
dramatically advanced their ability to restore the structure and
function of damaged bones and joints. Using new methods of internal
fixation, external fixation, and rehabilitation, they now successfully
treat even the most severe fractures and many severe joint injuries.
New biologic approaches to promoting tissue repair and regeneration
will further improve treatment of these injuries.11
Yet ultimately the result of the treatment of any musculoskeletal
injury depends on the skill of the surgeon in taking advantage of the
natural healing potential of the tissues that form the skeleton.
Surgeons can treat bone and joint injuries without extensive knowledge
of these tissues, but they are better able to select the optimal
treatment if they have this knowledge. Furthermore, they can treat, and
in some instances prevent, complications of musculoskeletal injuries or
problems of failed or inadequate healing more effectively when they are
as skilled in applying the knowledge of tissue healing as they are in
the use of surgical techniques.
Its tensile strength nearly equals that of cast iron, but it is three
times lighter and ten times more flexible. Yet bone is not a homogenous
inert material like iron, or the plastics and metals that form most
orthopaedic implants. Its matrix consists of organic and inorganic
components, and it is covered on its internal and external surfaces by
cells and cell processes.13 An
elaborate system of lacunae, canals or tunnels containing cells and
cell processes, blood vessels, lymphatics, and nerves permeates the
matrix, and various specialized cell populations responsible for
maintaining the tissue lie within the matrix lacunae and on the bone
surfaces.13 In most people bone
appears to remain unchanged for decades, but this appearance is
deceptive; it is constantly changing in response to mechanical and
hormonal signals.14 Over a lifetime, the skeleton is fully turned over multiple times, adjusting its alignment to altered loads.
marrow supported and surrounded by bone tissue and periosteum. Although
the three component tissues of bone differ in composition, structure,
and function, they are not independent. Marrow can serve as a source of
bone cells, marrow blood vessels form a critical part of the bone
circulatory system, and disorders or mechanical disruption of the
marrow can affect the activities of bone and periosteal cells.
The matrix contains mineral that gives the tissue great strength and
stiffness in compression and bending. The organic component of the bone
matrix, primarily type I collagen, and contributes to bone strength,
but also gives bone the plasticity that allows substantial deformation
without fracture. Bone matrix also contains various cytokines,
including growth factors that stimulate bone formation.13,14
These growth factors appear to have important roles in normal bone
metabolism and in fracture healing. The periosteum, consisting of two
layers—an outer fibrous layer and an inner, more cellular and vascular
cambium layer—covers the external bone surfaces and participates in the
healing of many types of fractures. The thicker, more cellular
periosteum of infants and children has a more extensive vascular supply
than that of adults. Perhaps because of these differences, the
periosteum of children is more active in fracture healing. Two types of
bone can be distinguished by their mechanical and biological
properties: woven or immature bone, and lamellar or mature bone.13 Woven bone forms the embryonic skeleton and is replaced by lamellar bone during development and growth.14
Woven bone also forms the initial fracture repair tissue and is
replaced by lamellar bone as the fracture remodels under mechanical
load. Compared with lamellar bone, woven bone has a more rapid rate of
deposition and resorption, an irregular woven pattern of matrix
collagen fibrils consistent with its name, approximately four times the
amount of osteocytes per unit volume, and an irregular pattern of
matrix mineralization. The frequent patchwork formation of woven bone
and the spotty pattern of mineralization create an irregular
radiographic appearance that distinguishes the woven bone found in
fracture callus from lamellar bone. Because of its lack of collagen
fibril orientation, irregular mineralization, and relatively high cell
content and water concentration, woven bone is less stiff and more
easily deformed than lamellar bone.
congruent articulating cartilaginous surfaces supported by subchondral
and metaphyseal bone, joint capsules and ligaments that link the bones
supporting the articular surfaces, and synovial membranes that cover
the inner surfaces of the joint except for the area of articular
cartilage. Some joints also have dense fibrous tissue menisci that lie
between the cartilaginous surfaces and attach to the joint capsule.
chondrocytes surrounded by an elaborate, highly organized
macromolecular framework filled with water.16
Three classes of molecules (collagens, proteoglycans, and
noncollagenous proteins) form the macromolecular framework. Type II
collagen fibrils give the cartilage its form and tensile strength, and
various quantitatively minor collagens help organize and maintain the
meshwork of type II collagen fibrils. The interaction of proteoglycans
with water gives the tissue its stiffness to compression and its
resiliency, thereby contributing to its durability.87
The noncollagenous proteins are less well understood than the
proteoglycans and collagens, but they appear to help organize and
stabilize the matrix, attach chondrocytes to the matrix macromolecules,
and possibly help stabilize the chondrocyte phenotype. Unlike the other
primary musculoskeletal tissues, cartilage lacks blood, nerve, and
lymphatic supplies.
application of forces to the skeleton that exceed the strength of the
tissues. Disruptions of bone tissue are called fractures.
Visible disruptions of articular cartilage also generally are referred
to as fractures when they involve both the articular cartilage and
subchondral bone. These are called osteochondral or intra-articular fractures, and when they involve only the cartilage they are called chondral fractures.
After repair has replaced the lost and damaged cells and matrix, a
prolonged remodeling phase ensues. The energy requirements of fracture
healing increase rapidly during inflammation and reach a peak during
repair, when the cells in the fracture callus are proliferating and
synthesizing large volumes of new matrix. These energy requirements
remain high until cell density and cell activity begin to decline as
remodeling starts.48
but also the surrounding soft tissues, including the periosteum and
muscle. A hematoma accumulates within the medullary canal, between the
fracture ends and beneath the elevated periosteum. The damage to the
bone blood vessels deprives osteocytes of their nutrition, and they die
as far back as the junction of collateral vascular channels, leaving
the immediate ends of the fracture without living cells (Fig. 4-2).
Severely damaged periosteum and marrow, as well as other surrounding
soft tissues, may also contribute necrotic material to the fracture
site.
dead and injured cells cause blood vessels to dilate and exude plasma
leading to the acute edema seen in the region of a fresh fracture.
Inflammatory cells migrate to the region, including polymorphonuclear
leukocytes, followed by macrophages and lymphocytes. These cells also
release cytokines that stimulate angiogenesis.53
As the inflammatory response subsides, necrotic tissue and exudates are
resorbed, and fibroblasts and chondrocytes appear and start producing a
new matrix, the fracture callus (Figs. 4-2 and 4-3).
include the chemotactic and growth factors released during inflammation
at the fracture site and bone matrix proteins, including growth factors
exposed by disruption of the bone tissue and the fracture hematoma.92
Although the inflammation caused by a fracture follows the same
sequence for almost every fracture, the amount and composition of
repair tissue and the rate of repair may differ depending on (i)
whether the fracture occurs through primarily cancellous bone or
through primarily cortical bone, (ii) the extent of the soft tissue
disruption surrounding the fracture, and (iii) other factors that are
discussed under the section titled “Variables That Influence Fracture
Healing.”
influences the repair process. The summaries of fracture repair and
remodeling that follow first describe healing of closed fractures that
are not rigidly stabilized; that is, fractures in which repair proceeds in the presence of motion at the fracture site (Fig. 4-4).
A closed clavicle fracture that is not treated by internal fixation
provides an example of repair and remodeling of an unstable fracture.
The second summary describes the healing of stable fractures; that is,
fractures in which repair proceeds at a rigidly stable fracture site
with the fracture surfaces held in contact. Transverse diaphyseal
fractures of the radius and ulna treated by open anatomic reduction and
rigid internal fixation provide examples of the repair and remodeling
of stable fractures.
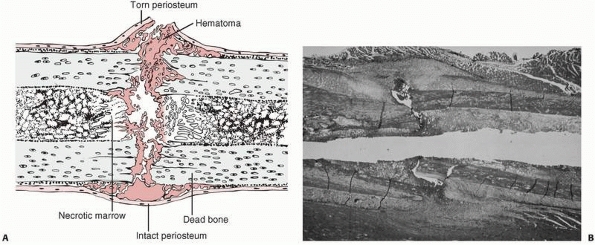 |
|
FIGURE 4-1 Initial events following fracture of a long bone diaphysis. A.
Drawing showing that the periosteum is torn opposite the point of impact, and may remain intact on the other side. A hematoma accumulates beneath the periosteum and between the fracture ends. There is necrotic marrow and cortical bone close to the fracture line. B. A photomicrograph of a fractured rat femur 3 days after injury showing the proliferation of the periosteal repair tissue. |
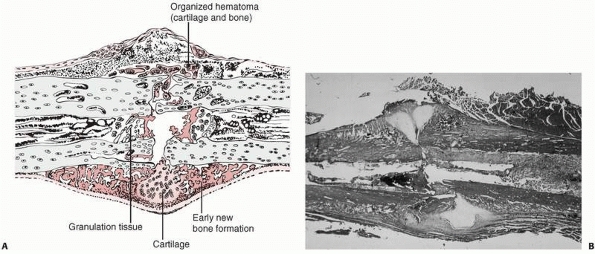 |
|
FIGURE 4-2 Early repair of a diaphyseal fracture of a long bone. A.
Drawing showing organization of the hematoma, early woven bone formation in the subperiosteal regions, and cartilage formation in other areas. Periosteal cells contribute to healing this type of injury. If the fracture is rigidly immobilized or if it occurs primarily through cancellous bone and the cancellous surfaces lie in close apposition, there will be little evidence of fracture callus. B. Photomicrograph of a fractured rat femur 9 days after injury showing cartilage and bone formation in the subperiosteal regions. (Reprinted from Einhorn TA. The cell and molecular biology of fracture healing. Clin Ortho 1998;335(Suppl):S7-S21, with permission.) |
periosteum, and surrounding tissue at the time of injury results in the
extravasation of blood at the fracture site and the formation
of a hematoma. Organization of this hematoma is usually recognized as the first step in fracture repair (see Fig. 4-2). Experimental work indicates that loss of the hematoma impairs or slows fracture healing,36,37
suggesting that the hematoma and an intact surrounding periosteal soft
tissue envelope that contains the hematoma may facilitate the initial
stages of repair. Open fractures or the treatment of fractures by open
reduction disrupts organization of the hematoma and may slow the repair
process. The precise reasons why a hematoma may affect fracture healing
remain uncertain. Presumably, the intact fracture hematoma provides a
fibrin scaffold that facilitates migration of repair cells. In
addition, growth factors such as parathyroid growth factors (PGGA) and
transforming growth factors (TGF-β) and other proteins released by
platelets and cells in the fracture hematoma mediate the critical
initial events in fracture repair. These include cell migration and
proliferation, and the synthesis of a repair tissue matrix.53,92 A breakdown product of thrombin provides strong stem cell attraction.6
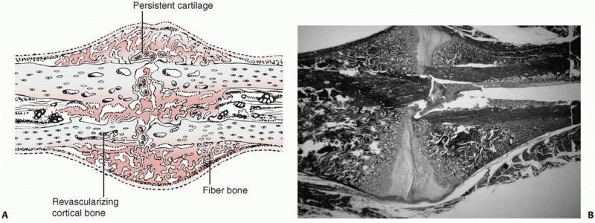 |
|
FIGURE 4-3 Progressive fracture healing by fracture callus. A.
Drawing showing woven or fiber bone bridging the fracture gap and uniting the fracture fragments. Cartilage remains in the regions most distant from ingrowing capillary buds. In many instances, the capillaries are surrounded by new bone. Vessels revascularize the cortical bone at the fracture site. B. Photomicrograph of a fractured rat femur 21 days after injury showing fracture callus united the fracture fragments. (Reprinted from Einhorn TA. The cell and molecular biology of fracture healing. Clin Ortho 1998;335(Suppl):S7-S21, with permission.) |
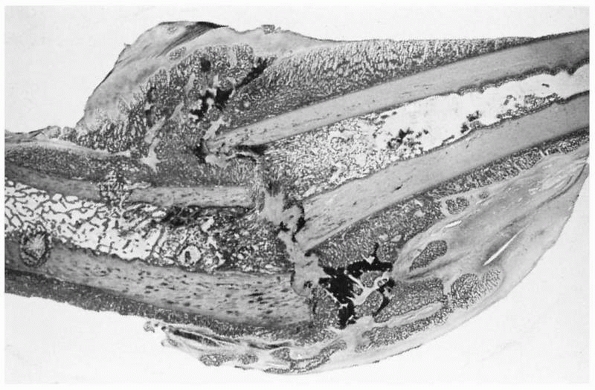 |
|
FIGURE 4-4
Light micrograph showing healing of a diaphyseal fracture under conditions of loading and motion. This femur fracture occurred in a pig that continued to use the limb for 3 weeks. Even though the fracture was not stabilized, it is healing. A large fracture callus consisting primarily of woven bone surrounds and unites the two fracture fragments. As the callus matures it progressively stabilizes the fracture. Notice that the fracture callus contains areas of mineralized and unmineralized cartilage. |
increases shortly after fracture, presumably because of vasodilatation,
vascular proliferation also occurs in the region of the fracture. It
appears that, under ordinary circumstances, the periosteal vessels
contribute most of the capillary buds early in normal bone healing,
with the nutrient medullary artery becoming more important later in the
process. The invading vessels carry along pericytes that provide a
large source of mesenchymal stem cells.7
Fibroblastic growth factors may be important mediators of the
angiogenesis in fracture healing, but the exact stimuli responsible for
vascular invasion and endothelial cell proliferation have not been
defined. When the surgeon interferes with the blood supply to the
fracture site, either by stripping the periosteum excessively or by
destroying the medullary system through reaming and the insertion of
intramedullary nails, repair must depend upon the remaining, intact
blood vessels.
blood supply, become necrotic and the surrounding bone is resorbed. In
some fractures this may create a radiographically apparent gap at the
fracture site several weeks or more after the fracture. The cells
responsible for this function, the osteoclasts, come from a different
cell line than the cells responsible for bone formation.13,14
They are derived from circulating monocytes in the blood and monocytic
precursor cells from the bone marrow, in which the osteoblasts develop
from the periosteum or from undifferentiated mesenchymal cells that
migrate into the fracture site.
origin, form the fibrous tissue, cartilage, and eventually bone at the
fracture site. Some of these cells originate in the injured tissues,
while others migrate to the injury site with the incoming
blood
vessels. Accompanying the neoangiogenesis are pericytes that provide a
large pool of undifferentiated mesenchymal stem cells capable of
differentiating into different tissue cell types. In addition, these
undifferentiated cells are productive sources of bone morphogenic
protein (BMP)—the growth factor that drives the differentiation process.2 Cells from the cambium layer of the periosteum form the earliest bone (see Fig. 4-1A).
Periosteal cells have an especially prominent role in healing fractures
in children because the periosteum is thicker and more cellular in
younger individuals. With increasing age, the periosteum becomes
thinner and its contribution to fracture healing becomes less apparent.
Osteoblasts from the endosteal surface also participate in bone
formation, but surviving osteocytes do not appear to form repair
tissue. Most of the cells responsible for osteogenesis during fracture
healing appear in the fracture site with the granulation tissue that
replaces the fracture hematoma.
differentiate, and produce the fracture callus consisting of fibrous
tissue, cartilage, and woven bone (see Fig. 4-3). Biological growth factors, notably members of the BMP family, drive the early differention process.6
The fracture callus fills and surrounds the fracture site, and in the
early stages of healing can be divided into the hard or bony callus and
the softer fibrous and cartilaginous callus. The bone formed initially
at the periphery of the callus by intramembranous bone formation is the
hard callus. The soft callus forms in the central regions, in which
there is relatively low oxygen tension, and it consists primarily of
cartilage and fibrous tissue. Bone gradually replaces this cartilage
through the process of endochondral ossification, enlarging the hard
callus and increasing the stability of the fracture fragments (see Fig. 4-4). This process continues until new bone bridges the fracture site, reestablishing continuity between the cortical bone ends.
The cells replace the fibrin clot with a loose fibrous matrix
containing glycosaminoglycans, proteoglycans, and types I and III
collagen. In many regions they convert this tissue to denser
fibrocartilage or hyaline-like cartilage. With formation of the
hyaline-like cartilage, type II collagen, cartilage-specific
proteoglycan, and link protein content increase. Newly formed woven
bone remodels to lamellar bone, and with remodeling the content of
collagen and other proteins returns to normal levels.
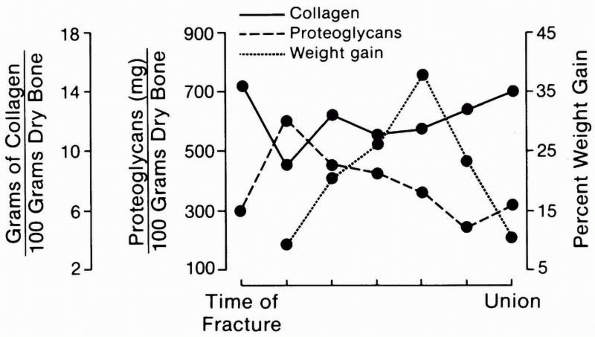 |
|
FIGURE 4-5
A schematic representation of the changing composition and mass of fracture callus. Collagen formation precedes significant accumulation of mineral. After an initial rise, proteoglycan concentration falls gradually as fracture healing progresses. The total mass of the fracture callus increases during repair and then decreases during remodeling. |
correlation between the activation of genes for blood vessel,
cartilage, and bone-specific proteins in the cells and the development
of granulation tissue, cartilage, and bone, respectively,82
demonstrating that fracture repair depends on the regulation of gene
expression in the repair cells. The simultaneous occurrence of
chondrogenesis, endochondral ossification, and intramembranous bone
formation in different regions of the fracture callus suggests that
local mediators and small variations in the microenvironment, including
mechanical stresses, determine what genes will be expressed and
therefore the type of tissue the repair cells form. Local mediators
that may influence repair cell function include growth factors released
from cells and platelets. BMPs influence this differentiation process.6
These factors are closely related to the WNT pathway. Acidic fibroblast
growth factor (FGF), basic FGF, and transforming growth factor beta
(TGF-β) may stimulate chondrocyte proliferation and cartilage
formation, osteoblast proliferation, and bone synthesis. TGF-β released
from platelets immediately after injury may initiate formation of
fracture callus. TGF-β synthesis is also associated with cartilage
hypertrophy and calcification at the endochondral ossification front.
Local oxygen tension is also an important factor. Hypoxia strongly
inhibits in vitro chondrogenesis and osteogenesis in mesenchymal stem
cells.52 Conversely, hypoxia
promotes chondrocytic differentiation and cartilage matrix synthesis
and suppresses terminal chondrocyte differentiation.40
These hypoxiainduced phenomena may act on chondrocytes to enhance and
preserve their phenotype and function during chondrocyte
differentiation and endochondral ossification. Proteoglycan synthesis
and aggregation in the various zones and stages of endochondral
ossification are differentially affected by the ambient oxygen
environment.23
ends gradually become enveloped in a fusiform mass of callus containing
increasing amounts of woven bone. Increasing mineral content is closely
associated with increasing stiffness of the fracture callus.3
The stability of the fracture fragments progressively increases because
of the internal and external callus formation, and eventually clinical union occurs—that is, the fracture site becomes stable and pain-free. Radiographic union
occurs when plain radiographs show bone trabeculae or cortical bone
crossing the fracture site, and this often occurs later than clinical
union. However, even at this stage healing is not complete. The
immature fracture callus is weaker than normal bone, and it only gains
full strength during remodeling.
repair tissue begins with replacement of the woven bone by lamellar
bone and resorption of unneeded callus. Radioisotope studies62
show increased activity in fracture sites long after the patient has
full restoration of function and plain radiographs show bone union,
demonstrating that fracture remodeling continues for years after
clinical and radiographic union. Remodeling of fracture repair tissue
after all woven bone has been replaced presumably consists of
osteoclastic resorption of superfluous or poorly placed bone and
formation of new bony trabeculae along lines of stress.
elaborate sequence of cellular and matrix changes, the important
functional result for the patient is an increase in mechanical
stability. The progressive increase in fracture stability consists of four stages.90
During stage I, a healing bone subjected to torsional testing fails
through the original fracture site with a low-stiffness pattern. In
stage II, the bone still fails through the fracture site, but the
characteristics of failure indicate a high-stiffness, hard-tissue
pattern. In stage III, the bone fails partly through the original
fracture site and partly through the previously intact bone with a
high-stiffness, hard-tissue pattern. Finally, in stage IV, failure does
not occur through the fracture site, indicating that new tissue at the
fracture site duplicates the mechanical properties of the uninjured
tissue. The sequence of repair depends upon the mechanical environment.
Loose connective tissue abundant in collagen can tolerate the marked
tensile demands. As the callus progresses, cartilage appears in
response to compression. The fracture repair process then moves toward
rigid stability by progressing through calcified cartilage, woven bone,
and ultimately lamellar bone. If the initial fracture environment is
stable, the initial repair matrix will be woven bone and then lamellar
bone, which is produced along lines of stress.
The clinical significance of these observations remains unclear, but
they suggest that fractures, and possibly the decreased loading of a
limb after fracture, may cause long-lasting changes in the tissues.
limits at a fracture site, fracture callus progressively stabilizes the
bone fragments and remodeling of the fracture callus eventually
produces lamellar bone. When the fracture surfaces are rigidly held in
contact, fracture healing can occur without any grossly visible callus.
This type of fracture healing has been referred to as primary bone healing, indicating that it occurs without the formation and replacement of visible fracture callus (Fig. 4-6).
bone ends directly apposed, there are regions of the fracture line
where the bone ends are in contact and other areas where there are
small gaps. Where there is contact between the bone ends, lamellar bone
can form directly across the fracture line by extension of osteons. A
cluster of osteoclasts cuts across the fracture line; osteoblasts
following the osteoclasts deposit new bone; and blood vessels follow
the osteoblasts. The new bone matrix, enclosed osteocytes, and blood
vessels form new haversian systems (Fig. 4-7).
Where gaps exist that prevent direct extension of osteons across the
fracture site, osteoblasts first fill the defects with woven bone.
After the gap fills with woven bone, haversian remodeling begins,
reestablishing the normal cortical bone structure. Cutting cones
consisting of osteoclasts followed by osteoblasts and blood vessels
traverse the woven bone in the fracture gap, depositing lamellar bone
and reestablishing the cortical bone blood supply across the fracture
site without the formation of grossly visible fracture callus. If a
segment of cortical bone is necrotic, gap healing by direct extension
of osteons still can occur but at a slower rate, and the areas of
necrotic cortical bone will remain unremodeled for a prolonged period.
body fractures, where cancellous and, in some regions, cortical bone
surfaces, interlock have sufficient stability to permit primary bone
healing where the bone surfaces are in direct contact. The same type of
cancellous bone healing can occur at rigidly stabilized osteotomies
through metaphyseal bone, intra-articular fractures, and surgical
arthrodesis sites. Most diaphyseal osteotomies, acute diaphyseal
fractures of long bones, and unstable metaphyseal fractures require the
use of devices that compress and rigidly stabilize the fracture site to
allow primary healing.
It is difficult to set the time when a given fracture should be united,
but when healing progresses more slowly than average, the slow progress
is referred to as delayed union. Watson-Jones89
described a condition he called slow union, in which the fracture line
remains clearly visible radiographically but there is no undue
separation of the fragments, no cavitation of the surfaces, no
calcification, and no sclerosis. This indolent fracture healing may be
related to the severity of the injury, poor blood supply, the age and
nutritional status of the patient, or other factors. It is not a
nonunion but rather a variation of normal healing. In contrast, failure
of bone healing, or nonunion, results from an arrest of the healing
process. This arrest of healing should be documented clinically and
radiographically over time. Most experts agree that there should be no
evidence of healing clinically or radiographically for at least 3
months before the term “nonunion” is used to describe the fracture.72
A nonunion that occurs despite the formation of a large volume of
callus around the fracture site is commonly referred to as a
hypertrophic nonunion (Fig. 4-8). This is in contrast to an atrophic nonunion (Fig. 4-9),
in which little or no callus forms and bone resorption occurs at the
fracture site. In some nonunions, cartilagenous tissue forms over the
fracture surfaces and the cavity between the surfaces fills with a
clear fluid resembling normal joint or bursal fluid creating a
pseudarthrosis, or false joint (Fig. 4-10).
Pseudarthroses may or may not be painful, but they almost uniformly
remain unstable indefinitely. In other nonunions the gap between the
bone ends fills with fibrous or fibrocartilaginous tissue.
Occasionally, dense fibrous and cartilaginous tissue firmly stabilizes
a fracture, creating a fibrous union. Although fibrous unions may be
painless and unite the fracture fragments, they fail to restore the
normal strength of the bone.
apparent cause, but in many instances injury, patient, and treatment
variables that adversely influenced fracture healing can be identified.
These variables include severe soft tissue damage associated with open
and high-energy closed fractures; infection; segmental fractures;
pathologic fractures; fractures with soft tissue interposition; poor
local blood supply; systemic diseases; malnutrition; vitamin D
deficiency; corticosteroid use; poor mechanical fixation; and
iatrogenic interference with healing. Many other variables have been
reported to retard bone healing.12
Some of them exert an adverse influence that can be measured in
experimental studies, but may not cause clinically significant
impairment of fracture healing. Others, like distraction of a fracture
site or interposition of soft tissues in the fracture
site
have not been examined systematically in experimental studies, but
clinical experience shows that they can impair fracture healing.
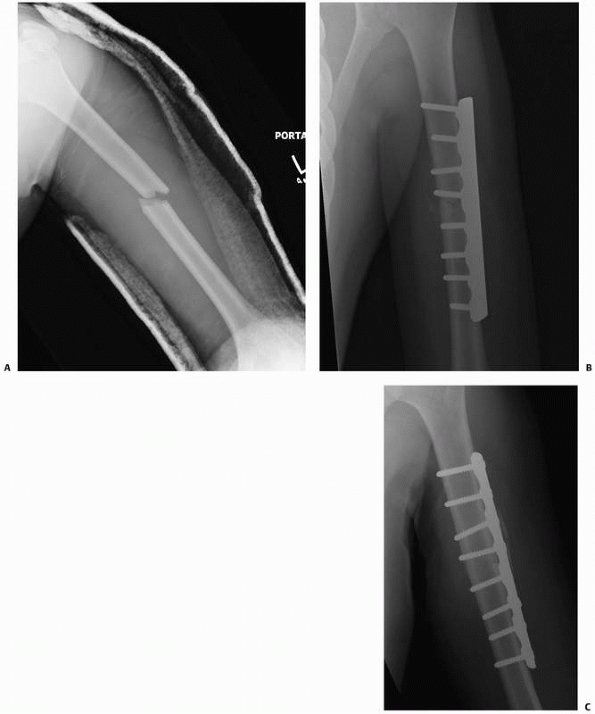 |
|
FIGURE 4-6 A humeral shaft fracture (A) was treated with rigid fixation using a 4.5-mm compression plate and interfragmentary screw (B). A radiograph 4 months later indicates healing without visible fracture callus. This represents primary bone healing (C).
|
disruption, fracture displacement, and, in some instances, significant
bone loss. Extensive tearing or crushing of the soft tissue disrupts
the blood supply to the fracture site, leaving substantial volumes of
necrotic bone and soft tissue, impeding or preventing formation of a
fracture hematoma and delaying formation of repair tissue (Fig. 4-11).
Exposed bone and soft tissue become desiccated, increasing the volume
of necrotic tissue and the risk of infection. Early use of vascularized
soft tissue flaps to cover
bone exposed by severe open fractures can prevent desiccation and facilitate healing of these injuries.72
In addition to the problems created by the soft tissue damage, open
fractures may become infected. Management of this complication usually
requires debriding infected bone and soft tissue along with providing
appropriate antibiotic treatment. Although infection compromises bone
healing, infected fractures can unite if they are stabilized and the
infection is suppressed. This may leave the patient with chronic
osteomyelitis, but in most instances bone union with a chronic
infection is a better result than an infected nonunion.
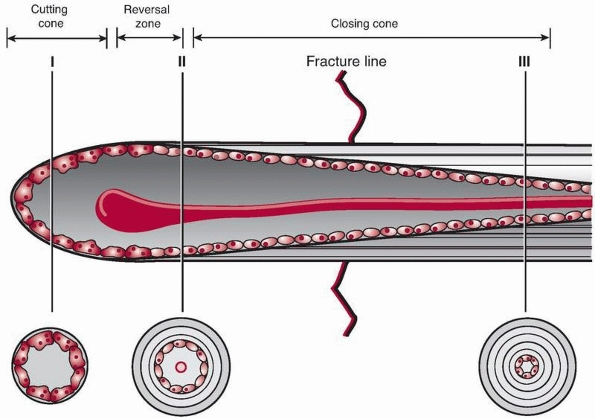 |
|
FIGURE 4-7 Primary bone healing utilizes an osteoclastic cutting cone crossing the fracture gap (I) followed by bone reconstitution by the trailing osteoblasts (II, III).
|
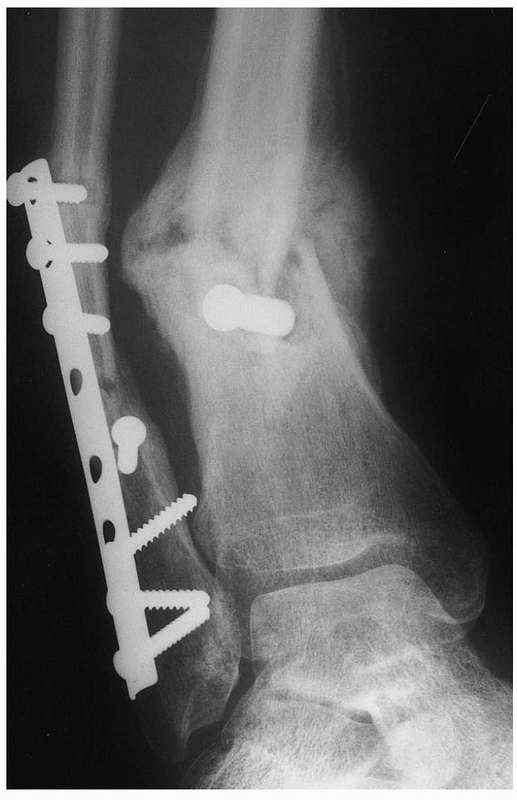 |
|
FIGURE 4-8
Hypertrophic delayed union of a distal tibial fracture 5 months after injury. Note the abundant callus but incomplete bridging of the fracture gap. |
 |
|
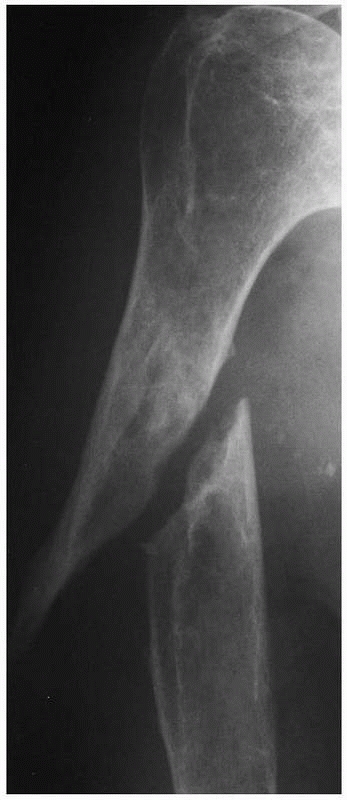
FIGURE 4-9 Atrophic nonunion of a humeral shaft fracture 18 months after fracture. Note the absence of callus.
|
 |
|
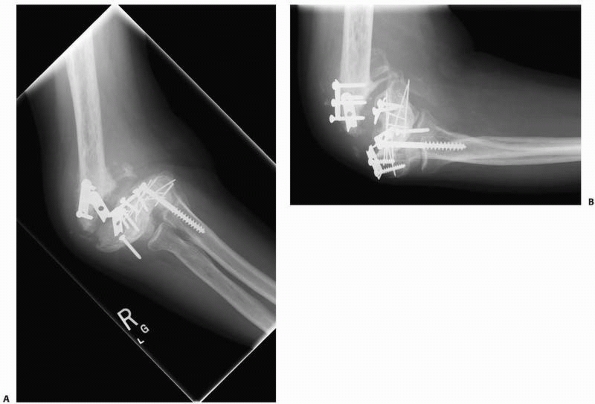
FIGURE 4-10 A,B.
A synovial pseudoarthrosis of the distal humerus is demonstrated in these radiographs taken after failed internal fixation. The nonunion is grossly mobile and the elbow joint is stiff. |
extensive local tissue necrosis. A severe fracture, open or closed, may
be associated with extensive soft tissue loss, displacement and
comminution of the bone fragments, loss of bone, and decreased blood
supply to the fracture site. Comminution of bone fragments generally
indicates that there is also extensive soft tissue injury. However,
some patients with osteopenic bone may sustain comminuted fractures
from low-energy injuries that have minimal soft tissue injury.
Displacement of the fracture fragments and severe trauma to the soft
tissues retard fracture healing, probably because the extensive tissue
damage increases the volume of necrotic tissue, impedes the migration
of mesenchymal cells, compromises vascular invasion, decreases the
amount of viable mesenchymal cells, and disrupts the local blood
supply. Less severe injuries leave an intact soft tissue envelope that
contains the fracture hematoma, provides a ready source of mesenchymal
cells, acts as a soft tissue tube to direct the repair efforts of these
cells, and serves as an internal splint that contributes to
immobilization of the fragments.
 |
|
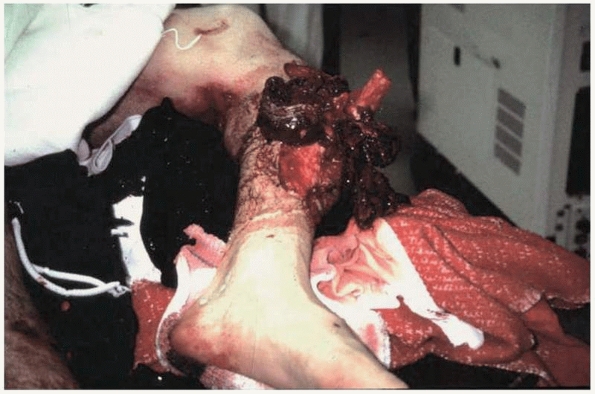
FIGURE 4-11
This severe open fracture of the tibia will certainly have delayed healing because of disrupted blood supply and the extensive amount of necrotic bone and soft tissue. |
joint surfaces and because joint motion or loading may cause movement
of the fracture fragments, intra-articular fractures can present
challenging treatment problems. Most intra-articular fractures heal,
but if the alignment and congruity of the joint surface is not
restored, the joint surface will be incongruous and the joint may be
unstable. In some instances, especially if the fracture is not rigidly
stabilized, healing may be delayed or nonunion may occur. However,
prolonged immobilization of a joint with an intra-articular fracture
frequently causes joint stiffness. For these reasons, surgeons usually
attempt to reduce and securely fix unstable intra-articular fractures.
This approach ideally restores joint alignment and congruity and allows
for at least some joint motion while the fracture heals. Unfortunately,
restoring joint alignment, congruity, and stability in patients with
severe intra-articular fractures may require extensive surgical
exposure that further compromises the blood supply to the fracture
site. Even after reduction and adequate initial stabilization,
intra-articular fractures may displace as a result of high
transarticular forces, failure of the stabilization, or collapse of the
subchondral cancellous bone. This late loss of reduction occurs most
frequently
after
comminuted fractures of the proximal and distal tibia and distal
radius. The creation of either a gap or a step-off exceeding 2
millimeters will lead to secondary osteoarthritis.60
implies that a large amount of energy was absorbed by this type of
injury and the two-level fracture pattern impairs or disrupts the
intramedullary blood supply to the middle fragment. If there is severe
soft tissue trauma, the periosteal blood supply to the middle fragment
may also be compromised. Possibly because of this, the probability of
delayed union or nonunion, proximally or distally, may be increased.
These problems occur most frequently in segmental fractures of the
tibia, especially at the distal fracture site.80,94
In contrast, segmental fractures of the femur less commonly develop
nonunions, presumably because of the better soft tissue coverage and
resulting better blood supply. When internal fixation of a segmental
fracture is performed, the soft tissue attachments of the middle
fragment should be preserved whenever possible.
including muscle, fascia, tendon, and occasionally nerves and vessels
between fracture fragments will compromise fracture healing. Soft
tissue interposition should be suspected when the bone fragments cannot
be brought into apposition or alignment during attempted closed
reduction. If this occurs, an open reduction may be needed to extricate
the interposed tissue and achieve an acceptable position of the
fracture fragments.
supply can significantly delay or prevent fracture healing in part
related to a deficiency of stem cells. An insufficient blood supply for
fracture healing may result from a severe soft tissue and bone injury
or from the normally limited blood supply to some bones or bone
regions. For example, the vulnerable blood supplies of the femoral
head, proximal scaphoid, and talar body may predispose these bones to
delayed union or nonunion, even in the absence of severe soft tissue
damage or fracture displacement. Extensive surgical dissection may also
compromise the vascular supply to a fracture site, especially in
regions of the skeleton with a vulnerable blood supply, or in fractures
with associated severe soft tissue injuries, or in regions with minimal
surrounding soft tissue, for example, the distal tibia.
healing both at the site of fracture and for the patient systemically.
Patients with Parkinson’s disease have a longer hospital stay and a
higher rate of referral to a nursing facility. Yet the rates of
complications, recovery of ambulation, and 1-year mortality are
comparable to those of non-Parkinson patient.42
Diabetes in animals and man results in impaired fracture healing in
part associated with increased rates of cartilage resorption and
diminished callus size.46 Fracture
healing is also impaired in HIV-positive populations. Apart from
directly impeding cellular function in bone remodeling, HIV infection
is reported to cause derangement in the levels of cytokines involved in
fracture repair.79
fracture healing. Infants have the most rapid rate of fracture healing.
The rate of healing declines with increasing age, and the ability to
achieve secure mechanical fixation of the fracture fragments decreases
with the development of osteoporosis. Multiple factors contribute to
age-related changes in fracture healing, including decreased number and
function of stem cells, decreased chondrogenic potential of the
periosteum, changes in the local signaling milieu at the fracture site,
and impaired vascularization.51
Biologic augmentation may partially remedy this phenomenon. The ability
to achieve stable fixation, however, declines in osteoporotic patients
resulting in bone fragment drift and malalignment.41,85,86
One possible reason for the greater healing potential of children may
be an increased availability of cells that produce repair tissue:
younger cells may differentiate more rapidly from the mesenchymal pool
and the pool of undifferentiated mesenchymal cells may be larger in
children.
synthesis necessary to heal a fracture requires substantial energy.
Furthermore, to synthesize large volumes of collagens, proteoglycans,
and other matrix macromolecules, the cells need a steady supply of the
components of these molecules: proteins and carbohydrates. As a result,
the metabolic state of the patient can alter the outcome of injury, and
in severely malnourished patients injuries that usually heal rapidly
may fail to heal. Although few surgeons in economically developed
countries see many severely malnourished patients, they may see
relatively large numbers of patients with milder forms of
protein-calorie malnutrition and other dietary deficiencies. Jensen and
associates44 found a 42.4% incidence
of clinical or subclinical malnutrition in patients undergoing
orthopaedic surgical procedures. A study of 490 patients with hip
fractures found that 87 of these patients (18%) suffered from
malnutrition and that the malnourished patients stayed in the hospital
longer, were less likely to recover their prefracture level of
activity, and were more likely to die within 1 year of their hip
fracture.47 Low serum albumin
levels, low iron-binding capacity, and low lymphocyte counts markedly
increase perioperative complications in fragility fracture patients.54
Both vitamin D insufficiency (<32 ng/mL) and deficiency (<20
ng/mL) occur in more than 60% of patients with low-energy fractures.37
reported that the adenosine triphosphate (ATP) content of a 2-week
rabbit fracture callus was a thousand times greater than the ATP
content of normal bone. Others have suggested that a single long-bone
fracture can temporarily increase metabolic requirements 20% to 25%,
and that multiple injuries and infection can increase metabolic
requirements by more than 50%.21,44
Failure to meet these increased nutritional demands may be associated
with increased mortality; more frequent surgical complications such as
infection, wound dehiscence, and impaired healing; and slower
rehabilitation. An experimental study of fracture healing demonstrated
that fracture callus does not achieve normal strength in states of
dietary deficiency, and that a dietary deficiency of protein reduces
fracture callus strength and energy storage capacity.33 For these reasons, the optimal treatment of injured patients
requires an assessment of their nutritional status and appropriate treatment, which may include nutritional support.
necessary for repair. Prolonged corticosteroid administration may also
decrease bone density and compromise the surgeon’s ability to achieve
stable internal fixation, leading to nonunion.1
The role of growth hormone in fracture healing remains uncertain. Some
experimental work suggests that growth hormone deficiency adversely
affects fracture healing and that growth hormone replacement can
improve healing.4,69 Other investigations indicate that excess amounts of growth hormone may have little or no effect22,70
and that normal alterations in the level of circulating growth hormone
have little effect on fracture healing. Thyroid hormone, calcitonin,
insulin, and anabolic steroids have been reported in experimental
situations to enhance the rate of fracture healing.12
Diabetes, hypovitaminosis D, and rickets have been shown to retard
fracture healing in experimental situations. However, clinical
experience shows that fractures heal in patients with hormonal
disturbances, although union may be slower than normal.
various other agents may adversely affect fracture healing. Clinical
experience suggests that cigarette smoking inhibits fracture healing,
and a study of tibial osteotomy healing in rabbits showed that animals
exposed to nicotine healed fractures more slowly and had a higher
percentage of nonunions.76 The
mechanism of the nicotine effect on bone healing remains unknown, but a
study of bone graft incorporation in rabbits showed that nicotine
inhibited vascularization of autogenous cancellous bone grafts.77 Agents used to treat malignancies may also inhibit bone healing.66
fractures in cancellous and cortical bone differs, probably because of
the differences in surface area, cellularity, and vascularity.
Wellapposed and impacted cancellous bone surfaces usually unite
rapidly, probably because the large surface area of cancellous bone
creates many points of bone contact rich in cells and blood supply and
because osteoblasts can form new bone directly on existing trabeculae.
Because woven bone forms across points of cancellous bone contact,
stable fractures located primarily in cancellous regions, especially
impacted fractures in which the trabeculae of the fracture fragments
have been forced together so that they interdigitate, form little or no
visible external callus, and rarely fail to heal. Where fractured
cancellous bone surfaces are not impacted, new bone spreads from the
points of contact to fill gaps. When a gap is excessively large, two
bone-forming fronts grow from the fracture fragments and eventually
meet, but if excessive motion occurs, external callus (including
cartilage) may develop. In contrast, cortical bone has a much smaller
surface area per unit volume and generally a less extensive internal
blood supply, and regions of necrotic cortical bone must be removed
before new bone can form.
sides of a fracture, but if one fracture fragment has lost its blood
supply, healing depends entirely on in-growth of capillaries from the
living side or the surrounding soft tissues. If one fracture fragment
is avascular the fracture can heal, but the rate is slower and the
incidence of healing is lower than if both fragments have a normal
blood supply.54 If both fragments are avascular, the chances for union decrease further (Fig. 4-12).
Traumatic or surgical disruption of blood vessels, infection, prolonged
use of corticosteroids, and radiation treatment can cause bone
necrosis. Irradiated bone, even when it is not obviously necrotic,
often heals at a slower rate than normal bone.75,91 Nonunion occurs in radiated bones44
probably because of radiation-induced cell death, thrombosis of
vessels, and fibrosis of the marrow. These changes may reduce the
population of cells that can participate in repair, increase the volume
of necrotic tissue, and interfere with the in-growth of capillaries and
the migration of fibroblasts into the fracture site.
diseased bone and therefore require less force than that necessary to
break normal bone. Commonly recognized causes of pathologic fractures
include osteoporosis, osteomalacia, primary malignant bone tumors,
metastatic bone tumors, benign bone tumors, bone cysts, osteogenesis
imperfecta, fibrous dysplasia, Paget disease, hyperparathyroidism, and
infections. Fractures through bone involved with primary or secondary
malignancies usually will not heal if the neoplasm is not treated.
Subperiosteal new bone and fracture callus may form, but the mass of
malignant cells impairs or prevents fracture healing, particularly if
the malignant cells continue to destroy bone. Fractures through
infected bone present a similar problem. Thus, the healing of a
fracture through a malignancy or infection usually requires treatment
of the underlying local disease or removal of the involved portion of
the bone. Depending on the extent of bone involvement and the
aggressiveness of the lesion, fractures through bones with nonmalignant
conditions like simple bone cysts and Paget disease will heal. The most
prevalent bone disease, osteoporosis, may impair fracture healing
beyond the simple consequences of aging.32,67,85,88
When there is diminished surface contact of apposing cortical or
cancellous bone surfaces due to decreased bone mass, the time required
to restore normal
bone
mechanical strength may be increased. Furthermore, decreased bone mass
reduces the strength and stability of the interface between the bone
and compromises internal fixation. This may lead to failure of internal
fixation and subsequent delayed healing or nonunion or malunion.85
 |
|
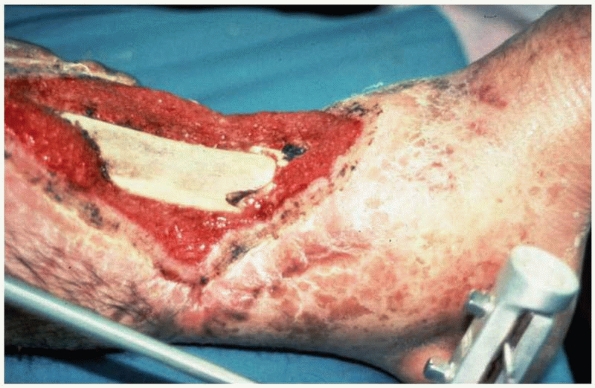
FIGURE 4-12
This photograph illustrates bone necrosis after a severe open tibia fracture with failure of soft tissue coverage. Fracture healing will not occur without aggressive intervention. |
For fracture healing to proceed at the maximum rate, the local cells
must be devoted primarily to healing the fracture. If infection occurs
following a fracture or if the fracture occurs as a result of the
infection, many cells must be diverted to wall off and eliminate the
infection and energy consumption increases. Furthermore, infection may
cause necrosis of normal tissue and thrombosis of blood vessels,
thereby retarding or preventing healing. Surgical debridement of
infected fractures may cause further tissue damage.
fracture gap decreases the volume of repair tissue needed to heal a
fracture. Restoring fracture fragment apposition is especially
important if the surrounding soft tissues have been disrupted or when
soft tissues are interposed between the fracture fragments. When a
significant portion of the periosteum and other soft tissue components
remains intact or can be rapidly restored, lack of bone fragment
apposition may not impair healing.
fracture healing include loading of the repair tissue. Based on the
available evidence it appears that loading a fracture site stimulates
bone formation while decreased loading slows fracture healing.15
In addition, experimental work and clinical experience have shown that
early or even almost-immediate controlled loading and limb movement,
including induced micromotion at long bone fracture sites, may promote
fracture healing.15 However, the
optimal timing, intensity, and pattern of loading for specific
fractures have not been defined and these factors probably vary not
only among fractures, but among patients.
 |
|
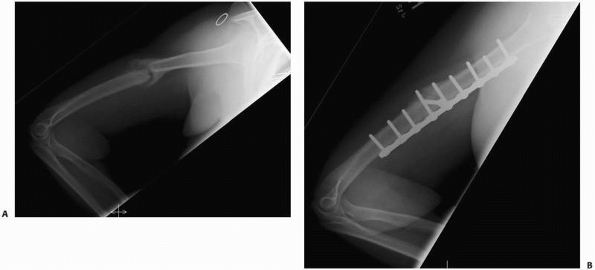
FIGURE 4-13 In this humeral shaft fracture, excessive motion led to a hypertrophic nonunion (A). Elimination of motion with a compression plate led to healing (B).
|
traction, cast immobilization, external fixation, or internal fixation
can facilitate fracture healing by preventing repeated disruption of
repair tissue. Some fractures (e.g., displaced femoral neck and
scaphoid fractures) rarely heal if they are not rigidly stabilized.
Fracture stability appears to be particularly important for healing
when there is extensive associated soft tissue injury, when the blood
supply to the fracture site is marginal, and when the fracture occurs
within a synovial joint. Excessive motion secondary to ineffective
stabilization, repeated manipulation, or excessive loading and motion
retards fracture healing and may cause nonunion (Fig. 4-13).
In these injuries it is probable that the repeated excessive motion
disrupts the initial fracture hematoma or granulation tissue, delaying
or preventing formation of fracture callus. A hypertrophied callus
response suggests impaired fracture fixation. If excessive motion
continues, a cleft forms between the fracture ends, and a
pseudarthrosis develops.
fractures, motion does not impair healing of all fractures. During the
early part of repair, motion occurs at most fractures except for those
treated by rigid internal fixation. Fractures with intact surrounding
soft tissues that provide some stability in a wellvascularized region
of bone may heal rapidly even though palpable motion of the fracture
site persists for weeks after injury. For example, closed rib,
clavicle, metacarpal, and metatarsal fractures heal even though the
fracture fragments remain mobile until the fracture callus stabilizes
them.
external fixation and internal fixation of the fractures with metallic
plates can rigidly stabilize a fracture (<3% strain). Although rigid
stabilization of a fracture makes possible primary bone repair without
cartilage or connective tissue intermediates, it does not accelerate
fracture healing. Rigid fixation of fractures makes it possible to
restore and maintain anatomic apposition of the fracture fragments.
This approach has proven especially beneficial in the treatment of
intra-articular fractures, diaphyseal fractures of the radius and ulna,
and other selected diaphyseal and metaphyseal fractures.
 |
|
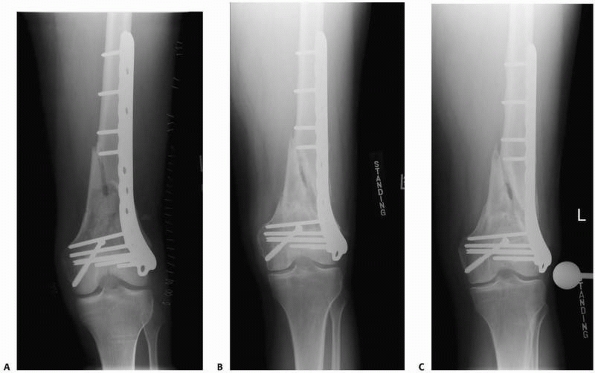
FIGURE 4-14 This distal femur fracture (A) was treated with a laterally based stainless steel locking plate. Radiographs 1 year (B) and 2 years (C)
after surgery demonstrate absence of medial callus and a delayed union or nonunion. Using a very stiff implant may have contributed to this complication. |
metallic implants has multiple advantages, it also has potential
disadvantages (Fig. 4-14). Rigid fixation can
alter fracture remodeling and decrease bone density because the
stiffness of most implants differs from that of bone. For example,
steel is more than 10 times as stiff as bone. When a fractured bone,
rigidly fixed with a stiff implant, is loaded, the bone is shielded
from normal stresses by the more rigid implant. Regional loss of bone
mass may occur, which increases the probability of refracture following
removal of the plate,39 although refractures following removal of plates may also be due the presence of screw holes that act as stress risers.
methods. Furthermore, the healing potential of many fractures,
especially those in children, can overcome less than optimal treatment,
but some surgical and nonsurgical interventions interfere with healing
and may cause delayed union or nonunion. Surgical exposure of a
fracture site can interfere with healing. The fracture hematoma is
disrupted, the blood supply to the fracture site and the surrounding
soft tissues may be damaged, and opening a closed fracture may lead to
infection. Inadequate immobilization of some fractures (for example,
scaphoid and femoral neck fractures), distraction of fracture fragments
by internal or external fixation devices or traction, and repeated
manipulations or excessive early motion of a fracture all may interfere
with healing.
fragility fractures. In the setting of an acute fracture these agents
can also affect the repair process. Osteoporotic therapy initially
consists of correcting vitamin D and calcium deficiency. Approximately
60% to 70% of fracture patients have either 25—(OH) vitamin D
insufficiency (<32mg/mL) or deficiency (<20ng/mL).34
Calcium (800 mg/day) and vitamin D (800 units/day) can decrease
fracture risk by 25%. Forty-five percent of women and 66% of men with
osteoporosis have secondary causes that require identification and
treatment. Osteoporosis therapy is either antiresorptive (estrogen,
calcitonin, selective estrogen receptor modulators, or bisphosphonates)
or anabolic (parathyroid hormone [PTH]).29,57 In the setting of an active fracture, PTH has been demonstrated in multiple animal studies to enhance the repair process.5
Both bisphosphonates and PTH increase the callus size. Bisphosphonates
delay cellular maturation while PTH speeds up callus maturation,
especially the chondroid stages. Hence, in fresh fractures and
osteoporosis, PTH not only treats the osteoporosis but can also enhance
the repair. In the steady nonfracture state, both the bisphosphonates
and PTH decrease the risk for subsequent fractures. However, after
prolonged bisphosphonate use (approximately 2 years), particularly if
the patient has not received adequate calcium and vitamin D
supplementation, the bone turnover becomes profoundly depressed and
subtrochanteric and femoral shaft stress fractures may occur in
diaphyseal bone.33,63,68
These fractures are best treated by correcting the calcium and vitamin
D deficiency, halting the bisphosphonate treatment, and initiating
anabolic PTH therapy.
cannot respond to injury with inflammation. However, injuries that
disrupt subchondral bone as well as the overlying cartilage initiate
the fracture healing process in the subcondral bone, and the repair
tissue from bone will fill an articular cartilage defect. Cartilage
healing then follows the sequence of inflammation, repair, and
remodeling like that seen in bone or dense fibrous tissue.9,12,19,20
Even after remodeling, the articular surface usually heals with
fibrocartilage that has weaker mechanical properties than the original
articular cartilage.
cartilage can sustain damage following disruption of the synovial
membrane exposing it to the outside environment. Because of these
special features, acute traumatic injuries to synovial joints can be
separated into the following two categories: disruption of the soft
tissues of the synovial joint without direct mechanical cartilage
injury, and mechanical injury of the articular cartilage.
surgical disruption of the joint capsule and synovial membrane, or by
blood in a hemarthrosis, can alter cartilage matrix composition by
stimulating degradation or suppressing synthesis of proteoglycans.18,91
A decrease in matrix proteoglycan concentration decreases cartilage
stiffness and may make the tissue more vulnerable to damage from impact
loading. Prompt restoration of the synovial environment by closure of
the synovial membrane will allow chondrocytes to repair the damage to
the macromolecular framework of the matrix, and the tissue may regain
its normal composition and function. However, prolonged exposure of the
articular surface to air can desiccate the tissue and kill chondrocytes.65
irreversible damage. The available evidence, based on animal
experiments, suggests that damage to the matrix macromolecular
framework may occur with any disruption of the synovial membrane,19
but clinical experience suggests that permanent or progressive damage
in human joints rarely occurs following temporary disruption of the
synovial cavity. Furthermore, articular cartilage can be restored to
its normal condition if the loss of matrix proteoglycans does not
exceed the amount the cells can replenish, if a sufficient number
chondrocytes remain viable, and if the collagenous meshwork of the
matrix remains intact.84
by decreasing the period of time that the cartilage is unprotected by
synovium or other soft tissues. If the cartilage must remain
unprotected, keeping the surface moist with a physiologic solution may
be helpful. Because cartilage that has sustained exposure injury may be
temporarily more vulnerable to mechanical injury, it seems advisable to
minimize immediate impact loading of cartilage that has experienced
this type of injury.
through several mechanisms. Osteochondral fractures mechanically
disrupt cartilage and bone tissue at the fracture site, but on the
other hand, the injury may be limited to the cartilage with abrasion of
the articular surface or the creation of chondral fractures.18,19,20
Alternatively, blunt trauma to a synovial joint may occur without an
associated bone or cartilage fracture. Therefore, acute articular
cartilage injuries can be separated into those caused by blunt trauma
that does not disrupt or fracture tissue and those caused by blunt
trauma or other mechanisms that mechanically disrupt or fracture the
tissue. Injuries that fracture or disrupt cartilage can be further
divided into those limited to articular cartilage and those affecting
both cartilage and subchondral bone.
cartilage have not been extensively studied clinically or
experimentally, blunt trauma to joints occurs frequently as an isolated
injury or in association with a fracture or dislocation. Among the
reasons for the limited amount of studies are the lack of clearly
defined clinically significant consequences of blunt trauma to
cartilage, the ability of cartilage to withstand large acute loads
without apparent immediate damage, the frequent lack of a clinically
detectable injury and repair response in cartilage following blunt
trauma, and difficulty in defining the relationship between the
intensity of blunt trauma and the extent of cartilage injury.18
Despite these limitations, current information suggests that acute
blunt trauma to articular cartilage may damage it even when there is no
grossly apparent tissue disruption, and these injuries may lead to
later degeneration of the articular surface.
demonstrated to produce cartilage injury, and clinical experience
suggests that acute impact loading considerably greater than
physiologic loading but less than that necessary to produce detectable
fractures rarely causes significant articular cartilage injury.
However, acute impact loading less than that necessary to produce
visible tissue disruption may cause chondrocyte necrosis, apoptosis,
release of matrix metalloproteinases, cartilage swelling, and altered
relationships between collagen fibrils and proteoglycans.17,92,93,94
This observation suggests that blunt trauma, under at least some
conditions, may disrupt the macromolecular framework of the cartilage
matrix and possibly injure cells without producing a detectable
fracture of the cartilage or bone. Presumably this tissue damage would
make cartilage more vulnerable to subsequent injury and progressive
deterioration if the cells could not rapidly restore the matrix. This
type of injury may help explain the development of articular cartilage
degeneration following joint dislocations or other types of acute joint
trauma that do not cause visible damage to the articular surface. Blunt
trauma may cause a bone bruise as evidenced by marrow edema on MRI (T2,
S71R sequence). This often occurs in conjunction with a cartilage
injury.49
traumatically induced splits of articular cartilage perpendicular to
the surface, or chondral fractures kill chondrocytes at the site of the
injury and disrupt the matrix. Viable chondrocytes near the injury may
proliferate, form clusters of new cells, and synthesize new matrix.15
They do not migrate to the site of the injury, and the matrix they
synthesize does not fill the defect. A hematoma does not form, and
inflammatory cells and fibroblasts do not migrate to the site of
injury. This minimal response may be due to the inability of
chondrocytes to respond effectively to injury,
the
inability of undifferentiated mesenchymal cells to invade the tissue
defect, and the lack of a clot that attracts cells and gives them a
temporary matrix to adhere to and replace with more permanent tissue.
Although the very limited response of chondrocytes to injury will not
heal a clinically significant cartilage defect, most traumatic defects
limited to small areas of articular cartilage do not progress.
surface tangential or parallel to the surface presumably follow a
similar course. Cells directly adjacent to the injury site may die and
others may show signs of increased proliferative or synthetic activity.
A thin acellular layer of nonfibrillar material may form over an
injured surface, but there is no evidence that the cell activity
stimulated by the injury restores the articular cartilage to its
original state.
also damages subchondral bone stimulates a fracture healing response in
the subchondral bone that includes inflammation, repair, and remodeling.10,17,55
Blood from ruptured bone blood vessels fills the injury site with a
hematoma that extends from the bony injury into the chondral defect.
The clot may fill a small chondral defect, generally one less than
several millimeters wide, but it usually does not completely fill
larger defects. Inflammatory cells migrate through the clot followed by
fibroblasts that begin to synthesize a collagenous matrix. In the
combined bone and chondral defect some of the mesenchymal cells assume
a rounded shape and begin to synthesize a matrix that closely resembles
the matrix of articular cartilage.
 |
|
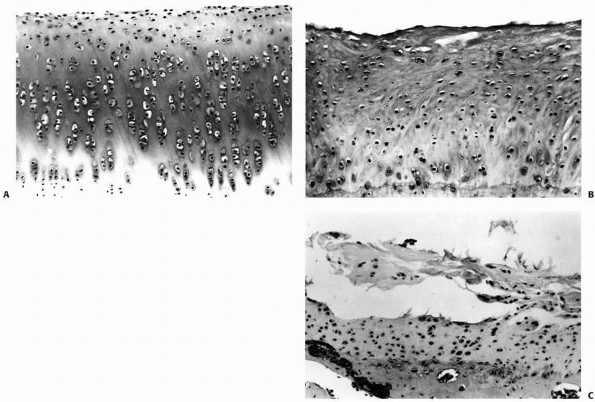
FIGURE 4-15 A.
Normal rabbit articular cartilage showing the homogenous extracellular matrix. The chondrocytes near the articular surface are relatively small and flattened, in which those in the middle and deeper zones of the articular cartilage have a more spherical shape. B. Wellformed fibrocartilaginous repair cartilage. Notice that the extracellular matrix is more fibrillar and the chondrocytes do not show the same organization as normal articular cartilage. Nonetheless, this repair cartilage does fill the defect in the articular surface. In most instances after osteochondral injury, this type of tissue forms within 6 to 8 weeks. C. Photomicrograph showing fibrillation and fragmentation of fibrocartilaginous repair tissue. Because fibrocartilaginous repair tissue lacks the mechanical properties of normal articular cartilage, it often degenerates over time. (Reprinted from Buckwalter JA, Mow VC. Cartilage repair and osteoarthritis. In: Moskowitz RW, Howell DS, Goldberg VM, et al eds. Osteoarthritis Diagnosis and Medical/Surgical Management. 2nd Ed. Philadelphia: WB Saunders, 1992:86-87, with permission.) |
chondral portion of the defect and the tissue forming in the bony
portion of the defect begin to differ. Tissue in the chondral defect
has a higher proportion of repair cells and matrix that resemble
hyaline cartilage, while the repair tissue in the bone defect has
started to form new bone. Within 6 weeks of injury repair tissue in the
two locations is distinguished by the new bone formed in the bone
defect, the absence of bone in the chondral defect, and the higher
proportion of hyaline cartilage repair tissue in the chondral defect.
usually follows a predictable course, subsequent changes in the
cartilage repair tissue vary considerably among similar defects. In
some chondral defects the production of a cartilaginous matrix
continues and the cells may retain the appearance and some of the
functions of chondrocytes, including the production of type II collagen
and proteoglycans. They rarely if ever restore the matrix to the
original state but they may succeed in producing a form of
fibrocartilaginous scar that maintains the integrity of the articular
surface and provides clinically satisfactory joint function for years.
Unfortunately, in many other injuries, particularly larger ones, the
cartilage repair tissue deteriorates rather than remodeling. It becomes
progressively more fibrillar, and the cells lose the appearance of
chondrocytes and appear to become more fibroblastic. The fibrous matrix
may begin to fibrillate and fragment, eventually leaving exposed bone (Fig. 4-15). The reasons why healing of some osteochondral injuries results in the formation of fibrocartilage that may provide at
least temporary joint function, while others fail to repair, have not been well defined.
degrees of disruption of joint congruity and stability can influence
joint healing (Fig. 4-16).9,19
Furthermore, clinical experience suggests that high-intensity joint
trauma may cause articular cartilage damage that is not detectable by
current imaging methods. Although it is generally accepted that more
severe joint injuries are more likely to lead to progressive joint
degeneration and osteoarthritis, the relationships between specific
injury variables including the intensity of force applied to the joint
surface, the degree of articular surface comminution and incongruity,
and the degree of joint instability and the risk of osteoarthritis have
not been defined.
That is, infants or young children have a greater potential to heal and
remodel chondral and osteochondral injuries than older individuals,
although this has not been thoroughly investigated. Other patient
variables such as weight, activity level, and systemic disease may be
clinically important, but their influence has not been demonstrated.
it seems reasonable to expect that treatments that decrease the volume
and surface area of a chondral defect, such as open reduction and
internal fixation of osteochondral fractures, will increase the
probability of successful cartilage repair. Experimental work indicates
that 1-mm or smaller defects tend to heal more successfully than larger
defects19,24
and eliminating the gap between articular fracture fragments results in
better anatomic restoration of an articular surface. Therefore,
decreasing the width of an osteochondral fracture gap should increase
the probability of a clinically successful result. However, depending
on the location of the chondral injury within the joint and the
presence or absence of other injuries to the joint, some persistent
separation of osteochondral fractures or loss of segments of the
articular surface may not produce clinically significant disturbances
of synovial joint function or rapid cartilage deterioration.55
 |
|
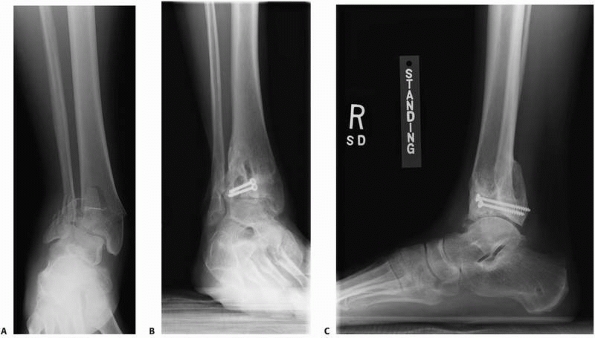
FIGURE 4-16 A severe tibial plafond fracture (A) has extensive cartilage injury and is at high risk to progress to posttraumatic arthritis despite surgical treatment (B,C).
|
fractures show that articular surfaces can sustain limited traumatic
loss of cartilage without immediate disturbance of joint function and
possibly without long-term consequences. However, the extent of
tolerable loss of the articular surface has not been defined and may
vary among joints.55
following osteochondral fractures can lead to significant adhesion
formation as well as deterioration of the uninjured cartilage,
resulting in poor synovial joint function. Early motion during the
repair and remodeling phases of healing decreases or prevents adhesions
and the immobilization-induced deterioration of uninjured cartilage.
However, loading and motion must be used carefully following injury,
because these measures alone
will
not predictably restore normal articular cartilage structure and
composition in clinically significant defects, and excessive loading
and motion may damage chondral repair tissue and displace fracture
fragments.
traumatically induced joint incongruity causes mechanical joint
dysfunction including instability, locking, catching, and restricted
range of motion, and may be associated with progressive deterioration
of the articular cartilage. It is not clear how much of the long-term
cartilage deterioration following injuries that cause joint incongruity
is secondary to the traumatic cartilage damage at the time of injury
and how much is related to the long-term effects of incongruity.
However, in most injuries restoration of acceptable joint congruity
avoids immediate problems with mechanical joint dysfunction and may
delay or decrease the severity and rate of cartilage deterioration.
be tolerated without causing long-term joint deterioration has not been
well defined. A study of contact stress aberrations following imprecise
reduction of experimental human cadaver tibial plateau fractures showed
that, in general, peak local cartilage pressure increased with
increasing joint incongruity (fracture fragment step-off), but the
results varied among specimens.8 In
most specimens, cartilage pressure did not increase significantly until
the fragment step-off exceeded 1.5 mm. When the step-off was increased
to 3 mm, the peak cartilage pressure averaged 75% greater than normal.
The authors estimated that the long-term “pressure tolerance level”’ of
cartilage may be much higher, probably about twice the normal level,
indicating that simple incongruities of several millimeters should not
cause immediate or long-term problems. However, they also found that in
some specimens even minor incongruities, as little as 0.25
mm, caused apparently deleterious peak local pressure elevations,
suggesting that results may vary even among individuals with the same
degree of articular incongruity. Experimental study of intra-articular
fractures stabilized with a step-off shows that the articular surface
can remodel, thereby decreasing the original incongruity.50
The long-term results of traumatically induced articular incongruity
may also depend on the age of the patient. Skeletally immature
individuals may have a greater capacity to remodel incongruities, and
age-related alterations in articular cartilage21 may decrease its capacity to repair injuries or withstand alterations in loading caused by joint incongruity.
osteochondral injury in an acceptable position increases the likelihood
of satisfactory healing by preventing disruption of the repair tissue
and restoring articular cartilage congruity. An equally important
benefit of stabilizing osteochondral fractures is that it allows early
controlled loading and motion. Continuous passive motion enhances soft
tissue healing, especially healing of the cartilage injury.71
and articular cartilage differ in their composition, structure, and
capacity for healing. Bone fractures initiate a response that begins
with inflammation (the cellular and vascular response to injury),
proceeds through repair (the replacement of damaged or lost cells and
matrices with new cells and matrices), and ends with remodeling
(removal, replacement, and reorganization of the repair tissue, usually
along the lines of mechanical stress). Injury to articular cartilage
does not trigger an inflammatory response, but the cells respond to
injury with an effort at cell proliferation and the synthesis of new
matrix. This effort rarely, if ever, restores a normal articular
surface. When injuries extend through articular cartilage into bone,
the repair tissue that forms in the bone extends into the region of the
chondral injury and produces a fibrocartilaginous tissue that in some
instances restores a functional articular surface.
include preventing further tissue damage, avoiding treatments that
compromise the natural healing process, and creating the optimal
mechanical and biological conditions for healing. This may include
removing necrotic tissue, preventing infection, rapidly restoring blood
supply when necessary, and in some circumstances providing apposition,
alignment, and stabilization of the injured tissues. One of the most
important recent advances in the treatment of bone and joint injuries
has been the recognition that early controlled loading and motion of
the repair and remodeling tissues improves healing of many injuries.
However, as with all treatments, this intervention must be used with
care, since uncontrolled or excessive loading and motion can adversely
affect or even prevent healing. At the tissue level, the effect of the
mechanical environment on repair and the function of the repair tissue
cells are not well understood, and at the clinical level the optimal
protocols for loading and motion of musculoskeletal tissue injuries
have not been well defined. Although future improvements in the
treatment of musculoskeletal tissue injuries, including controlled
motion and loading of repair and remodeling tissue, use of ultrasound
and electrical fields, and surgical restoration of apposition and
mechanical stability of injured tissue undoubtedly will advance the
practice of orthopedics, it is not likely that they will restore the
original state of the tissue for patients with the most severe
musculoskeletal tissue injuries. In particular, large segmental losses
of bone and many articular cartilage injuries will continue to present
especially difficult treatment problems. Future developments that may
help improve healing of these injuries include the creation and
implantation of synthetic matrices and the use of growth factors and
implanted mesenchymal cells to guide and promote regeneration of bone
and articular cartilage.
HC, Hodges PT, Agieilera XM, et al. Bone morphogenetic protein (BMP)
localization in developing human and rat growth plate methaphyses,
epiphysis, and articular cartilage. J Histochem Cytochem
2000;48:1493-1502.
HT, Wippermann BW, Hodgson SF, et al. Prediction of properties of
fracture callus by measurement of mineral density using microbone
densitometry. J Bone Joint Surg 1989;71A:1020-1030.
B, Jorgensen PH, Andreassen TT. The stimulating effect of growth
hormone on fracture healing is dependent on onset and duration of
administration. Clin Orthop 1991;264:295-301.
GL, Kakar S, Vora S, et al. Stimulation of fracture healing with
systemic intermitten parathyroid hormone treatment. JBJS Am
2008;90S:120-127.
GB, Einhorn TA. Current and future clinical applications of bone
morphogenetic proteins in orthopaedic trauma surgery. Int Orthop
2007;31:721-727.
CT, Hunt RM. Early histologic and ultrastructural changes in
microvessels of periosteal callus. J Orthop Trauma 1997;71:244-253.
TD, Anderson DD, Nepola JV, et al. Contact stress aberrations following
imprecise reduction of simple tibial plateau fractures. J Orthop Res
1988;6:851-862.
JA. Can tissue engineering help orthopaedic patients? Clinical needs
and criteria for success. In: Sandell LJ, Grodzinsky AJ, eds. Tissue
Engineering in Musculoskeletal Clinical Practice. Rosemont IL: American
Academy of Orthopaedic Surgeons, 2004:3-16.
JA, Einhorn TA, Bolander ME, et al. Healing of musculoskeletal tissues.
In Rockwood CA, Green D, eds. Fractures. Philadelphia: Lippincott,
1996:261-304.
JA, Glimcher MJ, Cooper RR, et al. Bone biology. Part I. Structure,
blood supply, cells, matrix, and mineralization. J Bone Joint Surg
1995;77A:1256-1275.
JA, Glimcher MM, Cooper RR, et al. Bone biology. Part II. Formation,
form, modeling, and remodeling. J Bone Joint Surg 1995;77A:1276-1289.
JA, Grodzinsky AJ. Loading of healing bone, fibrous tissue, and muscle:
implications for orthopedic practice. J Am Acad Orthop Surg
1999;7:291-299.
JA, Mankin HJ. Articular cartilage I. Tissue design and
chondrocyte-matrix interactions. J Bone Joint Surg 1997;79A:600-611.
JA, Martin JA, Olmstead M, et al. Osteochondral repair of primate knee
femoral and patellar articular surfaces: implications for preventing
posttraumatic osteoarthritis. Iowa Orthop J 2003;23:66-74.
JA. Mechanical injuries of articular cartilage. In: Finerman G, ed.
Biology and Biomechanics of the Traumatized Synovial Joint. Park Ridge
IL: American Academy of Orthopaedic Surgeons, 1992:83-96.
JA, Rosenberg LA, Hunziker EB. Articular cartilage: composition,
structure, response to injury, and methods of facilitation repair. In:
Ewing JW, ed. Articular Cartilage and Knee Joint Function: Basic
Science and Arthroscopy. New York: Raven Press, 1990:19-56.
JA, Rosenberg LC, R.Coutts, et al. Articular cartilage: injury and
repair. In Woo SL, Buckwalter JA, eds. Injury and Repair of the
Musculoskeletal Soft Tissues. Park Ridge, IL: American Academy of
Orthopaedic Surgeons, 1988:465-482.
JA, Woo SL-Y, Goldberg VM, et al. Soft tissue aging and musculoskeletal
function. J Bone Joint Surg 1993;75A:1533-1548.
JE, Hipp JA, Gerhart TN, et al. Failure of growth hormone to alter the
biomechanics of fracture-healing in a rabbit model. J Bone Joint Surg
1992;74A:359-367.
CC, Tolin BS, Brighton CT. The effect of oxygen tension on progeoglycan
synthesis and aggregation in mammalian growth plate chondrocytes. J
Orthop Res 1991;9:477-484.
FR, Akeson WH, Keown GH. The repair of large osteochondral defects: an
experimental study in horses. Clin Orthop 1972;82:253-262.
DT, Panepucci RA, Fortes AM, et al. Multipatent mesenchymal stromal
cells obtained from diverse human tissues share functional properties
and gene-expression profile with CD 146+ perivascular cells and fibroblasts. Exp. Mematol 2008;36:642-654.
DP. Further observations of the disturbance of metabolism caused by
injury, with particular reference to the dietary requirements of
fracture cases. Br J Surg 1936;23:505-520.
PA, Martins A, Minnich D, et al. Characterization of the systemic loss
of dendritic cells in murine lymph nodes during polymicrobial sepsis. J
Orthop Trauma 2003;17(9):635-641.
TA, Bonnarens F, Burstein AH. The contributions of dietary protein and
mineral to the healing of experimental fractures. A biomechanical
study. J Bone Joint Surg 1986;68A:1389-1395.
MJ, Demetrakopoulos D, Shindle MK, et al. Prevention and treatment of
osteoporotic fractures. Minerva Med 2005;96:343-352.
P, Tzioupis C, Almalki T, et al. Fracture healing in osteoporotic
fractures: is it really different? A basic science perspective. Injury
2007;38:590-599.
J, LeBoff MS, Kolatkar NS, et al. Importance of vitamin D in
hospital-based fracture care pathways. J Nutri Health Aging
2008;12:291-293.
SK, Yong K-Y, Koh JS, et al. Subtrochanteric insufficiency fractures in
patients on alendronate therapy: a caution. JBJS Br 2007;89:349-353.
O, Reikeras O. The role of the hematoma and periosteal sealing for
fracture healing in rats. Acat Orthop Scand 1993;64:47-49.
M, Tamai N, Tsumaki N, et al. Oxygen tension regulates chondrocyte
differentiation and function during endochondral ossification. J Biol
Chem 2006;281:31079-31092.
JO, Anikepe AO, MacKrell J, et al. Accelerated fracture healing in the
geriatric, osteoporotic rat with recombinant human platelet-derived
growth factor—BB and an injectable beta-tricalcium phosphate/collagen
matrix. J Orthop Res 2008;26;83-90.
NW, Roosendaal G, Bijlsma JW, et al. Exposure of human cartilage tissue
to low concentrations of blood for a short period of time leads to
prolonged cartilage damage: an in vitro study. Arthritis Rheum
2007;56(1):199-207.
MK, Nilsson BE, Obrant KJ. Bone mineral loss after lower extremity
trauma: 62 cases followed for 15-38 years. Acta Orthop Scand
1993;64:362-364.
RA, Tsatsas D, Bauer MA, et al. Diminished bone formation during
diabetic fracture healing is related to premature resorption of
cartilage associated with increased osteoclast activity. J Bone Miner
Res 2007;22:560-568.
KJ, Maurer SG, Su ET, et al. The effects of nutritional status on
outcome after hip fracture. J Ortho Trauma 1999;13:164-169.
X, MA BC, Bolbos RI, et al. Quantitation assessment of bone marrow
edema-like lesion and overlying cartilage in knees with osteoarthritis
and anterior cruciate ligament tear using MR imaging and spectroscopic
imaging at 3 Testa. J Magn Reson Imaging 2008;28:453-461.
A, McKellop HA, Marshall GJ, et al. Healing and remodeling of articular
incongruities in a rabbit fracture model. J Bone Joint Surg
1993;75:1508-1523.
P, Xu Y, Chiou M, et al. Effect of reduced oxygen tension on
chondrogenesis and osteogenesis in adipose-derived mesenchymal cells.
Am J Physiol Cell Physiol 2006;290:1139-1146.
H, Penington A, Nannmark U, et al. Microvascular invasion during
endochondral ossification in experimental fractures in rats. Bone
2004;35:535-542.
JL, Buckwalter JA, Evarts CM. Nonunion, delayed union, malunion, and
avascular necrosis. In: Epps CH, ed. Complications in Orthopaedic
Surgery. Philadelphia: Lippincott, 1994:183-211.
JL, Buckwalter J, Gelberman R, et al. Articular fractures: does an
anatomic reduction really change the result? J Bone Joint Surg Am
2002;84-A:1259-1271.
JA, Buckwalter JA. Telomere erosion and senescence in human articular
cartilage chondrocytes. J Gerontol A Biol Sci Med Sci 2001;56-B:172-179.
JA, Buckwalter JA. The role of chondrocyte senescence in the
pathogenesis of osteoarthritis and in limiting cartilage repair. J Bone
Joint Surg Am 2003;85-A(Suppl 2):106-110.
JA, Ellerbroek SM, Buckwalter JA. Age-related decline in chondrocyte
response to insulin-like growth factor-I: the role of growth factor
binding proteins. J Orthop Res 1997;15:491-498.
JA, Klingelhutz AJ, Moussavi-Harami F, et al. Effects of oxidative
damage and telomerase activity on human articular cartilage chondrocyte
senescence. J Gerontol A Biol Sci Med Sci 2004;59:324-337.
JM. Fracturing of the acetabulum: accuracy of reduction and clinical
results in patients managed operatively within 3 weeks after the
injuries. J Bone Jt Surg 1996;78A:1632-1645.
CG, Askanazi J. Current concepts review: the metabolic response to
injury: mechanism and clinical implantations. J Bone Joint Surg
1986;68A:782-787.
D, Rubel IF, Liew AS, et al. An in vivo rabbit model for cartilage
trauma: a preliminary study of the influence of impact stress magnitude
on chondrocyte death and matrix damage. J Orthop Trauma
2005;19(7):466-473.
JA, Gomez P, Stack J, et al. Effect of chemotherapy on segmental bone
healing enhanced by rhBMP-2. Iowa Orthop J 2004;24:36-42.
H, Appleyard R, Jansen J, et al. Osteoporosis influences the early
period of fracture healing in a rat osteoporotic model. Bone
2001;28:80-86.
As, Lane JM, Lenart BA, et al. Low-energy femoral shaft fractures
associated with alendronateuse. J Orthop Trauma 2008;22:346-350.
HM, Bak B, Jorgensen PH, et al. Growth hormone promotes healing of
tibial fractures in the rat. Acta Orthop Scand 1991;62:244-247.
MD, Wood MR, Meggitt BF. A biomechanical study of the effects of growth
hormone in experimental fracture healing. J Bone Joint Surg
1980;62B:391-396.
GE, Takara T, O’Neill JK, et al. Continuous passive motion applied to
whole joints stimulated chondrocyte biosynthesis of PRG4.
Osteoarthritis Cartilage 2007;15:566-574.
BM, Cornell CN, Carbone B, et al. Protein depletion and metabolic
stress in elderly patients who have a fracture of the hip. JBJS Am
1992;74:251-260.
SM, Landsman JC, Alexander VA, et al. Effect of nicotine on the rate
and strength of long bone fracture healing. Clin Ortho 1998;353:231-237.
GD, Boden SD, Whitesides TE, et al. The effect of nicotine on
incorporation of cancellous bone graft in an animal model. Spine
1995;20:2198-2202.
TM, Seldes R, Lunchetti W, Brighton CT. Similarities in the phenotype
expression of pericytes and bone cells. CORR 1998;346:95-103.
PM, Coosemans W, Broos PL. The difficult healing of segmental fractures
of the tibial shaft. Arch Orthop Traum Surg 1989;108:238-242.
JT, Sheller MR, Levine BP, et al. Thrombin peptide TP 508 stimulates
cellular events leading to angiogenesis, revascularization, and repair
of dermal and musculoskeletal tissues. JBJS Am 2006;88S:7:32-39.
DJ, Kunvin AS. Autoradiographic and biochemical investigations of the
effect of cortisone on the bones of the rat. Clin Orthop
1967;55:201-215.
KP, Callaghan JJ, Seaber AV, et al. The effects of exposure of
articular cartilage to air. A histochemical and ultrastructural
investigation. J Bone Joint Surg 1990;72A:1442-1450.
P, Sentuerk U, Riha T, et al. Influence of age and mechanical stability
on bone defect healing: age reverses mechanical effects. Bone
2008;42:758-764.
PA, Grigiene R, Borrelli J Jr, et al. Effect of impact load on
articular cartilage: cell metabolism and viability, and matrix water
content. J Biomech Eng 1999;121(5):433-441.
JW, Li W, Xu SW, et al. Osteoporosis influences the middle and late
periods of fracture healing in a rat osteoporotic model. Chin J
Traumatol 2005;8:111-116.
RF, Pelker RR, Friedlander GE, et al. Effects of prefracture
irradiation on the biomechanical parameters of fracture healing. J
Orthop Res 1993;11:422-428.
HE, Lips P, Nauta J, et al. Loss of bone in the proximal part of the
femur following unstable fractures of the leg. J Bone Joint Surg
1994;76A:230-236.