
VII – NEOPLASTIC, INFECTIOUS, NEUROLOGIC AND OTHER SKELETAL DISORDERS
> Tumors and Tumor-Like Conditions > CHAPTER 127 – BENIGN BONE
TUMORS
Orthopaedic Surgery, Columbia-Presbyterian Medical Center, College of
Physicians and Surgeons of Columbia University, New York, New York,
10032.
diverse group of clinical entities, varying greatly in their behavior
and thus treatment requirements. Some lesions may simply be observed
indefinitely, while others demand wide resection and reconstruction.
While benign bone tumors do not as a rule metastasize, they may at
times act very aggressively locally and be mistaken for malignant
tumors. On rare occasions, benign tumors may be multifocal or
associated with pulmonary metastases. It is incumbent upon the treating
orthopaedic surgeon to be familiar with the spectrum of benign bone
tumors and tumor-like conditions in order to provide the appropriate
management (Table 127.1). In this chapter, the
most common benign, primary tumors and tumor-like conditions of bone
across all age groups will be discussed as to their general and
surgical management.
 |
|
Table 127.1. Classification of Benign Bone Tumors
|
The stages are denoted by the Arabic numerals 1, 2, and 3, whereas
malignant bone tumors are classified by Roman numerals (I, II, III). As
will be detailed later, many benign bone tumors have the potential to
present at, and progress through, various stages during their disease
course.
 |
|
Table 127.2. Staging of Benign Bone Tumors
|
incidentally and usually do not progress. They may even spontaneously
resolve. These tumors are noninvasive with distinct margins. Latent
tumors can often be treated with observation alone. When surgical
intervention is necessary, intralesional excision or simple curettage
will suffice. Examples of stage 1 tumors are nonossifying fibroma,
enchondroma, unicameral bone cyst, osteochondroma, osteoid osteoma,
eosinophilic granuloma, and fibrous dysplasia.
that they usually are not self-limiting and tend not to resolve without
intervention. These lesions expand or deform the host bone but remain
contained in the bone. They have the potential to destroy the cortex,
invade the surrounding soft tissues, and recur if residual tumor is
left behind. Active lesions are most often treated with intralesional
curettage. The procedure may be augmented with some type of adjuvant
therapy to extend the margins if deemed necessary. Examples of stage 2
tumors are enchondroma, osteochondroma, osteoid osteoma, giant cell
tumor, chondromyxoid fibroma, fibrous dysplasia, eosinophilic
granuloma, aneurysmal bone cyst, osteofibrous dysplasia, and unicameral
bone cyst.
the adjacent soft tissues. These lesions can be so destructive that
they mimic malignant tumors. Because of their extensive cortical
destruction, it is not uncommon for stage 3 tumors to present with
pathologic fracture. Aggressive tumors are best treated with en bloc resection
when possible, or a very aggressive and thorough curettage. When these
tumors are treated by intralesional excision because of anatomic
restraints, they will almost always require some form of adjuvant
therapy and are associated with a fairly high recurrence rate. Examples
of stage 3 tumors are giant cell tumor, osteoblastoma, chondroblastoma,
and aneurysmal bone cyst.
refers to the persistence and subsequent growth of microscopic disease
left over from the initial procedure.
well-demarcated bone-forming lesion called a nidus, surrounded by a
radiodense, reactive zone of host woven or lamellar bone. The
surrounding secondary bony reactive zone represents a reversible change
that remodels and regresses after the nidus is removed. The nidus has
limited growth potential and rarely exceeds 1 cm in diameter. Lesions
larger than 1.5 cm in diameter have been arbitrarily designated
osteoblastomas. Osteoid osteoma accounts for 10% to 13% of all benign
bone tumors and 2% to 3% of all primary bone neoplasms (51,65,164).
It is most commonly seen in the second and third decades of life, with
a male to female predominance of approximately 3 to 1. Over half of
these lesions occur in the long bones of the lower extremity, with the
proximal femur being the most common location. In the spine, osteoid
osteomas almost exclusively occur in the posterior elements of the
lumbar vertebrae, namely the pedicular region of the posterior arch. In
addition, they may occur in juxta-articular bone within the confines of
the synovial cavity, where they are termed intra-articular osteoid
osteomas. Osteoid osteomas are most commonly located in the cortex of
the bone, but they may be located in the medullary cavity or periosteum
of the bone. They are usually diaphyseal or metaphyseal, but on rare
occasion they are epiphyseal.
pain, usually worse at night, dramatically relieved by aspirin or other
nonsteroidal anti-inflammatory drugs (NSAIDs). The duration of pain
prior to presentation can vary from weeks to years. The second most
common symptom is a limp. In lesions with relatively subcutaneous
locations, such as the tarsal bones or phalanges of the hand,
soft-tissue swelling may be present. Spine involvement may present as a
painful scoliosis and unilateral muscle spasticity, with the concave
part of the curve toward the side of the lesion (79).
When the lesions occur near the physis, leg-length discrepancies may be
observed with the affected extremity being longer. Other common signs
on physical exam include muscle atrophy and structural (bowing)
deformities.
characteristic and the diagnosis can often be made by plain
roentgenograms alone. Conventional radiographs reveal a small, round
radiolucent area within the cortex surrounded by a variable sclerotic
zone (Fig. 127.1A). The tumor may exhibit
different stages of mineralization and therefore display different
degrees of density. The reactive periosteal lamination may mimic
Ewing’s sarcoma. Epiphyseal lesions often demonstrate a lack of
sclerosis. Further imaging studies are indicated when typical plain
film findings are not yet evident, when the exact location of the nidus
is obscured because of extensive cortical thickening, when the lesion
arises in an atypical location, or in anatomic sites that are difficult
to image, such as the spine. The radionuclide bone scan is almost
always positive for osteoid osteoma. Cross-sectional studies such as a
computed tomography (CT) and magnetic resonance imaging (MRI) are
especially helpful for areas that are difficult to image on plain
films, such as the spine (Fig. 127.1B, Fig. 127.1C and Fig. 127.1D).
CT appears to be a more accurate diagnostic tool than MRI because this
modality displays the bony nidus and surrounding reactive bone in
contrast to the normal cortex more clearly (7).
The nidus is also less likely to be obscured by the edema associated
with the lesion on CT scans than with MRI. It is important to get thin
cuts through the area of a suspected nidus.
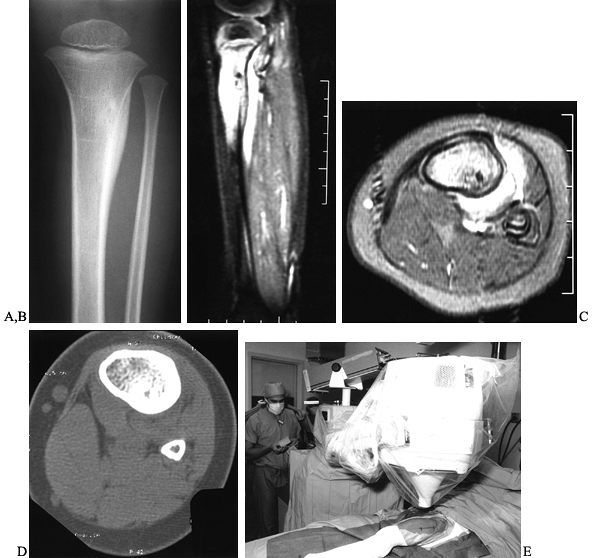 |
|
Figure 127.1. AP radiograph (A)
of the tibia in a patient with osteoid osteoma. Note that the nidus is not well visualized although there is thick, mature periosteal elevation and some medullary reactive bone in the lateral aspect of the proximal tibial metaphysis. The sagittal (B) and axial (C) MRI show extensive marrow edema and soft-tissue edema surrounding the proximal tibia. The edema extends well beyond the lesion. The nidus is visualized as a low signal (dark area) in the midst of the bright edema, which is present both in the soft tissues and in the medullary cavity. A CT scan (D) more clearly shows the radiodense nidus in the lateral aspect of the tibia. In this case, the nidus is in the medullary cavity and the thickness of the cortex is readily apparent. There is also thickening of the trabecular bone surrounding the lesion. The adjacent fibula is normal, and the soft tissues do not appear abnormal as they did on the MRI. The portable scanner used for intraoperative bone scans (E) allows precise intraoperative imaging of the area of abnormality and helps to plan the surgical approach. Disappearance of the area of increased uptake following excision of the nidus helps to confirm that it has been completely excised. |
Important aspects of the preoperative management include planning the
most appropriate surgical approach, selecting a method for
intraoperative localization of the nidus, and determining if a less
invasive method is a reasonable alternative to wide excision. A CT scan
is often useful in helping to plan the most direct operative approach.
The method by which intraoperative localization of the nidus is to be
accomplished must be determined prior to surgery. Intraoperative bone
scintigraphy, tetracycline labeling, CT guidance, and conventional
radiography have all been used (85,155).
Intraoperative nuclear scanning takes advantage of the lesion’s ability
to concentrate radioisotopes following preoperative administration. The
area in question is sequentially scanned intraoperatively until
activity is appreciably diminished (Fig. 127.1E) (85). This indicates that the lesion has been completely removed.
ability of the nidus to concentrate this compound. After oral
administration 24–48 hours preoperatively, an ultraviolet lamp is used
to demonstrate the presence of uptake in the resected specimen.
Tetracycline labeling should be reserved for patients over the age of 8
years because of its
affinity for developing dentin and associated tooth discoloration.
of the nidus but does not quantitate the adequacy of resection. Plain
radiographs can be used in conjunction with intraoperative markers for
immediate postresection imaging of the host or specimen. The exact
perioperative method to determine complete removal of the nidus is
chosen based on the surgeon’s experience and the imaging modalities
available.
osteoma may require excessive bone resection, or it may not be possible
because of anatomic considerations. Proper preoperative planning will
identify those patients in whom a significant amount of bone must be
removed to reach the lesion or to ensure adequate margins, and it will
allow the surgeon to anticipate the need for bone grafting and internal
fixation. Alternatively, a variety of percutaneous procedures for
osteoid osteoma removal have been described. Percutaneous treatments
have obvious advantages, including minimal invasiveness, relative
safety, cost-effectiveness, and earlier return to normal activities.
Bone trephination under CT guidance has been reported by several
investigators with varying degrees of success (163).
Percutaneous radiofrequency ablation has met with success and in a
number of tumor centers and has become the procedure of choice in some
institutions for lesions outside the spine (8,119,131). The technique involves introducing a radiofrequency probe over a biopsy needle, placed under CT control (Fig. 127.2).
A 1 cm area containing the nidus is subjected to thermal necrosis after
obtaining tissue for pathology. When comparing this approach to the
more traditional open procedure, recurrence rates are essentially
equivalent. The results from these percutaneous techniques appear
encouraging.
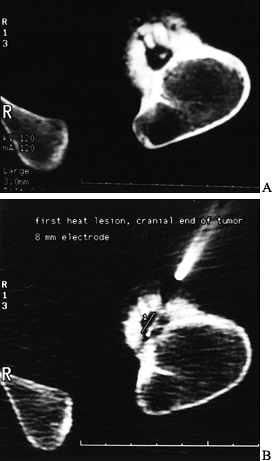 |
|
Figure 127.2. A:
The CT of another patient with an osteoid osteoma on the cortex of the proximal femur. The nidus is mostly radiolucent with a central radiodensity, and there is thick periosteal new bone around it. B: The trochar being placed into the lesion for radiofrequency heat ablation under CT control. The patient is under general anesthesia. |
Their experience argues that there are circumstances in which operative
treatment may cause complications and disability more severe than those
associated with the original condition. The medical management of
osteoid osteoma involves a prolonged course of NSAIDs. Patients treated
successfully by this regimen were administered medication for an
average of 33 months. Contraindications to medical management include
drug hypersensitivity, progressive deformities, and uncertain
diagnosis. While nonoperative management may have a role, the subset of
patients in which this treatment plan is preferred has yet to
identified.
marginal or wide excision. The goal of surgery is to remove the entire
nidus, as incomplete excision is associated with high recurrence rates
and persistent pain. The amount of the surrounding reactive bone that
is removed varies: The intent is to leave as much bone as possible to
reduce the risk of pathologic fracture. To limit the amount of bone
removed, one of the intraoperative imaging modalities just described
should be utilized. As mentioned, bone grafting and internal fixation
may be required to restore adequate structural integrity. This is
especially true of femoral and tibial diaphyseal lesions in which
extensive
cortex
may need to be excised. Excise lesions about the femoral neck using a
fracture table, or a radiolucent table to allow for fluoroscopic
imaging and internal fixation if needed.
possible complications of an extensive excision, which weakens the bone
and predisposes it to fracture and the morbidity associated with
autogenous or allogeneic bone graft.
-
After intraoperative localization,
identify the reactive cortex visually by its characteristic elevation
and roughening, and expose it subperiosteally. -
Use a high-speed burr or osteotomes in a successive sweeping motion over the reactive cortex until the nidus is encountered (164).
-
Remove the nidus of tissue by curettage
and submit it for histologic confirmation. Then burr the cavity in all
directions for 1–2 mm and smooth the edges. Bone grafting is usually
not necessary.
resection. Wide excisions often require prolonged periods of
immobilization, a longer hospital stay, and protected weight bearing.
Patients treated by the burr-down technique reportedly require fewer
activity restrictions. Percutaneous techniques are often performed as
an out-patient procedure and may not require weight-bearing precautions.
tumor accounting for less than 1% of all primary bone tumors. It has
been called by some authors giant osteoid osteoma, and these two
lesions are often histologically indistinguishable (44,76).
The microscopic appearance is characterized by benign-appearing
osteoblasts producing woven bone and osteoid in varying shapes and
amounts. Despite their similar microscopic patterns, their respective
growth potentials and hence clinical courses differ remarkably (76).
Osteoid osteomas rarely exceed 1.5 cm, whereas osteoblastomas often
exceed 2 cm. In addition, patients with osteoblastoma do not typically
describe night pain, a hallmark feature of osteoid osteoma.
Radiographically, osteoblastomas often lack the peripheral sclerosis
seen with osteoid osteoma. Despite these differences, it may still be
difficult to distinguish between these two entities. In such instances,
size greater than 1.5 cm has arbitrarily been chosen as the determining
factor to make the diagnosis of benign osteoblastoma. Osteoblastoma
typically presents during the second decade of life. Most patients are
between the ages of 5 and 45. There is a male to female ratio of 3 to
1, which is similar for the sex distribution encountered in osteoid
osteoma.
been identified. These tumors are borderline lesions whose radiographic
and histologic characteristics fall between benign osteoblastoma and
osteosarcoma. Radiographically, aggressive osteoblastomas extend beyond
the cortical margins of the bone, resembling an aneurysmal bone cyst.
Microscopically, areas of epithelioid osteoblasts are present. Neither
malignant transformation nor metastases of these lesions has been
reported. However, these tumors are more likely to recur following
incomplete or intralesional excision. The true incidence of this subset
of osteoblastomas is not known.
and distinguishes it from other benign bone lesions, including osteoid
osteoma. Osteoblastoma has a predilection for the spine, particularly
the posterior vertebral elements (84). In large
series, up to a third of cases were located in the spine with near
equal distributions in the cervical, thoracic, lumbar, and sacral spine
(112). The second most frequent site of
involvement is the long bones, followed by the bones of the hands and
feet. Pathologic fractures through the lesions have not been reported.
The pain increases in severity with time and has an unpredictable
response to anti-inflammatory agents. Swelling and atrophy of the
affected area may be present on physical exam. When the spine is
involved, the patient often presents with a painful scoliosis and
limitation of motion secondary to paraspinal muscular spasm. The lesion
is usually located at the apex of the concavity of the curve (112).
The scoliosis usually resolves following removal of the offending
lesion. Persistent structural deformity may ensue if there is a
significant delay in diagnosis. Neurologic deficit as the presenting
complaint is not uncommon with spinal lesions. While the symptoms are
typically mild, severe neurologic compromise has been reported (117).
osteoblastoma. Standard biplanar radiographs are typically capable of
detecting lesions outside the sacrum. They reveal a round osteolytic
lesion often greater than 2 cm in diameter, containing varying amounts
of internal mineralization and surrounded by reactive bone. The lesions
may be intramedullary, but the majority are intracortical. In addition,
osteoblastomas may arise in the diaphysis, metaphysis, or epiphysis.
When epiphyseal lesions do exist, they most often occur in the hands
and feet. The lesion may expand through the involved cortex, but it
seldom has an associated soft-tissue mass. In the spine, at least 90%
of the lesions involve the posterior elements, namely the spinous
process and arches. Although the
tumor
may extend into the vertebral body, it rarely arises in or exclusively
involves the anterior spine. Details of spine lesions, such as the
extent of mineralization and degree of rim sclerosis, are more easily
discernible on CT scan. As spine lesions are often expansile, CT scan
is superior to plain films in evaluating the relationship of the tumor
to adjacent neurologic structures and for the purposes of staging and
preoperative planning. If evidence of spinal cord or nerve root
compression is apparent, obtain an MRI, as it provides exceptional
imaging of the neural elements. Radionucleotide studies will
demonstrate intense focal accumulation of the isotope. This study is
perhaps most useful to help localize the origin of the lesion in
patients with back pain in whom the diagnosis of osteoblastoma is
suspected. Multicentric involvement has not been reported.
recurrence must be balanced against the complications associated with
aggressive resections, given the nonmalignant nature of the lesion.
Treatment of the osteoblastoma largely depends on its location and
behavior. When possible, perform a complete or marginal excision.
Recurrence following incomplete intralesional curettage occurs in up to
15% (75,98). Margins can be extended using cryotherapy or phenol as described in the section on management of giant cell tumors.
treatment protocols for managing aggressive benign lesions in the
extremities often are impractical given the anatomic restrictions and
potential neurologic complications associated with spinal surgery. Such
tumors may prove to be unresectable or even lethal in the spine. Employ
the methodical approach to all spine neoplasms as described by
Weinstein et al. (166).
-
Perform a complete clinical and imaging workup before biopsy.
-
Decide the most appropriate medical and
surgical treatments, as well as possible adjuvants depending on the
patient’s overall condition and tumor stage. -
Plan the surgery according to a standardized spinal surgical staging system (165).
-
Make every effort to preserve neurologic function.
-
Maintain spinal column stability. Treat
osteoblastoma according to its oncologic staging. Stage 2 lesions can
probably be managed adequately with intralesional curettage. Stage 3
lesions are best treated with marginal resection. When marginal
resection is not feasible, adjuvant radiation therapy may have a role,
although its efficacy has yet to be demonstrated (18).
excision is not possible because of anatomic considerations, such as
proximity to the physes, intralesional curettage is acceptable,
especially for stage 1 and 2 lesions.
-
Curet the tumor with conventional hand
curets, mechanically extending the margins to normal tissue with the
use of a high-speed motorized burr. -
For aggressive osteoblastomas, some authors advocate the use of adjuvants such as cryotherapy and phenol (65,157).
Use these agents judiciously as their complications have been well
documented and experience in treating osteoblastomas is limited. -
Small defects can be filled with bone
graft. Larger defects may require internal fixation to achieve adequate
structural integrity. -
In the spine, intralesional curettage is
often used, as complete excision is frequently not possible. In such
cases, perform an extralesional dissection as far as possible. As these
lesions are usually extremely vascular, ligate identifiable feeder
vessels as they are encountered. -
Then remove the tumor in piecemeal
fashion. Curet or burr the walls of the cavity. All of the perilesional
sclerotic reaction need not be removed. -
Take care to preserve stability of the spine; internal fixation may be required.
for only 4% to 5% of all primary bone tumors in the United States. In
Southeast Asian regions, GCT accounts for approximately 20% of all
primary bone tumors (156,176).
The peak age of incidence is in the third decade of life, with 70% of
the cases diagnosed in persons 20–40 years old. Although there have
been numerous reports of GCT arising in patients with open growth
plates, this presentation is the exception. When it occurs in children
with open growth plates, the lesion is metaphyseal. In skeletally
mature patients, it is found most often in the epiphyseal ends of long
bones, especially the distal femur, proximal tibia, and distal radius.
Approximately 60% of the cases occur about the knee. It is uncommon in
the axial skeleton with the exception of the sacrum (133).
When it does occur in the spine, the vertebral body is most frequently
involved. A slight female to male predominance exists. Multicentric
involvement has been reported but is rare.
active and locally aggressive tendency to increase pressure within the
bone or break through the cortex and stretch the periosteum. As such,
the predominant presenting symptoms are pain and swelling of variable
severity. In addition, patients may present with decreased joint range
of motion or pathologic fracture. On physical exam, a tender, hard mass
is typically found. The involved extremity may display muscle atrophy
secondary to disuse, joint effusion, and elevated temperature of the
overlying skin.
juxtacortical, expanding zone of radiolucency at the
epiphyseometaphyseal end of a long bone (Fig. 127.3A, Fig. 127.3B).
The tumor is a well-delineated lesion with irregular endosteal margins.
There is usually no reactive host bone at the periphery of the lesion.
According to the staging system for benign bone tumors, the lesion most
typically presents as an active (stage 2) or aggressive (stage 3)
tumor. The tumor frequently extends to the subchondral bone of the
articular surface and destroys the surrounding cortex, extending into
the soft tissue. Because of the rapid expansion, a periosteal reaction
is seldom seen. The combination of substantial destruction and poor
margination may suggest malignancy.
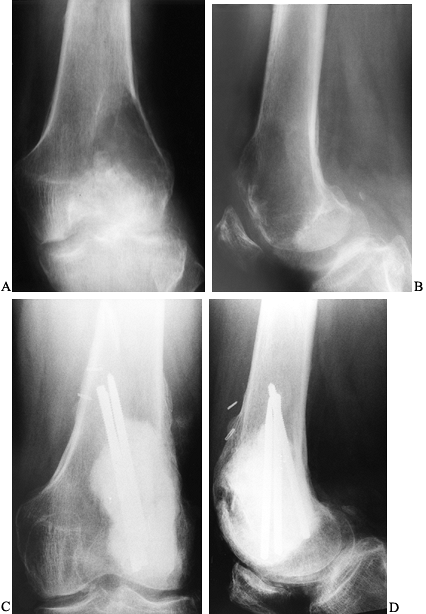 |
|
Figure 127.3. AP radiograph (A)
of a giant cell tumor (GCT) of bone shows a radiolucent lesion of the lateral distal femur involving the epiphysis and the metaphysis. It has destroyed the cortex although the periosteum is intact, and it is well delineated although not well marginated. In the lateral film (B), it is apparent that the lesion extends down to the subchondral bone distally and along the patellofemoral joint. This has the typical appearance of a GCT of bone. AP (C) and lateral (D) radiographs show the postoperative findings in this patient who was treated with extensive curettage, burring of the cavity, and packing with PMMA bone cement and reinforcing metal rods. |
of well-documented cases of pulmonary metastases have been reported in
the literature (22,29).
These so-called pulmonary implants are believed to occur in only 1% to
2% of patients with GCT. They are to be distinguished from true
metastases secondary to malignant degeneration of GCT. Malignant
degeneration of GCT is a phenomenon that usually occurs secondary to
treatment of the original lesion, most typically radiation therapy (22).
Pulmonary involvement in GCTs typically has a good prognosis, although
up to 25% of patients die from the disease. Occasionally, the
metastatic lesions may even spontaneously regress. The histology of the
primary lesions of patients with pulmonary involvement (unlike that of
patients without pulmonary involvement) fails to demonstrate any
significant predicative features (29). In fact,
histologic material from the lung is identical to that of the primary
lesion. Chest radiographs should be obtained at regular intervals as
part of the patient’s management. Early detection is important given
the unpredictable nature of pulmonary lesions. Complete resection is
recommended (29,78).
establish a firm diagnosis. Malignant lesions must be ruled out by
tissue biopsy prior to surgical treatment.
recurrence. Adequate removal of the tumor leads to the lowest risk of
recurrence (16). Aggressive resections, which
may necessitate allograft or prosthetic reconstruction with their
associated complications, must be weighed against the fact that GCT is
almost always a benign neoplasm (125). Several
methods for the treatment of GCT have been described and used with
varying degrees of success: curettage alone, curettage combined with
adjuvant therapies, en bloc resection, amputation, and radiation.
considering three factors: (a) the type of resection, (b) the use of
adjuvant therapy, and (c) the type of material to be used to fill the
resultant defect. The first of these considerations involves deciding
whether to do an intralesional or en bloc
resection. Most GCTs are treated by intralesional curettage. The
recurrence rate using curettage alone has been reported to be as high
as 50%. The risk of local recurrence is greatly diminished by en bloc resection, so this is advocated for expendable bones such as the proximal fibula or portions of the ilium (55). Other indications for en bloc
resection include circumstances in which reconstruction after
intralesional curettage is not possible, such as some patterns of
pathologic fracture and massive involvement with an incomplete cortical
shell that is insufficient to contain cement. In these instances,
reconstruction must be achieved with osteoarticular allograft or metal
prosthetic replacements.
following intralesional curettage. The most commonly used adjuvants
alone or in combination are cryotherapy, phenol, and
polymethylmethacrylate (PMMA). All have distinct advantages and
disadvantages. Cryotherapy using liquid nitrogen has the ability to
advance the resection margins because it creates a 1–2 cm zone of
tissue necrosis (96). Complications of liquid
nitrogen include fracture and local skin necrosis. Phenol also causes
tumor cell necrosis, and reduced penetration results in only 1–2 mm of
osteonecrosis, providing a theoretical advantage over liquid nitrogen
by reduction of fracture rates. Phenol, however, can cause local
chemical burns and exert systemic toxic effects on other organ systems.
Both of these agents must be used with considerable caution: Their
potential local toxicity can be difficult to control because of
leakage. PMMA causes local necrosis in as much as 2–3 mm of bone by its
thermal effects during polymerization (107). A major advantage of PMMA is its immediate structural stability; also, radiographic detection of local recurrence is easier (15,114).
The long-term risk of early degenerative changes in an adjacent joint
secondary to the stiffness induced by subchondral PMMA, while yet to be
substantiated, remains a concern.
cavity if the defect is of substantial size, which it will be in the
majority of cases. The two most commonly used
bone
fillers for this purpose are PMMA and bone graft. The results of
autogenous or allogenic bone graft, when used in conjunction with
phenol or liquid nitrogen, appear to be comparable to cementation (125).
The risks of donor-site morbidity for autogenous graft and the
possibility of disease transmission for allogenic graft must be
considered.
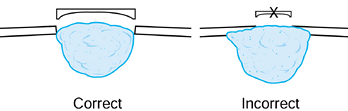
-
After exposing the bone over the tumor,
make a large cortical window the size of the longest longitudinal
dimension of the tumor to provide adequate visualization of the entire
tumor cavity (Fig. 127.4) (13).
Make the window elliptical, with its axis parallel to the long axis of
the bone to reduce the stress-riser effect. Incorporate an already
thinned or destroyed area of cortex to minimize additional bone loss.![]() Figure 127.4.
Figure 127.4.
The correct approach for creating a window for adequate curettage of an
aggressive lesion such as a giant cell tumor. It is important to be
able to directly visualize the entire cavity, so a small window such as
that shown on the right is inadequate. -
Remove the soft-tissue portion of the
tumor and thoroughly curet the walls, breaking down septae and getting
into all the nooks and corners. Then thoroughly remove any residual
tumor with a power burr. -
If the tumor extends into the soft
tissues, excise the entire pseudocapsule. Take care to minimize
contamination of surrounding tissues. Then thoroughly irrigate the
cavity with pulsed lavage using saline.
-
If cryotherapy is chosen as the adjuvant
of choice, before the introduction of liquid nitrogen, identify any
bony perforations and seal then to prevent damage to the surrounding
soft tissues and neurovascular bundles. -
Use a tourniquet. This maximizes the freezing by minimizing heat exchange due to 2° blood flow.
-
Use the direct pour technique described by Marcove (96). Place a stainless steel funnel in the bony defect, which is insulated with Gelfoam (Fig. 127.5). Pour liquid nitrogen (–196°C) through the funnel, filling the entire cavity.
 Figure 127.5.
Figure 127.5.
The technique of liquid nitrogen as an adjuvant for a GCT of the
lateral femoral condyle. Following thorough curettement of the tumor,
totally protect the soft tissues surrounding the cavity with a layer of
gelfoam. Do not let the gelfoam rest on the soft tissues. Instill
liquid nitrogen through a stainless steel funnel. -
Use a warm saline solution to irrigate the surrounding soft tissues to minimize thermal injury.
-
Leave the liquid nitrogen in the cavity
until it evaporates completely, which typically takes 1–2 minutes
followed by 3–5 minutes of spontaneous thawing. Repeat this freeze and
thaw cycle 3 times. Some authors advocate the use of liquid nitrogen in
a spray form, claiming better control over the chemical’s dispersal.
Irrigate the cavity with saline between cycles.
coagulation. It has the capacity to penetrate only 1 or 2 mm, behaving
more like a surface agent.
-
After thorough intralesional excision,
apply phenol to the bony surface using cotton-tip applicators. Usually,
a concentration of 12% to 50% phenol is used. Treat the entire surface,
including crevices. -
Wash the cavity with alcohol to dissolve the phenol and minimize its local adverse effects. Follow with pulsative lavage.
-
Following adjuvant treatment, reconstruct the defect using bone graft, bone graft substitute, or PMMA (130). Pack PMMA from the articular surface outward (Fig. 127.3C,D).
As the PMMA sets, use iced saline lavage to help control the
temperature at the subchondral bone–cement interface to prevent thermal
damage
P.3391
to
the articular surface. Use radiopaque cement to facilitate follow-up
examinations for recurrence. If cryotherapy has not been used
autogenous and/or allogenic bone graft may be used as an alternative to
PMMA (16).
may be necessary to protect the bone until bone healing and remodeling
occurs. Several standard internal fixation devices are available,
including plates and screws, Steinmann pins, or flexible intramedullary
devices. In one large, long-term series, fracture following cryosurgery
occurred only in those patients whose construct was not augmented with
internal fixation (93).
most often arise in stage 2 or 3 lesions, or in anatomic sites that are
difficult to treat aggressively. Small local recurrences can be treated
with repeat curettage. More extensive local recurrence is probably best
treated with wide excision, which often necessitates an extensive
reconstructive procedure, such as arthrodesis or total joint
replacement for a periarticular tumor.
cortical defect (FCD) and nonossifying fibroma (NOF) are common lesions
in childhood, perhaps representing 5% of benign bone tumors (159). These benign conditions have many designations. The World Health Organization (WHO) prefers the term metaphyseal fibrous defect
and describes the lesion as “a well-defined nonneoplastic bone lesion”
containing “spindle-celled fibrous tissue with a storiform pattern and
containing a variable number of multinucleated giant cells, hemosiderin
pigment, and lipid-bearing histiocytes” (138). Nonossifying fibroma is probably the most commonly applied term, but lesions confined to the cortex are called fibrous cortical defects. The terms histiocytic xanthogranuloma, histiocytic fibroma, and nonosteogenic fibroma
also have been used. The distinction between NOF and benign fibrous
histiocytoma is subtle, and some pathologists use the terms
interchangeably (11).
It may be a developmental defect or hamartoma, associated with a
vascular disturbance or related to intraosseous hemorrhage. The
relationship to the growth plate and metaphysis suggests an alteration
in the remodeling process (12,128). The lesion is usually solitary, but it may involve multiple bones (17). Most often they slowly involute and fill in with host bone over time. Some may enlarge in the adolescent years (177).
long bones, especially the distal femur and the proximal and distal
tibia, but they may also occur in the upper extremities (proximal
humerus, distal radius, and ulna). They occur primarily in the first
two decades and are rarely seen in adults. Boys are affected slightly
more than girls (138,159). Caffey (21)
found one or several of these lesions to be present in early childhood
on skeletal surveys in 36% of individuals studied. Although
occasionally persisting into adulthood, most of these lesions disappear
soon after late adolescence and generally produce no symptoms unless a
fracture occurs (5).
characteristic to make biopsy unnecessary. They are oval, elongated,
purely radiolucent areas that align themselves along the cortex of the
metaphysis and metadiaphysis (Fig. 127.6A, Fig. 127.6B) (24,38). They are well marginated by host reactive bone and frequently exhibit bony septations. Ritschl et al. (128,129)
observed that they are most frequently seen at sites of ligament,
tendon, or intraosseous membrane attachment to the bone. The cortex may
be thinned, but the periosteum is intact. However, axial imaging with
CT or MRI may show areas of incomplete periosteal containment. By
adulthood, the epiphysis has migrated away from the lesion and it
appears as a dense osseous cortical scar or occasionally involves the
medullary cavity (now called NOF). No metastases have ever been
reported, and spontaneous regression of these lesions is the rule.
 |
|
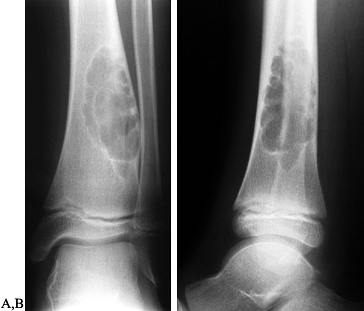
Figure 127.6. AP (A) and lateral (B)
radiographs of a typical nonossifying fibroma. It is a “bubbly” lesion aligned with the cortex, which is well marginated. The lateral cortex is thinned, but the periosteum is intact. The lesion itself is purely radiolucent, although septa of reactive bone transgress the lesion. |
injury to the ankle or knee in an otherwise asymptomatic child. It is
seldom the cause of pain, although in rare instances an NOF may be
painful for unknown reasons. In about 20% of the cases, the lesion
presents as a pathologic fracture.
whether the lesion is weakening the bone sufficiently to warrant
surgical intervention. For patients who present without a fracture,
decide whether or not to recommend surgical treatment. If left alone,
these lesions will regress and heal, usually at about the time of
skeletal maturity. In a young, athletically active child, however, the
risk of fracture may be significant. It is difficult to convince the
child to be careful for several years while the lesion heals.
lesions that appear to be significantly weakening the bone.
Unfortunately, there are no clear guidelines to make
this determination. One recommendation is to graft lesions that occupy more than half the diameter of the bone (5,21,122).
However, other authors have observed low fracture rates in NOFs of the
femur and tibia involving more than 50% of the long-bone diameter (45).
More recently, CT scans obtained by a special protocol using density
phantoms have been used. A biomechanical analysis of the CT data using
the density of the involved bone and the thickness of the cortex
compared to the uninvolved side yields a prediction of bone strength in
various loading conditions (68,104). If the bone appears to be significantly weakened, we are more likely to recommend operation.
recommend allowing the fracture to heal with cast immobilization,
followed by observation. In a fracture of a major long bone, such as
the femur, internal fixation may be necessary initially. At times, the
lesion will heal after fracture. If it does not, it seems prudent to
curet and graft the lesion to prevent subsequent fracture.
not have to be as aggressive as that for GCT, but the entire lesion
should be curetted. It may be useful to complete the curettage with a
high-speed burr.
fixation and grafting of the fracture may be indicated. The choice of
autograft or freeze-dried allograft to fill the defect is decided by
the patient and treating physician (39,120). Local recurrence is unusual.
unknown etiology that is common in the first two decades of life,
primarily between the ages of 5 and 15 years. Boys are affected three
times more commonly than girls (23,138,159).
The cysts most commonly occur in the proximal humerus (50% to 60%) or
proximal femur (25% to 30%) of a growing child, but other bones may be
affected (23,138). In older children and adults, cysts occur most commonly in the calcaneus or flat bones (147).
fracture. They may occasionally be discovered by serendipity on
radiographs obtained for other reasons. There is no mass or tenderness
unless there is a fracture. There may be angulation of the limb
secondary to the fracture, or shortening if the adjacent growth plate
is involved. Occasionally,
a cyst that has fractured will heal and disappear as the fracture heals, but this is the exception.
endothelial cells but rather by a thin fibrous lining of compressed
fibrous tissue and blood vessels. The fluid is either serous fluid
similar to extracellular fluid or blood filled if there has been a
fracture (33). The cause is completely unknown (26).
Many authors have proposed theories, but a definitive explanation has
not been forthcoming. Some presume it to be either a disorder of the
growth plate or a transient circulatory compromise resulting from a
developmental anomaly of the veins of an affected bone (31,33,34,67,77,138).
The role of trauma has been postulated but not proven. Increased
intraosseus pressure has been noted, and recently prostaglandins, free
radicals, and other bone-resorbing factors have been noted in the fluid
of the cyst (81,82,144).
This may explain the success of steroid injection in the treatment of
these lesions. The cysts may occasionally cross the growth plate and
destroy it over time, which leads to shortening (19,23,26).
In the humerus, this is not a major problem for function, but it may
produce a cosmetic problem if it occurs early in childhood. Angular
deformities are also rarely noted (109).
The cortex is thinned but intact, and the lesion is usually well
demarcated. The cavity is filled with a fluid similar to serum and
extracellular fluid, which may be bloody if fracture has occurred. Some
authors classify cysts as active when they are juxtaposed to the growth
plate, and latent when the growth plate has migrated away from the cyst
(23,26,110).
Presumably latent cysts are more likely to heal following treatment. A
“fallen fragment” sign may be present, indicating that the lesion is
fluid filled rather than solid. This sign is seen when a fragment of
cortical bone becomes dislodged and gravitates to the dependent portion
of the unicameral bone cyst. It
is seen in 10% to 20% of cysts and only in the presence of a fracture (86,102,126,153).
 |
|
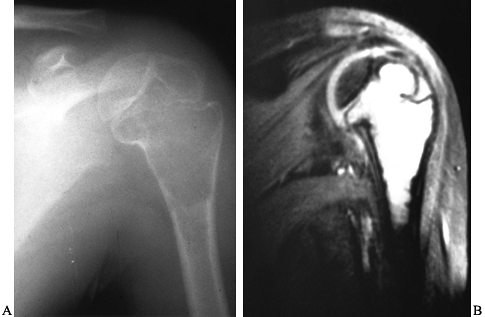
Figure 127.7. A:
A unicameral bone cyst of the left proximal humerus is purely radiolucent and thins the cortex. It is not wider than the width of the growth plate, although it extends up to it. There is a suggestion that it crosses the growth plate and enters the epiphysis, which is somewhat unusual. B: An MRI of this patient’s humerus shows the fluid-filled nature of the lesion and clearly demonstrates that it crosses the growth plate. This patient developed a limb-length inequality despite treatment of the cyst with injections. |
in unusual locations such as the pelvis. In such locations, a CT scan
or MRI can document the extent of the lesion and its cystic nature. The
MRI shows low signal on T1- and high signal on T2-weighted scans (Fig. 127.7B) (26).
It can be helpful in distinguishing unicameral cysts from aneurysmal
cysts, GCT, fibrous dysplasia, and other benign lesions. Unless there
has been a recent fracture, UBCs do not have fluid–fluid levels (20,89). A bone scan usually shows uptake at the periphery of the lesion, and a cold central area.
Despite this aggressive approach, recurrence rates from 20% to 45% have
been reported. Because of this, subtotal resection with grafting (with
recurrence rates in the 10% range) has been advocated by some, but at
the expense of a much larger operation (49,53,103). More recently, Scaglietti et al. (135,136)
in Italy proposed needle aspiration and instillation of
methylprednisolone acetate into these cysts. Although multiple
injections were often required, they reported success in more that 90%
of their cases.
not been performed, most reports have indicated that steroid injections
are as successful as curettage, if not more so (19,50).
Injection is associated with the least morbidity, so operative
treatment is reserved for those who do not respond to steroid
injection. It usually requires several steroid injections to achieve
healing of the cyst, and the mechanism of action of the steroid remains
uncertain. One suggestion has been that the steroids reduce the levels
of prostaglandin within the cyst fluid, thus retarding bone resorption (144). Campanacci et al. (23)
compared the results of curettage and bone grafting with steroid
injection in a large series of patients. They also defined radiographic
criteria for complete and incomplete healing. Complete healing rates
from curettage and grafting (46%) and steroid injection (42%) were
comparable. Partial healing was noted in another 26%. Multiple
injections led to complete healing in 50% and incomplete healing in
another 25%. Repeat curettage in the surgically treated group was
associated with a high recurrence rate. Risk of recurrence in the
curettage group was related to the proximity of the growth plate;
recurrence after steroid injection depended on the size of the cyst,
contiguity with the growth plate, and the multilocular appearance of
the cyst.
bone gel have been reported to be successful in preliminary,
unpublished reports (26). Other authors have employed high-porosity hydroxyapatite cubes (27,72) or tricalcium phosphate to fill the cavities (4,27),
with variable success. Another approach has been to inject the cavities
with a sclerosing agent similar to that used for soft-tissue
hemangiomas (1). Calcaneal cysts appear to respond better to curettage and grafting than they do to steroid injection (59) and therefore are usually treated with surgery.
-
Use standard surgical approaches to expose the wall of the cyst in the involved bone (see Chapter 1, Chapter 2 and Chapter 3).
-
Make a window in the bone and aspirate
the fluid. Completely curet the lining tissue, taking care to prevent
injury to the growth plate. Then pack the lesion with autograft or
allograft bone chips or other bone graft substitutes.
-
Inject methylprednisolone acetate into
the lesion under general anesthesia and fluoroscopic control. Place two
spinal or bone marrow biopsy needles into the cyst; aspirate the fluid
and send it for cytology. -
Inject radiographic contrast into the
cyst to ensure that the entire lesion has been filled. Sometimes
contrast will be seen to rapidly fill the draining veins. -
If there is relatively clear fluid
initially and the lesion fills with contrast, inject 80–200 mg (2–5 ml)
of methylprednisolone acetate (Depo-medrol) and remove the needles. -
Reinject the cyst at 2-month intervals until adequate healing is noted (19,23,26).
crest has been mixed with demineralized bone gel and injected into
these cysts. There are no published reports yet, but some surgeons
believe this is a more reliable method to treat the cysts. In our early
experience, no definite difference has been observed between this
method and steroid injection, but the technique deserves proper study
in a controlled study.
cyst, and in some instances to leave wires in place, to allow continued
drainage. The successful results suggest that venous obstruction may
play an important role in the etiology of UBCs, and drilling may
decrease the intracystic pressure (30,31 and 32,145). We have no experience with this technique. It is not yet used much in North America.
approximately 1.5% of primary bone tumors (141).
It is a multiloculated, radiolucent, eccentric lesion that expands the
bone, giving it a “blown out” appearance. Histologically, it shows
mesenchymal tissue–lined cysts containing blood. ABCs are believed to
be reactive lesions caused by some hemodynamic disturbance in the rich
capillary network of the host bone resulting in an expansile
destructive process. This hypothesis was based on the observation that
intracystic manometric pressure measurements were similar to arterial
pressure (14).
distinct pathologic entity or a secondary phenomenon superimposed on a
preexisting lesion (83). In up to about 50% of
cases, a preexisting lesion can be identified. The most common of these
is GCT, followed by osteoblastoma and chondroblastoma (99).
Other underlying lesions include fibrous dysplasia, NOF, chondromyxoid
fibroma, solitary bone cyst, eosinophilic granuloma, osteosarcoma,
fibrosarcoma, trauma, and metastatic carcinoma. Although the lesion is
benign, it is locally destructive with a high propensity for
recurrence. The reactive lesion can also be solid in the form of a
giant cell reparative granuloma, and then it is called a solid ABC (134).
ABC is diagnosed by exclusion, as many tumors both benign and malignant
can have a similar clinical and radiographic presentation.
life. They have been reported to arise in almost every bone of both the
axial and the appendicular skeleton, most often about the knee and in
the vertebral column. Patients complain of pain and swelling of
variable duration (weeks to years). Occasionally, a palpable mass may
be identified. In the spine, compression may cause radicular symptoms,
neurologic deficits, or even paraplegia. Patients may present with a
pathologic fracture through the cyst, but this is uncommon given the
eccentric nature of the lesion.
stage 3 disease. The radiographic appearance of an ABC is quite
distinctive and almost diagnostic (Fig. 127.8A).
The characteristic features include a subperiosteal lytic expansile
lesion inflating and thinning the cortex. In the early stages,
periosteal reaction is scarce, giving the appearance of a malignant
tumor. The lesion typically involves the metaphysis, and occasionally
the epiphysis, and it may even cross the physis. Because the periosteal
response may extend along the shaft of the bone beyond the lesion, it
may have the appearance of a finger in a balloon. Most often the lesion
is eccentric, but it can be central. In the
spine,
the lesion typically involves the posterior elements but may expand to
involve the vertebral body as well as adjacent vertebrae. CT and MRI
are useful imaging modalities, especially in the axial skeleton. They
are particularly helpful in confirming the diagnosis by demonstrating
the characteristic fluid levels within the cyst (Fig. 127.8B).
It should be stressed, however, that the presence of these fluid–fluid
levels is not pathognomonic for ABC. Other lesions, including GCT,
unicameral bone cyst with fracture, and osteosarcoma, may have this
finding.
 |
|
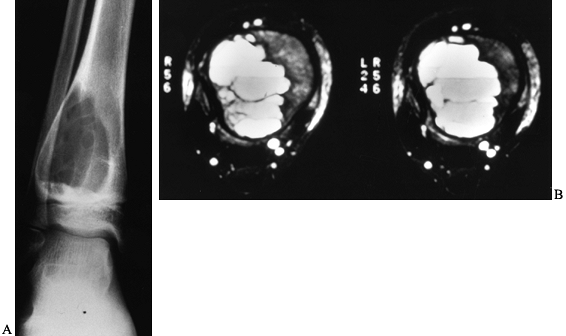
Figure 127.8. A:
AP radiograph of an aneurysmal bone cyst (ABC) shows an expanded radiolucent lesion in the metaphysis of the right distal tibia. The periosteum is intact and well marginated. There are some internal septations within the lesion. B: An MRI of this lesion shows the classic fluid–fluid levels. This was biopsied and proved to be an ABC. |
the management must be carefully considered prior to surgery. Although
the diagnosis of ABC can usually be made based on the imaging and
clinical findings, malignant conditions, namely telangiectatic
osteosarcoma, may mimic an ABC. As such, the initial surgical approach
must take into account the possibility of encountering a malignant
tumor and the need to perform a future limb-sparing procedure (see Chapter 126).
Because of the vascular nature of the lesion, a large blood loss should
be anticipated. Use tourniquets when possible and plan for blood
replacement. In addition, certain lesions may benefit from preoperative
selective arterial embolization. The extent of bony destruction must be
fully appreciated in order to restore adequate bony stability with or
without internal fixation. Because the majority of these cysts arise in
skeletally immature individuals, the proximity of the physis must also
be fully appreciated to prevent premature growth arrest.
be used in conjunction with intralesional excisions (curettage) as
discussed for GCTs. ABCs have a high incidence of recurrence after
incomplete excision. The recurrence rates after curettage alone have
been reported to be from 20% to 59% (14).
Numerous investigators have used bone graft, PMMA, or cryotherapy
(alone or in conjunction with bone grafting or PMMA) in an effort to
reduce the rate of recurrence (95,115,140).
include difficult accessibility, intraoperative bleeding, proximity of
the spinal cord and roots, necessity of removing the entire lesion to
prevent recurrence, and potential bony instability (118).
The posterior elements are usually involved, and the majority of these
lesions extend into the vertebral body. An axial imaging study such as
CT or MRI is essential in the preoperative evaluation of spinal
lesions. Most spinal ABCs can be treated through a posterior approach.
Some extensive lesions may require an anterior or combined anterior and
posterior approach (see Chapter 151 and Chapter 152).
amenable to surgery, such as when the extent of involvement is so great
that excision or curettage would risk neural compromise, or
reconstruction would be technically dangerous. Both radiation and
arterial embolization have been used with success in these instances.
While radiation has demonstrated efficacy, it is used infrequently
because of the concern for secondary radiation-induced sarcoma (146).
Arterial embolization was first explored as a neoadjuvant therapy to
decrease vascularity and hence intraoperative blood loss. However, its
usefulness as a definitive treatment method has been demonstrated (42,43).
Because spinal cord ischemia is a potential complication after arterial
embolization, perform angiography followed by a provocative arterial
infusion test with amobarbital sodium, with the patient awake or using
somatosensory evoked potential monitoring (118). If no adverse changes are observed, selective arterial embolization can be done using polyvinyl-alcohol particles.
marginal excision, or, rarely, wide excision. In bones deemed
expendable, such as ribs or the fibula, wide excision is recommended.
While marginal excision has been performed with low recurrence rates,
it is difficult to justify aggressive bone resection given that ABC is
a nonneoplastic lesion with the potential for spontaneous healing and
complete recovery after incomplete excision (141).
The most common method for treating ABCs is aggressive curettage
followed by reconstruction of the tumor cavity with bone graft or PMMA.
-
Expose the lesion under tourniquet
control whenever possible. Create a wide window by removal of part of
the expanded cortical shell. Send a biopsy specimen for frozen section
to confirm the diagnosis. -
Remove the cyst by thorough curettage
until all fibrous tissue has been excised. The cessation of bleeding
usually indicates that the lesion has been completely excised. The
margins of the excision may be further extended using a motorized burr
or a chemical adjuvant. Then assess the extent of the cavitary defect
and choose an appropriate method of reconstruction. Cancellous bone
grafting is preferred over PMMA for pediatric patients and small
contained defects.
far the most common benign tumor of bone, accounting for 35% to 50% of
benign bone neoplasms and 10% to
15% of all primary bone tumors (69).
The actual incidence is probably much higher, as most lesions are
asymptomatic and never discovered. They may occur as solitary lesions
or much less commonly as multiple hereditary exostoses (MHE), which is
most often inherited as an autosomal dominant trait although it can
also occur sporadically. MHE lesions show significant variability in
size, number, and distribution.
developmental enchondromatous hyperplasia resulting in the formation of
a cartilage-capped bony protrusion on the surface of a bone. They may
occur in any bone that develops by enchondral ossification, most often
in the distal femur, proximal tibia, and proximal humerus. In the axial
skeleton, they most commonly arise in the ilium. There is a male to
female predominance of up to 2 to 1, with most during the second and
third decades of life.
maturity should raise the suspicion of malignant transformation.
Malignant transformation almost never occurs in childhood. In adults,
consider malignant transformation of the cap when there is such growth,
and when the patient reports pain, especially in the setting of MHE.
The malignancy associated with osteochondromas is most often
chondrosarcoma, although malignant fibrous histiocytoma and
osteosarcoma have been reported (52,113,172).
It is estimated that the rate of sarcomatous change is approximately 1%
per osteochondroma, although it is probably less. Central lesions
(pelvis, scapula, ribs, spine) are at greatest risk for malignant
transformation. Chondrosarcomas arising from osteochondromas have a
better prognosis than other chondrosarcomas and rarely metastasize.
long duration on the involved bone. Pain may be present resulting from
impingement on or tethering of neighboring structures such as a tendon,
muscle, or nerve. Pain may also arise from overlying bursal
inflammation or a fracture through the stalk of the osteochondroma. On
physical examination, a palpable mass is usually the only finding.
Angular deformities or limb-length discrepancies may be present.
Juxta-articular lesions may cause limitations of joint motion. Spine
lesions may give rise to a variety of neurologic signs and symptoms
secondary to cord or root compression (2,143).
The most frequent sign of sarcomatous transformation is a sudden
enlargement of a preexisting lesion in an adult. Pain is also a common,
worrisome finding in these patients.
The roentgenographic picture of a pedunculated osteochondroma is so
characteristic that it is virtually pathognomonic. A pedunculated
lesion typically arises from the metaphysis of a long bone, with a
stalk that is continuous with the adjacent cortex and is oriented away
from the epiphysis. Sessile lesions demonstrate a flat, plateau-like
protuberance, and have a wider differential diagnosis. The outline of
the lesion is usually well demarcated, but it can vary from smooth to
irregular. Prominent cartilaginous caps produce surrounding areas of
calcification. CT and MRI are excellent imaging modalities to evaluate
any questionable lesion, as they very nicely establish continuity with
the cortex and underlying spongiosa of the host bone (Fig. 127.9C) (57).
This is an important finding to document, especially in sessile
osteochondromas that may be mistaken for parosteal osteosarcomas. When
considering chondrosarcomatous degeneration, suspicious radiographic
signs include loss of a distinctive bony margin, an adjacent
soft-tissue mass, and areas of variable mineralization within the
lesion. Historically, a cartilaginous cap larger than 1.5 cm in an
adult is considered suspicious, although there is no absolute thickness
specific for malignancy (52). While MRI and
ultrasound have comparable detection rates and measurement accuracies
for cap thickness determinations, MRI has the added ability to evaluate
the surrounding soft tissues (94).
 |
|
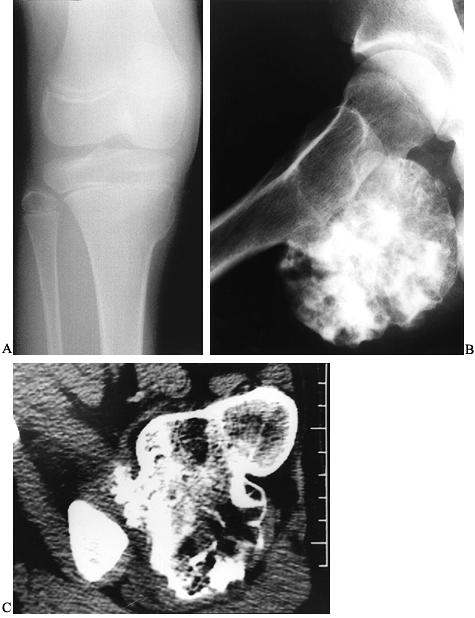
Figure 127.9. A:
AP radiograph of the tibia in a 10-year-old girl shows a small sessile osteochondroma of the right medial tibial metaphysis. It is adjacent to the growth plate. B: Another patient with a huge pedunculated osteochondroma arising from the posterior femur. At times, a lesion like this can be confused for a chondrosarcoma, which would be very rare in a child. C: The CT scan confirms that there is continuity between the cortex of the lesion and the cortex of the host bone and that the medullary cavities communicate. |
in patients with osteochondromas. Complications associated with
osteochondromas are listed in Table 127.3. In
general, osteochondromas need not routinely be removed. In one series
examining elective excision of these lesions, a 12% complication rate
following excision was reported (170). When
possible, it is best to avoid removing these lesions in skeletally
immature individuals when the lesion is in proximity to the physis. In
this circumstance, incomplete excision is likely to result in
recurrence. The primary indications for removal are pain, fracture,
nerve irritation, continued growth in a skeletally mature patient, and
concern about cosmetic appearance. Osteochondromas adjacent to major
vessels have on occasion led to pseudoaneurysm and may need to be
prophylactically removed in that instance. The important principle in
treating benign osteochondromas is to completely excise the cartilage
cap and perichondrium.
 |
|
Table 127.3. Osteochondroma: Associated Complications
|
imaging studies, such as those previously mentioned, to fully
appreciate the extent of the lesion. Treat these patients as you would
those with conventional chondrosarcoma, including staging studies (151).
A wide excision is the treatment of choice. Biopsy is usually not
performed preoperatively, as the risk of seeding the biopsy track
outweighs the possibility of obtaining a representative specimen,
considering that low-grade cartilage lesions are difficult to interpret
histologically. Most (85% in one series) of these secondary lesions are
grade 1 chondrosarcomas (see Chapter 128).
sometimes necessary to address functional impairment, angular
deformities, and limb-length inequality (121).
The three most common locations for involvement are the forearm, the
ankle, and the knee. The most common of these is forearm deformity,
resulting in ulnar deviation of the wrist associated with relative ulna
shortening, bowing of both the ulna and the radius, and late
dislocation of the radial head (6). These
deformities usually progress, leading to functional impairment and
cosmetic deformity. While a discussion of the exact surgical techniques
to correct these complex malalignments is out of the scope of this
chapter, it is worth mentioning that early recognition and intervention
may prevent progression and reduce disability. Structural ankle valgus
and genu valgum are not uncommon and benefit from corrective measures
such as hemiepiphyseal stapling (see Chapter 165, Chapter 169, and Chapter 180).
procedure performed is excision of the osteochondroma. Osteochondromas
are nearly always cured by complete excision. It is important to remove
the entire cartilaginous cap and overlying perichondrium (137).
Be careful not to violate the cartilaginous cap, as cutting into it
increases the rate of local recurrence. Remove the tumor flush with the
underlying bone. Incomplete excision of these elements is associated
with recurrence, although for benign lesions in adults this rate is
about 2%.
challenge. Fortunately, these lesions tend to cause fewer symptoms
necessitating surgery. For these lesions, it may occasionally be
necessary to leave behind part of the cortical stalk to avoid
compromising bony stability or the need to bone graft. Be certain to
remove the cartilage cap at the exostosis–host bone junction.
The majority of fractures through osteochondromas will heal with
activity restriction. Painful fibrous nonunions, particularly about the
knee, do occur. In such instances, the osteocartilaginous loose body
can be excised, usually without incident.
the possibility of sarcomatous degeneration. Use the surgical
principles for managing primary bone sarcomas (Chapter 128),
performing resection with a wide margin to minimize the risk of local
recurrence. In one large series from the Mayo clinic, intralesional
excision led to a 78% recurrence rate compared to only 15% in those who
underwent wide resection (52). For patients
with low-grade (grade 1) chondrosarcoma, wide resection is almost
always curative. Those with higher grades are at much higher risk for
local recurrence and distant metastasis. All patients, regardless of
grade, require long-term surveillance. For patients with MHE, although
they are at higher risk for malignant transformation than patients with
solitary lesions, the same criteria for excision apply.
for less than 1% of all primary bone tumors. They are composed of
immature cartilage, multinucleated giant cells, and areas of thin
calcifications with a “chicken wire” pattern. These tumors classically
arise in the epiphysis of a long bone, although they can appear in any
secondary center of ossification. Typical sites include the distal
femur, proximal humerus, and proximal tibia in tubular bones, and the
acetabulum and iliac crest in flat bones. There is a slight male to
female predominance. The majority of patients are in the second decade
of life at presentation. Although the lesion most commonly presents in
individuals
with
an open physis, tumors have occurred in skeletally mature patients.
Chondroblastoma is one of the few benign bone tumors that has the
potential to develop pulmonary metastases (152,160).
The pulmonary nodules have the same histologic characteristics as the
primary lesion, without evidence of malignant differentiation.
symptoms can be nonspecific. The majority of patients describe pain in
the affected extremity, usually of several months duration. The pain at
times may be quite severe and sharp, similar to that of osteoid
osteoma. Patients may also complain of swelling, joint stiffness, and
limp. On physical examination, approximately 50% of patients will have
tenderness to palpation over the lesion. Occasionally, a palpable
inflammatory mass will be found, raising the suspicion of a malignant
tumor. Decreased range of motion of the involved joint and muscle
wasting are also common. The lesion rarely presents with a pathologic
fracture.
Chondroblastomas commonly cross the physis and extend into the
metaphysis. Rarely is the lesion purely metaphyseal. The lesion may
also arise in an apophysis. A well-demarcated rim of sclerosis
surrounds the lytic area. Areas of calcification scattered throughout
the lesion are present in approximately one third. When these intrinsic
calcifications are not present on conventional radiographs, a CT scan
may confirm their presence, lending support to the diagnosis of
chondroblastoma. CT scans or MRI images are also of assistance in
determining the exact proximity of the tumor to the physis and
articular surface. An MRI will demonstrate the surrounding inflammatory
host response to the lesion (Fig. 127.10C,D).
Some chondroblastomas may exhibit an aggressive radiographic appearance
with erosion or even expansion of the adjacent cortex. Histologically,
these aggressive lesions often have an ABC component. Secondary ABC is
found in approximately 15% to 20% of chondroblastomas.
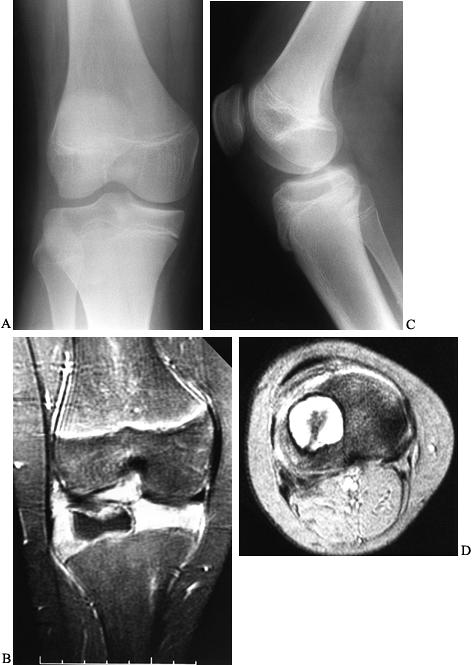 |
|
Figure 127.10. AP (A) and lateral (B)
radiographs of the right knee show a radiolucent lesion in the lateral tibial epiphysis, which extends from the growth plate to the subchondral bone. In this case, there is no stippled calcification, but this location is typical for a chondroblastoma in this age group, and biopsy confirmed this. Frontal plate (C) and cross-sectional (D) MRI images nicely delineate the extent of this lesion within the epiphysis. On the frontal plate image, surrounding edema of the epiphysis is evident. In the axial image, there is a central dark area that represents mineralization within the lesion. |
curative and the prognosis good. The standard treatment is biopsy
followed by curettage and bone grafting. With this method,
approximately 10% will recur within the bone or adjacent soft tissue,
although up to a 20% recurrence rate has been reported (71,150).
Biopsy is an important part of the treatment plan. While the
differential diagnosis of epiphyseal lesions in childhood and early
adulthood is somewhat limited (osteomyelitis and histiocytosis in rare
instances), osteosarcoma may originate in the epiphysis (epiphyseal
osteosarcoma) (158). In the adult, GCT, which
is more accurately described as epiphyseometaphyseal, is more likely;
less frequently is clear-cell chondrosarcoma seen. Clear-cell
chondrosarcoma presents as an epiphyseal lesion and looks very similar
to a chondroblastoma. The misdiagnosis of chondroblastoma in these
instances would clearly result in inadequate treatment. In general,
tumors involving the epiphysis in adolescents are usually
chondroblastomas, whereas in the adult, GCT is more likely.
surgical procedure is appreciating the close proximity of the tumor to
the physis and articular cartilage. It is important to avoid entering
the adjacent joint, as synovial membrane and articular recurrences may
follow transarticular spillage of the tumor. Most authors agree that if
the physis cannot be spared without compromising the margins of
resection, it should be sacrificed, as overly conservative curettage is
an invitation for tumor recurrence (71). Most of the time, the child is near skeletal maturity so that growth considerations are of lesser importance.
resection. For aggressive lesions, consider adjuvant therapy with
phenol or liquid nitrogen to minimize recurrence. Finally, decide if
autogenous or allogenic bone is necessary to fill the defect. Consider
the morbidity of harvesting autograft as compared to the risk of
disease transmission with allograft (58). For
small defects, autograft and allograft have similar healing rates. For
larger defects, autograft displays superior rates and completeness of
healing. This increased efficacy presumes that an adequate amount of
autogenous bone is available. Freeze-dried allograft bone is safe and
generally works quite well.
radiotherapy is contraindicated because of the average patient’s age
and the risk of secondary sarcoma. Irradiation of skeletally immature
bones leads to a reduction in bone growth, abnormal bone remodeling,
and increased propensity to fracture (60).
When possible, perform curettage in an extra-articular manner. For most
long-bone locations, standard surgical approaches can be utilized.
Intraoperative fluoroscopy is extremely helpful. Bone graft the
resultant cavitary defect. Avoid subchondral PMMA in children, as its
long-term effects on articular cartilage have yet to be determined. For
lesions not directly adjacent to subchondral bone, PMMA has been used
with success. In instances where the tumor has eroded through the
articular surface, reconstruction of the joint surfaces with an
osteoarticular allograft may be necessary. Recurrent lesions are
probably best treated by wide excision when possible.
While this lesion has historically been treated by anterior capsulotomy, an extra-articular approach has been described (152).
-
With the patient on a radiolucent or fracture table, make a lateral approach to the greater trochanter.
-
Under fluoroscopic guidance, introduce a
guide wire from the lateral femoral cortex, through the femoral neck,
into the center of the lesion in the epiphysis. -
Using a coring device or cannulated drill, drill a tunnel of sufficient diameter (approximately 1 cm) into the tumor.
-
Remove the tumor with long curets with narrow shanks and pituitary rongeurs.
-
Use an arthroscope to verify that all of
the tumor has been removed and to be certain that the articular surface
has not been violated. -
After thorough curettage, pack the cavity
with bone graft and replace the original autograft core back into the
metaphyseal window if possible. Prevention of fracture can be achieved
with interval fixation or spica case immobilization.
be one of two types depending on their location within a bone.
Intramedullary lesions are called enchondromas, and subperiosteal
lesions that form external to the cortex of a bone are termed
periosteal chondromas. Some authors prefer the term juxtacortical chondroma instead of periosteal chondroma, since these lesions can arise in areas not covered by periosteum such as the femoral neck.
medullary cavity of short, long, or flat bones and are the most common
bone tumor of the hand. From 35% to 58% occur in the hands and feet (159).
Common locations are the proximal humerus, the distal and proximal
femur, and the ribs. The lesions may occur at any age and are often
asymptomatic unless accompanied by pathologic fracture. They presumably
grow in size until maturity and then stabilize. In adults, an increase
in size, especially if associated with pain, may herald sarcomatous
change. Chondromas account for 25% of benign bone lesions, most of
which are enchondromas (69). Periosteal
chondromas are much rarer. Men and women are equally affected, most
commonly in the third or fourth decades of life. Periosteal chondromas
appear slightly earlier, probably because they are palpable and
detected by the patient.
complication of chondromas. Distal lesions, such as those of the
phalanges, almost never undergo malignant change, whereas more proximal
lesions, such as those of the pelvis or shoulder girdle, have a higher
propensity to do so (9). The rate of malignant transformation for solitary enchondromas for all locations combined is less than 1%.
Ollier’s disease is characterized by multiple skeletal
enchondromatoses. Maffucci’s syndrome is multiple enchondromatosis with
the additional component of hemangiomas. Both are nonheritable diseases
in which proliferating masses of benign cartilage afflict the bones
formed by enchondral ossification. These diseases tend to affect men
twice as often as women and are evident in childhood. The hands are
most commonly affected. Secondary chondrosarcoma is more common in both
Ollier’s and Maffucci’s syndrome. It has been estimated that
approximately 25% of patients with Ollier’s disease will develop
secondary chondrosarcoma, although this figure may be an overestimation
reflecting a bias of those patients presenting to a cancer center. It
is estimated that the incidence of malignant transformation of
enchondromas in Maffucci’s syndrome is higher, but the true incidence
is unknown because many of these patients die from complications
related to other malignancies (106).
are found incidentally as part of the workup for another condition or
coincidental local trauma. Patients may present with pain or fracture
through a weakened area of a longstanding tumor. Lesions in the
proximal humerus are often detected when the patient is being evaluated
for rotator cuff tears or tendinitis. This presents the difficult
dilemma of determining whether the symptoms are due to malignant
degeneration of the cartilage tumor or some other traumatic or
inflammatory cause. Distal phalanx lesions may lead to ruptures of the
flexor digitorum profundus tendon. Occasionally, painless swelling of a
digit may be the chief complaint. Few patients will complain of pain
related to the enchondroma in the absence of fracture. Pain in the
lesion that is sharp, immediate, and severe following some type of
strenuous or athletic activity is most likely a stress fracture through
the enchondroma. Pain referable to the lesion that is vague and of
longer duration is more likely to be associated with chondrosarcoma.
Although chronic pain is thought to be an important aspect of the
clinical history when assessing a patient for secondary chondrosarcoma,
up to 50% of patients with biopsy-proven chondrosarcoma did not
complain of pain (9).
swelling and often a palpable mass. The pain is usually described as
dull and aching, and it varies in duration from weeks to years. The
swelling tends to be localized and may gradually increase with time.
More frequently, these lesions are completely asymptomatic and
especially in children can be followed with periodic radiographic
evaluation (88).
manifestations include gradual enlargement of the fingers, bowing
deformities, shortening of the extremities, and limitations of joint
motion. Patients with Maffucci’s syndrome also present with
subcutaneous hemangiomas. The phenotypic penetrance of these two
entities is variable, with mild forms not detected until early
adulthood and severe forms obvious in the first year of life.
that of a centrally placed, lucent lesion within the metaphysis or
diaphysis, with mild forms not detected until early adulthood and
severe forms obvious in the first year of life.
that of a centrally placed, lucent lesion within the metaphysis or
diaphysis of the bone. Thinning or scalloping of the cortex may occur,
but there is usually a moderate amount of radiodense margination.
Rounded or stippled calcification is a frequent finding in adults (Fig. 127.11, Fig. 127.12).
Some lesions may show well-circumscribed areas of relative
radiolucency, with scattered amorphic calcifications centrally located
in the medullary cavity of the bone. This picture is common for an
enchondroma in the phalanges. Other lesions, such as those in the long
bones, show large granular or flocculent collections of calcifications,
with variable cortical thickening. Fractures through the thin cortices
of the lesions in phalanges may be evident. Most lesions show an
increased uptake on bone scans.
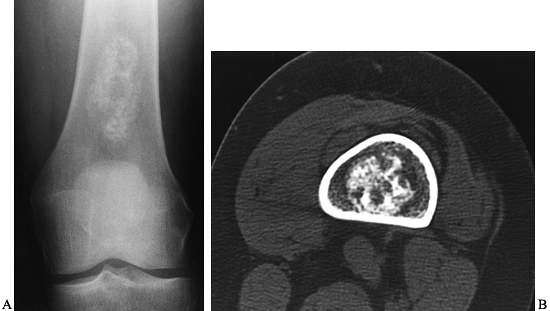 |
|
Figure 127.11. A:
Radiograph of the distal femur of a 30-year-old woman with an incidental finding of an enchondroma of the distal femur. Note the typical “popcorn” type of calcification with small rings and arcs. There is no evidence of cortical erosion. B: The CT scan from this patient confirms these findings. The cortex is intact and the mineralization of this lesion in arcs and amorphous rings is readily apparent within the medullary cavity. |
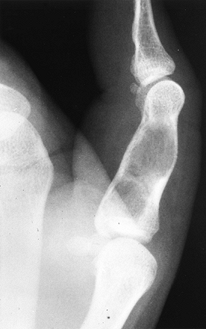 |
|
Figure 127.12.
A typical enchondroma in the proximal phalanx of the thumb of a 20-year-old man. It has enlarged the bone slightly, although the cortex is intact. There has been no mineralization, but the enchondroma has a characteristic appearance. |
juxtacortical radiolucent mass, surrounded by peripheral periosteal
host reaction at its base (Fig. 127.13). The
base of the lesion is framed by a layer of cortical reactive bone and
usually does not extend into the medullary cavity. There may be areas
of stippled calcifications throughout the cartilaginous portion of the
lesion.
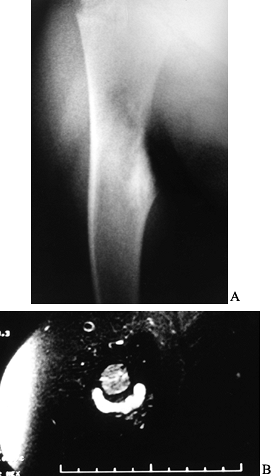 |
|
Figure 127.13. A:
AP radiograph of an 11-year-old boy shows a surface lesion of the right proximal humerus. Note “dish shaped” appearance and some periosteal elevation. B: MRI of this patient more clearly shows a cartilaginous mass sitting on the cortex of the bone. Unlike in osteochondroma, there is no communication of the medullary cavity with the lesion. |
The multiple enchondromas are expansive and bubbly. The cortical
margins are substantially thinned and may even be absent. The degree of
calcification is variable. In the long bones, the remodeling
deformities are manifested by bowing, shortening, and metaphyseal
widening.
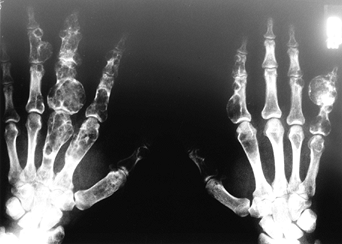 |
|
Figure 127.14.
AP radiograph of the hands shows findings characteristic of a patient with Ollier’s disease. Multiple enchondromas can be seen in the phalanges and metacarpals of the hands, with gross distortion of the bones and enlargement of portions of many of the bones. Also typical is the relative increase in severity of the disease on this patient’s left hand compared to his right. |
follow these lesions with serial radiographs and CT to monitor changes
in size and cortical destruction if the possibility of malignant change
is a concern. The frequency of radiographic evaluation, which is
necessary, has not been firmly established. We recommend that lesions
that have characteristics associated with higher rates of
chondrosarcoma, such as those in the axial skeleton, those larger than
5 cm, and those that involve more than 90% of the medullary cavity as
visualized on axial tomography, be reevaluated every 6 months (35,54). Smaller, more peripherally localized lesions can be followed yearly or biannually.
is distinguishing between benign enchondroma and low-grade
chondrosarcoma. There are no clear-cut ways to do this
radiographically, and even after histologic evaluation the distinction
may be difficult. Signs indicating possible malignancy include new
pain, soft-tissue mass, increase in size, cortical destruction,
periosteal reaction, an area of radiolucency within a previous area of
calcification, and marked increase in activity on bone scan (66,151). CT is a useful imaging modality for evaluating cortical scalloping, periosteal reaction, and soft-tissue mass (54). We feel that all adult patients with an enchondroma in a bone outside the hand should have a baseline CT scan or MRI.
-
Expose the lesion through standard
approaches. Treatment is principally by curettage and packing the
defect with autogenous or allograft bone. -
If you are concerned about malignancy, use recommended techniques for primary bone malignancies (see Chapter 128).
Make a direct and limited approach, allowing for a wide resection of
the tumor should it prove to be malignant. Make a small window in the
cortex to obtain tissue for diagnosis. -
Obtain tissue for a frozen section. If
there is doubt about the diagnosis, obtain enough to avoid a sampling
error—and wait for a permanent section diagnosis before definitive
treatment. It is advisable to plug the biopsy defect with PMMA to
prevent the spread of hematoma and tumor cells. -
For lesions that are clearly benign, make
a window in the cortex, outlining it with multiple drill holes and an
osteotome or a high-speed burr (Fig. 127.15).
Do a complete curettage to ensure complete removal. Submit all removed
tissue for histologic examination to rule out low-grade chondrosarcoma.![]() Figure 127.15. Intraoperative photograph of the patient whose radigraph appears in Figure 127.12.
Figure 127.15. Intraoperative photograph of the patient whose radigraph appears in Figure 127.12.
The lesion has been exposed through a cortical window that has been
removed. The glistening, glasslike appearance of the hyalin cartilage
that forms this lesion can be seen clearly. This lesion was completely
curetted and packed with bone graft, with a good result. -
Use adjuvants such as phenol, liquid
nitrogen, or PMMA if you are concerned about recurrence. Pack the
lesion with autograft or allograft chips. In locations where the bone
is expendable, complete local excision.
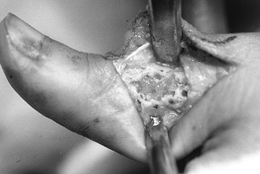
that more likely represents a developmental abnormality of bone than a
true neoplasm. No consistent familial or hereditary factors have been
identified, but recently, activating missense mutations of the Gs alpha gene leading to overactivity of adenylyl cyclase have been identified in patients with McCune-Albright syndrome (127).
This mutation can result in independent cell proliferation. The
mutation in this syndrome is a missense point mutation in exon 8 that
results in the substitution of either histidine
or
cysteine for arginine at position 201. Recently, four cases of
monostotic fibrous dysplasia were demonstrated to have such a missense
mutation, whereas other fibrous lesions and uninvolved tissue did not
contain a mutation (3). The presence of somatic activating mutations of Gs
may differentiate fibrous dysplasia from the other lesions and may be
responsible for the loss of control of local proliferation and growth
factor expression. Increased expression of the c-fos
proto-oncogene has recently been noted in patients with McCune-Albright
syndrome and may also be important in the pathogenesis of the bone
lesions in patients with fibrous dysplasia (25).
recognition may not occur until adult life. Three clinical syndromes
exist: monostotic fibrous dysplasia, polyostotic fibrous dysplasia, and
McCune-Albright syndrome (61). The monostotic
form occurs at any age, with the second and third decades
predominating. The sexes are equally involved, and the bone lesions are
most commonly located in the proximal femur, tibia, rib cage, and
facial bones (138,159).
Symptoms are usually related to deformity or pathologic fracture,
although cranial lesions may cause progressive visual impairment or
hearing loss. The polyostotic form usually presents at a younger age
because of the greater involvement of the skeletal systems, and unlike
the monostotic form, females outnumber males.
extremity (monomelic form), but 25% of patients have polyostotic
involvement of more than half of the skeletal system (64).
About 85% of patients with this form sustain pathologic fractures, and
deformities and limb-length inequality are common. Skin lesions may be
present. In McCune-Albright syndrome, polyostotic fibrous dysplasia is
associated with areas of cutaneous pigmentation and endocrine
dysfunction (40,87,91,100,167).
The skin lesions are flat and light to dark brown; sometimes, extensive
areas of pigmentation with irregular margins present as café-au-lait
lesions. Precocious puberty is the most common endocrinopathy, but
acromegaly, hyperparathyroidism, vitamin D–resistant rickets,
hyperthyroidism, and Cushing’s syndrome may be present. Short stature
secondary to premature physeal closure is common.
a lucent or “ground glass” appearance and cause thinning of the cortex
and endosteal scalloping (Fig. 127.16) (61).
The bone may be enlarged or deformed (e.g., “shepherd’s crook”
deformity of the proximal femur), and it may be involved from one end
to the other (Fig. 127.17). Tiny purposeless trabeculae are noted within the lesion.
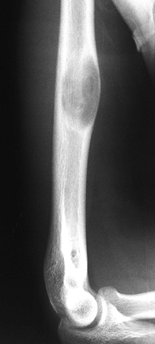 |
|
Figure 127.16.
Radiograph of an 18-year-old man with a typical lesion of fibrous dysplasia of his right humerus. He had monostotic fibrous dysplasia, which in this case involves a diaphysis with thinning of the cortex. There is a typical ground glass appearance and the lesion is relatively well marginated. |
 |
|
Figure 127.17. A:
A typical patient with polyostotic fibrous dysplasia and shepherd’s crook deformities of both femurs. The medullary cavities of both the proximal femora have been replaced with a radiolucent, ground glass lesion, which has thinned the cortex and led to the typical varus deformities seen in this condition. B: This AP pelvis radiograph is from another patient with polyostotic fibrous dysplasia. It shows more extensive involvement with gross distortion and enlargement of the bones of both the pelvis and the proximal femora. |
When malignant degeneration occurs, it is usually in fibrous dysplasia
that has received irradiation. Do not irradiate the bone lesions of
fibrous dysplasia.
bisphosphonates is currently under investigation. Intravenous
pamidronate has been shown to alleviate bone pain, reduce bone
turnover, and improve the radiographic appearance in one series of
patients with fibrous dysplasia. While these data are preliminary,
bisphosphonates look promising as an effective and well-tolerated
treatment (28).
disease in the absence of complete excision. Bone grafting is a
temporary measure, as grafts are eventually replaced by fibrous
dysplasia bone. Frequently, the goal of intervention is to maintain
bone strength and integrity, or to correct deformity, not to cure the
lesion. Symptomatic lesions can be treated by closed methods
(splinting), curettage and bone grafting, internal fixation, and wide
excision
(rarely including amputation). Asymptomatic lesions do not require treatment.
treated by closed methods. When operative intervention is necessary,
internal fixation is rarely required.
proximal femur, are particularly challenging and associated with
significant morbidity. The patient’s age at the time of symptomatic
presentation seems to play a significant role in outcome. In patients
older than 18 years, curettage and bone grafting appear to provide
satisfactory results. In contrast, patients less than 18 years old
treated by this method often sustain additional fractures, develop
progressive deformity, and require additional procedures. The use of
internal fixation in this group leads to a more favorable outcome (Fig. 127.18).
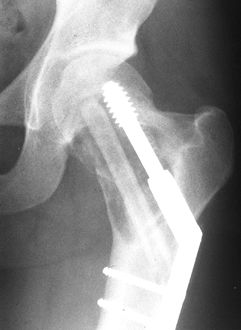 |
|
Figure 127.18.
AP radiograph of a patient with polyostotic fibrous dysplasia who had pain in his hip. To prevent a shepherd’s crook deformity, he was treated with internal fixation using a dynamic hip screw device and an allograft cortical strut to strengthen the proximal femur and femoral neck region. |
occasionally be necessary to distinguish fibrous dysplasia from
malignant lesions.
deformities (the shepherd’s crook deformity) that are difficult to
correct. In severe cases, multiple osteotomies may be necessary.
Patients with McCune-Albright syndrome are a special challenge and may
require multiple operative procedures to stabilize fractures, correct
deformity, and graft lesions (Fig, 127.19) (116).
It may be necessary to excise parts of the bone and replace them with
allograft segments, but we prefer to use fibular allograft struts
within the medullary cavity whenever possible.
 |
|
Figure 127.19. A:
This 21-year-old woman with precocious puberty, café-au-lait spots, and polyostotic fibrous dysplasia has classic McCune-Albright syndrome. She complained of bilateral hip pain and has had recurrent pathologic fractures through the large cystic lesion seen in the shaft of the left femur. This AP radiograph of the right femur shows classic findings of polyostotic fibrous dysplasia with a shepherd’s crook deformity. B: AP radiograph of the left femur shows a more severe deformity. C: AP radiograph of the right femur after correction of the varus deformity of the hip and femoral shaft with double osteotomies and fixation with a Zickel nail. D: Lateral radiograph right femur. E: AP radiograph of the left femur after a similar procedure done at the same time as the right side. F: Lateral postoperative radiograph of the left femur. Her osteotomies healed without difficulty and she experienced significant pain relief. Her wide perineum, caused by the bilateral hip deformities, returned to normal and her gait improved substantially. At the 17-year follow-up, she has had no refractures of the femurs, has married, and has had a family. Her current problems are mainly pain in the left hip due to degenerative arthritis. (Courtesy M. W. Chapman) |
histiocytosis of bone presents with bony lesions that may be confused
with benign bone tumors. It is a disorder of unknown etiology that
presents with a variety of manifestations, and the tumor cells are
believed to be derived from histiocytic cells resembling nonpigmented
dendritic cells in the epidermis known as Langerhans’ cells (162).
This group of disorders is now termed Langerhans’ cell histiocytosis,
or granulomatosis, although traditionally it was divided into three
distinct clinical entities.
self-limited disorder of bone seldom involving more than two or three
osseous sites (90,92,101,105).
It occurs most commonly in the first two decades of life, but it may be
seen up to the age of 50 years or beyond. Local pain or pathologic
fracture calls the lesion to the patient’s attention. Lytic, punched
out, irregular defects are seen on radiographs. In the spine, however,
a wafer-like appearance of the collapsed vertebral body is seen, which
accounts for the majority of cases of vertebra plana (Calvé disease).
Shortness of stature, diabetes insipidus, proptosis, exophthalmus, and
abnormal dentition (“floating teeth”) are part of this complex.
Lymphadenopathy, pulmonary infiltrates, and eczematoid weeping skin
lesions at the hairline, abdomen, and extensor surfaces of the
extremities are often seen. One third of the patients have involvement
of the liver and spleen. The occurrence of skull lesions, exophthalmus,
and diabetes insipidus is termed Christian’s triad and occurs in about
one third of the patients.
It is characterized by hepatosplenomegaly, lymphadenopathy, failure to
thrive, severe anemia, leukopenia, and thrombocytopenia. Marrow
replacement and pulmonary infiltrates often result in death.
They have a wide range of radiographic presentation and may mimic a
variety of other tumors, especially round cell tumors such as Ewing’s
sarcoma and lymphoma (Fig. 127.20A, Fig. 127.21).
In children under 5 years, exclude metastatic neuroblastoma and Wilms’s
tumor. It is wise to consider the diagnosis of histiocytosis for any
unexplained bone lesion in a child. This and fibrous dysplasia have a
wide variety of presentations. Often, histiocytosis is mistaken for
Ewing’s sarcoma. This can be made more confusing by the MRI appearance,
which can show large areas of inflammation and edema in the surrounding
marrow and soft tissues, mimicking the soft-tissue mass of Ewing’s
sarcoma (Fig. 127.20B,C). On careful
inspection, however, the distinction between the edema of histiocytosis
and the mass of Ewing’s sarcoma can usually be made. The pathologic
findings are similar in all involved sites and in all three variants.
 |
|
Figure 127.20. A:
AP radiograph of both femora of a 6-year-old boy with a punched-out radiolucent lesion of the left femur. There is periosteal elevation and thinning of the cortex. We were concerned about histiocytosis, Ewing’s sarcoma, or infection. B: MRI shows the extent of the lesion within the medullary cavity and considerable soft-tissue swelling on this coronal image. C: Axial T1-weighted MR image shows considerable edema in the surrounding tissues. This should not be mistaken for Ewing’s sarcoma, as histiocytosis commonly has significant edema surrounding the bone. After a biopsy proved that this was Langerhans’ cell histiocytosis, the patient was treated with curettage. Alternatively, this lesion could have been managed with a needle biopsy and an injection of corticosteroids. |
 |
|
Figure 127.21. A:
Lateral radiograph of the spine of a 10-year-old patient with typical vertebra plana involving T-10. This patient had a 20° kyphosis through the area. B: An MRI of this patient shows reduced height of the vertebral body at T-10 but maintenance of the disc space. There is no encroachment on the thoracic spinal cord. This patient was managed with a needle biopsy to confirm the diagnosis and treated in a brace with resolution of symptoms. |
with a thorough history and physical examination. Inquire about
polyuria and nocturia, eczematoid skin rashes, and otitis-like
symptoms. Evaluate the bone lesion(s) with plain radiographs, and
sometimes with MRI. Obtain a skeletal survey to search for other
lesions. A bone scan is also useful, but on rare occasions, the bone
scan may not detect lesions present on plain films. There is debate in
the literature over whether bone scans or skeletal surveys are best to
evaluate these patients (37,139,161,168,169).
Both are useful. Obtain a first-void urine sample for specific gravity
to exclude the possibility of diabetes insipidus. In a very young
child, urinary catecholamines and an abdominal CT scan may be necessary
to exclude the possibility of metastatic neuroblastoma or Wilms’s tumor.
leukemia, infections, and Ewing’s sarcoma. CT-directed needle biopsy is
frequently possible and avoids a surgical procedure.
seldom causes neurologic defects, and the compressed bony segment will
at least partially reconstitute in height with time (73).
Manage with bracing until the symptoms resolve. Opinion is divided as
to whether these lesions require low-dose radiotherapy (450–900 cGy) or
whether the same results might not be achieved by observation alone (63,108,142,171).
oncologist. Management is frequently by chemotherapy, usually starting
with vinblastine. In some patients, steroids and a variety of
chemotherapy regimens, rarely necessitating bone marrow
transplantation, are necessary over a protracted time to control the
disease (36,92,105,108,124,142,162,171).
eosinophilic granuloma will disappear if left alone. Open biopsy of
solitary lesions is indicated to establish the diagnosis, and this
alone is frequently curative, although
autogenous
or allograft bone graft of the curetted cavity is advisable if a large
bony defect is created. At times, internal fixation or a spica cast
will be necessary to avoid fracture until the lesion heals.
In our experience, this can be performed at the time of CT-directed
needle biopsy of long-bone lesions and is much easier for the patient.
scheme: *, classic article; #, review article; !, basic research
article; and +, clinical results/outcome study.
C, Kalifa G, Seringe R, Dubousset J. Direct Ethibloc Injection in
Benign Bone Cysts: Preliminary Report on Four Patients. Skeletal Radiol 1993;22:317.
S, Schwobel M, Pochon JP. Operative Treatment of Solitary Bone Cysts
with Tricalcium Phosphate Ceramic. A 1 to 7 Year Follow-up. Eur J Pediatr Surg 1992;2:180.
H, Brosjo O, Kreicsbergs A, Lindholm J. Low Risk of Recurrence of
Enchondroma and Low-grade Chondrosarcoma in Extremities: 80 Patients
Followed for 2–25 Years. Acta Orthop Scand 1995;66:283.
R, Manfrini M, Tigani D, Giunti A. Tricalcium Phosphate and
Hydroxyapatite Ceramics in the Surgery of Bone Tumors: Preliminary
Results. Chir Organi Mov 1991;76:245.
R, Sally P, Buckwalter K, Van Bastelaer F. MRI Assessment of Chondroid
Matrix Tumors. In: Limb Salvage: Current Trends. Proceedings of the 7th
International Symposium. 1993:89.
M, Robboy SJ, Kim S, et al. Cushing Syndrome, Sexual Precocity, and
Polyostotic Fibrous Dysplasia (Albright Syndrome) in Infancy. J Pediatr 1975;87:917.
R, Biagini R, Boriani S. Selective Arterial Embolization in the
Treatment of Aneurysmal Bone Cyst and Angioma of Bone. Skeletal Radiol 1992;21:523.
F, Waltrip R, Sponseller P, et al. Clinicopathologic Features of
Osteoid Osteoma and Osteoblastoma in Children and Adolescents. Orthop Clin North Am 1996;27:559.
M, Hermans J, Bloem J, et al. Usefulness of Radiography in
Differentiating Enchondroma from Central Grade I Chondrosarcoma. AJR Am J Roentgenol 1997;169:1097.
W, Dudley H, Barry R. The Natural History of Fibrous Dysplasia. An
Orthopaedic, Pathological, and Roentgenographic Study. J Bone Joint Surg Am 1962;44:207.
O, Ibaraki K, Shimabukuro H, Shingaki Y. Packing with High-porosity
Hydroxyapatite Cubes Alone for the Treatment of Simple Bone Cyst. Clin Orthop 1993;293:287.
T, Machinami R, Kojima T, Kikuchi F. Malignant Fibrous Histiocytoma and
Osteosarcoma in Association with Fibrous Dysplasia of Bone. Report of
Three Cases. Pathol Res Pract 1992;188:757.
R, Eckardt J, Seeger L, et al. Pulmonary Metastasis of Benign Giant
Cell Tumor of Bone. Six Histologically Confirmed Cases, Including One
of Spontaneous Regression. Clin Orthop 1994;302:219.
S, et al. Simple Bone Cyst. Treatment by Trepanation and Studies on
Bone Resorptive Factors in Cyst Fluid with a Theory of Its
Pathogenesis. Clin Orthop 1993;287:204.
D, Malawer M. Staging and Treatment of Primary and Persistent
(Recurrent) Osteoid Osteoma. Evaluation of Intraoperative Nuclear
Scanning, Tetracycline Fluorescence, and Tomography. Clin Orthop 1992;204:229.
J, Vande Berg B, Noel H, Maldague B. Benign Osteochondromas and
Exostotic Chondrosarcomas: Evaluation of Cartilage Cap Thickness by
Ultrasound. Skeletal Radiol 1992;21:33.
R, Weis L, Vagaiwalla M, et al. Cryosurgery in the Treatment of Giant
Cell Tumor of Bone: A Report of 52 Consecutive Cases. Cancer 1978;41:957.
V, Sissons H. Aneurysmal Bone Cyst: A Review of 123 Cases Including
Primary Lesions and Those Secondary to Other Bone Pathology. Cancer 1988;61:2291.
R, Springfield D, Motwani H, et al. Recurrence of Giant-cell Tumors of
the Long Bone after Curettage and Packing with Cement. J Bone Joint Surg Am 1994;76:1827.
P, Karnel F, Hajek P. Fibrous Metaphyseal Defects—Determination of
Their Origin and Natural History Using a Radiomorphological Study. Skeletal Radiol 1988;17:8.
D, Hornicek F, Wolfe M, et al. Percutaneous Radiofrequency Coagulation
of Osteoid Osteoma Compared with Operative Treatment. J Bone Joint Surg Am 1998;80:815.
N, Molt M, Roylance J. An Unusual Intraosseous Lesion with
Fibroblastic, Osteoclastic, and Fibromyxoid Elements: “Solid” Varient
of Aneurysmal Bone Cyst. Cancer 1983;51:2278.
O, Marchetti PG, Bartolozzi P. The Effects of Methylprednisolone
Acetate in the Treatment of Bone Cysts. Results of Three Years
Follow-Up. J Bone Joint Surg Br 1979;61:200.
O, Marchetti PG, Bartolozzi P. Final Results Obtained in the Treatment
of Bone Cysts with Methylprednisolone Acetate (Depo-medrol) and a
Discussion of Results Achieved in Other Bone Lesions. Clin Orthop 1982;165:33.
S, Sommelet D, Lascombes P, Prevot J. Treatment of Langerhans-cell
Histiocytosis in Children. Experience at the Children’s Hospital of
Nancy [Comments]. J Bone Joint Surg Am 1994;76:1513.
T, Arita S, Watanabe H, Chigira M. Simple Bone Cysts Treated by
Multiple Drill-Holes. 23 Cysts Followed 2–10 Years [Comments]. Acta Orthop Scand 1996;67:288.
KF Jr, Bright RW, Fitzgerald SP, Sell KW. Solitary Unicameral Bone
Cyst: Treatment with Freeze-dried Crushed Cortical-Bone Allograft. A
Review of One Hundred and Forty-four Cases. J Bone Joint Surg Am 1976;58:636.
KF, Sell KW, Brown RH. Solitary Bone Cyst: Treatment with Freeze-dried
Cancellous Bone Allograft. A Study of One Hundred Seventy-seven Cases. J Bone Joint Surg Am 1969;51:87.
M, Dorfman H. Epiphyseal Osteosarcoma: Distinguishing Features from
Clear Cell Chondrosarcoma, Chondroblastoma, and Epiphyseal Enchondroma.
Hum Pathol 1987;18:644.
Horn J, Vincent J, Wiersma-van Tilburg A, et al. Late Pulmonary
Metastases from Chondroblastoma of the Distal Femur: A Case Report. Acta Orthop Scand 1990;61:466.
SM Jr, Kenan S, Sissons HA, Lewis MM. Malignant Transformation of
Fibrous Dysplasia. A Case Report and Review of the Literature. Clin Orthop 1988;228:281.
AW, Fanning CV, Ayala AG, et al. Percutaneous Techniques for the
Diagnosis and Treatment of Localized Langerhans-Cell Histiocytosis
(Eosinophilic Granuloma of Bone). J Bone Joint Surg Am 1998;80:219.
JW, Levine AM, Dorfman HD. Case Report 293. Diagnosis: Nonossifying
Fibroma (NOF) of the Upper Tibial Diametaphysis, with Considerable
Increase in Size over a Three-Year Period. Skeletal Radiol 1984;12:294.