
that occurs primarily in males, resulting in clotting factors that are
nonfunctional or absent. Intraarticular hemorrhages in patients with
poorly controlled hemophilia can lead to progressive joint arthropathy.
The natural history of the disorder has changed significantly over the
past several decades, due to the development of factor replacement
therapy, but considerable orthopaedic morbidity still occurs.
-
The incidence of hemophilia is estimated to be 1 per 10,000 male births in the United States.
-
Approximately 95% of cases are caused by a variable lack of factors VIII or IX.
-
Hemophilia A (factor VIII deficiency) is the most common (about 80% of cases).
-
□ Several different mutations cause the disorder, which accounts for the variable clinical severity.
-
□ It is an X-linked recessive disorder that affects males.
-
□ Hemophilia A is the result of a new mutation in approximately 33% of patients.
-
-
Hemophilia B (factor IX deficiency), also known as Christmas disease, is the next most common.
-
□ Clinically indistinguishable from hemophilia A.
-
□ Also transmitted as an X-linked disorder.
-
-
The third most common inherited
coagulation disorder is von Willebrand disease, caused by a variable
lack not only of factor VIII coagulant activity but also of factor
VIII-related activity responsible for adhesion of platelets to exposed
vascular subendothelium.-
□ This form is characterized by abnormal
bleeding from mucosal surfaces, therefore major hemophilic arthropathy
is relatively uncommon.
-
-
The exact pathophysiology of hemophilic arthropathy remains controversial.
-
Both the inflammatory cascade and iron deposition have been implicated in cartilage destruction.
-
The degree of hemarthropathy does not always directly correlate with the number of joint bleeds.
-
Hemarthrosis probably starts as a subsynovial, intramural hemorrhage that then ruptures into the joint cavity.
-
Spontaneous hemorrhages without antecedent trauma commonly occur in patients with a severe deficiency.
-
With repeated exposure to blood, the
synovium of joint hypertrophies and becomes hypervascular, leading to a
progressive cycle of synovitis and more bleeding. -
Histology of the hypertrophic synovium is
characterized by villous formation, markedly increased vascularity, and
chronic inflammatory cells. -
Hemosiderin deposits accumulate in the
lining cells of the synovial villi, and the inflammatory cells
congregate around the vessels and hemosiderin deposits. -
In summary, the result of repeated hemorrhage into a joint with chronic synovitis
-
Proteolytic enzymes released by the inflamed synovium attack both cartilage and bone.
-
As the hypertrophied synovium continues to expand, and the articular cartilage erodes, the joint space narrows.
-
Osteoporosis occurs from disuse, and the joint becomes immobile.
-
Grading of articular involvement can be seen in Table 26.1-1.
-
In children, chronic synovitis may also
cause asymmetric physeal growth, or early physeal closure, leading to
angular deformities or leg length discrepancies.
|
TABLE 26.1-1 GRADING OF SYNOVITIS AND ARTICULAR INVOLVEMENT IN HEMOPHILIC PATIENTS
|
||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||||||||||
-
A history of similarly affected males on the maternal side of the family, or typical behavior
-
A history of hemorrhage from major surface wounds and musculoskeletal sites.
-
Hemophiliacs rarely experience bleeding during the first years of life in the absence of trauma or surgery.
-
□ Hemarthroses usually begin after the child starts to walk.
-
-
The frequency and severity of bleeding
episodes commonly increase as the child attends school and becomes more
physically active and socially interactive. -
Patients with severe hemophilia typically develop their initial joint pathology between the ages of 5 to 15 years.
-
Abnormal bleeding may occur in any area of the body, but joints are the most frequent sites of repeated hemorrhage (Table 26.1-2).
-
The weightbearing joints are the most
common sites of hemophilic arthropathy, with the frequency of
involvement being, in decreasing order, the knee, elbow, shoulder,
ankle, wrist, and hip. The vertebral column is rarely involved. -
Clinical findings depend on the severity of hemorrhage and whether the hemarthrosis is acute, subacute, or chronic (Table 26.1-3).
-
Other musculoskeletal problems in hemophilia include:
-
□ Pseudotumor (slowly progressive
hemorrhage that increases in size within a confined space, causing
pressure necrosis and erosion of the surrounding tissues) -
□ Neuropraxias (femoral, peroneal, sciatic, median, and ulnar)
-
□ Ectopic ossifications
-
□ Fractures
-
□ Ischemic contractures or compartment syndrome
-
-
After history and physical examination, laboratory tests should be ordered (Table 26.1-4).
If these screening procedures suggest a bleeding tendency, factor
assays must be carried out to establish not only the specific
deficiency but also its degree. -
Most patients with hemophilia present to
an orthopaedist with a known diagnosis. Clinical manifestations of
hemophilia A and B are similar and depend on the blood levels of factor
VIII or IX (Table 26.1-5).
|
TABLE 26.1-2 DIFFERENTIATING BLEEDING FROM SYNOVITIS
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
-
Distended capsule
-
Synovitis
-
Cartilage thinning
-
Widening and erosion of intercondylar notch
-
Enlargement of ossification centers (especially distal end of femur)
-
Widening of distal femoral epiphysis
-
Squaring of inferior pole of patella
-
Flattening of distal femoral condyles
-
Hemophiliac pseudotumor
-
Epiphyseal overgrowth with leg length discrepancy
-
Osteopenia
|
TABLE 26.1-3 CLINICAL STAGES OF HEMARTHROSIS
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
TABLE 26.1-4 ROUTINE LABORATORY TESTS FOR PATIENTS WITH COAGULOPATHY
|
||||||||
|---|---|---|---|---|---|---|---|---|
|
|
TABLE 26.1-5 CLINICAL MANIFESTATIONS OF HEMOPHILIA ACCORDING TO BLOOD LEVELS OF FACTORS VIII AND IX
|
||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
the orthopaedic advisory committee of the World Federation of
Hemophilia is shown Table 26.1-6. It includes functional status. An additional radiographic classification useful for bony changes is shown in Table 26.1-7.
has improved with the use of ultrasound and magnetic resonance imaging
(MRI), which can provide early, detailed information about the
synovium, cartilage, and the joint spaces.
-
Hemosiderin deposits can be seen on MRI (Fig. 26.1-2), as can subchondral and intraosseous cysts or hemorrhage (Figs. 26.1-3 and 26.1-4).
-
□ The limitation of MRI is that the cost may be prohibitive when serial exams are necessary.
-
-
Ultrasound has been shown to be a sensitive and reproducible technique to assess synovial proliferation in a variety of joints.
-
The activity of disease, in terms of acute synovitis, may further be assessed with power Doppler ultrasound.
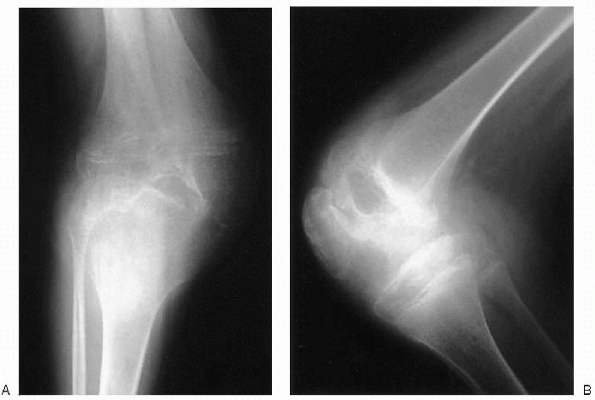 |
|
Figure 26.1-1 Anteroposterior (A) and lateral (B)
knee plain radiographs of a 10-year-old boy with hemophilia: loss of joint cartilage space, marked enlargement of the epiphysis, and extensive joint destruction (stage V according to Arnold and Hilgartner radiographic classification). |
|
TABLE 26.1-6 ROENTGENOGRAPHIC CLASSIFICATION OF HEMOPHILIC ARTHROPATHY
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
TABLE 26.1-7 RADIOGRAPHIC CLASSIFICATION OF HEMOPHILIC ARTHROPATHY
|
||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||||||||
hematologist, with input from a variety of other medical professionals
including orthopaedists, physical therapists, vocational therapists,
nurses, dentists, social workers, psychiatrists, and genetic
counselors. There are several factors that affect the choice of type of
therapy (Table 26.1-8).
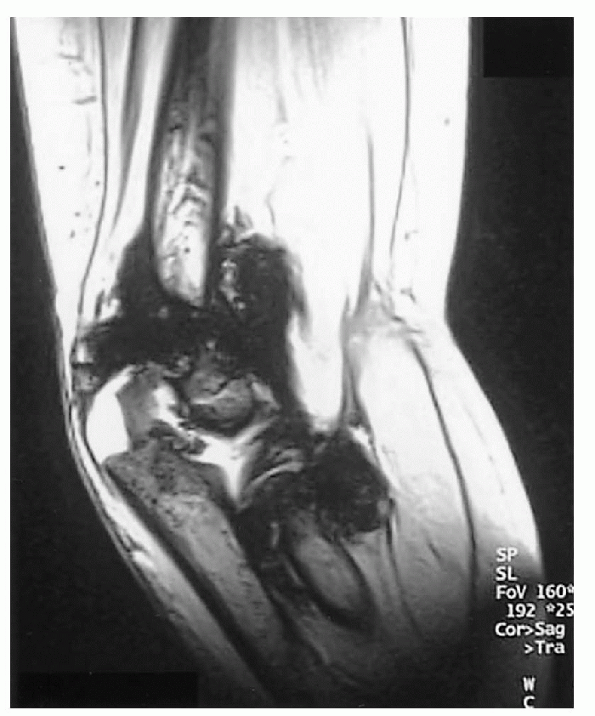 |
|
Figure 26.1-2
Right elbow magnetic resonance image of a 6-year-old boy with hemophilia. Synovial hypertrophy and large hemosiderin deposit are seen. |
America involves early intervention for bleeding episodes with
increased factor given at the onset of discomfort and, if possible,
even prior to the observation of joint swelling. This approach is often
referred to as an on-demand treatment. It involves the use of home
infusion of factor VIII without the need to be seen initially at a
hospital or a clinic. Factor levels of 30 to 50 IU/dL are optimal in
controlling an acute hemorrhage.
factor levels greater than 1%) prevents hemophilic arthropathy better
than on-demand therapy (Table 26.1-9).
It has been useful in preventing the spontaneous bleeding typically
seen in patients with severe disease. The amount of product necessary
to achieve a nonbleeding state with few breakthrough bleeding episodes
varies from patient to patient. The age of onset at which a patient may
begin to experience hemarthrosis also varies, but is usually before the
age of 4 years. Decreased hemophilic joint destruction and arthropathy
have been demonstrated in patients who have started prophylaxis at 2 to
3 years of age. In certain situations, factor VIII levels should be
increased for prophylaxis (Table 26.1-10).
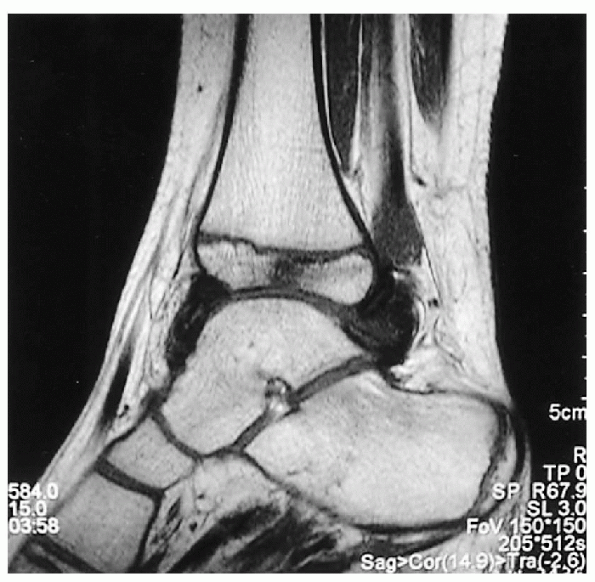 |
|
Figure 26.1-3 Left ankle sagittal magnetic resonance image of a 6-year-old boy with hemophilia. Subchondral bone cyst is seen.
|
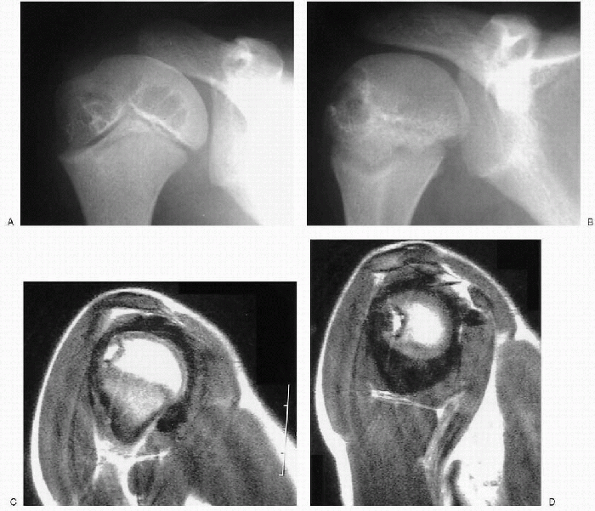 |
|
Figure 26.1-4 Eight-year-old boy with factor VIII deficiency. (A and B) Anteroposterior plain x-rays of the shoulder show cyst formation in the humeral head. (C and D) Coronal magnetic resonance images reveal marked synovial hyperplasia and hemosiderin deposits, called a blooming appearance.
|
|
TABLE 26.1-8 FACTORS THAT AFFECT THE CHOICE OF THERAPY FOR HEMOPHILIC ARTHROPATHY
|
||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||||||||||
|
TABLE 26.1-9 PROPHYLACTIC TREATMENT OF HEMOPHILIC ARTHROPATHY
|
||||||||
|---|---|---|---|---|---|---|---|---|
|
(HIV) transmission and hepatitis in human factor VIII replacement,
recombinant products are preferred and are available for factors VII,
VIII, and IX. The dosage required to replace a factor deficiency
depends on the patient’s weight and plasma volume. The hematologist
makes the calculation and is in charge of administering the factor. The
orthopaedic surgeon, however, should be aware of the fact that 20 to 30
minutes after administration of the antihemophilic factor, the plasma
level will rise.
platelet aggregation and prolong the bleeding time, should be avoided
in patients with hemophilia. Patients should be warned against the use
of any medication containing an aspirin compound, guaiacolate, or
antihistamine. Proxyphene, paracetamol, meperidine, codeine
amitriptyline, or methadone may be used. Narcotic analgesics are used
with care because in such a chronic disease, addiction can easily
become a problem.
|
TABLE 26.1-10 SITUATIONS IN WHICH FACTOR VIII LEVELS SHOULD BE INCREASED
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
and yields the best results. Adequately prescribed and correctly
supervised physiotherapy very rarely requires concurrent replacement
therapy, except after surgery. Every patient generally requires
individual exercise programs.
cushioned lining. Care should be taken to avoid muscle atrophy. There
are four distinct functions for orthoses used in hemophiliac
arthropathy:
-
Protection and stabilization of an unstable joint
-
Nocturnal use, in the immediate postoperative period or to complete casting
-
Prevention of some joint contractures, in particular flexion deformity
-
Braces, splints, and walking bandaging are useful to enhance joint stability without limiting movement.
hemophilic arthropathy are generally divided into two groups:
procedures such as synovectomy that are done to control repetitive
joint bleeding episodes and the more conventional orthopaedic
procedures designed to correct or reconstruct joint deformities.
careful technique using tourniquet control when feasible and securely
ligating all vessels insofar as possible. Cautery is a less
satisfactory method of hemostasis in hemophiliacs. Before the wound is
closed, the tourniquet must be released and bleeding surgically
controlled. Appropriate perioperative factor replacement is imperative.
Some experts recommend aspiration only for an extremely tense
hemarthrosis and avoid aspiration in ordinary cases, citing the risk of
introducing infection, the discomfort to the patient, and the
possibility that aspiration will incite more bleeding of the joint.
Aspiration of the joint should be performed under strict aseptic
conditions.
-
Severe recurrent hemarthrosis (2 or 3 major bleeding episodes/mo)
-
Hemarthrosis that does not respond to aggressive medical management maintained for ≥6 mo
-
Failure to respond to orthopaedic nonsurgical treatment consisting of physical therapy and protection with crutches and orthoses
-
Radiographic stage II or III hemophilic arthropathy
of hemophilic arthropathy. The general consensus of opinion is that
synovectomy relieves pain, decreases swelling, and diminishes the
number of bleeding episodes per year (Box 26.1-1). Synovectomy is ineffective and contraindicated patients with radiographic stage IV or V hemophilic arthropathy.
loss of range of motion of the affected joint. Arthroscopic synovectomy
is most useful when performed before severe degenerative changes have
developed. Contraindication for arthroscopy of hemophilic patients is
inhibitory antibodies to factor replacement. As complete a synovectomy
as possible should be performed. After surgical synovectomy the knee
should be immobilized in a Jones bandage for 3 days and active movement
encouraged. Postoperative factor replacement supervised by a
hematologist is required. Weidel has shown with a 10- to 15-year
follow-up that the procedure was effective in stopping the bleeding
episodes, and by maintaining the range of motion, joint deterioration
continued to progress at a slower rate.
tissue by intraarticular injection of a radioactive agent (yttrium-90,
gold-198, phosphorus-32, and rhenium-186). It requires only one
session, but has a higher cost (Box 26.1-2). A
possible concern is that the damaging effects of the radioactivity are
not strictly limited to the synovium but may also affect the articular
chondrocytes. In addition, there is a theoretic concern of future
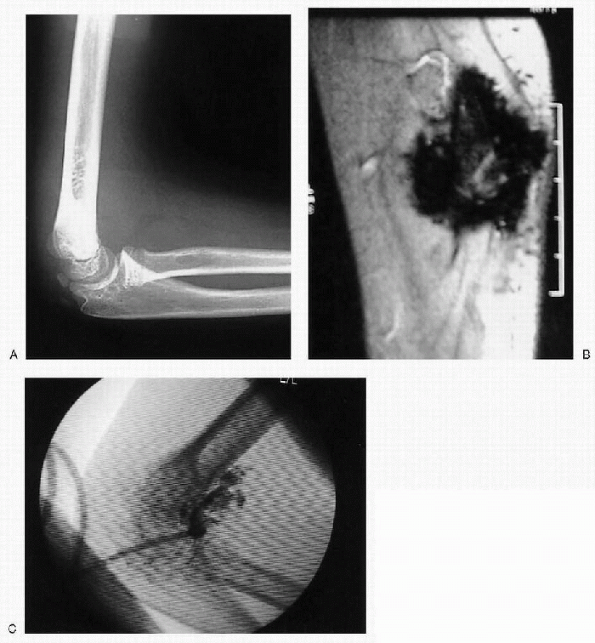
oncogenesis (Fig. 26.1-5).
-
A minimal requirement of antihemophilic factor
-
Performance on an outpatient basis
-
Concurrent treatment of several joints
-
Low risk of hemorrhage in patients with inhibitors
-
Administration of local anesthetic only,
which is beneficial in patients who are not candidates for surgery
because of systemic illness -
Low cost
D-penicillamine, and osmic acid. Rifampicin is commonly used with
successful results for chemical synovectomy in developing countries
where there is a lack of radioactive materials. However, it is quite
painful during injection and must be injected weekly, with the number
of injections ranging from 5 (in ankles and elbows) to 10 (in knees).
Osmic acid is another agent for using chemical synovectomy, but its
results are no better than those of rifampicin.
total joint replacement. Disabling pain with advanced destruction of
articular cartilage is the prime indication for surgery. It has been
done routinely for knees, hips, and shoulders and sporadically for
elbows and ankles. The problems of arthroplasty in hemophiliacs are
considerable and include difficulties with lack of bone stock,
deformities of the joint, muscle contractures, adhesions, and soft
tissue contractures. The most common cause of failure is infection. It
is difficult to salvage prostheses complicated by infection. However,
the life expectancy of hemophilic patients is lower than that of the
general population of patients treated with total knee arthroplasty,
and the improvement in the quality of life after knee arthroplasty for
hemophilic arthropathy may outweigh the risk of failure.
V hemophiliac arthropathy when pain is persistent with severe
disability not relieved by conservative measures. The most common
problem is aseptic loosening of cemented components, probably because
of microhemorrhages at the bone-cement surface. Studies suggest that
arthroplasty, particularly of the hip and knee, can be a valuable
option in the management of severe hemophilic arthropathy although
complications are commonly described and the surgery is technically
demanding.
joints in which arthroplasty has failed or become infected. Arthrodesis
of the ankle, subtalar, and midtarsal joints in the foot, elbow, or
shoulder may be indicated when these joints are destroyed.
possible, fractures are treated by closed reduction and immobilization
in a cast. External fixators should be avoided. Open reduction and
internal fixation are carried out when closed methods are not
appropriate.
accessible. Prior to surgical intervention, angiography, computed
tomography, and nuclear MRI should be performed to provide accurate
anatomic detail of adjacent vessels. Rarely, radiotherapy may be
considered in surgically inaccessible sites.
 |
|
Figure 26.1-5 Right elbow lateral plain radiograph (A) and coronal magnetic resonance image (B) of an 8-year-old boy with hemophilia A and recurrent bleeding approximately eight times a year despite factor treatment. (C) Right elbow P-32 radioisotope synovectomy was performed.
|
F, Rivas S, Viso R, et al. Synovectomy with rifampicine in hemophiliac
haemarthrosis. Haemophilia 2000;6:562-565.
WB, Wilson FC. The management of musculoskeletal problems in
hemophilia. Part II (Pathophysiologic and roentgenographic changes in
hemophilic arthropathy). AAOS Instr Course Lect 1983;32:217-233.
G, Gilbert MS, Abdelwahab IF. Hemophilia: evaluation of musculoskeletal
involvement with CT, sonography, and MR imaging. AJR Am J Roentgenol
1992;158:119-123.
EC, Wiedel JD. General principles and indications of synoviorthesis
(medical synovectomy) in haemophilia. Haemophilia 2001;7(Suppl 2):6-10.
F. Hematologic disorders. In: Pediatric orthopaedic deformities—basic
science, diagnosis, and treatment. San Diego: Academic Press,
2001:909-933.
HJ, Luck JV Jr, Siegel ME, et al. Phosphate-32 colloid radiosynovectomy
in hemophilia: outcome of 125 procedures. Clin Orthop 2001;392:409-417.
hemoglobinopathies, which cause chronic hemolytic anemia,
immunosuppression, and pain and organ damage secondary to vascular
occlusion. Variants include sickle cell anemia (SCA), hemoglobin SC
disease, and hemoglobin S β-thalassemia. SCA is the most common form,
and can present with numerous musculoskeletal manifestations and
complications. The diagnosis and management of the musculoskeletal
problems associated with SCA can pose a considerable challenge to the
orthopaedist.
inherited genetic mutation that codes for the substitution of normal
adult hemoglobin (HbA) with sickle hemoglobin (HbS), or less commonly,
with another variety of abnormal hemoglobin. Normal HbA consists of two
α-polypeptide chains and two β-polypeptide chains. In HbS, a mutation
on chromosome 11 substitutes valine for glutamine in the sixth amino
acid position of the β-globin chain. Children who are homozygous for
the mutation have SCA; heterozygotes have sickle cell trait (SCT).
people) have SCT, and approximately 1 in 400 has SCA. In regions where
malaria is endemic, the incidence of SCT can be as high as 40%, since
heterozygotes have slightly increased resistance to malaria.
Individuals of a variety of other ethnicities (Mediterranean, Asian,
Hispanic, and Middle Eastern) may also inherit the trait or disease.
The total number of persons in the United States with a clinically
manifest variant of sickle cell disease is about 72,000.
|
TABLE 26.2-1 CLASSIFICATION OF SICKLE CELL ANEMIA
|
||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
which changes it from a liquid to a gel. The orientation of the
hemoglobin S molecules into longitudinal fibers, combined with their
decreased plasticity due to increased viscosity, is what causes the
characteristic sickling of the erythrocytes. The presence of other
forms of hemoglobin mitigates the severity of the sickling. That is why
heterozygotes, whom have a significant proportion of HbA, are
clinically asymptomatic, except under conditions of severe hypoxia.
Similarly, sickle cell disease does not clinically manifest itself in
children under the age of 1 year, since fetal hemoglobin (HbF), which
persists until that time, is protective. Even other abnormal
hemoglobins, such as those with the thalassemia mutation, which
decreases the amount of β-globulin produced, generally are less
severely affected than HbSS homozygotes.
polymerization include dehydration, increased temperature, and falling
pH. Though sickling is reversible, repeated episodes cause cell
membrane damage that leads to permanently sickled cells. These cells
are hemolyzed intravascularly or in the spleen. The average life span
of a red blood cell in a patient with SCA is only 10 to 30 days (120
days is normal). The relative inelasticity of the sickled cells (and
even nonsickled cells with increased amounts of polymerized HbS) causes
increased blood viscosity, and decreased blood flow rate; this
contributes to occlusion and infarction of the microvasculature.
disease is a hemoglobin electrophoresis. The different disease variants
have characteristic electrophoresis patterns. A 5-minute solubility
test, called a Sickledex, can be used in initial screening, or in an
emergency setting, but if it is positive, an electrophoresis needs to
be done to differentiate between a carrier and disease state. The
Sickledex also has a relatively high false-negative rate.
result of vascular occlusion, hemorrhage, infarction, and ischemic
necrosis secondary to increased blood viscosity caused by sickling.
They can be summarized by use of the mnemonic HBSS PAIN CRISIS (problems for which an orthopaedist is frequently consulted are highlighted in italics):
-
Affects short tubular bones of hands and feet
-
Age 6 to 12 months (when HbF is replaced by HbS)
-
Distal extremities become swollen, tender, and painful
-
Triggered by cold weather
-
Lasts 1 to 2 weeks
-
Incidence: 45%
-
Recurrence: 41%
-
Rare after 6 years of age (when marrow activity in hands/feet ceases)
-
Dactylitis
-
Avascular necrosis
-
Osteomyelitis
-
Septic arthritis
-
Growth retardation
-
Leg ulcers
-
Bone marrow hyperplasia
-
Increased tendency to fracture
-
Age between 3 and 4 years
-
Lasts 3 to 5 days
-
Localized bone marrow or muscle infarction secondary to sequestration of sickled cells
-
Commonly: humerus, tibia, femur
-
Clinically: swelling, decreased range of motion, increased temperature
-
Clinically: fever, pain, swelling
-
Polyostotic: 12% to 47%
-
Erythrocyte sedimentation rate unreliable (usually low) secondary to sickled red blood cells
-
Staphylococcus aureus and MRSA common, but patients also susceptible to encapsulated organisms secondary to decreased splenic function:
-
□ Streptococcus pneumoniae
-
□ Haemophilus influenzae
-
□ Salmonella
-
□ Meningococcus
-
□ Klebsiella
-
-
Septic and reactive arthritis are less common than osteomyelitis but do occur
-
Joint fluid in reactive arthritis will show less than 20,000 white blood cells/mm3
-
Can lead to destruction of the physis, growth arrest, and limb deformity
-
Incidence: 8%
-
May be hemorrhagic or infarct
-
Greater than 50% recurrence rate, unless chronic life-time transfusion therapy instituted
-
May result in spastic hemiplegia requiring orthopaedic management
-
May be secondary to bone marrow hyperplasia, osteomyelitis, or disuse osteopenia
-
Attempt to minimize immobilization during treatment to prevent recurrence
-
Femoral head most common, followed by humeral head
-
Incidence: approximately 10%; bilateral: 54%
-
Usually diagnosed on plain x-ray, but magnetic resonance imaging may be useful to show extent of head involvement
by a hematologist, the systemic nature of the illness and the clinical
manifestations in various organ systems practically guarantee that most
patients will also be treated by a variety of other subspecialists. The
various acute crises and complications common to the disease often
necessitate presentation in the emergency department as well. Treatment
of patients with SCA can be thought of in three groups: preventive,
problem management, and systemic.
-
Daily supplemental folate, to keep up with red blood cell production demand
-
Periodic complete blood count check, to catch changes/problems early
-
Use supplemental iron sparingly; iron overload can become a problem in older patients
-
Maintain immunizations
-
Encourage adequate oral hydration
-
Encourage patients to seek early medical attention when ill
-
Discourage smoking and alcohol intake
-
Discourage excessive physical exertion
-
Avoid extremes of temperature
|
TABLE 26.2-2 DIFFERENTIAL DIAGNOSIS OF SICKLE CELL ANEMIA
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||||
|
TABLE 26.2-3 DIAGNOSIS BASED ON SEQUENTIAL BONE MARROW AND BONE SCANS RESULTS
|
|||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||||||||
-
Nonsteroidal antiinflammatory drugs (NSAIDs) for chronic pain
-
Analgesia with NSAIDs/narcotics
-
Hydration
-
Bed rest
-
Identify and correct underlying precipitators (especially infection)
-
Administration of oxygen, and monitoring, if the patient has documented hypoxemia
-
Antibiotic coverage for S. aureus and Salmonella
-
Surgical drainage for chronic
osteomyelitis with sequestrum formation, or for patients who are
systemically septic or not responding to antibiotic therapy
-
Arthrotomy and drainage, in conjunction with antibiotics
|
TABLE 26.2-4 PROBLEM MANAGEMENT IN AVASCULAR NECROSIS
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
-
Dose 1 to 35 mg/kg/day (see Table 26.2-5 for pros and cons)
-
Simple transfusion to hemoglobin level of 10 g per dL now preferred over exchange transfusions for preoperative optimization.
-
Use of antigen-matched blood and adequate
hydration/oxygenation helps prevent complications during transfusion
therapy and postoperatively. -
Acute transfusions are indicated for life-threatening complications of SCA such as stroke and aplastic anemia.
-
Chronic transfusion therapy for life is indicated to prevent recurrent strokes.
|
TABLE 26.2-5 PROS AND CONS OF ORAL HYDROXYUREA THERAPY
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||||
-
By use of transcranial Doppler ultrasound or magnetic resonance imaging/magnetic resonance angiography
-
15% of patients have asymptomatic CNS ischemic injury
-
Trials ongoing to establish role of transfusion therapy for prevention of CNS ischemic injury
AR, Roberson JR, Eckman JR, et al. Total hip arthroplasty in patients
who have sickle-cell hemoglobinopathy. J Bone Joint Surg (Am)
1988;70:853-855.
WW. Pathological fracture complicating long bone osteomyelitis in
patients with sickle cell disease. J Pediatr Orthop 1986;6:177-181.
CA, Hughes JL, Abrams RC, et al. Osteomyelitis in the patient with
sickle-cell disease. J Bone Joint Surg (Am) 1971;53:1-15.
P, Bachir D, Galacteros F. Avascular necrosis of the femoral head in
sickle-cell disease: treatment of collapse by the injection of acrylic
cement. J Bone Joint Surg (Br) 1993;75:875-880.
SL, Cotran RS, Kumar V. Sickle cell disease: pathologic basis of
disease, 3rd ed. Philadelphia: WB Saunders, 1984:618-622.
DL. Differentiation between bone infarction and acute osteomyelitis in
children with sickle-cell disease with use of sequential radionuclide
bone-marrow and bone scans. J Bone Joint Surg (Am) 2001;83:1810-1813.
MC, Padwick M, Serjeant GR. Observations on the natural history of
dactylitis in homozygous sickle cell disease. Clin Pediatr
1981;20:311-317.
MC, Patience M, Leisenring W, et al. Marrow transplantation for sickle
cell disease: results of a multicenter collaborative investigation. N
Engl J Med 1996;355:369-376.