
|
|

Orthopaedic training programs often provide little education for the
orthopaedic resident in the area of skeletal dysplasia, except perhaps
teaching a few facts that may appear on intraining or
board-certification examinations. Many orthopaedists relegate this
subject to the arena of orthopaedic trivia. But, when a child or adult
with skeletal dysplasia shows up in your office, the orthopaedist’s
confusion as to what to look for or what to ask about often surfaces.
With the advent and growth of support groups and Internet sites for
unusual disorders, the skeletal dysplasia patient or parents often know
the latest facts about this disorder, putting the orthopaedist at a
further disadvantage. This chapter is designed to try to help a bit in
this regard—to suggest a way to approach the evaluation and treatment
of your patients with skeletal dysplasias and to help you appear
smarter than the last orthopaedist they saw.
skeletal dysplasias can be separated into those with short limbs and
those with short trunks. Most of these can be appropriately identified
at birth, but some metabolic disorders that mimic skeletal dysplasias
(e.g., Morquio syndrome) are usually not diagnosed until at least a
year and often not until a few years of age. If a child has a short
trunk, there is some type of spinal involvement, which can range from
the most common platyspondyly to a significant spinal deformity. If
short limbs predominate, try to ascertain if the shortening is proximal
(rhizomelia), mid-segment (mesomelia), or in the hands and feet
(acromelia), as this will help to separate one syndrome from another.
Other physical features that may be helpful in diagnosis determination
are the facial features and angular deformity of the lower extremities.
 |
|
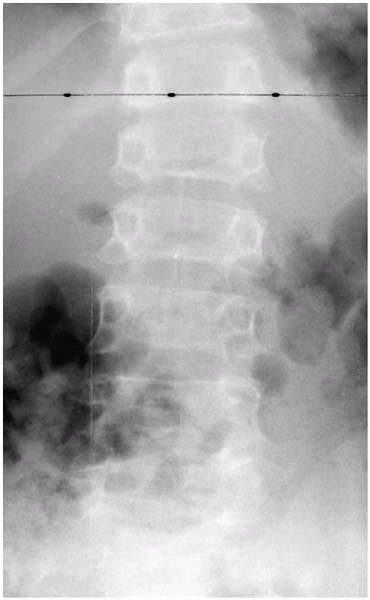
▪ FIGURE 18-1
AP spine radiograph demonstrates the finding of platyspondyly (height of vertebrae less than normal in relation to width) in a teenager with spondyloepiphyseal dysplasia congenita. |
established by anyone else, it is helpful to do a skeletal survey,
which includes radiographic evaluation of the spine and all
extremities. Long-bone radiographs will establish whether there is
epiphyseal or metaphyseal involvement, or both—which in turn will help
to narrow down the possible diagnoses to consider. If there is
platyspondyly, the term “spondylo” appears somewhere in the name of the
condition (Fig. 18-1). If there is primarily
epiphyseal involvement, early joint degeneration is likely to occur. If
there is primarily metaphyseal involvement, angular deformity with
growth is often seen.
may seem characteristic for a given skeletal dysplasia, it is useful to
obtain the opinion of a geneticist for confirmation of diagnosis and
counseling of the family with regard to risks of future children having
a similar condition. Once you and the geneticist agree on a diagnosis,
the next step is to evaluate the orthopaedic conditions associated with
that dysplasia and provide ongoing orthopaedic care as needed. Remember
that there is a paucity of literature on many of these dysplasias as
far as effective treatment is concerned, at least in part due to the
small numbers seen in most places. Keeping in mind what is known about
the natural history of orthopaedic problems in a particular skeletal
dysplasia condition, the best orthopaedic approach often used here is
to employ tested orthopaedic principles for joint problems, spine
deformity or instability, and limb realignment in the average-sized
person, and then apply them to the dysplastic skeleton. Examples would
be decompression for spinal stenosis, custom-made implants for total
joint replacement, and osteotomies to regain mechanical axis alignment.
There are specific pitfalls with each of these procedures that can
occur more often in many patients with skeletal dysplasia, but the
underlying orthopaedic principle stays pretty much constant.
dysplasias and is recognizable at birth. Rhizomelia is present in all
extremities and the facial appearance includes frontal bossing and
nasal bridge depression. Head size is large in proportion to body size
and, while hydrocephalus may be suspected, this is usually not present.
An anteroposterior spinal radiograph will show interpediculate
narrowing in the lumbar spine, a finding characteristic of
achondroplasia (Fig. 18-2). The genetic defect in fibroblast growth factor receptor 3 leads to the growth inhibition here.
 |
|
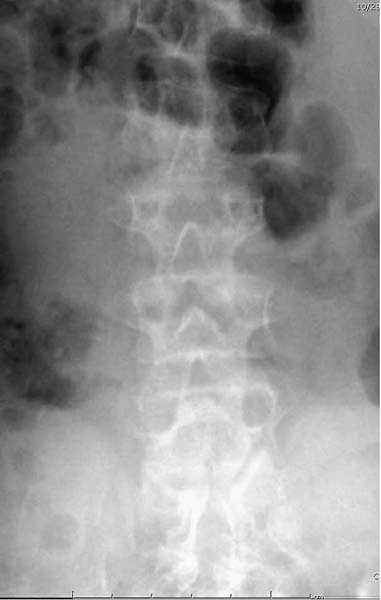
▪ FIGURE 18-2
AP spine radiograph of a child with achondroplasia demonstrates the interpediculate narrowing, most obvious in the lumbar spine. |
bowed legs and spinal stenosis. Neither are problematic at birth.
Initially, the main medical concern usually centers around sleep apnea,
which may be due to foramen magnum narrowing to compress the upper
cervical spinal cord (Fig. 18-3). Sleep apnea
monitors are often used to detect and follow this. If the sleep apnea
is severe enough, neurosurgical decompression of the foramen magnum is
considered. While published reports have noted improvement in
respiratory function after foraminal decompression, there also is a
substantial morbidity associated with this procedure in infancy. Over
the first few years of life, there is more of an enlargement of the
foramen magnum than of the spinal cord so that sleep apnea becomes less
of a problem by age 3 or so. Unless there is clear-cut and
life-threatening respiratory compromise from this foraminal stenosis,
it may be wiser to wait to see if mild sleep apnea resolves with time
without surgery.
 |
|
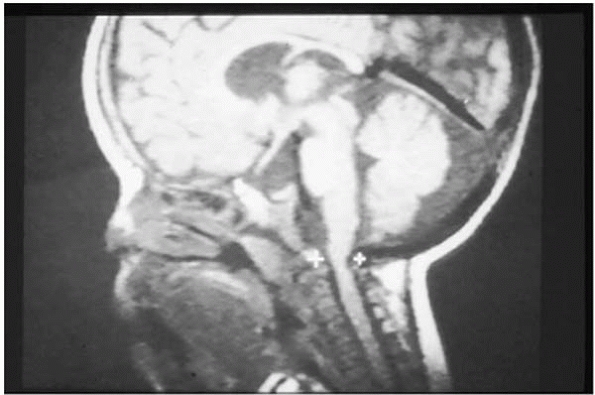
▪ FIGURE 18-3
Sagittal plane MRI of the upper cervical spine demonstrates narrowing of the spinal canal at the foramen magnum and C1, often seen in young children with achondroplasia. |
childhood, especially due to the midface hypoplasia present in
achondroplasia. Respiratory function can be compromised not only by the
foramen magnum stenosis, but also by a small (<third percentile)
thoracic cage and a narrowed pharyngeal area. Because of the midface
hypoplasia, drainage of the Eustachian tubes is impaired and frequent
otitis media occurs. If unrecognized or not treated aggressively
enough, these ear infections can result in significant permanent
hearing loss and delay in speech development.
progressive bowing of the legs. In achondroplasia, the fibula is longer
than the tibia and some degree of bowing is present in all with this
condition. It is unclear why some get worse and
some
do not, but it is expected that about 50% of the tibiae in
achondroplasia will at some point need corrective osteotomy. It is a
curious finding that older adults with achondroplasia and mild genu
varum do not seem to develop medial compartment degenerative arthritis
at the same rate as an average-sized person with mild genu varum will.
The primary reasons for surgical correction, which usually is done
between the ages of 4 and 12, are medial knee pain with walking or
running activity and the physical finding of a lateral thrust of the
knee in early-and mid-stance. Surgery may be needed either on one tibia
or on both. Gait analysis studies have shown excellent improvement and
normalization of gait parameters from this relatively simple proximal
tibial/fibular valgus osteotomy. The need to repeat the tibial
osteotomy at a later age is uncommon (Fig. 18-4).
 |
|
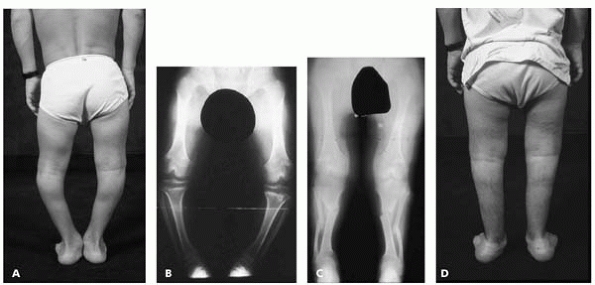
▪ FIGURE 18-4 (A) This 9-year-old boy with achondroplasia has genu varum frequently seen in this condition. (B) Standing AP radiograph of the lower extremities confirms genu varum due to fibulae being longer than the tibiae. (C)
Standing AP radiograph of the lower extremities following corrective tibial and fibular osteotomies demonstrates reestablishment of normal mechanical axis. (D) Postoperative clinical photograph confirms improvement in genu varum. |
the possibility and the wisdom of doing limb-lengthening surgery on
their child’s short bones. It is important to provide accurate
information so that the parents can make this decision with the older
child. Of all the skeletal dysplasia conditions, achondroplasia is the
only one in my mind for which limb lengthening should even be
considered. There is redundancy of soft tissue here, as seen in femoral
arteriography in which the femoral artery is S-shaped within the thigh,
so soft-tissue tightness is less of a problem during bone lengthening
than in other dysplasias. In Europe and Russia, there is great
enthusiasm for limb lengthening in achondroplasia, in which the femora
and tibiae are lengthened up to 35 cm. In these instances, the humera
also require lengthening so that the children can reach their feet.
Keep in mind that limb lengthening generally takes about 1 month per 1
centimeter of lengthening, so that the whole process usually takes
about 2 to 3 years of lengthening treatment. The Little People of
America, the primary support group for patients with
skeletal
dysplasia, have not generally supported limb lengthening in
achondroplasia, but have rather pushed for societal accommodations to
the short stature. The decision for or against limb lengthening is one
the individual family and patient has to make, but the orthopaedist
should be able to present the pros and cons objectively to help them
form this decision. It should be remembered that the quality of life of
achondroplastic individuals is the same as that of average-sized people
up to about age 40, at which time the quality of life in achondroplasia
deteriorates due to a higher incidence of back pain. It is not the
short stature that adversely affects quality of life.
infancy, there is nearly always a thoracolumbar kyphosis present, with
the kyphosis accentuated clinically when the child is sitting.
Independent walking generally does not take place until 18 to 24 months
of age in achondroplasia, but the standing position seems to lead to
resolution of this thoracolumbar kyphosis in about 90% of the children.
As lumbar lordosis increases with standing and walking, sagittal plane
balance occurs with improvement in the thoracolumbar kyphosis. If the
thoracolumbar kyphosis persists past age 6, surgical treatment is
needed to prevent progression and protect against the development of
earlier-onset spinal cord compression and resultant lower extremity
neurologic problems. Spinal fusion and instrumentation with pedicle
screw fixation, without any implants in the spinal canal, is needed and
correction is usually <50% due to a high risk of neurologic deficits
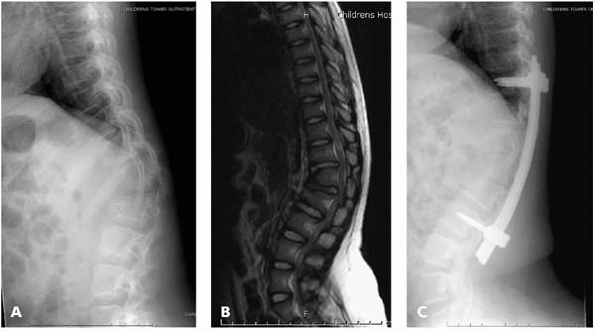
and evoked potential changes if greater correction is attempted (Fig. 18-5).
 |
|
▪ FIGURE 18-5 (A)
Lateral standing radiograph of the spine in an 8-year-old child with persistent thoracolumbar kyphosis associated with increased difficulty in walking. (B) Sagittal MRI of thoracolumbar spine confirms wedging of the apical vertebra and early spinal cord and cauda equina compression at this kyphosis. (C) Following anterior strut graft and posterior pedicle screw instrumentation and fusion, there is improvement in the thoracolumbar kyphosis that was associated with clinical improvement in walking and other activities. |
It is most severe in the lumbar spine. There is characteristic
narrowing of the interpediculate distances with less diminution in the
anteroposterior diameter of the spinal canal. Spinal claudication signs
and symptoms can occur at any age but are uncommon before the midteen
years. Spinal stenosis tends to become increasingly a problem as
degenerative disc changes occur, further narrowing the spinal canal and
compressing either the cauda equina or the spinal cord. Multiple levels
of compression may be present so it is very important to evaluate lower
extremity symptoms of spinal stenosis with a magnetic resonance image
(MRI) of both the thoracic and lumbar spines.
commonly T11 to S1, to relieve the lower extremity neurologic problems.
Using a high-speed burr to
transect
the lamina adjacent to the facet and then lifting the lamina dorsally
appears to be a safer technique of laminectomy than placing ronguers
initially within the tight spinal canal, but increased neurologic
deficit as a complication of laminectomy is not rare. Fusion is not
needed after multilevel laminectomy unless there is persistent
thoracolumbar kyphosis. If thoracolumbar kyphosis is present in the
area of laminectomy, this will worsen postlaminectomy. Since the
pedicles are of adequate size to accept screw placement, pedicle screw
instrumentation and posterior fusion at the time of the laminectomy is
required to treat this kyphosis adequately. In the most severe cases of
persistent thoracolumbar kyphosis, anterior vertebrectomies are
combined with posterior laminectomy and fusion to have the best chance
of relieving the lower extremity neurologic deficits.
 |
|
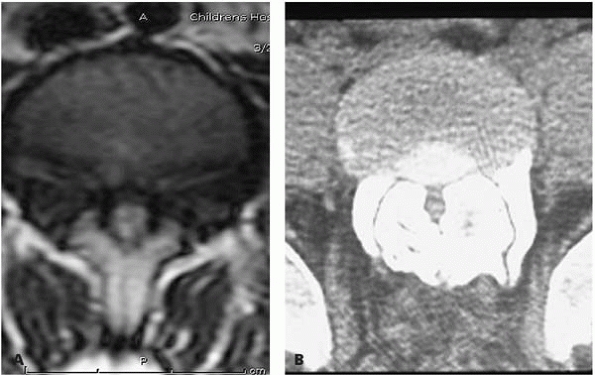
▪ FIGURE 18-6 (A)
Transverse cut of MRI of the lumbar spine shows a small spinal canal resulting from interpediculate narrowing in achondroplasia. (B) Transverse cut of CT scan of L5 demonstrates marked diminution of the spinal canal in a teenager with achondroplasia. |
manifestations, is recognizable at birth and results from a defect in
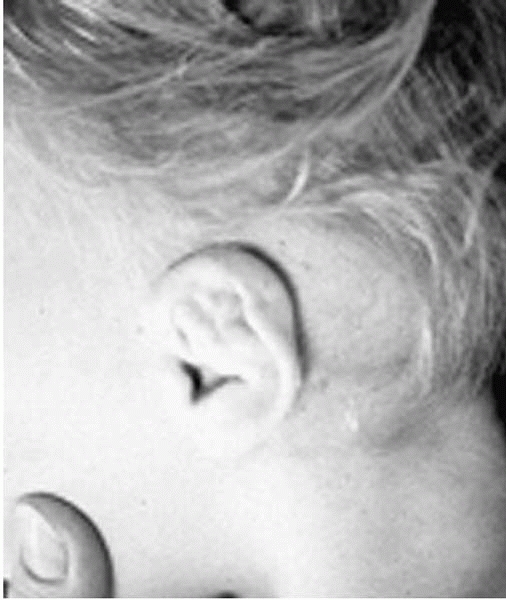
diastrophic dysplasia sulfate transportase. Characteristic features
include very short extremities, hand defects with hitchhiker’s thumb
and stiff interphalangeal (IP) joints, severe club feet, and, within a
few weeks of birth, external ear cysts which develop into “cauliflower
ears” (Fig. 18-7). About 25% have an associated cleft palate. Intelligence is normal.
 |
|
▪ FIGURE 18-7
Clinical photograph of a young child with diastrophic dysplasia illustrates the typical “cauliflower ear” seen in this condition. |
abnormalities. Starting at the top, the cervical spine often has
cervical kyphosis at a young age, though the majority will resolve with
growth and age. If this kyphosis is progressive, this can be fatal if
untreated and requires aggressive treatment and fusion if progression
is proven. Flexion/extension MRI can be useful to assess the cervical
kyphosis on a serial basis and to evaluate possible need for surgical
treatment. In the thoracic spine, the primary problem is the
development of severe progressive mid-thoracic kyphoscoliosis,
simulating a severe congenital scoliosis, which occurs in about 30% of
these children and always is noted before the age of 4. These severe
deformities are best managed with early anterior and posterior fusion
before the severe deformity is allowed to progress too much. Another
30-40% develop lesser degrees of scoliosis, many of which do not
require much
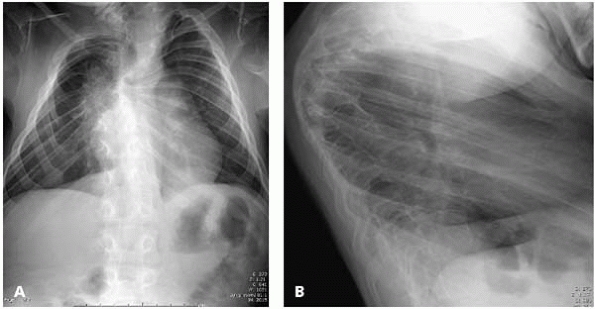
treatment, since spine growth seems to be complete in this condition by about 8 or 9 years of age (Fig. 18-8).
 |
|
▪ FIGURE 18-8 (A)
AP radiograph of spine demonstrates mid-thoracic scoliosis that is common in diastrophic dysplasia and initially presents before the age of 4. (B) Lateral spinal radiograph of the same child illustrates the severe angular midthoracic kyphosis usually associated with scoliosis in diastrophic dysplasia. |
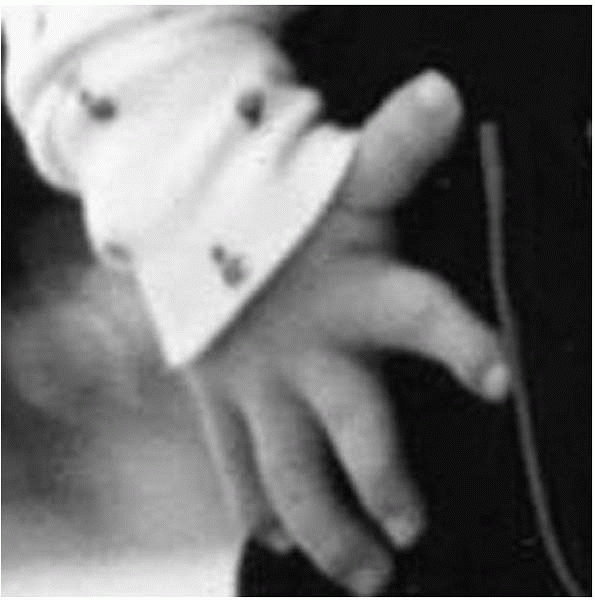
present. The IP joints of all fingers are stiff and usually immobile,
but the metacarpophalangeal (MP) joints move normally. Hand function is
adequate but without good power grip, due both to the IP stiffness and
the thumb deformity (Fig. 18-9).
 |
|
▪ FIGURE 18-9
Clinical photograph of the hand of a child with diastrophic dysplasia demonstrates the “hitchhiker’s thumb” and stiff interphalangeal joints characteristic of this condition. |
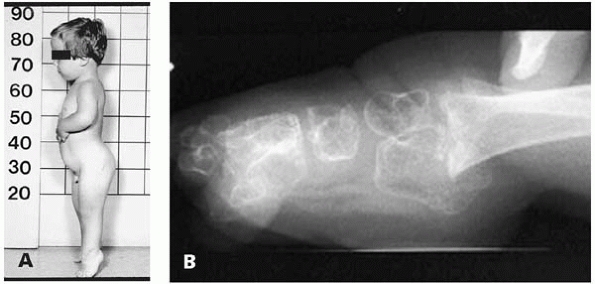
more striking than the varus deformity. Surgical treatment is needed,
which includes not only the standard extensive tendon lengthening and
capsulotomies, but also frequently removing some of the unossified
cartilage of the talus and calcaneus and splitting the syndesmotic
ligament to allow for talus reduction in the ankle joint. At the end of
the procedure the foot must be plantigrade and bracing is needed
usually to prevent equinus deformity recurrence (Fig. 18-10).
 |
|
▪ FIGURE 18-10 (A) Young child with diastrophic dysplasia with typical body habitus, including severe foot equinus deformity. (B) Lateral foot and ankle radiograph demonstrates the severe foot equinus deformity common in diastrophic dysplasia.
|
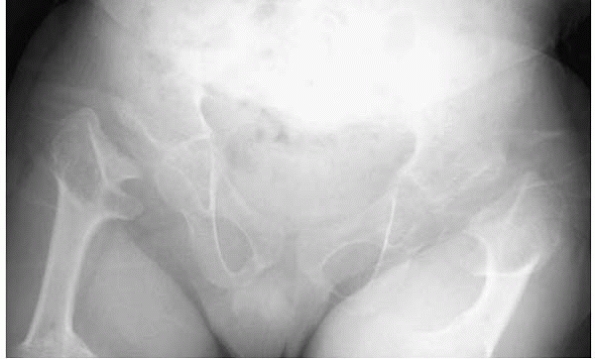
from birth. Congenital lateral patella dislocation is present in many
with diastrophic dysplasia; if present, this needs to be evaluated and
treated with surgical relocation to improve knee extension
substantially. Hip flexion contractures can usually be accommodated and
tendon release shouldn’t be needed. By early adult life, lateral hip
subluxation and degenerative arthritis of the hip may be improved
markedly with customsized total hip replacements (Fig. 18-11).
 |
|
▪ FIGURE 18-11
AP radiograph of pelvis and hips in a 13-year-old with diastrophic dysplasia demonstrates the hip deformity and subluxation common in this condition and often leading to joint replacement in early adult life. |
although some require use of a walker, depending on the position of the
feet, knees, and hips. Although most eventually use power wheelchairs
as long-distance transport, the maintenance of household ambulation
improves the independence for these individuals; this should be a goal
in the orthopaedic treatment of these patients.
(SED); these types are very distinct from one another. SED tarda
affects only males and is usually not diagnosed until preadolescence
when degenerative arthritis of the hips begins to develop, usually
leading to total hip arthroplasty in early adult life for pain relief.
Those with SED tarda are commonly over 5 feet tall and this diagnosis
is usually delayed, while those with SED congenita are very short and
customarily have the diagnosis made in infancy.
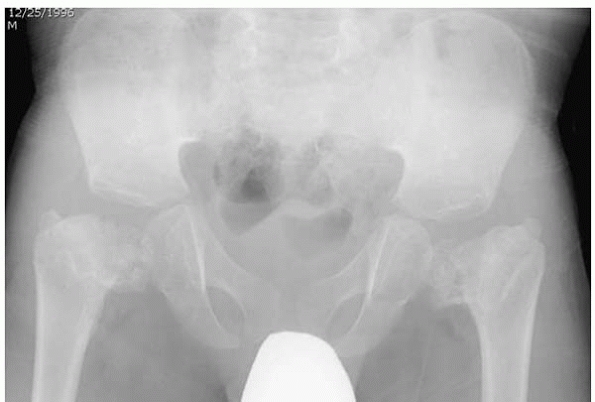
congenita. The trunk is short and radiographs show delayed epiphyseal
ossification throughout, as well as associated marked coxa vara.
Angular deformity of the femur and tibia may develop with growth.
Walking will occur but may be somewhat delayed and characteristically
is associated with an external foot progression angle due to the
excessive external rotation of the hips. Osteotomy of the proximal
femur to correct the varus, excessive external rotation, and flexion is
often very useful in late childhood to improve the gait and trunk
position (Fig. 18-12).
 |
|
▪ FIGURE 18-12
AP radiograph of the pelvis and hips in a 6-year-old boy with spondyloepiphyseal dysplasia congenita demonstrates the marked coxa vara and delayed epiphyseal ossification typical of this condition and commonly requiring proximal femoral valgus extension-internal rotation osteotomy. |
treatment is upper cervical spine instability secondary to odontoid
hypoplasia. Odontoid hypoplasia occurs to some degree in essentially
all children with SED congenita, and instability may be seen as young
as one year of age. Since vertebral ossification is delayed in this
condition, the use of flexion/extension MRIs of the cervical spine is
useful periodically over the first 10 years of life to assess most
accurately the possible need for upper cervical fusion to protect the
spinal cord from injury. If instability is demonstrated, posterior
occiput to C2 fusion, with the child immobilized in a halo, is
necessary and effective treatment (Fig. 18-13).
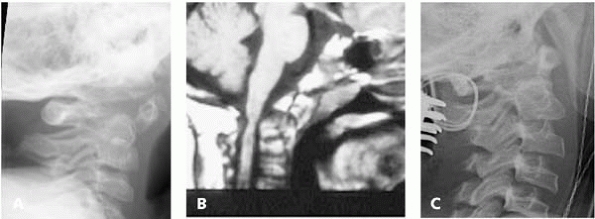 |
|
▪ FIGURE 18-13 (A)
Lateral radiograph of upper cervical spine in a child with spondyloepiphyseal dysplasia congenita demonstrates odontoid hypoplasia common in this condition. (B) Sagittal MRI view of upper cervical spine in older child with spondyloepiphyseal dysplasia congenita demonstrates odontoid hypoplasia and signal changes within the upper cervical spinal cord characteristic of significant C1-C2 instability requiring posterior fusion stabilization. (C) Lateral cervical spine radiograph obtained intraoperatively in a teenager with spondyloepiphyseal dysplasia congenita with instability illustrates one technique to provide stabilization and fusion for this condition. In younger children, a halo is routinely applied to facilitate fusion. |
same as with achondroplasia, the genetic defect is different and
clinical manifestations differ. The genetic defect here is in cartilage
oligomeric matrix protein. Clinically, the face is normal and angular
deformity that develops with growth differs from that seen in
achondroplasia. While kyphosis and scoliosis can occur, spinal stenosis
is not seen in pseudoachondroplasia. The
most common cervical spine problem is upper cervical instability, which
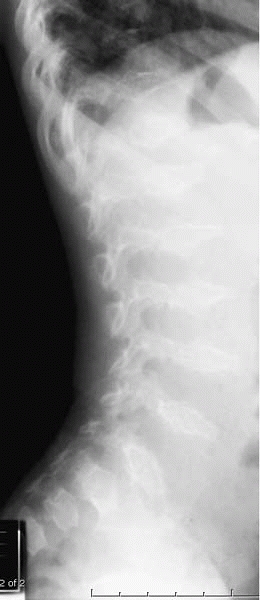
may require fusion in some. On a lateral spinal radiograph, there is a
characteristic vertebral body shape with a mid-vertebral anterior
projection, which is essentially diagnostic for this condition and is
absent in achondroplasia (Fig. 18-14).
 |
|
▪ FIGURE 18-14
Lateral radiography of thoracolumbar spine demonstrates the vertebral shape, with anterior mid-vertebral projections, characteristic of pseudoachondroplasia. |
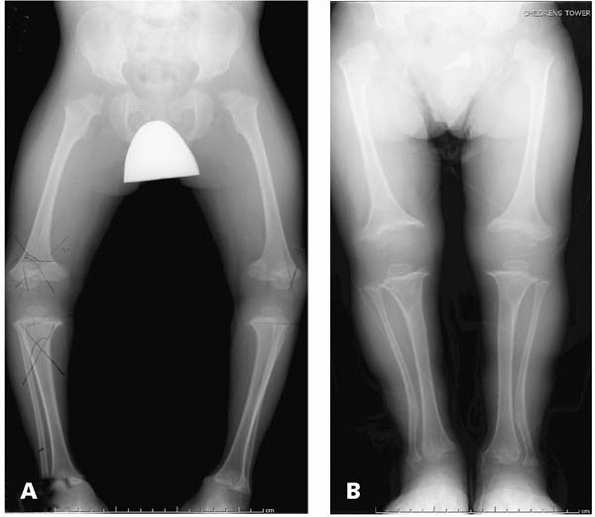
the epiphyses and metaphyses, apparently the source of the angular
deformities that occur with growth and that often recur after
realignment osteotomy (Fig. 18-15). Realignment
surgery is difficult due to the joint laxity in this condition and to
the delayed ossification of the epiphyses, which makes the joint level
difficult to see without concomitant arthrography. The hips tend to
slowly subluxate and commonly require total hip arthroplasty in early
adult life.
 |
|
▪ FIGURE 18-15 (A)
Standing AP radiograph of lower extremities of a child with pseudoachondroplasia illustrates typical changes in both the epiphyses and metaphyses, resulting in angular deformity. (B) Following femoral and tibial/fibular osteotomies bilaterally, the standing AP radiograph of the lower extremities demonstrates significant improvement in alignment. |
seen, all with a different genetic defect. The most common type seen by
orthopaedists is Morquio syndrome (type IV). In the past, Hurler
syndrome (type I) was not treated orthopaedically, since most of these
children died by age 5, but with recent enzymatic or bone marrow
treatment, these children are now surviving much longer and now require
periodic orthopaedic followup as well as potential surgical treatment.
is to fail to recognize and diagnose the condition correctly. These
infants look normal at birth. Developmental milestones are somewhat
delayed, which may be a reason the orthopaedist is consulted. If the
diagnosis of Hurler syndrome can be made before the age of one, the
results of enzyme replacement or bone marrow transplant treatment to
block further progression of the disorder are much better than if the
diagnosis is not made until the child is 2 or 3 years of age. However,
even though the systemic effects on many organ systems can be greatly
reduced by early treatment, many of the orthopaedic abnormalities of
this condition still often need ongoing followup and even surgical
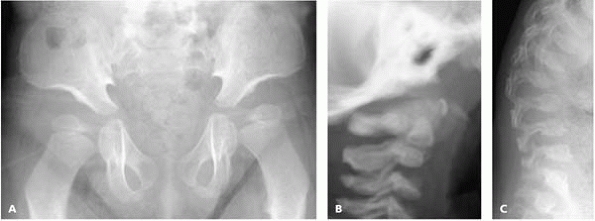
treatment (Fig. 18-16). Some of the early signs
of Hurler syndrome are developmental delay, generalized joint stiffness
even in the fingers, corneal clouding, herniae, and facial dysmorphism
(remember, if the child does not look at all like either parent, think
of a genetic disorder).
 |
|
▪ FIGURE 18-16
Radiographs of a 4-year-old boy who has undergone treatment for Hurler syndrome demonstrate that even though the underlying defect can be treated, there is still need for ongoing orthopaedic followup. (A) AP radiograph of pelvis shows mild hip subluxation. (B) Lateral radiograph of the upper cervical spine shows odontoid hypoplasia. (C) Lateral thoracolumbar spine radiograph shows persistent thoracolumbar kyphosis with vertebral changes typical of Hurler syndrome and some of the other mucopolysaccharidoses. |
have an opportunity to be the first to make this diagnosis. The other
MPS syndromes include Hunter (type II), Sanfilippo (type III), Morquio
(type IV), Scheie (type V or I-S), Marateaux-Lamy (type VI), and Sly or
beta-glucuronidase deficiency (type VII). While all of these have
individual differences that are beyond the scope of this chapter, many
do have odontoid hypoplasia, thoracolumbar kyphosis, and early
degenerative hip disease. Morquio syndrome is probably the most common
(though still uncommon) of these conditions to come to see the
orthopaedist.
syndrome may present to the orthopaedist because of a “bump” in the
back and some delay in development. A lateral spinal radiograph in
these children will show a thoracolumbar kyphosis that can easily be
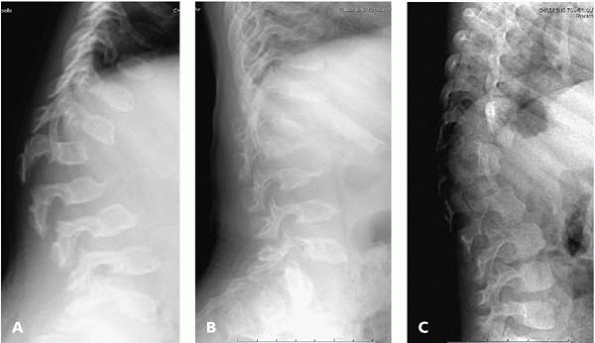
confused with congenital kyphosis (Fig. 18-17).
However, in Morquio syndrome, the apical vertebra of the kyphosis—as
well as one or two adjacent vertebrae—will have a vertebral body
projection that
extends
out anteriorly from the inferior aspect of the vertebra; this is not
seen in congenital kyphosis from failure of vertebral formation. It is
important for the orthopaedist to differentiate these two conditions
for several reasons:
 |
|
▪ FIGURE 18-17
This series of lateral thoracolumbar spine radiographs illustrates the difference between thoracolumbar kyphosis seen in the spine in Morquio syndrome (A,B) with anterior inferior vertebral projections at several levels compared with congenital kyphoscoliosis (C), with abnormality at usually only one vertebral level. |
-
If there is congenital kyphosis, early fusion is usually needed, whereas in MPS early fusion is rarely done.
-
Recognizing MPS syndrome early will allow
genetic counseling for the family before there is another sibling born
with the same MPS syndrome. -
If MPS is diagnosed, there are other associated problems that need to be evaluated.
sulfate that can be diagnosed by one or more tests. The simplest is a
urine assay, which is often able to confirm the diagnosis. If it does
not, several molecular tests are available in special centers to
determine whether Morquio syndrome is the diagnosis. Radiographic
findings are also helpful in establishing this Morquio diagnosis—
odontoid hypoplasia is common, femoral head ossification is delayed and
hip subluxation occurs in early childhood, genu valgum is common.
not be as short as those with more extensive involvement, further
delaying diagnosis. In later childhood, the presentation to the
orthopaedist is usually for the progressive genu valgum and declining
walking ability. In this setting, it is important to suspect Morquio
syndrome enough to obtain flexion/extension lateral cervical spine
radiographs to see if upper cervical instability is the cause of the
declining walking ability, not the genu valgum. If upper cervical
instability is present, posterior fusion is necessary to protect the
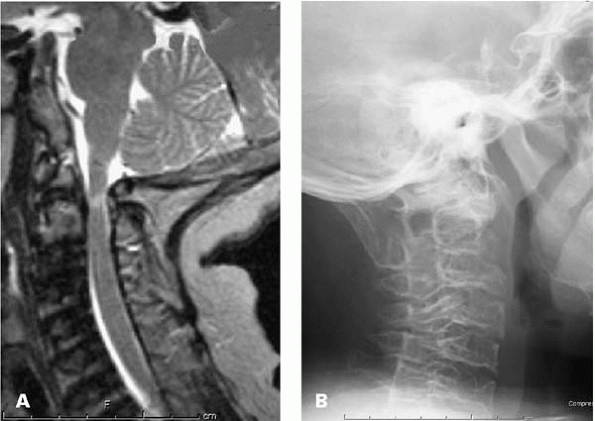
cervical spinal cord (Fig. 18-18). If the
cervical spine is stable and walking is still a problem, realignment
osteotomy of the distal femur (plus sometimes the proximal tibia) is
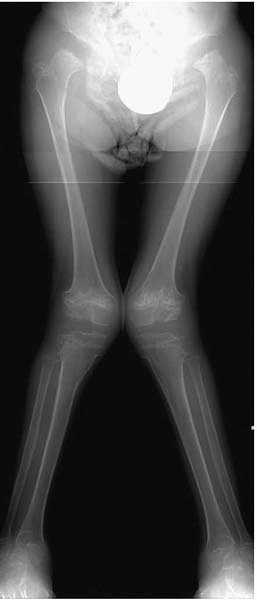
useful to improve the genu valgum and the walking (Fig. 18-19).
Hip subluxation is also relatively common and may require surgical
treatment in childhood or custom total hip arthroplasty in adulthood.
 |
|
▪ FIGURE 18-18 (A)
Sagittal cut of MRI in a 12-year-old boy with Morquio syndrome and declining walking ability shows spinal cord compression and signal change associated with upper cervical instability resulting from his odontoid hypoplasia. (B) Postoperative lateral radiograph of upper cervical spine illustrates solid occipital-C2 posterior fusion 6 months following his surgery. |
in infancy may be the first reason for orthopaedic consultation,
surgical treatment is not always needed for this, as this kyphosis may
never progress from that seen at birth. Bracing has not been proven to
be effective and, in my opinion, if there is no kyphosis progression,
no treatment is needed. If there is kyphosis progression, particularly
with MRI evidence of spinal cord or cauda equina compression, spinal
instrumentation (beware of hooks inside the somewhat small spinal
canal) and fusion is effective.
 |
|
▪ FIGURE 18-19
Standing AP radiograph of both lower extremities in this 9-year-old with Morquio syndrome demonstrates the genu valgum and hip subluxation commonly seen in children with this condition. It is important to evaluate the child for upper cervical instability before doing long bone realignment surgery. |
-
Skeletal dysplasias generally will present with height <third percentile.
-
Physical exam can determine if limbs or spine or both are involved most.
-
Radiographic survey
is used to determine if epiphysis, metaphysis, and spine are
involved—which often allows the establishment of a tentative diagnosis. -
Diagnosis is established by the orthopaedist, confirmed by the geneticist.
-
It’s useful for the
orthopaedist to be familiar with the natural history of the specific
condition and what to expect from the orthopaedic abnormalities over
time. -
Early recognition of
genetic skeletal disorders helps both the family to obtain genetic
information and counseling and the child to receive appropriate care. -
Because of the
rarity of these conditions, it is usually best to refer these children
to a pediatric orthopaedic center for ongoing care.