
VII – NEOPLASTIC, INFECTIOUS, NEUROLOGIC AND OTHER SKELETAL DISORDERS
> Tumors and Tumor-Like Conditions > CHAPTER 128 – MALIGNANT BONE
TUMORS
an aggressive pattern of local growth and a marked propensity to
metastasize (39,129,155).
It is the second most common primary malignant bone tumor after
myeloma, and accounts for 20% of all bone sarcomas. The majority of
patients are in their second decade, and men and women are affected
equally. Although this tumor can occur in any bone, the appendicular
skeleton is most frequently involved; approximately 50% of lesions
occur about the knee (138). In long bones, it
tends to occur in the metaphysis. Multifocal involvement is extremely
rare. Most patients present with a short history of pain and swelling
in the involved limb. The pain may be intermittent at first, becoming
constant later, and often accentuated at night. Because these patients
are generally young and active, pain around the knee is often mistaken
as a sports injury and is not recognized until the tumor has grown.
Patients may present with a pathologic fracture. The majority of
hematogenous metastases occur in the lungs and develop in approximately
half of the patients.
important to classify them in order to predict prognosis and design
treatment (27) (Table 128.1).
The classic lesion is a high-grade intramedullary osteosarcoma, which
occurs in 85% of cases. Approximately 1% of cases are low-grade,
well-differentiated central osteosarcomas, which have a better
prognosis (9). A variant known as small-cell osteosarcoma may be difficult to distinguish histologically from Ewing’s sarcoma (140). There are three types of surface osteosarcomas, including high-grade, periosteal, and parosteal lesions (10,18,108,124,130).
Parosteal osteosarcoma accounts for 3% of tumors, and it has a tendency
to arise on the posterior aspect of the distal femur. Its prognosis is
distinctly better than the conventional lesion. Rarely, a parosteal
osteosarcoma can dedifferentiate into a higher grade tumor if it is
neglected or incompletely excised. The telangiectatic subtype is a
high-grade, intramedullary lesion with a separate histologic appearance
that must be differentiated from benign lesions such as aneurysmal bone
cyst (94,120). It occurs in 3% of cases and has a prognosis similar to that of conventional osteosarcoma.
 |
|
Table 128.1. Classification of Osteosarcoma
|
but secondary lesions may occur after irradiation, in Paget’s disease,
or as the highly sarcomatous portion of a dedifferentiated
chondrosarcoma (44,45,157).
Transformation should be suspected in patients with known Paget’s
disease who develop a sudden increase in pain. Rarely, osteosarcoma
develops at the site of a pre-existing benign lesion such as fibrous
dysplasia or a bone infarct. Primary osteosarcoma can also occur in the
soft tissues (77).
may be present in any given tumor. The cortex is frequently destroyed
by the tumor, with extension into the surrounding soft tissues.
Telangiectatic osteosarcoma is a large blood-filled tumor that may
grossly appear similar to an aneurysmal bone cyst (ABC). Depending on
the predominant pattern, three histologic variants of conventional
osteosarcoma exist: osteoblastic, chondroblastic, and fibroblastic.
However, all of these histologic subtypes of high-grade osteosarcoma
have a similar prognosis. The small cell variant can be mistaken for
Ewing’s sarcoma under light microscopy, but current immunohistochemical
and cytogenetic studies can help differentiate the two lesions.
Periosteal osteosarcoma has a chondroblastic histologic pattern,
whereas the parosteal variant tends to be heavily ossified and have a
well-differentiated fibroblastic appearance. Telangiectatic
osteosarcoma has large blood-filled spaces, but a careful search
reveals the characteristic malignant cells producing osteoid that
differentiates it from an ABC.
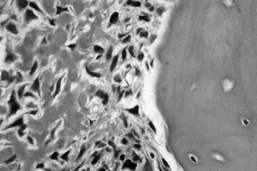 |
|
Figure 128.1. (See COLOR FIG. 128.1.) Osteoblastic osteosarcoma showing malignant spindle cells with lacelike osteoid. Trabeculum of normal bone is on the right.
|
A subset of patients with osteosarcoma have mutations in the p53 and
retinoblastoma tumor suppressor genes, although there is no consensus
on whether having the mutation affects the prognosis (96).
The tumor is aggressive with indistinct margins and evidence of
cortical destruction. An extensive extraosseous mass is common. New
bone formation occurs in a typical sunburst appearance of the
periosteum. Some lesions may be purely lytic, particularly the
telangiectatic subtype. Other lesions may be heavily ossified, such as
the parosteal type, which must be differentiated from myositis
ossificans. Magnetic resonance imaging (MRI) is the most useful
modality for determining local soft-tissue boundaries and proximity to
adjacent structures. An MRI is particularly helpful in determining
the
intraosseous extent of a lesion when planning surgery. The low
intramedullary signal seen on T1-weighted images corresponds closely to
the gross tumor extent (146). Radionuclide scans can be helpful in identifying skip lesions.
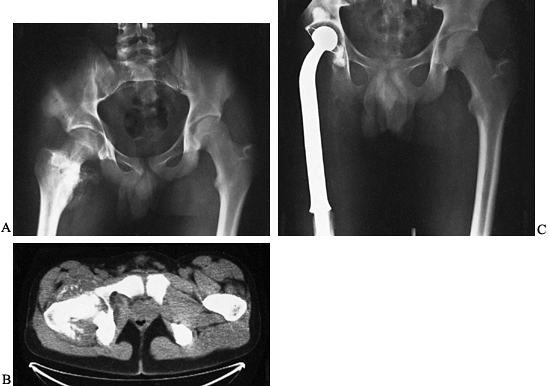 |
|
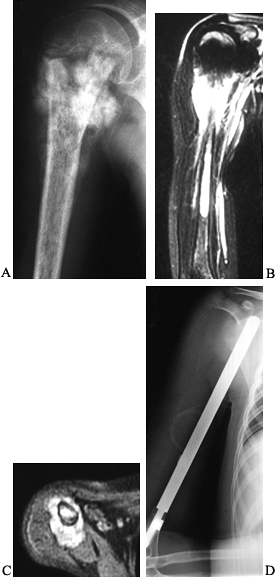
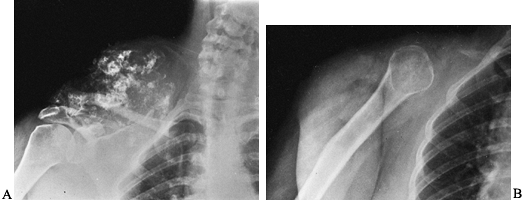
Figure 128.2. A:
AP radiograph of the pelvis in a patient with osteosarcoma demonstrating a bone-producing lesion in the right proximal femoral metaphysis. B: CT scan showing the extraosseous extension of the osteosarcoma. C: Neoadjuvant chemotherapy was administered, followed by wide resection and reconstruction using a proximal femoral replacement prosthesis. (From Sim FH, Bowman WE Jr, Wilkins RM, Chao EYS. Limb Salvage in Primary Malignant Bone Tumors. Orthopedics 1985;8:574, with permission.) |
osteosarcoma was amputation. This treatment was associated with an
overall 5-year survival of approximately 20%. The amputation was
usually performed 8 to 10 cm proximal to the most proximal extent of
the lesion, as determined by local staging studies. Despite the
excellent local control achieved with amputation, pulmonary metastases
developed in many patients (68,138). More effective treatment for systemic disease was needed to improve survival.
the early 1970s with reports of 5-year survival increasing to between
45% and 60% (76). Other controlled trials showed no apparent survival benefits compared with surgery alone (34).
However, the benefits of adjuvant chemotherapy soon became apparent,
and there is now a strong consensus that it improves survival.
Chemotherapy is now a component of all major osteosarcoma treatment
protocols (3,82,91,98,163).
The most effective agents have been methotrexate, cisplatin,
doxorubicin, and ifosfamide. All standard protocols use preoperative
(neoadjuvant) chemotherapy. This was historically done to allow time
for the manufacture of a custom prosthesis, but preoperative
chemotherapy is now used to allow more patients to undergo limb-salvage
surgery and because it serves as an in vivo
efficacy test of the chemotherapy itself. The extent of tumor necrosis
at the time of definitive resection is used as a measure of the tumor’s
response to chemotherapy, and this has been correlated with patient
survival (3,116,164).
nonmetastatic osteosarcoma included high-dose methotrexate,
doxorubicin, cisplatin, with or without ifosfamide and the
immunostimulant MTP/PE (74). Future
chemotherapy trials will incorporate the use of carboplatin. Of note,
parosteal osteosarcoma is a well-differentiated low-grade tumor with a
good prognosis after wide resection alone; therefore, chemotherapy is
generally not used in its treatment protocol. However, periosteal
osteosarcoma is intermediate in its biologic behavior, and in general,
chemotherapy is included in the treatment protocol of this tumor.
tumor response to chemotherapy before surgical resection. Then the
operation can be tailored for the individual patient. Studies using
various imaging modalities such as dynamic MRI and thallium scans have
shown promising results (40,107). In addition, work continues on the molecular level to identify predictive biologic markers (158).
improved techniques of oncologic reconstruction have allowed the
majority of patients with osteosarcoma to undergo limb salvage, as
outlined in Chapter 126. A surgical margin can
often be achieved with a limb-sparing resection that is comparable to
the margin achieved with an amputation. A large multi-institutional
study compared amputation with limb salvage for lesions of the distal
femur and found no significant difference in local recurrence rates, as
long as a wide surgical margin was achieved (141). Other investigators confirmed a low incidence of local recurrence (approximately 5%) after limb salvage (3,33).
The functional results achieved with limb salvage are generally
superior to those after an amputation. Most medical centers active in
the management of osteosarcoma report that as many as 85% of their
patients are now being treated with limb-salvage surgery (3,33), compared with fewer than 20% before 1980.
the patient with osteosarcoma. Careful preoperative staging, including
a well-planned, adequate biopsy, forms the basis of the overall
treatment plan. Despite significant advances in limb salvage surgery,
there remains a role for amputation. Extremely large lesions with
obvious neurovascular involvement, pathologic fracture with associated
contamination of multiple tissue compartments, lesions of the distal
portions of the extremities, local recurrences, and lesions in the very
young are situations in which the best treatment may be amputation.
depend on the location of the tumor and the resulting limb function
expected by the patient. The alternatives after resection of all or
part of a major joint include custom prosthesis, osteochondral
allograft, allograft prosthetic composite, or an arthrodesis (41,134,138).
Limb salvage is important for lesions of the pelvis because of the
severe functional impairment after hemipelvectomy, and because the
surgical margin that is achieved often may be the same as that achieved
by amputation. The use of current techniques depends on the location
and extent of pelvic resection. With complete iliosacral joint
resection, fusion of the remaining ilium to the sacrum can be achieved
by hinging the pelvis on the symphysis pubis. After periacetabular
resection, the proximal femur can be fused to the ilium or pubis. This
construct is enhanced by the use of vascularized fibular grafts or
intercalary allograft segments. The resultant large discrepancy in limb
length can be corrected by bone transport of the femur in selected
cases. A saddle prosthesis is an option if enough ilium remains to
obtain a stable attachment (1). Some patients function quite well
after periacetabular resection without reconstruction by developing a
painless pseudarthrosis. Allograft reconstruction of the pelvis
combined with prosthetic hip replacement has been successful, but this
complicated technique is often fraught with postoperative complications
such as allograft fracture, prosthetic loosening, or infection.
in restoring function of the hip joint after resection of a proximal
femoral osteosarcoma (135) (Fig. 128.2).
A custom prosthesis or an allograft prosthetic composite are good
choices, with an allograft prosthetic composite used more often in
younger patients to restore bone stock for future needs.
resection of an osteosarcoma around the knee joint. Extra-articular
resection and arthrodesis provide a durable limb and are an option in a
very young patient (128). Techniques can be used to lengthen the limb after completion of chemotherapy to avoid a large limb-length discrepancy (112).
Segmental arthrodesis can be accomplished by various methods. An
effective method is use of hemicylindrical femoral turn-down or tibial
turn-up grafts combined with ipsilateral fibular grafts and a long
intramedullary rod with small antirotational plates at the host–graft
junction for stabilization. At present, more surgeons use an
intercalary allograft segment to span large defects and facilitate the
arthrodesis. Use of an osteoarticular allograft has the advantage of
being a biologic solution in a young patient; however, there is a high
likelihood of complications in the first several years after surgery (89) (Fig. 128.3).
At present, our preferred limb-salvage technique is a modular total
knee replacement using a rotating hinge design. This provides immediate
functional restoration and long-term durability, which has been
improved with modern implants (19,23,33,86).
Difficulty arises when the proximal tibia is replaced with a metal
prosthesis because there is still no ideal method for reattaching the
extensor mechanism to the prosthesis. (See Chapter 126
for some alternatives.) An allograft prosthetic composite is an
effective procedure to provide restoration of bone stock and allow
improved extensor function (62). The extensor
mechanism is reattached to the allograft tendon and a rotational
gastrocnemius flap is used to cover the allograft bone and tendon.
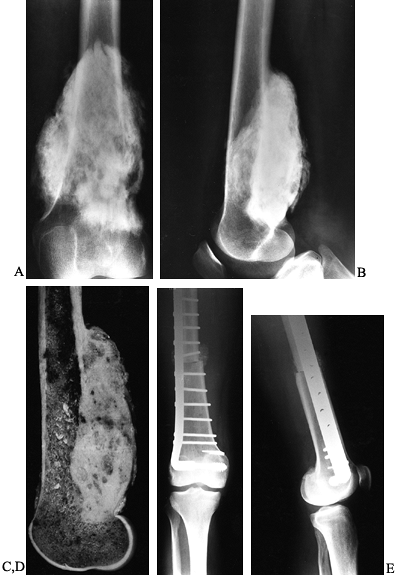 |
|
Figure 128.3. A, B:
AP and lateral views of the right distal femur in a 35-year-old woman demonstrating a heavily mineralized lesion consistent with a parosteal osteosarcoma emanating from the posterior cortex. This particular subtype of osteosarcoma is less aggressive than its conventional counterpart. C: Sagittal section through the gross specimen. The majority of the medullary cavity is free of tumor. D, E: AP and lateral radiographs of the distal femur after wide resection and reconstruction using an osteoarticular allograft. |
the shoulder. Resection of the proximal humerus often requires
sacrifice of the rotator cuff muscles, overlying deltoid, and axillary
nerve (85,90).
Reconstructive options include prosthetic replacements that use a
modular design with a porous coating for potential bone ingrowth.
Arthrodesis using dual fibular grafts, vascularized fibular
grafts, or allografts has also been successful (89) (Fig. 128.4).
If the axillary nerve and a portion of the deltoid can be maintained,
an osteoarticular allograft or allograft prosthetic composite allows
the patient to achieve reasonable function. If the scapula is involved
by the tumor along with the proximal humerus, a Tikoff-Lindberg
procedure is required, with placement of a metal spacer attached to the
remaining chest wall to provide shoulder stability (see Chapter 126).
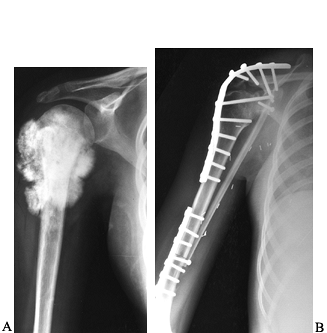 |
|
Figure 128.4. A:
AP view of the proximal end of the right humerus in a 15-year-old girl with an extensive osteosarcoma. She has no evidence of metastatic disease. B: Postresection arthrodesis in which an intercalary allograft was combined with a vascularized fibular graft. The soft-tissue extent of the tumor necessitated sacrifice of the axillary nerve. |
of a limb involves the physeal plate, the limb-length discrepancy at
adulthood may be substantial. Options that allow limb salvage for young
patients include the use of an expandable prosthesis or a
rotationplasty (81,152,162).
The Ilizarov technique for limb lengthening and bone transport may have
a role in the management of skeletal defects in young patients (112).
Innovative methods of physeal distraction to improve the marginal
resection and preserve the articular surface have been described.
the outlook for patients with osteosarcoma continues to improve. The
increase in limb-sparing surgery has not adversely affected survival.
The multi-institutional study that looked at lesions in the distal part
of the femur found no difference in survival among patients treated by
limb salvage and those treated by amputation (141). Sim and colleagues (139)
also found no adverse effects of limb-sparing surgery when compared
with amputation. Current 5-year survival rates after a
multidisciplinary approach are between 65% and 70%. The survival rate
is as high as 80% in patients with extremity lesions who have an
apparent good response (greater than 90% necrosis) to preoperative
chemotherapy (3). In patients using multiple chemotherapeutic agents, one recent study showed a 10-year survival of 68% (95).
The Mayo Clinic pilot study using ifosfamide-based chemotherapy showed
an actuarial survival rate of greater than 90% at 2.5 years (98).
For patients with metastases at diagnosis, only 40% remained free of
progression of their disease at 2 years. The outlook in patients with
systemic disease remains poor, although there are reports of long-term
survival for patients treated with an aggressive surgical approach,
including multiple thoracotomies and systemic chemotherapy (21) (Fig. 128.5). Patients who develop local recurrence of their osteosarcoma have a poor prognosis (119,141,149).
 |
|
Figure 128.5. A:
AP radiograph of the right shoulder and humerus in a 10-year-old girl with osteosarcoma. Note the mineralized lesion in the proximal humeral metaphysis. B, C: Coronal and axial MRI views showing the extensive soft tissue and intramedullary component of the osteosarcoma. There was some decrease in the extraosseous component after neoadjuvant chemotherapy. D: Extraarticular resection of the proximal humerus, followed by placement of an intercalary metal spacer. This patient had systemic metastases at presentation, which were resected via bilateral thoracotomies. |
It accounts for approximately 10% of malignant bone tumors and tends to
occur in older adults; the peak incidence is in the fifth to sixth
decades of life. More than 75% of these lesions are located in the
trunk or proximal portions of the femur or humerus. The inner wall of
the acetabulum is a particularly common site. When it is found in the
extremities, chondrosarcoma occurs centrally within the intramedullary
portion of the bone. The lesions tend to grow slowly, and pain may be
experienced for months or years before a mass or swelling is detected.
Metastasis is generally to the lung but is rare in low-grade lesions
and often occurs late in the course of the disease. However, the tumor
has a tendency to recur locally (50,121).
 |
|
Table 128.2. Classification of Chondrosarcoma
|
It tends to occur in patients who are in their second or third decade.
Two thirds of the lesions occur within the bone, and the remainder
arise in the soft tissues. Clear cell chondrosarcoma is the least
common of the chondrosarcoma subtypes. It is a low-grade malignant
tumor with a predilection for the ends of long bones, particularly the
proximal femur and humerus (11). Patients are
usually in the third or fourth decades of life, and symptoms of vague
pain can be present for years before discovery of the lesion.
Dedifferentiated chondrosarcoma is a highly malignant variant that
accounts for approximately 10% of chondrosarcomas. It is a high-grade
noncartilaginous sarcoma that arises within a pre-existing low-grade
chondrosarcoma (45,93).
as a primary lesion, or it may occur secondarily at the site of a
previous benign lesion such as an osteochondroma or enchondroma (47,103).
More than 75% are primary tumors. Patients with multiple benign
cartilage tumors, such as in Ollier’s disease, Maffucci’s syndrome, or
multiple hereditary osteochondromas, are more likely to develop a
secondary chondrosarcoma than are patients with solitary lesions.
The center of the lobules often undergoes myxoid degeneration. Cortical
destruction with soft-tissue extension is common, and calcific
densities are frequently found within the chondroid matrix. This tumor
has been divided into three histologic grades. Approximately 90% are
well-differentiated (grade 1 or 2) tumors. In general, the grade of the
tumor correlates with its cellularity (87,88).
It is important to differentiate high-grade chondrosarcoma from
chondroblastic osteosarcoma. Chondroblastic osteosarcoma has marked
cytologic atypia and osteoid production. It is also often difficult to
differentiate a low-grade chondrosarcoma from an enchondroma.
Therefore, it is extremely important to correlate the clinical,
radiographic, and histologic presentations before rendering a final
diagnosis. Usually, a chondrosarcoma has more pleomorphic cells and may
have myxoid changes within the chondroid matrix. Binucleation of the
cells is not a distinguishing feature because it can be seen in both
lesions. Chondrosarcoma of the small bones of the hand and foot is
rare, and clinical and radiographic correlation is necessary to
differentiate it from a benign cartilage lesion. Enchondromas
in
these sites are very common, but they have a much more aggressive
histologic appearance than their long-bone counterparts. This makes the
diagnosis difficult based on histology alone.
 |
|
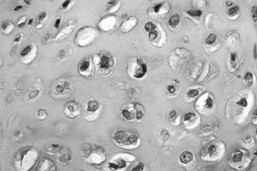
Figure 128.6. (See COLOR FIG. 128.6.)
Grade I chondrosarcoma. The lesion is quite cellular, the nuclei are enlarged and irregular, and double nucleated cells are present. |
dedifferentiated lesion. Grossly, there is an abrupt transition from
the centrally located hyaline component to the spindle cell portion.
Histologically, it is characterized by sheetlike regions of highly
anaplastic spindle cell sarcoma immediately adjacent to lobules of
well-differentiated chondrosarcoma. The spindle cell component may show
features of osteosarcoma, fibrosarcoma, or malignant fibrous
histiocytoma. Another subtype, clear cell chondrosarcoma, has a
cartilagenous matrix and is variably lobulated under low power. There
are areas of mononuclear and multinucleated giant cells. The tumor
cells have abundant clear cytoplasm and distinct boundaries.
Mesenchymal chondrosarcoma is grossly well defined, with calcific foci
scattered throughout the tumor. The low-power pattern is bimorphic,
with islands of benign-appearing hyaline cartilage among highly
cellular areas of uniform, small, round, or spindled cells. There are
numerous branching vessels with a pattern reminiscent of
hemangiopericytoma.
that arises in the medullary cavity but usually involves the cortex as
well. (See Figure 128.7, Figure 128.8 and Figure 128.9.)
Eventually it extends through the cortex and forms a large soft-tissue
mass. Matrix calcification is present in the majority of tumors and
appears as rings of mineral. Endosteal scalloping and cortical
expansion are characteristic of a chondrosarcoma, as opposed to a
benign enchondroma. In long bones, the tumor is usually metaphyseal or
metadiaphyseal in location. Periosteal chondrosarcoma is a rare variant
that arises de novo on the surface of the
bone. A tumor arising in the pelvis is less distinct on plain films but
is often associated with a characteristic large soft-tissue mass and
mottled mineralization. Computed tomography (CT) and MRI can help
define soft-tissue margins and cortical destruction in pelvic tumors.
Both are useful in detection of recurrent lesions. If the cartilage cap
on an
osteochondroma exceeds 2 cm on MRI or CT scan in a patient with clinical symptoms, consider secondary chondrosarcoma.
 |
|
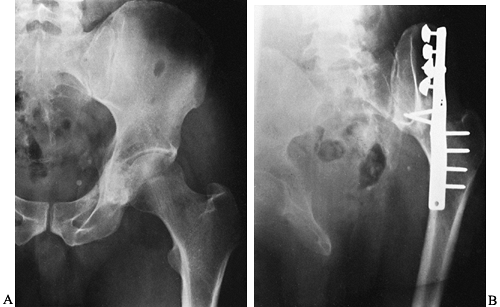
Figure 128.7. A: AP radiograph of the left hemipelvis showing a chondrosarcoma of the ilium and acetabulum. Note the stippled calcification. B: En bloc
resection, followed by iliofemoral arthrodesis. (From Sim FH, Bowman WE Jr, Chao EYS. Limb Salvage and Reconstructive Techniques. In: Sim FH, ed. Diagnosis and Treatment of Bone Tumors: A Team Approach. Thorofare, NJ: Slack, 1983:75, with permission of the Mayo Foundation.) |
 |
|
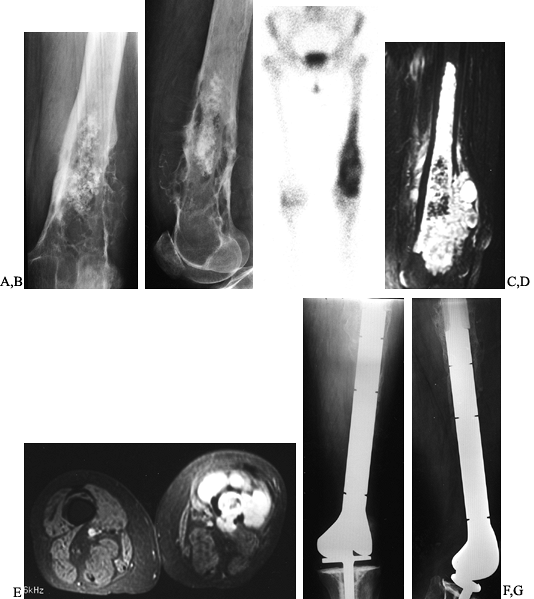
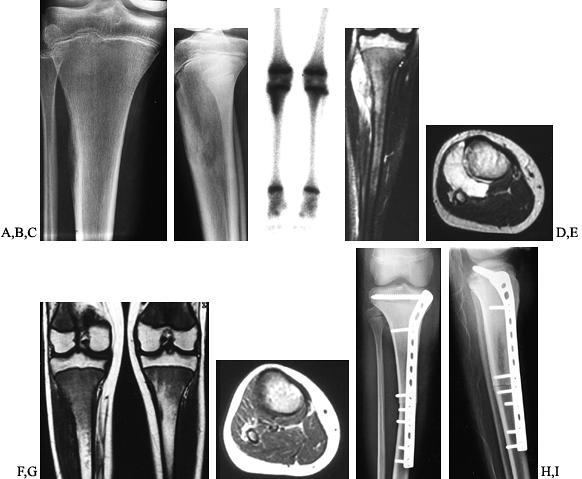
Figure 128.8. A, B:
AP and lateral radiographs of the left distal femur in a 61-year-old woman with chondrosarcoma showing a calcified intramedullary lesion with destruction of the surrounding cortex. C: Bone scan showing the extent and activity of the tumor. D, E: Coronal and axial MRI images further define the soft tissue and intramedullary extent to assist in preoperative planning. F, G: Wide resection of the distal femur followed by reconstruction using a custom rotating hinge knee prosthesis. |
 |
|
Figure 128.9. A: Anteroposterior view of right shoulder showing an extensive chondrosarcoma arising from the upper scapula. B: After scapulectomy.
|
appearance of a conventional chondrosarcoma. However, 25% have an
aggressive destructive area adjacent to the cartilage lesion. Clear
cell chondrosarcoma has an appearance similar to chondroblastoma, with
sharp margins and a sclerotic rim at the end of a long bone. There may
be some mineralization and slight expansion of the cortex. Late in the
course of the disease, there is extensive cortical destruction.
Mesenchymal chondrosarcoma has the appearance of a conventional
chondrosarcoma when it arises in bone. Matrix calcification is present
when it occurs in the soft tissues.
Two important principles apply in surgical management. First, a
well-planned and adequate biopsy that is representative of the entire
tumor is necessary. Second, a wide margin must be achieved at initial
resection to ensure the best chance of cure for this tumor, which is
notorious for local recurrence. Accurate preoperative staging with
determination of the histopathologic grade and regional tumor extent
dictates the aggressiveness needed at operation. For lesions of higher
grade with extensive soft-tissue and neurovascular involvement,
amputation may be required to ensure local control. Even lower grade
lesions that have recurred in the pelvis often involve the
neurovascular bundle and necessitate external hemipelvectomy. When a
limb-sparing procedure is feasible, the reconstructive techniques
described for osteosarcoma apply equally well for chondrosarcoma (1,19,23,35,41,62,85,90,128,134) (Fig. 128.7, Fig. 128.8, and Fig. 128.9).
Internal hemipelvectomy and iliofemoral fusion for pelvic lesions have
yielded satisfactory results. For lesions involving the proximal femur,
a custom prosthetic replacement or an allograft prosthetic composite is
an effective reconstructive option after proximal femoral resection. In
general, chondrosarcoma is resistant to chemotherapy and radiation.
chondrosarcoma is curable after wide resection. Mesenchymal
chondrosarcoma also necessitates an aggressive surgical approach to
achieve a wide resection margin. One report found radiation and
chemotherapy to be beneficial in a subset of these tumors (66). Dedifferentiated chondrosarcoma has a dismal prognosis, despite radical surgical resection and adjuvant therapy.
Because local recurrence affects survival adversely and late
recurrences are common, 5-year survival rates are unreliable indicators
of long-term results. A 10-year survival rate of 77% for grade 1, 59%
for grade 2, and 36% for grade 3 lesions was documented in another
study (121). Local recurrence occurs in 20% and pulmonary metastases in 15% (12).
curable. Patients treated with intralesional curettage have an 80% risk
of local recurrence, with metastases to the lung and other skeletal
sites (11). Patients with mesenchymal chondrosarcoma have a survival rate of 50% at 5 years and 28% at 10 years (66,102).
Results of treatment of highly malignant, dedifferentiated
chondrosarcoma are poor despite radical surgery. The 5-year survival
rate is approximately 10% (45,93).
Ewing’s sarcoma is the second most common primary malignant bone tumor
in children and the fourth most common malignant bone tumor in all
ages. It occurs slightly more often in male patients and almost never
in the black population. The tumor occurs typically in the first and
second decades of life; more than 90% of patients are younger than 30
years of age. It can arise in any bone of the body, but more than 60%
of the lesions occur in the pelvic girdle and lower extremities. When
it occurs in the long bones, the diaphysis is usually involved. Ewing’s
sarcoma can also occur primarily in the soft tissues (126).
 |
|
Table 128.3. Small Round Cell Tumors of Bone
|
may exist for several months before diagnosis. Occasionally, there are
systemic signs and symptoms such as low-grade fever, increased
erythrocyte sedimentation rate, leukocytosis, and anemia, which may be
difficult to differentiate from an infection. Only 1% of lesions occur
in the mobile spine, and these frequently present with a neurologic
deficit (56). Pathologic fractures are the
presenting feature in approximately 10% of patients. Fifty percent of
the metastatic lesions are found in the lungs, although other skeletal
sites, including the skull and vertebral column, are common.
Microscopically, it is a cellular sheetlike proliferation of small
round cells without matrix production. However, fibrous strands may be
identified traversing the lesion. The cells have indistinct cytoplasmic
borders with round or oval nuclei. It may be difficult to differentiate
Ewing’s sarcoma from other round cell tumors such as lymphoma,
embryonal rhabdomyosarcoma, metastatic neuroblastoma, and small cell
osteosarcoma by light microscopy alone. In recent years, great strides
have been made using immunohistochemical and molecular genetic methods
to differentiate Ewing’s sarcoma from other small round cell tumors (Table 128.4). Cytogenetic studies have revealed a reciprocal translocation (11;22) (q24;q12) found in both Ewing’s
sarcoma and primitive neuroectodermal tumor (PNET). Reverse
transcriptase polymerase chain reaction (RT-PCR) assays can be used to
look for this translocation even if traditional cytogenetic testing is
unsuccessful (28,131).
Other translocations have also been identified. In addition, these
tumors have a cell surface glycoprotein called p30/32 MIC2, which is a
product of the MIC2 gene located on the shorearms of the X and Y
chromosomes. This glycoprotein can be recognized by commercially
available monoclonal antibodies.
 |
|
Table 128.4. Ewing’s Sarcoma—Ancillary Studies
|
 |
|
Figure 128.10. (See COLOR FIG. 128.10.) Small uniform nuclei with marked hyperchromasia typical of Ewing’s sarcoma.
|
cavity and commonly involves an extensive portion of the diaphysis of a
long bone (Fig. 128.11). Lytic destruction in a
mottled or moth-eaten pattern is the most common radiographic
appearance. The margins are indistinct and the actual bone involvement
seen on plain films may be subtle. Cortical destruction with periosteal
elevation and multiple layers of subperiosteal new bone gives the
classic “onion skin” appearance; however, this is not pathognomonic for
Ewing’s sarcoma. Radiating spicules of new bone in a sunburst pattern
can also occur, making radiographic differentiation from osteosarcoma
difficult. In rare lesions with little or no intramedullary involvement
(e.g., subperiosteal Ewing’s sarcoma), saucer-shaped destruction of the
exterior cortex is a fairly characteristic feature. As in other
malignant tumors, MRI is most helpful in defining the soft-tissue
extension that is common in Ewing’s sarcoma (140).
 |
|
Figure 128.11. A, B:
AP and lateral radiographs of the right tibia in a 13-year-old girl with Ewing’s sarcoma. Note the periosteal elevation along the lateral tibial cortex. C: Bone scan demonstrates increased activity throughout the metadiaphysis. D, E: Coronal and axial MRI images before neoadjuvant chemotherapy. Note the large soft-tissue mass extending to the surface of the fibula. F, G: Coronal and axial MRI images after chemotherapy. The soft-tissue mass has resolved. H, I: Transepiphyseal resection of the tibia followed by reconstruction using an intercalary allograft. The extensor mechanism was reconstructed from allograft tissues. |
multiagent chemotherapy. Over the past 20 years, multi-institutional
chemotherapy studies have greatly enhanced our ability to increase the
long-term survival rates in patients with this disease (54,156).
In 1981, the first Intergroup Ewing’s Sarcoma Study Group (IESS-I)
demonstrated that adding doxorubicin (Adriamycin) to the standard
regimen of vincristine, cyclophosphamide, and actinomycin-D increased
survival (104). IESS-II showed that dose
intensification of the four-drug regimen gave a significant improvement
in disease-free and overall survival. Seventy-three percent of patients
with nonmetastatic, nonpelvic disease were relapse free at a median
follow-up of 5.6 years (16,142).
IESS-III compared the intensified four-drug regimen with a similar
protocol with the addition of ifosfamide and etoposide. There was an
overall improvement in disease-free survival with the six-drug regimen
that was most striking in the young age group and in patients with
pelvic tumors (55,99).
Current national cooperative studies are comparing the standard
six-drug regimen with use of these agents over a shorter time course
with intensification of ifosfamide and cyclophosphamide.
considered to be radiosensitive. In the past, radiotherapy protocols
consisting of 4000 to 6000 cGy were delivered to the affected bone to
provide the primary means of local control along with chemotherapy.
However, radiation inconsistently controlled the primary tumor, and
local recurrence rates were 15% to 20% after treatment. Microscopically
viable tissue was found in 65% of these patients in autopsy studies (156).
The late morbidity associated with irradiation includes limb-length
inequality, pathologic fracture, fibrosis, and ankylosis. The increased
possibility of radiation-induced sarcoma led to a renewed interest in
the role of surgical treatment for local control of Ewing’s sarcoma (51,77,106,153).
resection of expendable bones, such as the proximal fibula or rib
lesions. The role of surgery expanded dramatically with reports of
increased survival associated with a wide surgical margin (111,161).
The current philosophy is use of intense neoadjuvant chemotherapy to
decrease the size of the primary lesion, followed by wide resection (Fig. 128.11).
Radiation is added postoperatively if the surgical margin is
intralesional or marginal. The use of modern imaging techniques such as
MRI has improved the surgeon’s ability to define the lesion and make
better decisions about the feasibility of resection with and without
reconstruction. MRI scans are performed before and after completion of
neoadjuvant chemotherapy. Many studies have shown a survival advantage
for patients treated with surgical excision of their primary tumor
compared with those treated with radiation for local control. Studies
looking specifically at surgical resection of pelvic tumors reveal
mixed conclusions (37,42,132,161).
In general, these studies are retrospective with limited numbers of
patients, and thus, the results must be evaluated with caution.
However, there does seem to be a trend for prolonged survival as well
as a decreased recurrence rate for patients who undergo surgery for
local control of their tumor. Surgery should be considered in all cases
in which the surgeon believes that the primary tumor can be completely
removed with a wide margin.
Reconstruction options exist similar to those discussed for
osteosarcoma. The same challenges of limb salvage in pediatric patients
exists in Ewing’s sarcoma. Limb lengthening by an expandable prosthesis
or through bone transport are options for young patients when wide
resection necessitates loss of the physis (see Chapter 126).
Complicated surgical reconstructions that may necessitate a prolonged
chemotherapy-free interval should be avoided, or else the entire course
of chemotherapy should be given before surgery. The latter approach is
encouraged in the management of patients with Ewing’s
sarcoma of the spine, in whom surgical procedures are frequently staged and often complicated.
sarcoma was considered one of the most lethal sarcomas, with a 5-year
survival rate of less than 20%. With a modern, multidisciplinary
approach to treatment, the overall 5-year survival rate has improved to
greater than 70% (4,16,69,104). As with osteosarcoma, a favorable response to preoperative chemotherapy is a good prognostic sign (118).
Survival rates as high as 80% to 90% have been reported for patients
with extremity lesions who have more than 90% necrosis after
neoadjuvant treatment (69). In addition,
patients with distal tumors have an improved prognosis compared with
those with central lesions in the pelvis or sacrum. Poor prognostic
variables include large tumor size, pelvic location, metastatic disease
at initial presentation, and poor response to preoperative chemotherapy
(4,36,69,161).
In a review of 140 patients at the Mayo Clinic, 25% presented with
metastatic disease. The 2-year survival rate of this subset was 39%
compared with those with nonmetastatic disease, who had a 2-year
survival rate of 69% (161).
by the proliferation of spindle cells without any discernible matrix
production (39,129,155).
This tumor accounts for less than 4% of primary osseous malignancies
and occurs in patients from the second through sixth decades of life.
Patients with fibrosarcoma have an age distribution different from
those with fibroblastic osteosarcoma, although the tumors may be
similar histologically. Men and women are affected equally. The
skeletal distribution is similar to that of osteosarcoma, with more
than 50% occurring in long bones, usually the femur or tibia. Most of
these lesions arise de novo, but 25% can
be considered secondary, arising at sites of pre-existing disease or
after irradiation. Symptoms of fibrosarcoma include pain and swelling,
which may exist for a long time before diagnosis. Fifteen percent of
patients present with a pathologic fracture. Fibrosarcoma metastasizes
to the lungs in a high proportion of cases.
Some well-differentiated lesions may be firm, dense, and seemingly well
circumscribed. Poorly differentiated lesions tend to be soft and
friable with regions of myxoid degeneration. Areas of hemorrhage and
necrosis are frequently evident. Cortical destruction is common, and
soft-tissue involvement may be extensive. Fibrosarcoma has the same
histologic features as its soft-tissue counterpart; however, it
infiltrates through and destroys the bone (8).
It is characterized by spindle cells arranged in a herringbone pattern.
The cells form interlacing bundles of collagen fibers. They show
varying degrees of cytologic atypia and stroma production. Some lesions
are so well-differentiated that they can be confused with benign
fibrous lesions such as desmoplastic fibroma (155).
Other tumors are highly anaplastic, with little or no recognizable
collagenous stroma. Approximately 68% of the lesions are grade 3 or 4.
 |
|
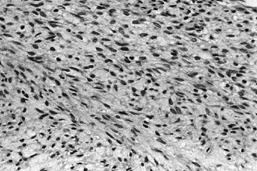
Figure 128.12. (See COLOR FIG. 128.12.) Well-differentiated fibrosarcoma with fascicles of spindle cells without pleomorphism.
|
There is osteolytic bone destruction with cortical and soft-tissue
extension suggesting a malignant process, but it is difficult to
differentiate from other malignancies such as osteosarcoma (Table 128.5).
The absence of calcification or ossification can be helpful in ruling
out other lesions. Periosteal new bone formation is uncommon. Most
lesions are eccentrically located, with permeative or moth-eaten
patterns of destruction evident well beyond the principal area of
lysis. A fibrosarcoma can originate on the periosteal surface, but most
lesions appear to arise from the intramedullary portion of the bone,
with frequent extension into the adjacent soft tissues. MRI aids in
determining osseous extent and in defining regional margins.
 |
|
Table 128.5. Fibrosarcoma of Bone—Differential Diagnosis
|
 |
|
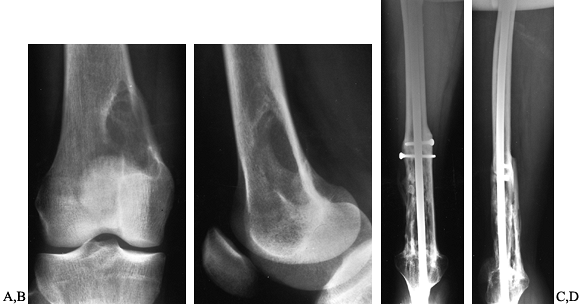
Figure 128.13. A, B:
AP and lateral radiographs of the right distal femur in a 25-year-old woman with fibrosarcoma. There has been cortical destruction along the medial femoral cortex. C, D: Extra-articular resection, followed by allograft arthrodesis, which is solidly healed three years later. |
it is apparent that a wide margin cannot be obtained to achieve local
control, an amputation is required. The principles and techniques of
limb-sparing surgery to preserve function yet achieve local tumor
control have been previously discussed (19,23,134). The reconstructive choices are similar to those for osteosarcoma and chondrosarcoma (Fig. 128.13).
may have a palliative role in lesions deemed unresectable. Neoadjuvant
multidrug chemotherapy programs similar to those used for osteosarcoma
are being recommended for treatment of systemic disease, although
long-term results are not available (8,65).
It behaves similar to malignant fibrous histiocytoma of bone. In recent
studies, the 5-year survival rate for fibrosarcoma is approximately 45%
with or without adjuvant treatment (113). This reflects the improved preoperative staging and sophisticated imaging modalities compared with those of prior eras (65).
As with other spindle cell sarcomas, the prognosis relates to the
histology with low-grade, well-differentiated lesions having a
distinctly better prognosis than high-grade tumors (65,122). Other factors associated with a poorer prognosis for survival include age greater
than 40 years and location of the tumor in the axial skeleton.
Metastases, primarily to the lung, are reported in up to 75% of
patients with this rare tumor. Therefore, development of effective
adjuvant chemotherapy protocols is a priority. Treatment of the
metastatic lesions with aggressive thoracotomy has been reported (147).
malignant neoplasm. It has previously been difficult to differentiate
from fibrosarcoma and fibroblastic osteosarcoma, but it is now
recognized as a distinct clinicopathologic entity. The histiogenesis of
this tumor is debated, but many believe that it is derived from
primitive mesenchymal cells. It is similar to its soft-tissue
counterpart but much less common, accounting for approximately 5% of
malignant bone tumors. Arising in patients of any age, it reaches its
peak incidence in the fourth and fifth decades, and it occurs slightly
more frequently in male patients (20,39,67,129,155).
Malignant fibrous histiocytoma occurs as a metaphyseal lesion in the
appendicular skeleton, particularly around the knee, but it can also be
found in the pelvis and axial skeleton. Patients present with pain with
or without swelling. Occasionally, a pathologic fracture is noted at
initial presentation (Fig. 128.14).
 |
|
Figure 128.14. A, B:
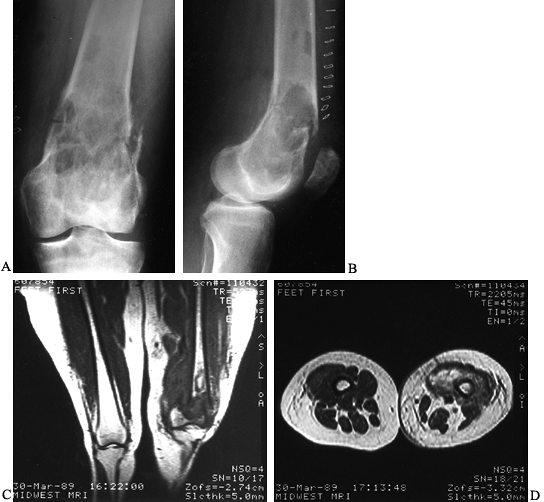
AP and lateral radiographs of the left distal femur in a 66-year-old woman with malignant fibrous histiocytoma of bone. Note the extensive metaphyseal involvement, with thinning of the cortices and a pathologic fracture. C, D: Coronal and axial MRI images reveal the large soft-tissue component of the tumor, along with the hematoma from the pathologic fracture. This patient was treated with an above-knee amputation. |
arise as secondary tumors in pre-existing conditions such as bone
infarcts or Paget’s disease (26,46,157).
They also occur as the predominant histologic type in postirradiation
sarcomas and the sarcomatous portion of dedifferentiated
chondrosarcomas (45,64).
They are typically gray to yellow, with areas of necrosis and
hemorrhage. Cortical destruction and soft-tissue extension are
prominent features. Malignant fibrous histiocytoma has a pleomorphic
pattern at low magnification. The tumor contains both malignant spindle
cells and malignant histiocytic cells. The spindle cell regions have a
matted or storiform pattern reminiscent of primary soft-tissue lesions.
Considerable cytologic variation is identifiable at high magnification.
Histiocytic appearance is characterized by indentation of nuclei and
large, well-defined cytoplasm. Other cells show a spindle cell
arrangement with ovoid to ellipsoid nuclei. The vast majority of
lesions are high grade. Osteoid or
chondroid
matrix production by the tumor cells precludes a diagnosis of malignant
fibrous histiocytoma. Differentiation from fibrosarcoma may be
difficult. The particular histologic features of this tumor have no
prognostic significance.
 |
|
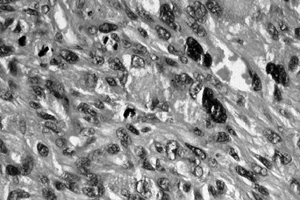
Figure 128.15. (See COLOR FIG. 128.15.) High-grade malignant fibrous histiocytoma. There is marked pleomorphism.
|
characterized by geographic lysis, but occasionally, there is a
permeative picture. Cortical destruction and indistinct margins suggest
an aggressive malignancy. There is a wide zone of transition from
normal to abnormal bone. Little reactive periosteal or sclerotic bone
is seen. Radionuclide scans are helpful in detecting other lesions. CT
and MRI scans confirm the cortical destruction and aid in determining
soft-tissue margins and the relation of the tumor to adjacent
neurovascular structures.
similar to that of other high-grade bone tumors. Surgery is the
mainstay of treatment, and the goals of adequate surgical control with
maximal preservation of limb function determine the feasibility of
limb-sparing surgery (134). Often, a pathologic
fracture or extremely large soft-tissue mass that comprises
neurovascular function will necessitate amputation (Fig. 128.14).
Lesions that cannot be adequately excised or present in problematic
axial skeleton locations may be treated with adjuvant or primary
radiotherapy. Occasional long-term survivors have been reported after
treatment with radiation only (105).
report an increased disease-free survival rate of 67% at median 7 years
using a neoadjuvant chemotherapy protocol similar to that used for
osteosarcoma. However, effective tumor necrosis was seen in only 25% of
patients. The overall survival rate for patients treated surgically in
the Mayo study is approximately 52% at 5 years and 41% at 10 years (105). Inadequate surgical margins, metastatic disease at presentation, age
greater than 40 years, and axial tumor location have been correlated with a poorer prognosis (105).
Local recurrence is increased after inadequate surgical margins. There
is debate on whether the prognosis is worse in patients with a
secondary malignant fibrous histiocytoma (20,64,105).
Metastasis occurs hematogenously, usually to the lungs but also to
other osseous sites. Patients occasionally can be cured with aggressive
resection of pulmonary lesions.
It has a wide clinical spectrum of disease, accounting for the variety
in its presentation. It is most commonly manifested by widespread
skeletal involvement with marked overproduction of monoclonal
immunoglobulins. Myeloma is the most common primary malignancy of bone,
accounting for approximately half of all bone tumors. Most patients are
between 50 and 70 years old. The most common tumor locations are in
bones that contain active hematopoietic elements; more than 85% of the
lesions involve the axial skeleton and proximal portions of the femur
and humerus.
 |
|
Table 128.6. Plasma Cell Proliferative Disorders
|
a pathologic fracture occurs. Systemic symptoms of weakness and
fatigue, along with abnormal laboratory findings, are helpful in
establishing a diagnosis. Systemic amyloidosis occurs in 15% of
patients with multiple myeloma. Patients have a characteristic
immunoglobulin profile that can be demonstrated with
immunoelectrophoresis. This particular finding is one part of a set of
specific criteria necessary to diagnose myeloma accurately. Diagnosis
of multiple myeloma is based on one major and one minor, or three
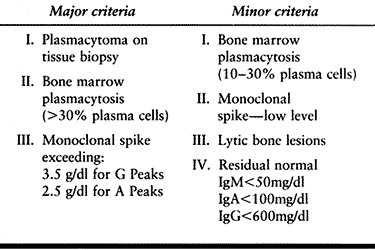
minor, diagnostic criteria, as outlined in Table 128.7.
It is important to differentiate this tumor from metastatic disease,
which occurs in a similar age group and causes similar clinical
symptoms. The classic staging system of Durie and Salmon (31) identifies patients at risk for shortened survival.
 |
|
Table 128.7. Multiple Myeloma—Diagnostic Criteria
|
(plasmacytoma), which is defined as the concurrent finding of a
positive biopsy of an isolated lesion, a negative bone marrow aspirate,
and a negative skeletal survey, occurs in approximately 25% of patients
(43). Subsequent dissemination of the disease
occurs in more than 50% of these patients by 10 years. More than half
of the solitary osseous lesions are located in the vertebral column (78).
Solitary extramedullary plasmacytomas, often located in the upper
airway passages, can occur alone or in patients in whom multiple
myeloma later develops.
 |
|
Table 128.8. Multiple Myeloma—Poems Syndrome
|
The gross boundaries are indistinct, and often the medullary and
extraosseous involvement is greater than anticipated. Microscopically,
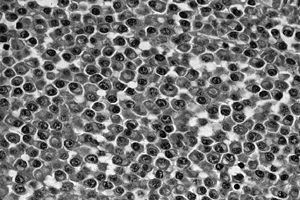
the tumor consists of sheets of closely packed plasma cells. The cells
are homogenous, with eccentric nuclei having a
clumped
chromatin pattern. There is abundant granular cytoplasm, and
multinucleated cells are occasionally seen. A variable amount of pink,
amorphous amyloid is present in 10% to 15% of myelomas (144).
 |
|
Figure 128.16. (See COLOR FIG. 128.16.) Sheets of plasma cells typical of myeloma. There is abundant pink cytoplasm, and the nuclei are structured eccentrically.
|
The lesions are small and uniform. Typically, there is no reactive zone
of sclerosis. A balloonlike expansion of bone may occur, particularly
in rib lesions. Osteosclerotic lesions are either osteoblastic or mixed
in appearance (123). In 15% to 25% of patients,
no discrete lysis occurs, and diffuse osteopenia and osteoporosis are
the only skeletal manifestations. MRI can help define the lesion when
there is nothing definite on plain films. Technetium 99m bone scans are
less reliable in identifying myeloma lesions because of a lack of
osseous response to the tumor. A skeletal survey with plain tomography
or CT is more helpful in finding additional lesions or defining the
extent of osseous destruction. Sixty percent of patients develop
pathologic fractures, with the majority being vertebral compression
fractures.
 |
|
Figure 128.17. A:
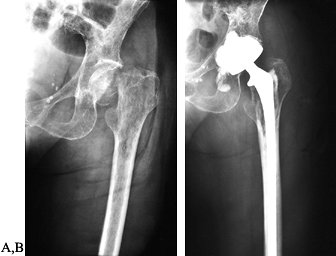
AP radiograph of the left hip in a 66-year-old woman with multiple myeloma. The patient has widespread lytic lesions throughout the proximal femur with a pathologic fracture of the femoral neck. B: A cemented long-stemmed hip replacement was quickly able to restore the patient’s ambulatory function. |
of the lesion; complete cure is rare. The primary systemic modality for
patients with multiple myeloma is chemotherapy. Most protocols include
the use of melphalan (L-phenylalanine mustard) and prednisone (52).
Combination chemotherapy has not been definitively shown to prolong
overall survival compared with the above-mentioned agents in randomized
trials (109,115). A
positive response to treatment can be monitored objectively by
observing a decrease in M-type proteins, the rate of hematologic
recovery, and occasionally, resolution of the skeletal disease. No
single test can be reliably used to follow multiple myeloma, but serial
serum protein electrophoresis or test for quantitative immunoglobulins
is probably the most reasonable. Recent work using interventions such
as interferon and peripheral stem cell transplantation is promising (5). In addition, biologic substances such as interleukin-6 are being targeted in cytokine growth factor–directed treatment (53).
Despite this and other exciting new molecular work, there has been only
minimal improvement in the overall survival of the patient in the last
20 years.
are causing disabling pain or limitation of activity. Radiation tends
to slow the growth of the tumor, which allows microfractures to heal.
Occasionally, the lesion reossifies with a return of structural
integrity. This, along with occasional bracing, is the standard
treatment for patients with vertebral involvement. A more aggressive
approach to therapy is warranted for patients with solitary myeloma. A
radiation dose of 4500 cGy to the solitary lesion has been shown to
prevent local recurrence (43). Localized extramedullary disease of the upper respiratory tract can be cured with radiation (58).
for patients with vertebral involvement who have compressive paraplegia
and for patients with impending or actual pathologic fractures.
Decompressive laminectomy is indicated if radiation does not cause
resolution of symptoms, and moderate to excellent return of function
can be expected. Spinal stabilization is occasionally warranted, and
recent advances in the techniques of anterior and posterior
instrumentation have improved the effectiveness in restoring stability (79).
The indications for prophylactic internal fixation of an impending
pathologic fracture are the same as those used in metastatic disease (57,79,97,136,137).
The principles of open reduction and internal fixation, often using
supplemental methylmethacrylate, can achieve skeletal stability in most
of these fractures (167). For those involving the femoral neck, resection and prosthetic replacement are effective (114,135).
For subtrochanteric lesions, our preferred technique is the use of a
reconstruction nail with methylmethacrylate augmentation. If there is
extensive involvement of the proximal femur, a standard sliding hip
screw and side plate has been shown to fail with progression of the
disease; therefore, more extensive reconstruction techniques, including
proximal femoral replacement, may be indicated. A more aggressive
surgical approach is warranted in a patient with a solitary
plasmacytoma because survival may be prolonged.
is poor because patients tend to die within 3 years of diagnosis. The
5-year survival rate is 18% for multiple myeloma, with a median
survival time of 20 months and a 1-year survival rate of 66% (78).
Chemotherapy improves the median survival time to 3 years in the 50% to
60% who respond to treatment. Poor prognostic variables include the
presence of M-proteins, pancytopenia, hypercalcemia, diffuse skeletal
lesions, and renal failure. New biologic assays are emerging that
identify patients who are likely to have long-term survival. Young
patients with a low β2M level and a low plasma cell labeling index have
a better prognosis, according to recent studies (51).
plasmacytoma is 74% at 5 years and 45% at 10 years. Fifty percent of
patients progress to multiple myeloma (6,43).
reticuloendothelial system. It accounts for approximately 3% of primary
osseous malignancies; an equal number of cases represent secondary
osseous involvement. Non-Hodgkin’s lymphoma is three times more likely
to have bone involvement compared with Hodgkin’s disease. There are
well-described and useful classification and staging systems for both
types of lymphoma (17). Any age group can be
affected, but it is rare in the very young and peaks in the fifth to
seventh decades of life. In primary lymphoma of bone, the lesions tend
to be diaphyseal or metadiaphyseal, occurring in the proximal femur,
proximal humerus, and distal femur. In disseminated disease, the bony
involvement is commonly in the axial skeleton, including the pelvis and
spinal column (15,155).
Pain, often present for many months or years, is a constant feature.
Neurologic symptoms are common with spinal lesions, as are pathologic
fractures. Systemic disease is either via hematogenous spread or
direct, contiguous extension from the nearby soft tissues or lymph
nodes.
The tumor is generally soft if it has completely destroyed the bone but
is firm if residual osseous trabeculae exist. Areas of necrosis may be
present. Extraosseous masses are usually soft and friable.
Microscopically, the tumor consists of an admixture of large and small
cells amid a fine reticulin meshwork. It is composed of a proliferation
of histiocytes and lymphoid cells at various stages of differentiation.
Most common is the diffuse large-cell, B-cell phenotype. Varying
degrees of fibrosis may be present. Histologically, primary malignant
lymphoma of bone is indistinguishable from a lymphoma originating
elsewhere. Immunohistochemical markers can help identify and
subclassify these tumors.
 |
|
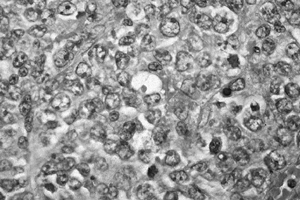
Figure 128.18. (See COLOR FIG. 128.18.) Malignant lymphoma demonstrating proliferation of large cells with irregular nuclei. There is no matrix.
|
metadiaphyseal region, although 25% to 100% of the bone may be involved
in a mottled or patchy fashion on plain radiographs. The margins are
indistinct, with marked cortical destruction and soft-tissue extension.
In some lesions, there is cortical thickening. There is usually no
matrix mineralization. At times, plain radiographic changes may be
subtle, but MRI scans show diffuse marrow signal change.
Differentiation from Ewing’s sarcoma, metastatic carcinoma,
osteomyelitis, and Paget’s disease may be difficult.
staging of the patient with lymphoma. In the past, inadequate staging
has confused the analysis of clinical outcome. Routine blood counts and
chemistries, chest radiograph, chest/abdomen/pelvis CT scan, and
bilateral bone marrow biopsies are required. The primary treatment of
lymphoma of bone is radiation (Fig. 128.19).
The entire bone is treated with 4000 cGy, with a boost to the affected
region of 4500 to 5000 cGy. Local control up to 85% has been reported (30).
Chemotherapy is generally advocated for systemic disease, and different
regimens are used for Hodgkin’s and non-Hodgkin’s lymphoma (92).
The use of chemotherapy in the treatment of lymphoma with a single bony
focus (stage 1E) is debated but appears to show some benefit in small
series (2,38). Surgery
is reserved for impending or actual pathologic fractures that cannot be
managed reasonably by other means. Surgery is also indicated for
isolated, uncontrolled lesions of an extremity that have failed to
respond to radiation treatment.
 |
|
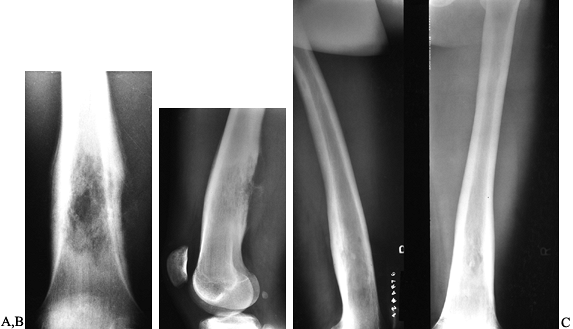
Figure 128.19. A, B:
AP and lateral radiographs of the left distal femur in an 18-year-old man with lymphoma of bone. Note the lytic destruction with extensive periosteal reaction. C: Radiographs of the left distal femur 5 years after treatment with radiation and chemotherapy. |
Metastasis or recurrence may occur many years later, making 5-year
survival values less reliable. The clinical staging of lymphoma has
important prognostic significance (148).
It is a lesion that originates from primitive notochord remnants and
accounts for 1% to 4% of primary osseous malignancies. It arises
primarily at the cephalad and caudad regions of the spine, with the
remainder within the cervical, thoracic, and lumbar vertebral bodies.
Sacrococcygeal lesions comprise 50% of the tumors. The vast majority of
sacral lesions present in patients between 50 and 70 years of age and
are extremely unlikely in those younger than 30 years of age (155). Chordomas are slow-growing, locally invasive tumors, but they metastasize in 10% of patients (22).
Symptoms depend on the location. The lesions arising at the base of the
skull present a decade earlier than their sacral counterparts because
they have less space to grow before causing symptoms. Vertebral
chordomas cause symptoms as a result of pressure on nerve roots or the
spinal cord (13). Patients may have numbness in
an extremity, followed by pain. Many develop motor weakness, and
paralysis is a late complication. The sacrococcygeal lesions can become
quite large before discovery and may be associated with a long history
of vague lower back pain. This may be referred to the hip or knee with
further neoplastic progression. The mass usually displaces but does not
invade the rectum and may cause constipation. Bladder symptoms such as
urinary frequency are common, and incontinence is a late finding. If
the chordoma originates caudal to the S-1 level, there is rarely any
sensory or motor disturbance to the lower extremities. Late in the
course, pain may become severe and intractable. Sacral lesions often
arise as a fixed, palpable presacral mass noted on rectal examination.
deceptively well circumscribed, but the tumorous tissue often extends
beyond visible boundaries (Fig. 128.20; see also COLOR FIG. 128.20).
It is soft, gray, and has a gelatinous consistency. Translucent areas
may give it the appearance of a chondrosarcoma or mucinous carcinoma.
The tumor can be focally cystic or hemorrhagic. The periosteum of the
affected bone may be elevated, and a large soft-tissue mass is common.
 |
|
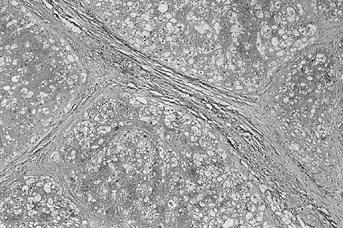
Figure 128.20. (See COLOR FIG. 128.20.)
Typical appearance of chordoma. The tumor is divided into lobules with fibrous septa. Tumor cells float in a blue myxoid background. |
Lobules are separated by fibrous septa. Abundant basophilic
extracellular matrix contains mucin and stains positively for glycogen.
The cells are arranged in cords rather than being isolated in
individual lacunae. Occasional islands of bone or cartilage are
visible. On higher magnification, the cells are noted to be of various
sizes and shapes with indistinct boundaries. The most classic type,
called a physaliforous cell, has a round nucleus with multivacuolated
cytoplasm, giving it a bubbly appearance. The vacuoles may displace the
nucleus to the periphery of the cell. The differential diagnosis
includes liposarcoma, metastatic carcinoma, and myxoid chondrosarcoma.
(Fig. 128.21).
The anterolateral mass is usually more extensive than the bony
involvement. The lesions are poorly marginated and may be difficult to
discern in the sacrococcygeal region. In the vertebral body, the
chordoma is lytic, centrally located, and slowly expansile. Areas of
sclerosis due to reactive bone formation are seen. Adjacent vertebral
bodies and the intervening disc space can be involved.
 |
|
Figure 128.21. A, B:
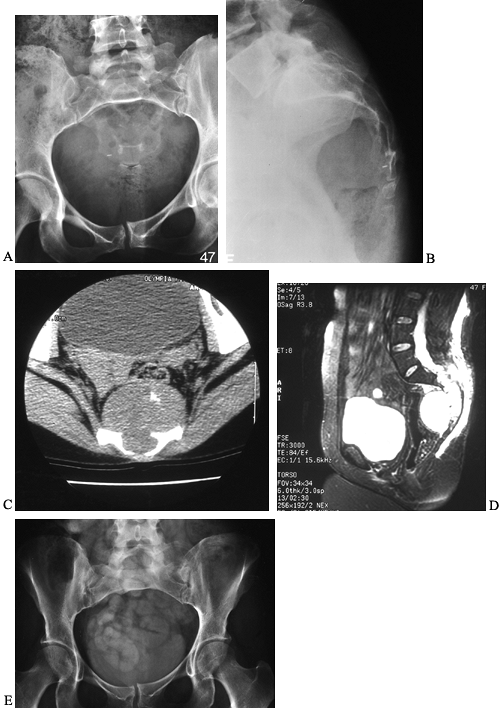
AP and lateral radiographs of the sacrum in a 47-year-old woman with a chordoma. It is often difficult to see the lucency on plain radiographs. C: CT scan through the distal sacrum reveals a large presacral mass with characteristic flecks of calcification. There is midline destruction of the sacrum. D: Sagittal MRI scan with a T2-weighted sequence reveals the large mass to be pressing on the rectum and extending proximally to the S2–S3 junction. E: Postoperative radiograph of the sacrum after en bloc resection at S2–S3. |
scan, and accumulation of isotope in the bladder can obscure the sacral
area. CT scans and MRI have been extremely helpful in determining the
extent of the lesion and determining its proximity to vital structures.
This is essential in preoperative planning. CT scans identify calcified
areas that are not evident on plain films. Along with myelography, CT
is helpful in planning resection of a vertebral lesion. MRI is useful
in discovering recurrent nodules after surgical resection. Angiography
is only occasionally indicated to identify the proximity of a cervical
chordoma to the vertebral arteries. The radiographic differential
diagnosis includes metastatic disease, multiple myeloma, giant cell
tumor, and neurogenic tumors.
to be a chordoma, perform a biopsy after all staging studies are
completed. A posterior needle biopsy can be used in collaboration with
an experienced bone pathologist. On the other hand, most biopsies are
performed through an open posterior approach. Under no circumstances
should a transrectal biopsy be performed due to contamination of
intervening tissue planes.
This resection is easier to perform in sacrococcygeal lesions because
the anatomy of the skull base often precludes complete excision (48,165).
The sacral nerve roots must be sacrificed, if necessary, to obtain an
adequate margin. Given the large size, poorly accessible location, and
tendency to adhere to the bowel, a wide margin is difficult to achieve,
so a marginal resection is often the best that can be done. There is a
high recurrence rate with inadequate resection, and recurrent lesions
carry a poor prognosis because they often infiltrate the surrounding
tissues.
intravenous antibiotic prophylaxis to decrease the chance of wound
infection. Intraoperatively, suture the anus to prevent possible fecal
contamination of the surgical field. Close the wound primarily only if
this can be done without undue tension. The operative time can be
prolonged with difficult resections; therefore, attention to blood loss
is critical. Greater blood loss can be expected in higher sacral
lesions from the middle sacral vessels and branches of the internal
iliac veins. Control hemorrhage by ligation of one or both of the
internal iliac vessels, particularly in lesions above S-3.
Postoperatively, administer antibiotics and provide prophylaxis for
deep venous thrombosis. Resection of lesions below the third sacral
level can be accomplished through a posterior approach, as described by
MacCarty and colleagues (84; see also references 25 and 83).
Lesions more cephalad often need a combined anterior and posterior
approach in the same operative setting. For high sacral lesions,
sacrifice of involved nerve roots and plans for a colostomy should be
anticipated. Surgical resection in this location is associated with
high morbidity. Full sacral resection can be performed when necessary
to achieve a wide margin. Current advances in skeletal reconstruction
can maintain stability after extensive resections (see Chapter 126).
Thoracic lesions should be approached via thoracotomy, although
combined anterior and posterior reconstruction and stabilization is
often required after vertebral body excision. A retroperitoneal
approach is usually adequate for midlumbar to lower lumbar lesions.
treatment has been used for contaminated surgical margins or surgically
inaccessible lesions. Given the increased recurrence rate of large
lesions, attention has focused on new preoperative and postoperative
irradiation approaches to improve the chance of local control (125,145).
The amount of irradiation is limited by the sensitivity of the spinal
cord in the cranial and cervical regions and by the pelvic organs,
colon, rectum, and overlying sacral skin in more caudal lesions. To
facilitate the use of high local radiation doses without structural
damage, proton beam and photon beam radiation techniques have been
successful, but their long-term effectiveness is yet to be established (145).
Wound healing problems contribute to high morbidity after radiation
therapy. In addition, there have been reports of postirradiation
high-grade sarcomas after treatment of a chordoma (147).
Although this is a rare occurrence, radiation therapy should not be
routinely prescribed to all patients with this tumor without careful
consideration. Chemotherapy has no current role in the management of
chordoma.
the adequacy of the initial surgical resection. Multiple studies have
shown increased rates of recurrence with intralesional margins compared
with wide excision. One report showed an increased recurrence rate from
28% after en bloc resection of a chordoma to 64% if the tumor was exposed during resection (70).
resection and on which sacral roots remain. If the S-1 level is
preserved, loss of motor function is minimal (71). Stener
and Gunterberg (143)
reported on extirpation of high sacral tumors that no deficit of
urogenital or anorectal function occurs if there is only unilateral
sacrifice of all sacral nerves. If only the first sacral roots are
preserved, no sphincter control will remain, and the patient will need
routine bladder self-catheterization. If bilateral S-2 roots are
maintained, up to 50% of patients may retain partial bowel and bladder
control with need for catheterization. If at least one S-3 root is
saved, sphincter control will most likely be retained. Conversely, if
the tumor is left untreated, 100% of patients will eventually have
complete incontinence. Reconstruction is not needed if pelvic
continuity can be maintained through preservation of half of the first
sacral body (143).
All lesions were approached posteriorly, and 14 had adjuvant
radiotherapy. Four patients died, and 15 of the remaining 17 were
disease-free at average 4.5-year follow-up. There was a 19% local
recurrence rate, which compares well with the available literature,
especially because a combined operative approach was not used. It was
suggested that preoperative or postoperative radiation therapy allowed
a better result after a marginal resection. There was a 5% incidence of
metastases at 5 years, which increased to 50% at 10 years.
Eighty-one percent were approached posteriorly, and only 19% were
resected with a wide margin. Fifty-two percent received adjuvant
postoperative irradiation. The cumulative probability of local
recurrence was 51% at 5 years and 75% at 10 years. When the lesion
extended to the S-1 level, there was 100% recurrence in seven cases.
There was a 24% probability of metastases at 5 years and 58% at 10
years. All but one patient with distant metastases also had local
recurrence.
likelihood ranging from 5% to 40% in the literature. They can be found
from 1 to 16 years after initial diagnosis. This finding is limited to
sacral, or less likely vertebral, chordomas; the intracranial types
rarely metastasize. Accurate prediction of which tumors will
metastasize is not yet possible. The spread is to the regional lymph
nodes, as well as the skin, lung, liver, and bone. Almost all patients
die as a result of complications from local treatment failure rather
than metastases, which are commonly asymptomatic. Studies show survival
of patients with chordoma to be 45% to 77% at 5 years and 28% to 50% at
ten years.
Half of the lesions occur in the second and third decades, and the
remainder present throughout life. There is a slight male predominance.
Ninety percent are found in the diaphysis of the tibia. Ten percent of
patients have an ipsilateral fibular adamantinoma. The etiology of
these tumors is not clear (59). They may have
their origin in cells capable of differentiating into either epithelial
or mesenchymal components, or else they are primarily an epithelial
malignancy with a reactive fibrous stroma.
pain. It is a slow-growing lesion, and symptoms may be present from a
few weeks to 50 years. More than one third of patients have symptoms
for more than 5 years before diagnosis. A mass is the only finding on
physical examination. An associated local traumatic event occurs in
half of the patients, but no causal effect has been documented. There
is slow but progressive bony destruction throughout adulthood.
Metastases occur late, most often to the lungs, and account for 70% of
the reported deaths from adamantinoma (100).
dysplasia, a benign tumor that presents in a similar location. However,
the clinical presentation of osteofibrous dysplasia is in the first
decade and generally asymptomatic. It is a self-limited process that
does not progress or metastasize (75,101).
It is grayish white and may have areas of necrosis, hemorrhage, or
cystic spaces. It is well marginated and easily distinguishable from
the surrounding normal tissue. Most are greater than 5 cm in length and
can often involve the entire tibial shaft.
 |
|
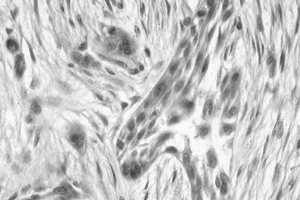
Figure 128.22. (See COLOR FIG. 128.22.) Clusters of epithelial cells surrounded by fibrosis diagnostic of adamantinoma.
|
appearances of the epithelial components in the adamantinoma; however, all of the epithelial cells stain strongly for keratin (7,159).
Disorganized bony fragments may be seen within the stroma. The stroma
can look strikingly similar to fibrous dysplasia. The nuclei are bland
with minimal atypia in only 15%. Usually, no mitotic figures are seen,
but they can be present without causing alarm. Electron microscopy and
immunohistochemical staining for cytokeratin or other epithelial
markers will confirm the lesion as an adamantinoma. The histologic
differential diagnosis includes metastatic carcinoma, vascular
neoplasms, synovial sarcoma, and osteofibrous dysplasia.
A “soap bubble” appearance is classically described, with the tumor
centered in the diaphysis of the tibia and asymmetrically expanding the
anterior cortex.
The
lytic areas are sharply defined and may extend into the metaphysis but
never occur in the epiphysis. Penetration of the cortex occurs in 15%
of cases. Although there is a dominant central lesion, the eccentric
lucent areas can occur throughout the shaft, separated by areas of
sclerosis and having a multiloculated appearance. There may be bowing
of the tibia in longstanding lesions. A bone scan is usually intensely
positive because of the extensive bony reaction, but no uptake is seen
past the edges of the lesion as defined on plain films. CT scans
demonstrate whether or not the cortical lesion has penetrated into the
soft tissues or medullary canal. MRI scans do not aid in the diagnosis
but are valuable for preoperative planning. The radiographic
differential diagnosis includes fibrous dysplasia and osteofibrous
dysplasia (14).
 |
|
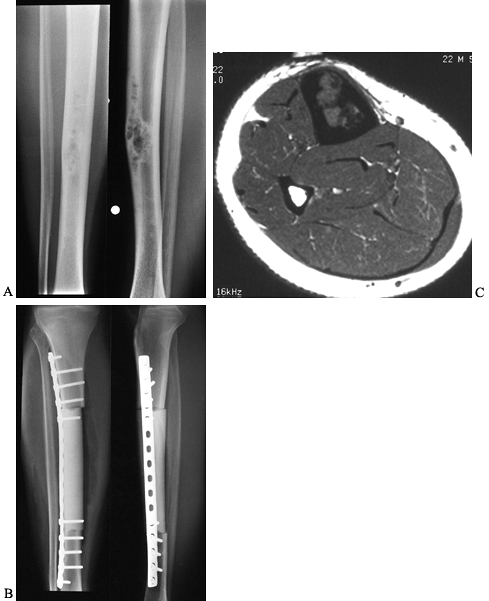
Figure 128.23. A:
AP and lateral radiographs of the tibia in a 23-year-old man with adamantinoma. Note the characteristic bubbly appearance of the anterior cortex in the tibial diaphysis. B: Axial MRI scan reveals medullary involvement of the tibia with expansion of the cortex but no soft-tissue mass. C: Following wide resection of the adamantinoma and reconstruction with an intercalary allograft. |
adamantinoma. In the past, the malignant potential of this tumor was
underestimated, and numerous cases were treated by local excision or
curettage. This approach is associated with a 60% chance of local
recurrence (72). It is now recognized that a wide margin is required by resection or amputation (49).
All of the satellite lesions must be removed to minimize recurrence.
Reconstruction after resection of a tibial lesion can be achieved by
using intercalary allografts or vascularized fibular bone grafts (Fig. 128.23).
If the entire shaft is involved, wide resection may not be practical,
and amputation may be necessary. Amputation is also considered for
recurrent lesions or those with extensive soft-tissue involvement when
a wide margin cannot be obtained. Mankin and colleagues (49)
described excellent early results after treatment by large segmental
tibial resection followed by intercalary allograft replacement.
Adjuvant radiation or chemotherapy has no role in the initial
management of adamantinoma.
the mortality rate was 18%, and known metastases were present in 70% of
the patients who died. Metastases develop in 15% to 20% of patients and
are more common following an inadequate initial resection with
recurrent disease. Given the slow-growing nature of this tumor, an
aggressive approach to pulmonary or lymph node metastases is warranted.
Local recurrence and metastases may occur months to decades after
treatment of the primary lesion; therefore, long-term follow-up is
necessary (60). It is important not to
underestimate the malignant potential of this tumor. Strict adherence
to the principles of oncologic surgery ensure an optimal result.
scheme: *, classic article; #, review article; !, basic research
article; and +, clinical results/outcome study.
AJ, Buch R, Mathews J, et al. Reconstruction Using the Saddle
Prosthesis Following Excision of Primary and Metastatic Periacetabular
Tumors. Clin Orthop 1995;314:203.
G, Picci P, Ferrari S, et al. Primary Chemotherapy and Delayed Surgery
for Nonmetastatic Osteosarcoma of the Extremities: Results in 164
Patients Preoperatively Treated with High Doses of Methotrexate
Followed by Cisplatin and Doxorubicin. Cancer 1993;72:3227.
M, Campanacci L, Gamberi G, et al. Cytokeratin Expression and
Distribution in Adamantinoma of the Long Bones and Osteofibrous
Dysplasia of Tibia and Fibula. An Immunohistochemical Study Correlated
to Histogenesis. Histopathology 1994;25:71.
F, Bacchini P, Fabbri N, et al. Osteosarcoma, Low Grade Intraosseous
Type, Histologically Resembling Parosteal Osteosarcoma, Fibrous
Dysplasia, and Desmoplastic Fibroma. Cancer 1993;71:338.
J, van der Heul R, Schuttevaer H, Kuipers D. Fibrous Dysplasia vs
Adamantinoma of the Tibia: Differentiation Based on Discriminate
Analysis of Clinical and Plain Film Findings. AJR Am J Roentgenol 1991;156:1017.
EO Jr, Nesbit EM, Garnsey LA, et al. Multimodal Therapy for the
Management of Nonpelvic Localized Ewing’s Sarcoma of Bone: Intergroup
Study IESS-II. J Clin Oncol 1990;8:1517.
R, Bertoni F, Bacchini P, et al. Malignant Fibrous Histiocytoma of
Bone: The Experience at the Rizzoli Institute: Report of 90 Cases. Cancer 1984;54:177.
PFM, Sim FH, Pritchard DJ, et al. Megaprostheses after Resection of
Distal Femoral Tumors. A Rotating Hinge Design in 30 Patients followed
for 2–7 Years. Acta Orthop Scand 1996;67:345.
S, Emmanuel R, Unni K, Sim F. Treatment of Sacrococcygeal Chordoma: A
Review of 47 Cases. Presented at the AAOS meeting February, 1995,
Orlando, Florida.
O, Zucman J, Melot T, et al. The Ewing Family of Tumors—A Subgroup of
Small Round Cell Tumors Defined by Specific Chimeric Transcripts. N Engl J Med 1994;331:294.
BGM, Salmon SE. A Clinical Staging System for Multiple Myeloma.
Correlation of Measured Myeloma Cell Mass with Presenting Clinical
Features, Response to Treatment, and Survival. Cancer 1975;36:842.
JH, Green SJ, Ivins JC, et al. A Controlled Pilot Study of High-Dose
Methotrexate as Postsurgical Adjuvant Treatment for Primary
Osteosarcoma. J Clin Oncol 1984;2:152.
R, Nesbit M, Askin F, et al. Local Recurrence, Rate and Sites of
Metastases, and Time to Relapse as a Function of Treatment Regimen,
Size of Primary and Surgical History in 62 Patients Presenting with
Nonmetastatic Ewing’s Sarcoma of the Pelvic Bones. Int J Radiat Oncol Biol Phys 1985;11:129.
RG, Nesbit ME, Gehan EA, et al. Multimodal Therapy for the Management
of Localized Ewing’s Sarcoma of Pelvic and Sacral Bones: A Report from
the Second Intergroup Study. J Clin Oncol 1991;9:1173.
BD, Hanna SL, Fairclough DL, Gronemyer SA. Pediatric Musculoskeletal
Tumors: Use of Dynamic, Contrast-enhanced MR Imaging to Monitor
Response to Chemotherapy. Radiology 1992;184:243.
FJ, Sim FH, Frassica DA, et al. Survival and Management Considerations
in Postirradiation Osteosarcoma and Paget’s Osteosarcoma. Clin Orthop 1991;270:120.
FJ, Unni KK, Beabout JW, Sim FH. Dedifferentiated Chondrosarcoma: A
Report of the Clinicopathologic Features and Treatment in Seventy-Eight
Cases. J Bone Joint Surg 1986;68-A:1197.
SJ, Weintraub HP, Proppe KH. Malignant Fibrous Histiocytoma and
Pleomorphic Sarcoma in Association with Medullary Bone Infarcts. Cancer 1978;41:607.
M, Lord F, Rosenberg A, Mankin H. The Treatment of Adamantinoma of the
Tibia by Wide Resection and Allograft Bone Transplantation. J Bone Joint Surg 1987;69-A:1177.
H, Krailo M, Link M, et al. Improved Outcome in Nonmetastatic Ewing’s
Sarcoma and PNET of Bone with the Addition of Ifosfamide and Etoposide
to Vincristine, Adriamycin, Cyclosphosphamide, and Actinomycin: A
Children’s Cancer Group and Pediatric Oncology Group report.
(Abstract.) Proc Am Soc Clin Oncol 1994;13:421.
KD, Sim FH, Enis JE, et al. Methylmethacrylate as an Adjunct in
Internal Fixation of Pathological Fractures: Experience with Three
Hundred and Seventy-Five Cases. J Bone Joint Surg 1976;58-A:1047.
AG, Rosen G, Dabska M, Marcove R. Mesenchymal Chondrosarcoma: A
Clinicopathologic Analysis of 35 Patients with Emphasis on Treatment. Cancer 1983;51:1230.
AG, Woodard HQ, Hellweil M. Postradiation Malignant Fibrous
Histiocytoma of Bone: A Clinicopathologic Study of 20 Patients. Am J Surg Pathol 1986;10:9.
N, Patel SR, Benjamin RS. Chemotherapy in Osteosarcoma. Basis for
Application and Antagonism to Implementation; Early Controversies
Surrounding its Implementation. Hematol Oncol Clin North Am 1995;9:825.
H, Exner U, Gadner H, et al. Multidisciplinary Treatment of Primary
Ewing’s Sarcoma of Bone: A 6-Year Experience of a European Cooperative
Trial. Cancer 1988;61:23.
M, Ertel I, Makley J, et al. A Randomized Study Comparing High-Dose
Methotrexate with Moderate-Dose Methotrexate as Components of Adjuvant
Chemotherapy in Childhood Nonmetastatic Osteosarcoma: A Report from the
Children’s Cancer Study Group. Med Pediatr Oncol 1987;15:69.
JF, Wexler LH, Marcus RB, et al. Second Malignancies After Ewing’s
Sarcoma: Radiation Dose-Dependency of Secondary Sarcomas. J Clin Oncol 1996;14:2818.
MM. The Use of an Expandable and Adjustable Prosthesis in the Treatment
of Childhood Malignant Bone Tumors of the Extremity. Cancer 1986;57:499.
MP, Goorin AM, Horowitz M, et al. Adjuvant Chemotherapy of High-Grade
Osteosarcoma of the Extremity: Updated Results of the
Multi-institutional Osteosarcoma Study. Clin Orthop 1991;270:8.
MM, McHale KA. Limb-Sparing Surgery for High-Grade Malignant Tumors of
the Proximal Tibia: Surgical Technique and a Method of Extensor
Mechanism Reconstruction. Clin Orthop 1989;239:231.
HJ, Cantley KP, Lippiello L, et al. The Biology of Human
Chondrosarcoma. I. Description of the Cases, Grading, and Biochemical
Analyses. J Bone Joint Surg 1980;62A:160.
HJ, Cantley K, Schiller AL, Lippiello L: The Biology of Human
Chondrosarcoma. II. Variation in Chemical Composition Among Types and
Subtypes of Benign and Malignant Cartilage Tumors. J Bone Joint Surg 1980;62A:176.
P, Leonard R, Skarin A, et al. Improving Survival Following Combined
Radiation Therapy and Chemotherapy for Unfavorable Prognosis Stage I–II
non-Hodgkin’s Lymphoma. J Clin Oncol 1985;3:1301.
P, Heller G, Vlamis V, et al. Osteosarcoma of the Extremities:
Chemotherapy Experience at Memorial Sloan-Kettering. Cancer Treatment Res 1993;62:309.
J, Arndt C, Smithson W, et al. Long Term Follow-Up of High Grade
Osteosarcoma Treated with Preoperative Ifosfamide, Adriamycin and High
Dose Methotrexate with Cisplatin Added Postoperatively for Poor
Histologic Responders. (Abstract 2054.) Proc Am Soc Clin Oncol 1998;17:535a.
JS, Krailo M, Myers P, et al. Metastatic Ewing’s Sarcoma and Primitive
Neuroectodermal Tumor of Bone; Failure of New Regimens to Improve
Outcome. (Abstract.) Proc Am Soc Clin Oncol 1996;15:467.
H, Yamamoto S, Hiramatsu K, et al. Adamantinoma of the Tibia:
Ultrastructural and Immunohistochemical Study with Reference to
Histogenesis. Clin Orthop 1984;190:299.
ME, Gehan EA, Burgert EO, et al. Multimodal Therapy for the Management
of Primary, Nonmetastatic Ewing’s Sarcoma of Bone: A Long Term
Follow-Up of the First Intergroup Study. J Clin Oncol 1990;8:1664.
T, Hillmann A, Hoffmann C, et al. Significance of Surgical Margin on
the Prognosis of Patients with Ewing’s Sarcoma. A Report from the
Cooperative Ewing’s Sarcoma Study. Cancer 1996;78:892.
P, Galanis E, Greipp PR, Sim FH. Prosthetic Hip Replacement for
Pathologic or Impending Pathologic Fractures in Myeloma. Clin Orthop 1997;341:192.
D, Deicher H, Coldewey R, et al. Induction and Maintenance Therapy in
Multiple Myeloma: A Multicenter Trial of MP versus VCMP. Eur J Cancer Clin Oncol 1988;24:1061.
P, Bacci G, Campanacci M, et al. Histologic Evaluation of Necrosis in
Osteosarcoma Induced by Chemotherapy: Regional Mapping of Viable and
Nonviable Tumor. Cancer 1985;56:1515.
P, Bacci G, Ferrari S, Mercuri M. Neoadjuvant Chemotherapy in Malignant
Fibrous Histiocytoma of Bone and in Osteosarcoma Located in the
Extremities: Analogies and Differences between the Two Tumors. Ann Oncol 8:1107; 1997.
P, Rougraff BT, Bacci G, et al. Prognostic Significance of
Histopathologic Response to Chemotherapy in Nonmetastatic Ewing’s
Sarcoma of the Extremities. J Clin Oncol 1993;11:1763.
P, Sangiorgi L, Rougraff BT, et al. Relationship of
Chemotherapy-induced Necrosis and Surgical Margins to Local Recurrence
in Osteosarcoma. J Clin Oncol 1994;12:2699.
G, Bacci G, Picci P, et al. Telangiectatic Osteogenic Sarcoma of the
Extremities: Results in 17 Patients Treated with Neoadjuvant
Chemotherapy. Clin Orthop 1991;270:99.
D, Greenway GD, Bardwick PA, et al. Plasma-Cell Dyscrasia with
Polyneuropathy, Organomegaly, Endocrinopathy, M-Protein, and Skin
Changes. The POEMS Syndrome. Radiology 1981;140:17.
K, Serra M, Manara MC, et al. Immunostaining of the p30/32 MIC2 Antigen
and Molecular Detection of EWS Rearrangements for the Diagnosis of
Ewing’s Sarcoma and Peripheral Neuroectodermal Tumor. Hum Pathol 1996;27:408.
MA, Aschliman MA, Thomas N, Mankin HJ. Limb-Salvage Treatment Versus
Amputation for Osteosarcoma of the Distal End of the Femur. J Bone Joint Surg 1986;68-A:1331.
MA, Ungerleider RS, Horowitz ME, et al. Influence of Doxorubicin Dose
Intensity on Response and Outcome for Patients with Osteogenic Sarcoma
and Ewing’s Sarcoma. J Natl Cancer Inst 1991;83:1460.
JG, Audeh MW, Copenhaver CM, Warnke RA. Immunophenotypic Differences
Between Plasmacytoma/Multiple Myeloma and Immunoblastic Lymphoma. Cancer 1988;61:1782.
HD, Goitein M, Munzenrider J, et al. Definitive Radiation Therapy for
Chordoma and Chondrosarcoma of Base of Skull and Cervical Spine. J Neurosurg 1982;56:377.
M, McGuire MH, Herbold DR, et al. Magnetic Resonance Imaging in
Planning Limb Salvage Surgery for Primary Malignant Tumors of Bone. J Bone Joint Surg 1986;68-A:809.
DL, Mass DP, Simon MA, Shapiro CM. Histiocytic Lymphoma (Reticulum-Cell
Sarcoma) of Bone: Current Strategy for Orthopaedic Surgeons. J Bone Joint Surg 1981;63-A:79.
M, Kalifa C, Rodary C, et al. Osteosarcoma Recurrences in Pediatric
Patients Previously Treated with Intensive Chemotherapy. J Clin Oncol 1994;12:2614.
WS, Gill-Murdoch C, McKeown PP, Conant P. Bilateral Thoracoscopy and
Limited Thoracotomy. A Combined Approach for the Resection of
Metastatic Fibrosarcoma. Chest 1994;106:935.
KL, Bolander ME, Rock MG, et al. Evidence for the Upregulation of
Osteogenic Protein-1 mRNA Expression in Musculoskeletal Neoplasms. J Orthop Res 1998;16:8.
S, Dorfman, H. Adamantinoma of Long Bone. An Analysis of Nine New Cases
with Emphasis on Metastasizing Lesions and Fibrous Dysplasia-like
Changes. Hum Pathol 1977;8:141.
LH, DeLaney TF, Tsokos M, et al. Ifosfamide and Etoposide plus
Vincristine, Doxorubicin, and Cyclophosphamide for Newly Diagnosed
Ewing’s Sarcoma Family of Tumors. Cancer 1996;78:901.
K, Bielack S, Delling G, et al. Effect of Intraarterial versus
Intravenous Cisplatin in Addition to Systemic Doxorubicin, High-Dose
Methotrexate, and Ifosfamide on Histologic Tumor Response in
Osteosarcoma (Study COSS-86). Cancer 1990;66:1703.
K, Bieling P, Bielack S, et al. Local Control and Survival from the
Cooperative Osteosarcoma Study Group Studies of the German Society of
Pediatric Oncology and the Vienna Bone Tumor Registry. Clin Orthop 1991;270:79.
T, Toguchida J, Yamamuro T, et al. Allelotype analysis in
osteosarcomas: Frequent allele loss on 3q, 13q, 17p, and 18q. Cancer Res 1992;52:2419.
Y, Frassica FJ, Chao EYS, et al. Metastatic Bone Disease. A Study of
the Surgical Treatment of 166 Pathologic Humeral and Femoral Fractures.
Clin Orthop 1990;251:213.