
|
|
presenting in children is intimidating and interesting. Most of this
information is generally towards the periphery of most orthopaedic
surgeons’ knowledge. Stay out of trouble by knowing a few common
threads in these conditions that can help you identify a child with a
potential neuromuscular condition.
an orthopaedic surgeon because the child walks funny, falls down, wants
to be carried when walking far, or some other seemingly innocuous
complaint. Ninety-nine percent of the time it is age appropriate, but
this chapter may help with the other 1%. Delay in diagnosis can have
serious implications, e.g., before Duchenne muscular dystrophy is
diagnosed, 20% of families already have conceived a second child. Do
not hesitate to examine the parents (subtle cavovarus feet,
café-au-lait spots, etc.) and ask for a family history. Many parents
have unrecognized subtle forms of the same disease of their child. This
aids in diagnosis of the child.
-
Complains of tiring easily when walking
-
Tripping or falling if new onset or worsening
-
Delayed walking
(consider up to 18 months for achievement of independent ambulation
“normal”—one must often clarify to the parents that this means the
child walks without the child holding on to anything) -
“Floppiness” as an infant
-
No complaints of pain
-
Hypo- or hypertonia (though this seems to be overdiagnosed these days)
-
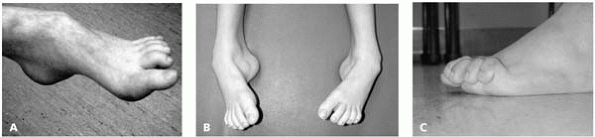
Changing or progressive deformity of feet (Fig. 15-1)
-
Involvement is symmetrical (except for hemiplegia; if asymmetrical, think spine pathology)
 |
|
▪ FIGURE 15-1 (A)
This child with a cavus foot has an underlying neuromuscular disorder. In children with unilateral foot deformity, consider spinal pathology as more likely; with bilateral foot deformity, consider underlying neuromuscular disease as more common. (B) A child with bilateral cavo-varus feet with Charcot-Marie-Tooth. (C) Claw toes are also an indication of an underlying neuromuscular disease. |
neuromuscular conditions have multiple orthopaedic implications. For
example, a child with Charcot-Marie-Tooth (CMT) disease may present to
you with a foot deformity, but do not forget there is an increased risk
of scoliosis1 and hip dysplasia.2,3
This raises an important point even for pediatric orthopaedic surgeons
who feel comfortable seeing children with neuromuscular conditions.
Each condition has its own unique pitfalls, and it is probably
worthwhile to quickly review literature on a child’s condition prior to
instituting treatment. For example, children with various muscular
dystrophies have widely variable prognoses. Know the natural
history
and expected function of the condition prior to planning surgery as
many of these children may never be expected to walk, or even live very
long, which may influence whether surgery is indicated. If a muscle
biopsy is required, biopsy the weak muscle—i.e., the deltoid, for
shoulder girdle weakness.
 |
|
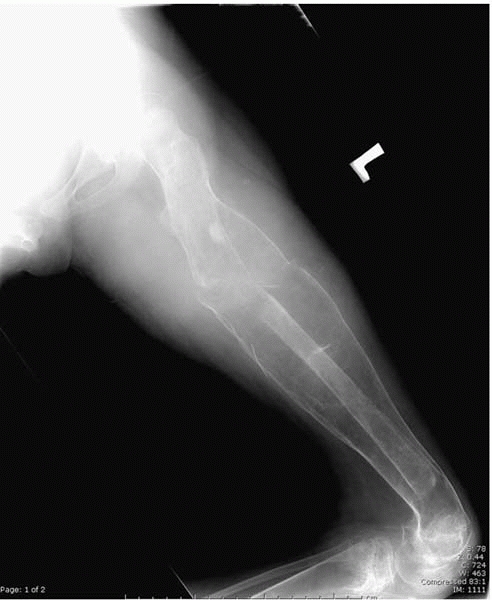
▪ FIGURE 15-2
Radiograph of a femur of a nonambulator with cerebral palsy who has sustained multiple femur fractures. The clinical and radiographic appearance of this fracture may be confused with infection. |
variety of neuromuscular disorders (including cerebral palsy) have had
multiple exposures to latex (ventriculoperitoneal shunt,
catheterization, previous surgeries, etc.) and may be considered at
risk for latex allergy and intraoperative anaphylaxis.4
ambulation are at an increased risk for osteopenic fractures. In an
insensate area, fractures are often not appreciated at the time of
injury, and present late as a swollen, red, and warm limb that is often
mistaken for infection by the inexperienced. To make things even more
confusing, the child may present with a fever and abnormal laboratory
studies. This scenario unfortunately occurs in children who have severe
communication challenges (Figs. 15-2, 15-3 and 15-4).
 |
|
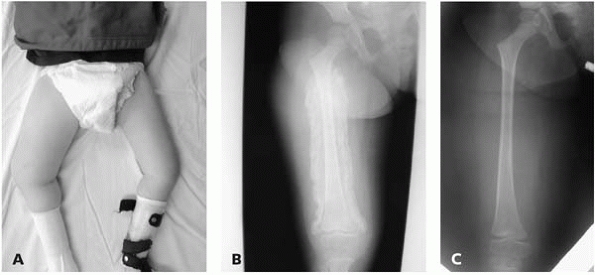
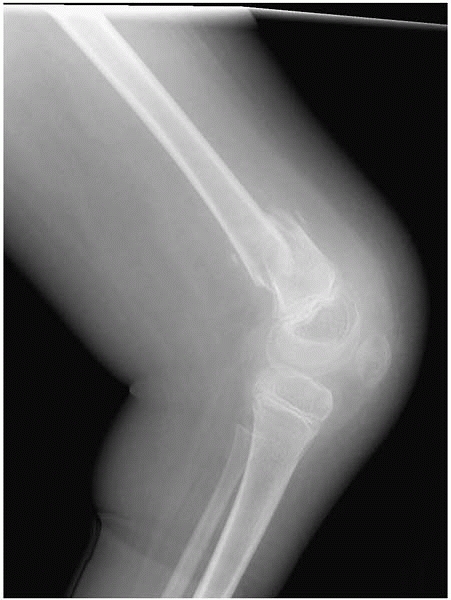
▪ FIGURE 15-3 (A)
This boy with thoracic level spina bifida presented with a chief complaint of (painless) leg swelling. The leg was warm to touch. (B) Radiographs demonstrate copious new bone formation. (C) To make matters even more confusing, when children with spina bifida fracture through the growth plate, there may be no callus formation. Clinically, this young girl with thoracic level spina bifida had motion through the Salter I fracture of the distal femur. |
 |
|
▪ FIGURE 15-4
Nonambulators with osteopenia frequently have fractures of the distal femoral metaphysis. Have a high index of suspicion when children who were in a hip spica cast for hip surgery present with increasing pain shortly after the spica cast was removed. |
in otherwise normal, active children. First, mobilization should begin
as early as possible. If a child with progressive weakness and
increasing difficulty in walking (e.g., 11-year-old boy with Duchenne)
is in a long leg cast that prevents walking for 6 weeks, the child may
lose so much muscle strength that he never
walks independently again. You do not want to contribute to that.
Second, immobilize for as little a time period as possible.
Immobilization leads to localized osteopenia. These children already
have some degree of osteopenia to begin with; following immobilization,
fractures at other areas in the limb are quite possible. One of us
(DLS) assumed care of a spina bifida patient who spent 11 months in
lower extremity casts for
multiple
fractures at different locations. Third, these children often have
abnormal sensation; soft-tissue problems are more likely to occur, and
less likely to be recognized, than in the general population.

technique for fracture care in nonambulatory children with
neuromuscular conditions. This can be accomplished with semi-rigid
fiberglass and/or significant padding, such as polyurethane foam. This
decreases osteopenia secondary to immobilization, lessens the chance of
a secondary fracture at the edge of a rigid cast, and decreases skin
problems from poorly sensate soft tissues rubbing against the edge of a
hard cast.
rapid and overwhelming callus formation, and the child’s demands
usually do not require anatomic reduction.
-
In a child with cavo-varus feet, look at the hands; if there is intrinsic muscle weakness think CMT.
-
If the child shakes your hand, then can’t relax the grip, think myotonic dystrophy.
-
If the primary
complaint is ataxia, think Friedreich’s ataxia. This looks obvious in
print, but not always as obvious when a markedly ataxic child presents
for the first time. -
Tongue fasciculations = spinal muscular atrophy
-
Dermatomyositis: acute onset of weakness and a malar rash
diagnosis of Duchenne muscular dystrophy was missed in every case
referred to an orthopaedic surgeon (37 patients). Including visits to
other specialists, there was a mean delay of diagnosis of 2 years. The
authors of the original survey believe this delay has not changed since
then, and recommend that a serum creatine phosphokinase should be
carried out in all boys presenting with nonspecific neuromuscular
complaints.5,6
and many other types of muscular dystrophy are less severe. The level
of creatine phosphokinase (CPK) may be 20-200X normal in children with
Duchenne, but can be elevated for patients with other forms of muscular
dystrophy as well as many other types of muscle disease. The CPK may be
elevated by birth in normal children, but should return to normal
quickly. Duchenne muscular dystrophies are not that rare, occurring in
1/3,500 births, so there is a good chance most orthopedic surgeons will
encounter a new patient at some point in their career. While Duchenne
is X-linked and thus occurs only in boys, beware that it can occur in a
child with Turner syndrome (XO), and other forms of muscular
dystrophies can occur in girls. Dr. Sarwark points out that Duchenne
can occur in girls in the setting of the Lyon Hypothesis, in which
there is inactivation of one of the two X chromosomes. Mental
retardation may be present—just because a boy has mental retardation
doesn’t mean he doesn’t have muscular dystrophy. Perhaps the greatest
way to stay out of trouble in Duchenne is to make it a practice to do
Gower’s test on all 3- to 6-year-old boys with gait abnormalities other
than torsion (see Fig. 2-1).
-
3- to 6-year-old boy
-
Normal birth history
-
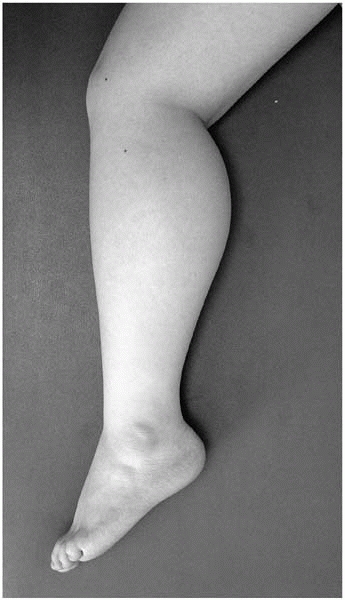
Huge calves—pseudohypertrophy of calves, on palpation a hard, “rubbery” feeling (Fig 15-5)
-
Ankle equinus
-
Waddling gait secondary to weak hip abductors
-
Hyperextension of knee during standing phase
-
Partially in reaction to ankle equinus
-
Partially to augment weak quadriceps
-
 |
|
▪ FIGURE 15-5 Pseudohypertrophy of the calf in the setting of weakness suggests Duchenne muscular dystrophy.
|
be done for these children to prolong strength. While still early, it
appears that steroids significantly increase muscle strength,
performance, and pulmonary function, as well as slowing the progression
of weakness. Refer these children to a practitioner with experience in
this area. Minimize periods of immobility; once children are in a
wheelchair for 3 to 6 months, they are unlikely to ever walk again. In
general, minimize surgery. For most children, a percutaneous
tendoachilles lengthening (TAL), and possibly a posterior tibial tendon
release or transfer is all that is ever needed aside from possible
spine surgery. Do not be too aggressive in treating mild equinus in the
ambulatory child, as this may be protective against knee flexion
contractures. After a child is nonambulatory, foot surgery is unlikely
to improve quality of life and is performed only for comfort.
As the disease progressively weakens the cardiac and pulmonary systems
with time, stay out of trouble by performing a spinal fusion early, at
20 to 30 degrees of scoliosis, before cardiopulmonary deterioration
becomes severe. There is an estimated 4% loss of forced vital capacity
(FVC) for every 10 degrees of spinal curvature.8
Some have suggested that a FVC of 35% is the minimum for safe spinal
fusion, though with approval and support from the pulmonologist,
cardiologist, and anesthesiologist, children with even worse pulmonary
function undergo surgery safely and successfully. Make certain the
family understands that although there is a realistic possibility of
death, especially in children with small FVCs, the more likely outcome
is the child needing a tracheotomy (which means a loss of verbal
communication), as well as the possible need for permanent mechanical
ventilation. It is also wise to discuss preoperatively with parents and
patient what to do if loss of spinal cord function occurs during the
surgery, such as leaving implants in place to maintain sitting balance.
The indications to fuse to the pelvis have been debated. In our
experience, and that of others, fuse to the pelvis during the primary
surgery.9 We believe the risk of
fusing to the pelvis during primary surgery is much less than extending
the fusion down to the pelvis years later following further
cardiopulmonary decline.
over 150 specific conditions, there are common clinical
characteristics. Children with the most common
type,
amyoplasia, are generally of normal intelligence and the majority will
be able to ambulate. The joints (including bones, cartilage, muscle
units, ligaments, and skin) have never had the necessary movement in
utero for normal development, and will never have normal motion. This
is important for the surgeon and family to understand. Long-term
physical therapy is of little use, and may be psychologically
counterproductive when significant effort leads to little if any gains.
The goal of treatment is not
to create normal motion of joints, as this is a setup for failure. The
goal is to align the extremities and joints to maximize the ability to
stand, ambulate, and sit.

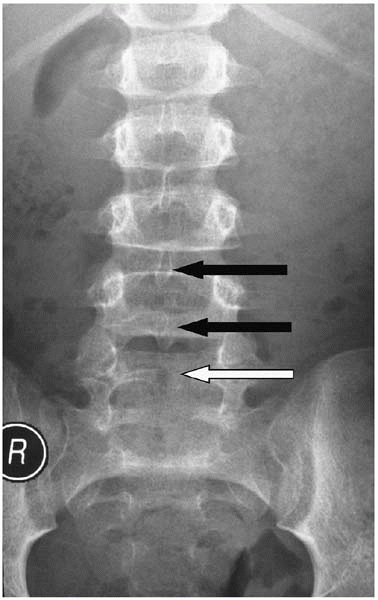
bifida occulta, a lack of complete formation of the posterior arch
(usually of L5 and present in about 10% of the population) is not spina bifida (Fig. 15-6). By definition it is a normal variant and there are no sequelae, other than parental distress.
important to remember that the abnormality is not just at the end of
the spine—it’s the whole central nervous system. Problems such as
hydrocephalus, Chiari malformations, and others are an expected part of
spina bifida.
 |
|
▪ FIGURE 15-6 Spina bifida occulta. Spinous processes of L3 and L4 are visible (black arrows). An absent spinous process at L5 (white arrow) is consistent with spina bifida occulta in an otherwise normal child.
|
-
Shunt malfunction leads to hydrocephalus and is quite common. This may happen multiple times during childhood.
-
Can be evaluated by CT of brain or CSF pressure, and/or neurosurgical consultation.
-
Symptoms may range
from subtle behavior changes or difficulty concentrating, to subtle
gait changes, to progressive scoliosis, to permanent brain damage.
problems, are largely determined by the level of motor involvement,
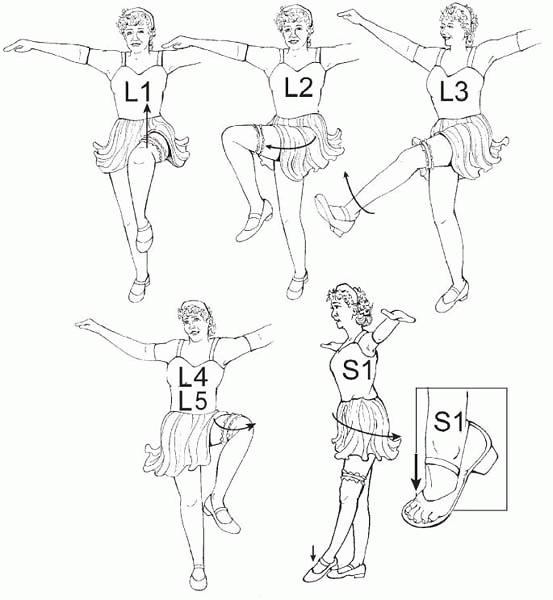
defined as the lowest level with 4/5 strength or better. The Can-Can
dance can help one determine levels (Fig. 15-7).
Document the level of involvement on a regular basis to help recognize
deterioration of function and, if needed, begin a search for the
underlying cause.
or better, the child will be a walker, if there is no significant
cognitive pathology. This is just not true in most cases. Walkers are
most often L4 (grade 4/5 hamstring) or better, though even many
children with low-lumbar and sacral lesions will not be long-term
walkers. In one study looking at patients between 10 and 30 years of
age, half of those with sacral lesions are not community ambulators.10
While the congenital lesion may not change, deterioration in ambulation
can be expected for a variety of factors, including CNS problems
(shunting and tethering) and increased mass outstripping poor muscle
strength. While outcome varies by center and population, Rancho Los
Amigos Medical Center reviewed 35 patients with sacral level lesions
who
were initially community ambulators. At an average age of 29 years they
found a decline in the ability to walk in 11 of 35 patients, and over
half had ostomyelitis.11
Keep out of trouble by sharing with families early on that
deterioration in ambulation is an expected part of the disease and not
necessarily anyone’s fault.
 |
|
▪ FIGURE 15-7
Can-Can Dance: Spina Bifida Levels for Everyone. Need 4/5 strength to make the level. These may not be exact, but they are easy to remember, easy to do, and makes it easy to notice when something is changing. L1, hip flexion (iliacus, psoas, sartorius); L2, hip adduction (hip adductors); L3, knee extension (quadriceps); L4, knee flexion (medial hamstrings); L5, hip abduction (tensor fascia lata, gluteus medius, gluteus minimus); S1, hip extension and ankle plantar flexion (gluteus maximus, gastrocnemius, soleus). |

by positioning and range of motion exercises. For overall mobility the
importance of weight control cannot be overstressed to parents—before
it becomes a problem. Up to one third of children with spina bifida are
latex sensitive, thus treat all children with this condition as if they
are latex sensitive. There are no definitive tests to prove they are
not, and the stakes of anaphylaxis are too high to risk when prevention
is so simple.4
universally present on MRI in children with spina bifida. One should
only consider surgical release of a tethered cord in the setting of
signs and symptoms (new-onset lumbar, sacral or buttock pain,
lower-extremity loss of function or spasticity) or in accordance with
surgical scoliosis correction that will significantly lengthen the
spine. Beneficial effects of surgical release of a tethered cord are
often underwhelming. Detethering has variable outcomes and can lead to
neurologic worsening in many areas.12
secondary to underlying or changing CNS pathology (syrinx,
Arnold-Chiari type II, tethered cord, shunt malfunction). Investigate
with an MRI of the entire spine and neurosurgical assessment of shunt
function. Scoliosis >45 degrees is very uncommon in L5 and sacral
level patients, and if present is usually due to underlying CNS
pathology. Beware that congenital scoliosis is also frequently present;
go with traditional treatment such as early fusion of a unilateral bar
opposite a hemivertebrae, as applicable.
aorta and vena cava are too short to allow full correction of the
deformity, thus vertebral bodies are removed to shorten the spine.
There are anecdotes of acute vascular insufficiency leading to loss of
legs following an overly aggressive correction. Consider an arterial
line with blood pressure monitoring in the lower extremities or pulse
oximetry when planning significant corrections of a sharp kyphosis.
-
Where posterior
elements are lacking, a posterior fusion alone is often not adequate,
as there is simply not enough posterior bone present for a fusion. -
One can often do
anterior fusions from the back in the lumbar spines, analogous to a
posterior lumbar interbody fusion (PLIF), without worrying about
damaging nerve roots—what a pleasure. -
Pedicle screws are
often essential when laminae are absent, but beware that the pedicle
anatomy may be quite distorted. Pedicles may be facing almost straight
lateral. -
Consider having a
plastic surgeon close with lateral flaps and excision of midline
skin—the tissue is almost always compromised from the original spinal
closure. -
Assess shunt
function preoperatively as per your neurosurgeon’s routine. If the
shunt is not functioning, and CNS drainage through the spine is
abruptly stopped intraoperatively, the child can die within minutes. -
These children are likely to need spine imaging in the future (MRI); titanium instrumentation may be preferable.
-
Prevent incontinence
from soiling wound. Ioban dressing with benzoin applied in the OR
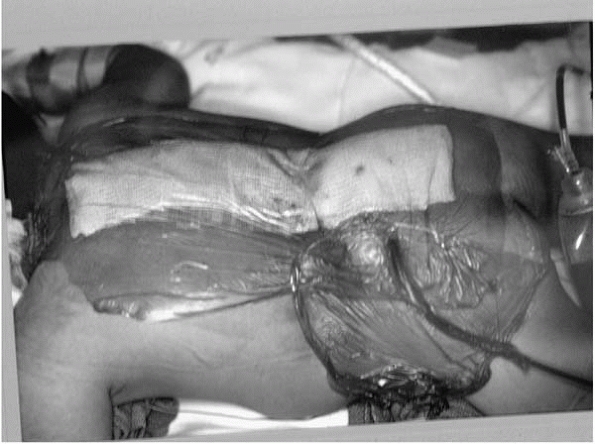
between the incision and anus is helpful (Fig. 15-8). -
Pelvic obliquity may lead to decubitus ulcers over an insensate pelvis—correct this aggressively.
-
Fusing in a position of excessive lordosis may make it impossible for females to selfcatheterize.
-
If the soft tissue
appears to be too damaged to cover any instrumentation, spinal rods can
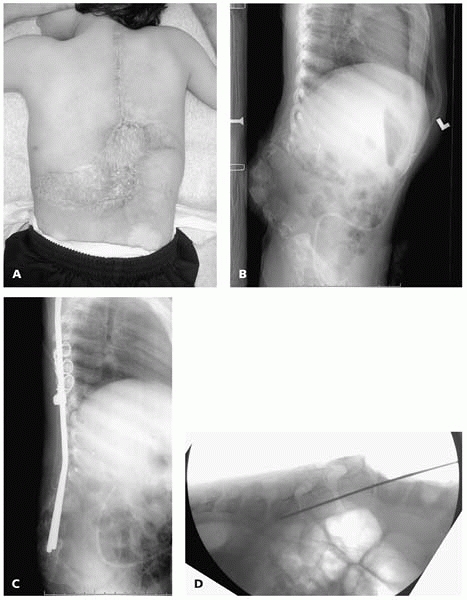
be placed directly into vertebral bodies in cases of kyphosis (Fig. 15-9).

Surgical treatment of dislocated hips in ambulatory children has been
advocated. A comprehensive study of ambulatory children with L3 and L4
level spina bifida who underwent surgical hip reduction found that
benefits were marginal at best, and patients with failed operations had
worse function than those who were not operated on.13
A frequent and unfortunate outcome of attempts to reduce a dislocated
hip in spina bifida is a stiff hip, which may interfere with
positioning and transfers. In our experience we have never seen a child
whose hip we wished we’d reduced, but have seen many attempts at hip
reduction we wished hadn’t been done. Dr. Sarwark says the principle is symmetry and mobility free of contractures.
 |
|
▪ FIGURE 15-8
Placing iodineimpregnated surgical drape and skin adherent between the perineum and wound helps keep the wound from becoming soiled. Children with spina bifida have a much higher incidence of gram-negative and mixed floral infections, possibly due to wound contamination from incontinence. |
 |
|
▪ FIGURE 15-9 (A)
Child with thoracic level spina bifida with skin graft directly adherent to bone over the progressive kyphotic gibbus with frequent ulceration. Plastic surgical consultation concluded that there was no means of providing soft-tissue coverage for traditional posterior spinal instrumentation. (B) Preoperative lateral supine radiograph demonstrates rigid nature of kyphosis. (C) Surgery was performed with no incision distal to L3, by placing the rods directly into the vertebral bodies of L-4-S1. (D) Intraoperative imaging with preliminary K-wires assists in correct placement of rods. |
knee (and hip) contractures to develop over time. Physical therapy and
sleeping in knee immobilizers may help. As many children with spina
bifida have weak quadriceps to begin with, the addition of knee flexion
contractures makes walking even more difficult. The use of ground
reaction ankle-foot orthoses (AFOs) help prevent knee flexion
contractures, as well as accommodate for quadriceps weakness. Children
with absent or very weak quadriceps strength may benefit from
knee-ankle-foot orthoses (KAFOs) to assist in standing, transfers, and
ambulation. This is one of the few remaining indications for bracing
above the knee in contemporary pediatric orthopaedics.
orthopaedic care in spina bifida feet is a braceable, supple,
plantigrade foot. Fusions are generally contraindicated, as a stiff,
insensate foot is a setup for ulcers.
all tendons to help minimize recurrence and minimize future surgeries.
Although this sounds extreme, a flexible, braceable foot is far
superior to a rigid deformed foot prone to ulcers. Even in children
with thoracic level involvement, in which the muscles to the foot are
not normally innervated, the muscle tendon units can act as deforming
forces.
quickly and aggressively, often starting with total contact casting. An
infected ulcer in a child with spina bifida can lead to osteomyelitis
and amputation if not cared for appropriately. Educate parents to
inspect feet every day and to seek medical care at the first signs of
more than transient redness or ulcers. Adjustment of a pressure spot on
an orthosis may prevent a frank ulcer when redness is identified early.
-
Do a Gower’s test on a young boy who seems to tire easily, falls, or is “doing worse.”
-
Think of the possibility of neuromuscular disease when parents report that falling or tiring when walking are worsening.
-
Treat all children with spina bifida as if they are latex allergic.
-
For almost any problem in spina bifida that does not have an obvious cause, your first thought should be “It’s the shunt, Stupid!”
SJ, Marks HG, Bowen JR, et al. Hip dysplasia associated with
Charcot-Marie-Tooth disease in the older child and adolescent. J Pediatr Orthop. 1985;5(5):511-514.