermobility syndrome:
-
Laxity
-
Joint dislocations
-
Mitral valve prolapse
-
Positive family history
-
-
Type IV: Ecchymotic:
-
Thin skin
-
Normal joints
-
Aneurysms
-
Viscus rupture
-
-
Type V: X-linked:
-
Intramuscular hemorrhagia
-
Floppy baby characteristics
-
-
Type VI: Ocular-scoliotic
-
Type VII: Arthrochalasis multiplex congenital:
-
Extreme joint laxity
-
Short stature
-
Hip dislocations
-
-
Type VIII: Periodontosis (progressive periodontal disease)
-
Type IX: Occipital horns and skeletal dysplasia
-
Type X: Platelet dysfunction
-
Type XI: Familial joint laxity (patellar and hip dislocation)
-
General Prevention
-
Prevention of cardiovascular and bleeding emergencies should be the goal of treatment.
-
Reduction in frequency of joint dislocations also may be possible.
Epidemiology
Incidence
-
Overall, males and females are affected equally (2,3).
-
Incidence is impossible to calculate
accurately because of the numerous forms of this disorder and their
varying degrees of severity.
Risk Factors
A positive family history of the syndrome or of its major manifestations is a risk factor.
Genetics
-
Types I, IV, VIII, and XI are autosomal dominant.
-
Types V and IX are X-linked.
-
The remainder are autosomal recessive in transmission.
-
Many patients present as having a new mutation without a family history.
Etiology
-
Type IV, the ecchymotic variety, is secondary to a disorder of type III collagen.
-
Type VI (ocular-scoliotic) is the best characterized.
-
It is caused by a defect in lysine hydroxylase that affects collagen.
-
This change results in decreased collagen cross-linking.
-
-
Type VII (arthrochalasis multiplex congenital) is secondary to a defect in type I collagen.
-
Type X (with platelet dysfunction) also results from a defect in type I collagen.
The diagnosis is made by a medical geneticist on a clinical basis, with verification in some types by use of molecular testing.
Signs and Symptoms
-
Signs:
-
Lax skin (Fig. 1)
-
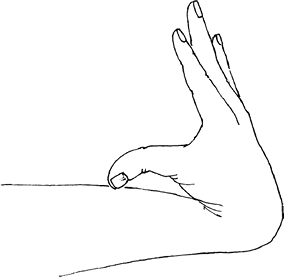
Joint hypermobility (Fig. 2)
-
Joint instability (1)
-
Scoliosis (4,5)
-
Ability of some affected persons to perform skeletal contortions impossible for nonaffected persons
-
-
Symptoms:
-
Multiple joint pains
-
Vague musculoskeletal pains
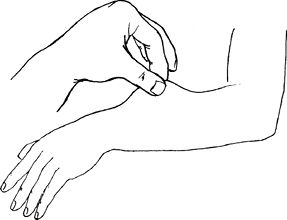 Fig. 1. Ehlers-Danlos syndrome is one of several conditions characterized by cutaneous laxity.
Fig. 1. Ehlers-Danlos syndrome is one of several conditions characterized by cutaneous laxity.
-
Physical Exam
-
Record height.
-
Observe the proportions of the skeleton.
-
Systematically measure joint ROM.
-
Note the ability to hyperextend the fingers and the knees.
-
Check the shoulders, elbows, and knees for stability.
-
Feel the quality of the skin.
-
Note any bruising.
-
Pursue an ocular examination if any symptoms of deficit are present.
-
Observe the spine for kyphosis.
-
Conduct a forward-bend test for scoliosis.
Tests
-
Molecular testing is available to confirm many, but not all, types of Ehlers-Danlos syndrome.
-
An experienced genetics laboratory should be consulted.
Imaging
-
Imaging of the heart and aorta should be obtained periodically for patients with type I and type IV disorders.
-
Plain radiographs should be obtained when physical examination suggests scoliosis, kyphosis, or spondylolisthesis.
Pathological Findings
-
Light microscopic examination of fibroblasts of the skin shows irregular collagen fibers.
-
Gross examination of the aorta may show
dissecting aneurysms in type I, myxomatous changes in the cardiac
valves, and redundant chordae tendineae.
Differential Diagnosis
-
Marfan syndrome also is characterized by
laxity of major joints, but it is rarely symptomatic, and it has
well-defined diagnostic criteria. -
Larsen syndrome also presents with
multiple joint dislocations, but contractures also are present, and
cervical kyphosis is common. -
Cutis laxa and pseudoxanthoma elasticum also should be ruled out in patients with predominant skin findings.
![]() Fig. 2. Hypermobile joints are characteristic of Ehlers-Danlos syndrome.
Fig. 2. Hypermobile joints are characteristic of Ehlers-Danlos syndrome.
P.109
General Measures
-
Specialist referral for the systems listed earlier is indicated when problems are manifested by the patient.
-
One should use caution when recommending surgery for joint instability because the failure rate is higher than normal.
-
Surgical treatment should not be undertaken in Ehlers-Danlos syndrome unless symptoms are severe.
-
Fusion of joints may be necessary to provide stability.
Special Therapy
Physical Therapy
-
Muscle conditioning may ameliorate some of the symptoms of joint instability, even if these symptoms are not eliminated.
-
Physical therapy also should be helpful in educating patients about how to decrease the frequency of joint dislocations.
Surgery
-
Fusion for scoliosis is indicated if
curves are >45° (approximately) and the patient’s medical condition
is otherwise satisfactory (4,5). -
Physical activity usually is encouraged, but should be tailored to the patient and focused on low-impact sports.
Disposition
Issues for Referral
The best specialist for the routine follow-up of patients with Ehlers-Danlos syndrome is usually a medical geneticist.
Prognosis
The listed complications lead to a moderate decline in the mean life expectancy.
Complications
-
Sudden death from cardiovascular complications
-
Osteoarthritis of joints
-
Visual deficits
References
1. Badelon O, Bensahel H, Csukonyi Z, et al. Congenital dislocation of the hip in Ehlers-Danlos syndrome. Clin Orthop Relat Res 1990;255:138–143.
2. McKusick
VA. Ehlers-Danlos syndrome. In: Heritable Disorders of Connective
Tissue, 4th ed. St. Louis: CV Mosby Co., 1972:292–371.
VA. Ehlers-Danlos syndrome. In: Heritable Disorders of Connective
Tissue, 4th ed. St. Louis: CV Mosby Co., 1972:292–371.
3. Beighton P, De Paepe A, Steinmann B, et al. Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Am J Med Genet 1998;77:31–37.
4. Akpinar S, Gogus A, Talu U, et al. Surgical management of the spinal deformity in Ehlers-Danlos syndrome type VI. Eur Spine J 2003;12:135–140.
5. Vogel LC, Lubicky JP. Neurologic and vascular complications of scoliosis surgery in patients with Ehlers-Danlos syndrome. A case report. Spine 1996; 21:2508–2514.
Codes
ICD9-CM
756.83 Ehlers-Danlos syndrome
Patient Teaching
-
Genetic counseling should be offered.
-
Understanding the nature of any cardiovascular abnormality should be taught, in case of medical emergency.
-
Contact or high-impact sports should be discouraged.
Activity
Activity is encouraged but should be limited to noncontact sports that do not cause pain.
FAQ
Q: Are braces helpful in Ehlers-Danlos syndrome?
A: They may be in some cases, but in others they produce more inefficient movement. A trial can help determine applicability.
Q: Is any medication available that can improve the tissue laxity?
A: No, not at this time.
Q: How does one deal with the pain felt by some patients with Ehlers-Danlos syndrome?
A: If standard measures fail, referral to a pain specialist may be helpful.