
Editors: Tornetta, Paul; Einhorn, Thomas A.; Cramer, Kathryn E.; Scherl, Susan A.
Title: Pediatrics, 1st Edition
Copyright ©2004 Lippincott Williams & Wilkins
> Table of Contents > Section III: – Specialty Clinics > 27 – SKELETAL DYSPLASIAS
27
SKELETAL DYSPLASIAS
Michael Craig Ain
George S. Naseef III
Skeletal dysplasias are a diverse group of disorders
characterized by developmental abnormalities of the structure of bone
and cartilage. They may be classified according to their specific
deficiency (i.e., protein deficiency) or by the anatomic region they
predominately affect (i.e., epiphysis, metaphysis, or spine). Skeletal
dysplasias typically result in short stature, which is defined as
height below the third percentile. Short stature may be proportionate,
with symmetric decreases in both truncal and limb lengths.
Disproportionate statures (dwarfing conditions) may be divided into
shortened trunk, shortened limb, or shortened trunk and limb types.
Short limb types may be subdivided by the region of limb involvement.
Rhizomelic refers to shortening of the proximal portion of the limb;
mesomelic, to the middle segment; and acromelic, to the distal segment.
Over 300 skeletal dysplasias are now recognized, presenting myriad
orthopaedic abnormalities. This chapter reviews six of the most common
skeletal dysplasias, emphasizing orthopaedic evaluation and treatment.
characterized by developmental abnormalities of the structure of bone
and cartilage. They may be classified according to their specific
deficiency (i.e., protein deficiency) or by the anatomic region they
predominately affect (i.e., epiphysis, metaphysis, or spine). Skeletal
dysplasias typically result in short stature, which is defined as
height below the third percentile. Short stature may be proportionate,
with symmetric decreases in both truncal and limb lengths.
Disproportionate statures (dwarfing conditions) may be divided into
shortened trunk, shortened limb, or shortened trunk and limb types.
Short limb types may be subdivided by the region of limb involvement.
Rhizomelic refers to shortening of the proximal portion of the limb;
mesomelic, to the middle segment; and acromelic, to the distal segment.
Over 300 skeletal dysplasias are now recognized, presenting myriad
orthopaedic abnormalities. This chapter reviews six of the most common
skeletal dysplasias, emphasizing orthopaedic evaluation and treatment.
ACHONDROPLASIA
PATHOGENESIS
-
Most common type of short limb disproportionate dwarfism
-
Incidence is estimated at 1 per 25,000 live births.
-
Incidence correlates with greater paternal age.
-
Achondroplasia is an autosomal dominant condition with 100% penetrance.
-
It arises from a point mutation on the
short arm of chromosome 4 at nucleotide 1138 of the fibroblast growth
factor receptor 3 (FGFR3) gene (Table 27-1), resulting in a single amino acid change (glycine to arginine) in the transmembrane domain of the receptor. -
FGFR3 is expressed in the cartilaginous
precursors of bone, where it is believed to decrease chondrocyte
proliferation in the proliferative zone of the physis and to regulate
growth by limiting endochondral ossification. -
80% of new cases arise from a spontaneous mutation
|
TABLE 27-1 GENETIC DISORDERS OF DYSPLASIAS
|
|||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||||||||||||||||||||
DIAGNOSIS
PHYSICAL EXAMINATION AND HISTORY
Clinical Features (Table 27-2 and Fig. 27-1)
-
Enlarged head with frontal bossing
-
Midface hypoplasia
-
Appearance of mandibular protrusion
-
Flattened or depressed nasal bridge
-
Normal trunk length
-
Proximal limb shortening (rhizomelic)
-
Mild elbow flexion contractures
-
Trident hands with inability to approximate the long and ring fingers
-
Genu varum (bow leg) is most common; genu valgum may occur
-
Internal tibial torsion and ankle varus may be present
P.308
|
TABLE 27-2 ORTHOPAEDIC MANIFESTATIONS OF DYSPLASIAS
|
||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Children with achondroplasia are typically delayed in reaching motor milestones during the first few years of life.
-
This is secondary to their enlarged head size, relative hypotonia, joint laxity, and the presence of foramen magnum stenosis.
-
The timeframe of motor milestones in achondroplasia is:
-
□ Head control at 4 months
-
□ Sitting up at 10 months
-
□ Standing and ambulation begin between 17 and 20 months
-
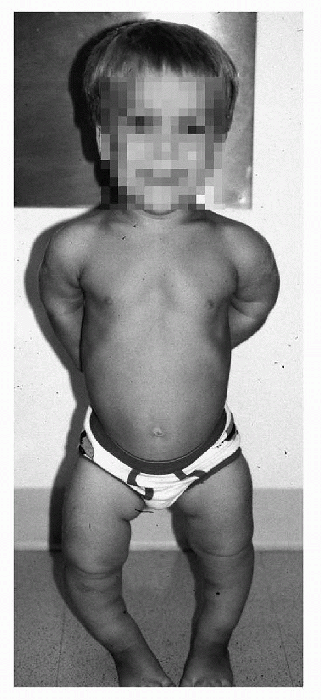 |
|
Figure 27-1 Typical clinical appearance in achondroplasia.
|
The foramen magnum is narrowed in achondroplasia.
-
This occasionally causes cervical cord compression at either the posterior portion of the foramen or at the atlas.
-
Cord compression is best visualized using magnetic resonance imaging (MRI).
-
Symptoms of cord compression include respiratory difficulties (apnea), hypotonia, hyperreflexia, and clonus.
Radiographic Features
The lumbar spine of achondroplasts has unique anatomic findings:
-
The interpedicular distance consistently
decreases from upper to lower lumbar levels (best seen on
posteroanterior radiograph of lumbar spine), which is opposite to what
is found in unaffected individuals. -
The pedicles are approximately 30% to 40% thicker than unaffected individuals.
-
These findings contribute to an overall
decrease in the area of the lumbar spinal canal [best seen on computed
tomography (CT) scan].
TREATMENT
GENU VARUM
-
Present in 40% of patients with achondroplasia, but clinically significant in only 25%.
-
Fibular overgrowth has been suggested as the source of this deformity.
-
Several operations based on this
assumption, including proximal fibular epiphysiodesis, have been
reported, but no long-term satisfactory results have been published. -
Indications for operative intervention
include pain, progressive deformity, and presence of a fibular thrust
(lateral subluxation of the knee joint) while ambulating. -
Surgical options:
-
□ Proximal tibial osteotomy with fibular osteotomy with or without fibular epiphysiodesis
-
□ Distal femoral osteotomy
-
□ Supramalleolar osteotomy
-
THORACOLUMBAR KYPHOSIS (FIG. 27-2)
-
Centered between T10 and L4.
-
Most pronounced when the child begins to sit up.
-
90% spontaneously regress when the child begins to ambulate.
-
Small, wedge-shaped, slightly recessed apical vertebrae predict persistent deformity.
-
Kyphosis beyond the age of 3 years should be treated with extension bracing.
-
Anterior and posterior spinal fusion may
be considered for significant kyphosis (greater than 50 degrees) beyond
the age of 5 years.
P.309
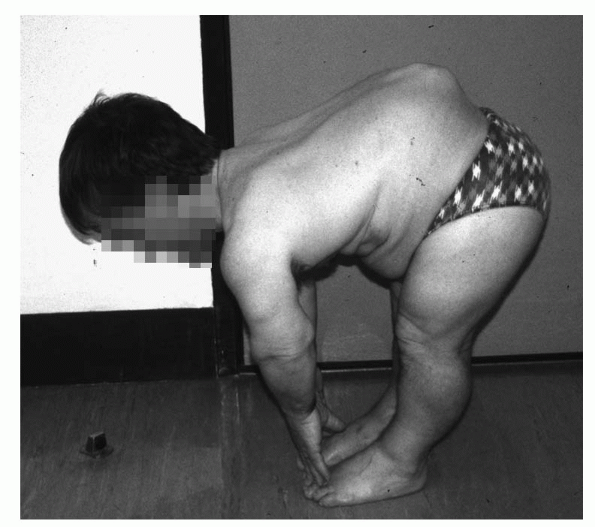 |
|
Figure 27-2 Achondroplasia with thoracolumbar kyphosis.
|
RESPIRATORY DEPRESSION
-
May contribute to cases of sudden infant death
-
Central or obstructive in nature
-
Treatment based its source and the severity of symptoms
-
Severe central apnea treated with foramen magnum decompression
-
Milder symptoms usually resolve with normal growth.
LUMBAR STENOSIS
-
During the second or third decade of
life, degenerative changes of the intervertebral discs further
compromise the lumbar canal leading to development of lumbar stenosis. -
Patients with achondroplasia will
typically present with complaints of lower back pain, leg pain, and
progressive weakening of the extremities. -
Symptoms are decreased with squatting or bending over, which reduces lumbar lordosis.
-
CT scan myelogram through cisternal puncture or MRI is useful to visualize the areas of stenosis (Fig. 27-3).
-
Surgical treatment includes a wide multilevel laminectomy with care to preserve the facet joints.
-
If thoracolumbar kyphosis is present, posterolateral fusion with instrumentation is recommended.
LIMB LENGTHENING
-
Controversial within the Little People community
-
Femur, tibia, and humerus are most often lengthened.
-
Multiple techniques described; most involve osteotomy and distracting ex-fix
-
Limb lengthening of 40% to 50%
-
Complications include infections,
hardware failures, nerve injuries, osteomyelitis, soft tissue
deformities, bone or joint deformities, high fracture rate, and
progression of spinal stenosis. -
Requires up to 3 years to obtain desired results
-
Decision best made by the well-informed and mature patient and not by the parents
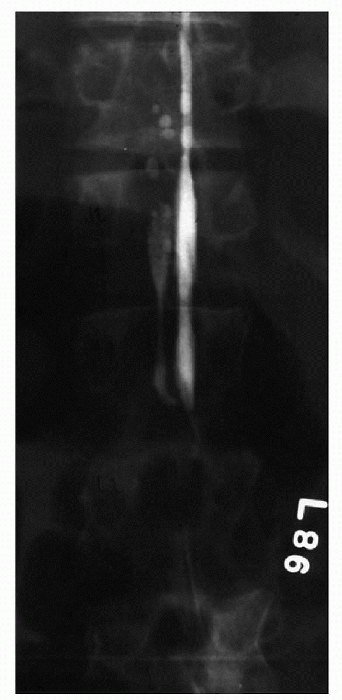 |
|
Figure 27-3 Myelogram showing lumbar spine stenosis.
|
PSEUDOACHONDROPLASIA
PATHOGENESIS
-
Second most common short limb dwarfism
-
Incidence is estimated to be 4 per million live births.
-
Autosomal dominant condition
-
Most cases are secondary to a spontaneous mutation.
-
Arises from a mutation occurring on chromosome 19, affecting the gene coding for cartilage oligometric matrix protein (COMP)
-
□ COMP is a large extracellular matrix protein normally found surrounding chondrocytes in developing bone.
-
DIAGNOSIS
PHYSICAL EXAMINATION AND HISTORY
Clinical Features (Table 27-2)
-
Normal faces, head sizes, and stature at birth
-
Rhizomelic shortening recognized around the age of 2 or 3 years
-
Thoracolumbar kyphosis, scoliosis, or lumbar lordosis.
-
Windswept knees, genu valgum, or genu varum are common.
-
Shortening of the digits of the hands and feet
-
Pes planus and marked ligamentous laxity
-
Patients with hypoplasia or aplasia of the odontoid must be monitored for the development of atlantoaxial instability.
-
Myelopathic symptoms, including increasing fatigability and hyperreflexia, are cause for further studies.
Radiographic Features
-
Flattened vertebrae (platyspondyly) with anterior wedging
-
Odontoid hypoplasia in 50%, warranting flexion-extension radiographs of the cervical spine
-
Delayed epiphyseal ossification and flaring of the metaphysis
-
Articular deformities in weightbearing joints (precocious osteoarthritis)
TREATMENT
ATLANTOAXIAL INSTABILITY
-
Indication for posterior cervical fusion:
atlantoaxial translation greater than 5 mm on flexion-extension
radiographs in conjunction with progressive myelopathic symptoms or
translation greater than 8 mm are the absolute numbers
SPINAL DEFORMITIES
-
Kyphosis and scoliosis are usually
flexible and compensatory to pelvic obliquity arising from lower
extremity malalignment and hip flexion contractures. -
Bracing can be used to prevent lesser curves (25 to 45 degrees) from progressing.
-
Curves greater than 50 degrees typically require posterior spinal fusion.
-
Younger patients and those with greater kyphotic deformities often require anterior and posterior spinal arthrodesis.
LOWER EXTREMITY DEFORMITIES
-
Hip, knee, and ankle radiographs for planning corrective osteotomies
-
Indications for operative intervention
include progressive deformity, pain, ligamentous laxity, and
prophylaxis to delay the onset of osteoarthritis. -
Corrective osteotomies commonly include:
-
□ Proximal femoral
-
□ femoral
-
□ Proximal tibial
-
□ Supramalleolar osteotomies
-
-
Operative goals:
-
□ Correct overall limb alignment
-
□ Obtain joint surfaces that are horizontal to the floor
-
-
Lower extremity deformities often recur due to severe ligament laxity.
HIP JOINT DEFORMITIES
-
Subluxation, hinged abduction, or lateral
extrusion of the hip joint may occur secondary to delayed ossification
of the femoral epiphyses. -
Intraoperative arthrography and dynamic
fluoroscopic evaluation of the affected hip help to evaluate articular
deformity and joint stability. -
Operative intervention, consisting of
proximal femoral osteotomy with or without pelvic augmentation, is
based upon the fluoroscopic findings.
DIASTROPHIC DYSPLASIA
PATHOGENESIS
-
Rare disorder resulting in short limb dwarfism
-
Autosomal recessive condition secondary to a mutation occurring in the diastrophic dysplasia sulfate transporter (DTDST) gene.
-
This gene is found on chromosome 5 and is believed to regulate the intracellular concentration of sulfate.
-
Proteoglycans, a major constituent of
cartilage extracellular matrix, contribute to the flow-dependent
viscoelastic properties of cartilaginous tissues. -
Mutation of DTDST gene impairs the
sulfation of proteoglycans, decreasing their net negative charge and
lowering their affinity for water, which impairs the flow-dependent
viscoelastic properties of cartilage, ultimately decreasing the ability
of cartilage to function successfully as a shock absorber and
predisposing the cartilaginous precursors of bone to deformity and
precocious osteoarthritis.
DIAGNOSIS
PHYSICAL EXAMINATION AND HISTORY
Clinical Features (Fig. 27-4)
-
Rhizomelic shortened stature
-
Flattened nasal bridge
-
High incidence of cleft palate deformities
-
Hitchhiker thumb deformity (short, proximally based, and abducted)
-
Characteristic cauliflower ear appearance
-
Not associated with an increased risk of atlantoaxial instability
-
Subluxated shoulders and radial heads
-
Ankylosis of the proximal interphalangeal joints of the index, middle, and ring fingers
-
Clinodactyly
-
Lower extremity deformities:
-
□ Severe hip and knee flexion contractures
-
□ Genu valgum
-
□ Dislocated patella
-
□ Complex deformities of the ankles and feet
-
P.311
 |
|
Figure 27-4 Clinical appearance in diastrophic dwarfism.
|
Early childhood screenings in the form of comprehensive
neurologic examinations and a lateral cervical spine radiograph for the
presence or progression of cervical kyphosis are warranted.
neurologic examinations and a lateral cervical spine radiograph for the
presence or progression of cervical kyphosis are warranted.
Radiographic Features
Cervical spina bifida and kyphosis, midthoracic kyphoscoliosis, and lumbar lordosis are frequently present.
TREATMENTS
CERVICAL KYPHOSIS
-
Occurs in over 30% of affected patients
-
Associated with quadriplegia and even death
-
Kyphosis angle greater than 60 degrees or
a round or triangular-shaped, posteriorly displaced, apical vertebrae
suggests progressive nature. -
Anteroposterior cervical fusion for progressive deformity or cord compression
HIP JOINT DEFORMITY
-
Examination often reveals decreased hip range of motion with an increased tendency to subluxate or dislocate.
-
These changes further stress the susceptible hyaline cartilage, leading to progressive deformities and epiphyseal flattening.
-
Arthrography is indicated to evaluate the proximal femoral epiphysis.
-
A contracted hip joint without evidence of epiphyseal deformity is treated with soft tissue releases.
-
Conservative treatment is indicated once epiphyseal flattening is identified.
-
Precocious arthritis can be treated with custom hip implant arthroplasty.
LOWER EXTREMITY DEFORMITIES
-
Talipes equinovarus is the classic foot deformity associated with diastrophic dysplasia.
-
However, the most common deformity may be that of tarsal valgus and metatarsal adductus (43% of foot deformities).
-
Deformity is rigid and often refractory to treatment.
-
A combination of serial casting and soft tissue releases is required to improve the deformity.
-
Interventions need to be individualized based on the patient’s response to treatment and the surgeon’s experience.
MULTIPLE EPIPHYSEAL DYSPLASIA
PATHOGENESIS
-
Multiple epiphyseal dysplasia (MED) is a common form of short limb dwarfism.
-
Incidence is estimated at 1 per 100,000.
-
Several different disorders with similar manifestations.
-
Primarily inherited as an autosomal dominant condition, but autosomal recessive transmission has been reported.
-
MED1 and MED2 and their corresponding genetic defects have been identified.
-
□ The genetic mutation associated with MED1 occurs on chromosome 19 and affects the gene coding for COMP.P.312
-
□ COMP is an extracellular matrix protein
normally found surrounding chondrocytes in developing bone. However,
the specific mechanisms of this genetic disorder remain unclear. -
□ A similar defect in COMP is associated with pseudoachondroplasia.
-
-
-
The genetic mutation associated with MED2 affects the α2-polypeptide chain of type IX collagen.
-
□ Type IX collagen is a trimer found on the surface of type II collagen, where it functions as a stabilizer.
-
□ Although the exact mechanism is
unclear, it is hypothesized that a defect in type IX collagen
predisposes hyaline cartilage to early degeneration.
-
DIAGNOSIS
PHYSICAL EXAMINATION AND HISTORY
Clinical Features
-
Manifestations are variable.
-
Mild decrease in stature
-
Head, face, and spine are normal.
-
“Stubby” hands and feet: stunted growth of metacarpals and metatarsals
-
Decreased range of motion, pain, and angular deformities of hips, knees, and ankles
-
Patients typically present with complaint of lower extremity pain or limp at about age 7.
-
The hips and knees, secondary to their weight-bearing nature, are the most frequently involved joints.
Radiographic Features
-
The principal radiographic findings can be witnessed in the epiphysis of long bones.
-
These epiphyseal centers initially demonstrate delayed ossification, and later appear mottled once ossification has begun.
-
Eventual flattening and deformation of joint surfaces occurs, leading to precocious arthritis.
-
The key to remember is that MED is bilateral with symmetric joint involvement.
-
A symptomatic hip may be mistakenly diagnosed as Perthes disease.
-
Plain radiographs revealing symmetric hip, knee, and ankle joint involvement suggest the diagnosis of MED over Perthes disease.
TREATMENT
HIP AND LOWER EXTREMITY DEFORMITIES
-
Primary indications for surgical intervention are pain and deformity.
-
Corrective osteotomies to restore normal joint alignment with respect to the ground
-
Precocious arthritis often necessitates joint arthroplasty.
SPONDYLOEPIPHYSEAL DYSPLASIA
PATHOGENESIS
ETIOLOGY
-
Spondyloepiphyseal dysplasia (SED) is several different disorders with similar manifestations.
-
Two distinct forms are recognized: SED congenita and SED tarda.
-
□ SED congenita is an autosomal dominant condition.
-
□ Most cases arise from spontaneous mutations.
-
□ Estimated incidence of 3 or 4 cases per million
-
-
-
SED tarda is an X-linked recessive condition.
-
The SED genetic defect occurs in the α-1 chain of type II collagen.
DIAGNOSIS
PHYSICAL EXAMINATION AND HISTORY
Clinical Features
-
SED congenita
-
□ Motor milestones are often delayed secondary to mild hypotonia.
-
□ Most affected individuals are ambulatory by 2 years of age.
-
□ Short trunk and limb dwarfism
-
□ Normal intelligence
-
□ Odontoid hypoplasia
-
□ Thoracic kyphoscoliosis
-
□ Pectus carinatum
-
□ Lumbar lordosis
-
□ Coxa vara
-
□ Hip flexion contractures
-
□ Equinovarus foot deformity
-
□ Ocular ailments: retinal detachment, cataracts, myopia, and glaucoma
-
-
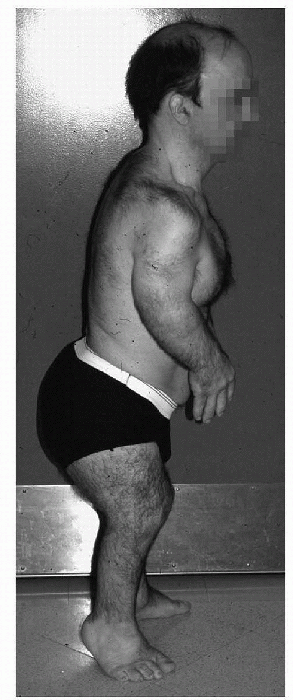
SED tarda (Fig. 27-5)
-
□ Normal at birth, mild symptoms develop in childhood
-
□ Mildly decreased stature
-
□ Back pain
-
□ Lower extremity pain
-
□ Odontoid hypoplasia, scoliosis, and coxa magna may occur
-
Radiographic Features
-
Plain radiographs may reveal odontoid hypoplasia.
-
Flexion-extension views of the cervical spine.
-
□ Evaluate for atlantoaxial instability (Fig. 27-6).
-
-
Lateral spine radiographs reveal the
characteristic biconcave vertebral end plates and flattened vertebral
bodies (platyspondyly). -
MRI in flexion and extension may reveal spinal cord encroachment.
P.313
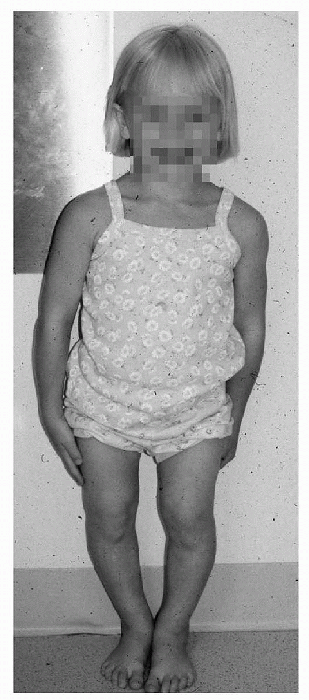 |
|
Figure 27-5 Clinical appearance in spondyloepiphyseal dysplasia tarda.
|
TREATMENT
ODONTOID HYPOPLASIA
-
Monitor regularly for the development of instability or myelopathy.
-
Myelopathy develops in up to 35% of children.
-
Atlantoaxial arthrodesis with or without
decompression is indicated for progression of myelopathy or when
instability exceeds 8 mm on flexion-extension radiographs.
 |
|
Figure 27.6 Flexion (A) and extension (B) radiographs of the cervical spine demonstrating atlantoaxial instability.
|
THORACIC KYPHOSCOLIOSIS
-
Bracing when curves are less than 40 degrees
-
Posterior spinal fusion plus or minus anterior release/fusion is indicated for severe curves or progressive deformity.
LOWER EXTREMITY DEFORMITIES
-
Corrective osteotomies are indicated for deformities and pain.
-
Unfortunately, progressive deformities and precocious arthritis are common.
-
Foot deformities may be responsive to stretching and serial casting.
OCULAR AILMENTS
-
Regular ophthalmologic examinations are encouraged.
SPONDYLOEPIPHYSEAL DYSPLASIA TARDA
-
Scoliosis is usually mild and can be managed with bracing.
-
Surgery is indicated for curves greater than fifty degrees or continued progression.
-
Valgus osteotomy of the proximal femur may improve hip joint congruity and delay the onset of degenerative arthritis.
MORQUIO SYNDROME
PATHOGENESIS
-
The mucopolysaccharidoses (MPS) are a
group of inherited lysosomal storage disorders resulting from specific
lysosomal enzyme deficiencies. -
At least 12 types have been identified.
-
Morquio syndrome (type IV) is the most common of these disorders.
-
□ Estimated incidence of 3 per 1 million
-
□ Three different subtypes of Morquio syndrome are now recognized.
-
□ All are autosomal recessive conditions
characterized by an inability to metabolize keratan sulfate, which
accumulates in cell lysosome, altering the metabolic activities of
lysosomes and ultimately leading to defective cartilage and connective
tissue formation and function.
-
P.314
DIAGNOSIS
PHYSICAL EXAMINATION AND HISTORY
Clinical Features (Fig. 27-7; Table 27-2)
-
Normal at birth
-
Normal intelligence
-
Short trunk dwarfism is appreciated within 2 to 3 years of life
-
Odontoid hypoplasia
-
Pectus carinatum
-
Thoracolumbar kyphosis with platyspondyly
-
Hip, ankle, and wrist laxity leading to subluxation and degenerative changes
-
Genu valgum
-
Cloudy corneas
-
Hearing loss
-
Coarsened facial characteristics
Radiographic Features
-
Plain radiographs may reveal odontoid hypoplasia.
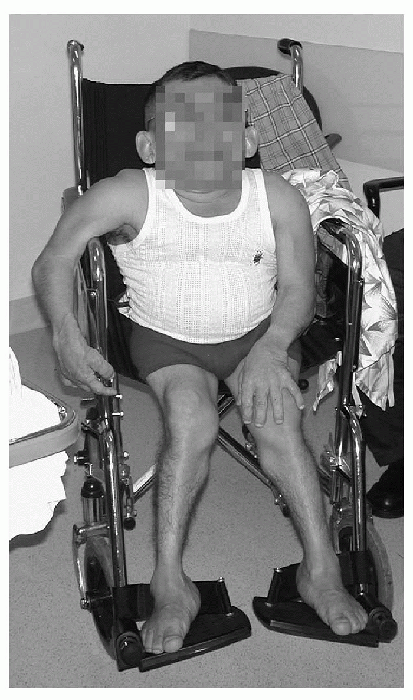 |
|
Figure 27-7 Clinical appearance in Morquio syndrome.
|
TREATMENT
ATLANTOAXIAL INSTABILITY
-
Atlantoaxial instability is found in
nearly all patients with Morquio syndrome and is the result of odontoid
hypoplasia and increased joint laxity. -
Children typically present with decreased endurance, hyperreflexia, clonus, and up-going Babinski sign at about 6 years of age.
-
Evaluation with flexion-extension cervical radiographs is helpful but may be misleading.
-
Further evaluation using
flexion-extension MRI often reveals the accumulation of extradural soft
tissue limiting the space available for the cervical spinal cord. -
Posterior spinal fusion with or without decompression of surrounding soft tissue is indicated when myelopathy is apparent.
ORTHOPAEDIC MANIFESTATIONS
-
Generally managed with bracing or observation
-
Genu valgum: corrective osteotomies once atlantoaxial instability is stabilized
-
Thoracolumbar kyphosis is best observed.
SUGGESTED READING
Aldegheri
R. Distraction osteogenesis for lengthening of the tibia in patients
who have limb-length discrepancy or short stature. J Bone Joint Surg Am
1999;81:624-634.
R. Distraction osteogenesis for lengthening of the tibia in patients
who have limb-length discrepancy or short stature. J Bone Joint Surg Am
1999;81:624-634.
Bassett GS. Lower extremity abnormalities in dwarfing conditions. Instr Course Lect 1991;39:389-397.
Bassett GS. Orthopedic aspects of skeletal dysplasias. AAOS Instr Course Lect 1991;39:381-387.
Bethem
D, Winter RB, Lutter L. Disorders of the spine in diastrophic dwarfism.
A discussion of nine patients and review of the literature. J Bone
Joint Surg (Am) 1980;62:529-536.
D, Winter RB, Lutter L. Disorders of the spine in diastrophic dwarfism.
A discussion of nine patients and review of the literature. J Bone
Joint Surg (Am) 1980;62:529-536.
Crossan
JF, Wynne-Davis R, Fulford GE. Bilateral failure of the capital femoral
epiphysis: bilateral Perthes’ disease, multiple epiphyseal dysplasia,
pseudoachondroplasia, and spondyloepiphyseal dysplasia congenita and
tarda. J Pediatr Orthop 1983;3:297-301.
JF, Wynne-Davis R, Fulford GE. Bilateral failure of the capital femoral
epiphysis: bilateral Perthes’ disease, multiple epiphyseal dysplasia,
pseudoachondroplasia, and spondyloepiphyseal dysplasia congenita and
tarda. J Pediatr Orthop 1983;3:297-301.
Dietz
FR, Murray JC. Update on the basis of disorders with orthopaedic
manifestations. In: Buckwalter JA, Einhorn TA, Simon SR, ed.
Orthopaedic basic science. Biology and biomechanics of the
musculoskeletal system. Rosemont, IL: American Academy of Orthopaedic
Surgeons, 2000:111-131.
FR, Murray JC. Update on the basis of disorders with orthopaedic
manifestations. In: Buckwalter JA, Einhorn TA, Simon SR, ed.
Orthopaedic basic science. Biology and biomechanics of the
musculoskeletal system. Rosemont, IL: American Academy of Orthopaedic
Surgeons, 2000:111-131.
Kopits SE. Orthopedic complications of dwarfism. Clin Orthop 1976;114:153-179.
Mankin
HJ, Mow VC, Buckwalter JA, et al. Articular cartilage structure,
composition, and function. In: Buckwalter JA ET, Simon SR, ed.
Orthopaedic basic science. Biology and biomechanics of the
musculoskeletal system. Rosemont, IL: American Academy of Orthopaedic
Surgeons, 2000:443-470.
HJ, Mow VC, Buckwalter JA, et al. Articular cartilage structure,
composition, and function. In: Buckwalter JA ET, Simon SR, ed.
Orthopaedic basic science. Biology and biomechanics of the
musculoskeletal system. Rosemont, IL: American Academy of Orthopaedic
Surgeons, 2000:443-470.
McKusick VA. Heritable disorders of connective tissue. St. Louis: Mosby, 1972.
Mikles M, Stanton RP. A review of Morquio syndrome. Am J Orthop 1997;26:533-540.
Poussa M, Merikanto J, Ryoppy S, et al. The spine in diastrophic dysplasias. Spine 1991;16:881-887.
Scott CI. Achondroplasia and hypochondroplastic dwarfism. Clin Orthop 1976;114:18-30.
Sponsellor
PD. The skeletal dysplasias. In: Morrissey RT, ed. Lovell and Winter’s
pediatric orthopaedics, 4th ed. Vol 1. Philadelphia: Lippincott
Williams & Wilkins, 2001:244-285.
PD. The skeletal dysplasias. In: Morrissey RT, ed. Lovell and Winter’s
pediatric orthopaedics, 4th ed. Vol 1. Philadelphia: Lippincott
Williams & Wilkins, 2001:244-285.
Tolo VT. Spinal deformity in short-stature syndromes. AAOS Instr Course Lect 1990;39:399-405.