
Editors: Tornetta, Paul; Einhorn, Thomas A.; Damron, Timothy A.
Title: Oncology and Basic Science, 7th Edition
Copyright ©2008 Lippincott Williams & Wilkins
> Table of Contents > Section III – Specific Soft Tissue Neoplasms and Simulators > 12 – Soft Tissue Sarcomas
12
Soft Tissue Sarcomas
Carol D. Morris
Soft tissue sarcomas are malignant neoplasms that arise
in nonepithelial extraskeletal tissue (e.g., fat, muscle, fibrous
structures, etc.) of mesenchymal origin. They account for less than 1%
of all cancers, with approximately 8,000 to 9,000 diagnosed annually in
the United States. Benign soft tissue tumors outnumber malignant ones
by a factor of at least 100. More than 50 histologic subtypes of soft
tissue sarcoma are recognized (Table 12-1).
in nonepithelial extraskeletal tissue (e.g., fat, muscle, fibrous
structures, etc.) of mesenchymal origin. They account for less than 1%
of all cancers, with approximately 8,000 to 9,000 diagnosed annually in
the United States. Benign soft tissue tumors outnumber malignant ones
by a factor of at least 100. More than 50 histologic subtypes of soft
tissue sarcoma are recognized (Table 12-1).
Soft tissue sarcomas can occur anywhere in the body,
though the majority occur in the extremities (50% to 75%). Ten percent
occur on the trunk. As with other malignancies, soft tissue sarcoma
tends to occur in older individuals, with a median age of 65, although
there are subtype-related variations in peak age. For example,
embryonal rhabdomyosarcoma occurs almost exclusively in children
(<10 years old), synovial sarcoma occurs in young adults (20 to 40
years old), and pleomorphic sarcoma and liposarcoma occur in older
adults (>60 years old). Approximately 10% of patients have
clinically detectable metastases at presentation, usually in the lung.
though the majority occur in the extremities (50% to 75%). Ten percent
occur on the trunk. As with other malignancies, soft tissue sarcoma
tends to occur in older individuals, with a median age of 65, although
there are subtype-related variations in peak age. For example,
embryonal rhabdomyosarcoma occurs almost exclusively in children
(<10 years old), synovial sarcoma occurs in young adults (20 to 40
years old), and pleomorphic sarcoma and liposarcoma occur in older
adults (>60 years old). Approximately 10% of patients have
clinically detectable metastases at presentation, usually in the lung.
The management of soft tissue sarcoma requires a
multidisciplinary approach that includes surgery, radiation therapy,
and chemotherapy. Treatment plans are best coordinated and administered
when possible by specialty centers with expertise in treating the
disease. The overall 5-year survival for patients with soft tissue
sarcoma is largely dependent on the stage of disease. This, in turn, is
determined by a combination of factors that include the grade, size,
and location of the tumor. Using the four-tiered staging system of the
American Joint Committee on Cancer (AJCC), 5-year survival rates are
approximately 90% for stage I, 70% for stage II, 50% for stage III, and
10% to 20% for stage IV.
multidisciplinary approach that includes surgery, radiation therapy,
and chemotherapy. Treatment plans are best coordinated and administered
when possible by specialty centers with expertise in treating the
disease. The overall 5-year survival for patients with soft tissue
sarcoma is largely dependent on the stage of disease. This, in turn, is
determined by a combination of factors that include the grade, size,
and location of the tumor. Using the four-tiered staging system of the
American Joint Committee on Cancer (AJCC), 5-year survival rates are
approximately 90% for stage I, 70% for stage II, 50% for stage III, and
10% to 20% for stage IV.
This chapter will review the rationale and outcomes for
the management of soft tissue sarcoma with current treatment paradigms.
In addition, the clinical and histopathologic presentation of the more
common soft tissue sarcomas likely to be encountered in practice and on
examinations will be discussed.
the management of soft tissue sarcoma with current treatment paradigms.
In addition, the clinical and histopathologic presentation of the more
common soft tissue sarcomas likely to be encountered in practice and on
examinations will be discussed.
Pathogenesis
The etiology of soft tissue sarcoma is largely unknown.
While numerous genetic aberrations continue to be identified, the
clinical significance of these is still being elucidated. The more
consistent genetic findings are outlined in connection with individual
tumor types.
While numerous genetic aberrations continue to be identified, the
clinical significance of these is still being elucidated. The more
consistent genetic findings are outlined in connection with individual
tumor types.
Etiology
-
Largely unknown
-
Chemical carcinogens
-
Increased incidence reported after exposure to dioxins (herbicides)
-
Controversial
-
-
Radiation
-
Termed “post-radiation” or “radiation-induced” sarcoma
-
More common in women, reflecting the
distribution of conditions for which radiation is widely used: breast
cancer, genitourinary cancers -
Risk increases with dose, with most patients having received at least 50 Gy.P.295Table 12-1 Who Classification of Malignant Soft Tissue Tumors
Adipocytic tumors Atypical lipomatous tumor/well-differentiated liposarcoma
Dedifferentiated liposarcoma
Myxoid liposarcoma
Round cell liposarcoma
Pleomorphic liposarcoma
Mixed-type liposarcoma
Liposarcoma, not otherwise specifiedFibroblastic/myofibroblastic Solitary fibrous tumor and hemangiopericytoma
Inflammatory myofibroblastic tumor
Low-grade myofibroblastic sarcoma
Myxoinflammatory fibroblastic sarcoma
Infantile fibrosarcoma
Adult fibrosarcoma
Myxofibrosarcoma
Low-grade fibromyxoid sarcoma
Hyalinizing spindle cell tumor
Sclerosing epithelioid fibrosarcomaFibrohistiocytic tumors Undifferentiated pleomorphic sarcoma
Pleomorphic malignant fibrous histiocytoma (MFH)
Giant cell MFH
Inflammatory MFH
Not otherwise specifiedSmooth muscle tumors Leiomyosarcoma Skeletal muscle tumors Embryonal rhabdomyosarcoma (including spindle cell, botryoid, anaplastic)
Alveolar rhabdomyosarcoma (including solid, anaplastic)
Pleomorphic rhabdomyosarcomaVascular tumors Retiform hemangioendothelioma
Papillary intralymphatic angioendothelioma
Composite hemangioendothelioma
Kaposi sarcoma
Epithelioid hemangioendothelioma
Angiosarcoma of soft tissueChondro-osseous tumors Mesenchymal chondrosarcoma
Extraskeletal osteosarcomaTumors of peripheral nerves Malignant peripheral nerve sheath tumor (MSNST) Tumors of uncertain differentiation Synovial sarcoma
Epithelioid sarcoma
Alveolar soft part sarcoma
Clear cell sarcoma of soft tissue
Extraskeletal myxoid chondrosarcoma (“chordoid” type)
Primitive neuroectodermal tumor/extraskeletal Ewing tumor
Desmoplastic small round cell tumor
Extrarenal rhabdoid tumor
Malignant mesenchymoma
Neoplasms with perivascular epithelioid cell differentiation (PEComa)
Clear cell myomelanocytic tumor
Intimal sarcoma -
Median time between exposure and tumor development is ~10 years.
-
More common in patients with germline mutations
P.296 -
-
Viral and immunological factors
-
Increased incidence of sarcomas in immunocompromised individuals
-
Immunodeficiency syndromes
-
Therapeutic immunosuppression associated with organ transplantation
-
Stewart-Treves syndrome: an acquired
“regional” immunodeficiency of the edematous upper extremity in breast
cancer patients following radical mastectomy associated with
lymphangiosarcoma
-
-
Oncogenic viruses
-
Human herpes virus 8 associated with Kaposi sarcoma
-
Epstein-Barr virus associated with leiomyosarcomas
-
-
-
Genetic predisposition
-
Neurofibromatosis-1 associated with malignant peripheral nerve sheath tumors (MPNST)
-
Li-Fraumeni syndrome: germline mutation in p53 suppressor gene
-
Hereditary retinoblastoma: germline mutation of RB1 locus
-
Epidemiology
-
Approximately 8,700 new cases diagnosed annually in the United States
-
Annual incidence is 1.5 per 100,000 individuals
-
8 per 100,000 in individuals greater than 80 years old
-
-
Slight male predominance
-
No proven racial variation
Classification
Soft tissue sarcomas are a highly heterogeneous group of
tumors that are most commonly classified on a histological basis
according to the tissue they most resemble. The most widely recognized
classification system is that of the World Health Organization (WHO),
which was first published in 1969 and most recently updated in 2002
(see Table 12-1).
tumors that are most commonly classified on a histological basis
according to the tissue they most resemble. The most widely recognized
classification system is that of the World Health Organization (WHO),
which was first published in 1969 and most recently updated in 2002
(see Table 12-1).
Staging
-
Staging systems incorporate histological and clinical information for prognostic value.
-
The staging system used throughout this chapter is the AJCC staging system (Table 12-2).
-
75% of soft tissue sarcomas are high grade.
-
One third of soft tissue sarcomas are superficial and two thirds are deep.
-
Diagnosis
-
The diagnosis of soft tissue sarcoma is
made with a combination of a good history and physical examination,
appropriate radiology imaging, and biopsy. -
The pertinent components of the history
and physical examination as well as the clinical and radiologic
features are detailed in Chapter 2.
Clinical Findings
-
Summary of clinical features and examination findings:
-
Most soft tissue sarcomas are painless.
-
Masses that are suspicious for sarcoma:
-
>5 cm regardless of location
-
Deep to fascia
-
Firm or fixed
-
Enlarging
-
-
Clinical features of tumors with advanced size
-
Distal edema
-
Nerve compression
-
Bladder symptoms (pelvic sarcomas)
-
-
Metastatic disease
-
10% of patients present with metastatic disease.
-
Lung is the most common metastatic site.
-
Bone (6%) and lymph node (3%) metastases are less common.
-
-
P.297
Radiologic Findings
-
Necessary imaging
-
Chest x-ray
-
Computed tomography (CT) of chest: preferred for detection of metastases
-
Magnetic resonance imaging (MRI) of primary site
-
CT with contrast substituted in patients with contraindication for MRI
-
CT often preferred for intra-abdominal tumors
-
-
Role of positron emission tomography (PET) scan unclear
-
Other Diagnostic Tests
-
Histologic analysis of tissue is required
for staging and should be performed prior to initiating any treatment,
with rare exceptions. -
A good biopsy is the first step in a successful limb salvage operation.
-
Diagnostic tissue can be obtained by the following methods:
-
Needle
-
Fine-needle aspiration (FNA)
-
Core
-
-
Open incision
-
Open excisional
-
-
The advantages and disadvantages of each type of biopsy are discussed in Chapter 3, Biopsy of Musculoskeletal Tumors.
Diagnostic Tools
-
Numerous investigative tools are available to the pathologist to assist in the diagnosis of specific sarcoma subtypes (Tables 12-3 and 12-4).
|
Table 12-3 Common Immunohistochemistry Stains and Tissue Distribution
|
|||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Treatment
Soft tissue sarcoma is treated with an interdisciplinary
approach that incorporates surgery, radiation, and chemotherapy. The
details, rationale, and outcomes for each of these modalities are
reviewed in Chapter 4, Treatment Principles. The following is a summary.
approach that incorporates surgery, radiation, and chemotherapy. The
details, rationale, and outcomes for each of these modalities are
reviewed in Chapter 4, Treatment Principles. The following is a summary.
Surgery
-
Complete surgical excision is the main cornerstone of treatment.
-
Often curative for localized disease
-
Limb salvage is the preferred method.
-
Amputation is ultimately required in 5% to 10% of patients.
Radiation
-
Methods of delivery
-
External beam (pre-, post-, and intraoperation)
-
Brachytherapy
P.298Table 12-4 CHROMOSOMAL TRANSLOCATIONS IN SOFT TISSUE SARCOMATumor Type Translocation Involved Genes Ewing/primitive neuroectodermal tumor 11;22 FLI1, EWS Clear cell sarcoma 12;22 ATF1, EWS Extraskeletal myxoid chondrosarcoma 9;22 CHN, EWS Synovial sarcoma X;18 SSX1 or SSX2, SYT Myxoid liposarcoma 12;16 CHOP, TLS Alveolar rhabdomyosarcoma 2;13 PAX3, FKHR Alveolar soft part sarcoma X;17 TFE3, ASPL Dermatofibrosarcoma protuberans (DFSP) 17;22 COL1A1, PDGFB1 -
-
Typical dose ~6,000 cGy
-
Primarily indicated for:
-
High-grade tumors (unless margins are very wide)
-
Intermediate-grade tumors with close margins
-
Large tumors
-
Recurrent disease
-
-
Improves local control by 20% to 35%
Chemotherapy
-
Indicated for patients at the highest risk of developing metastatic disease or patients with metastatic disease
-
Best administered in the setting of a clinical trial
-
Doxorubicin-based therapy is associated with a minimal improvement in overall survival (<10%).
-
Ifosfamide-based therapy is associated
with moderately improved survival at intermediate follow-up; long-term
results are unknown.
Results and Outcome
The outcome of patients with soft tissue sarcoma is
multifactorial but largely dependent on the stage of disease. The
overall survival for all patients with soft tissue sarcoma is
approximately 70%. Using the four-tiered staging system of the AJCC,
5-year survival rates are approximately 90% for stage I, 70% for stage
II, 50% for stage III, and 10% to 20% for stage IV.
multifactorial but largely dependent on the stage of disease. The
overall survival for all patients with soft tissue sarcoma is
approximately 70%. Using the four-tiered staging system of the AJCC,
5-year survival rates are approximately 90% for stage I, 70% for stage
II, 50% for stage III, and 10% to 20% for stage IV.
-
Negative prognostic factors
-
Metastatic disease
-
High histologic grade
-
Size >10 cm
-
Bone and neurovascular invasion
-
Advanced age
-
Retroperitoneal and visceral location
-
Positive microscopic surgical margins
-
Presentation with locally recurrent disease
-
Adipocytic Tumors
Well-Differentiated Liposarcoma
-
Synonym: atypical lipomatous tumor
Pathogenesis
-
No potential for metastasis unless it undergoes dedifferentiation (<2% in the extremity, ~20% in retroperitoneum)
-
Locally aggressive nature can cause
symptoms and even death when complete excision is not possible, as in
the retroperitoneum and mediastinum. -
Genetics: ring chromosome 12 with amplification of the MDM2 gene
Epidemiology
-
Frequency: accounts for 40% to 45% of all liposarcomas
-
Age: incidence peaks during sixth decade of life
-
Site: most commonly occurs in the thigh
Classification
-
Subtypes: lipoma-like, sclerosing, inflammatory, and spindle cell
Diagnosis
-
Histology: mature adipocytic tissue with slight atypia and scattered to rare lipoblasts (Fig. 12-1)
Myxoid Liposarcoma
-
Synonyms: round cell liposarcoma, myxoid/round cell liposarcoma
Pathogenesis
-
Tendency for extrapulmonary metastases (soft tissue and bone, especially the spine)
-
Multifocal presentation not uncommon
-
Genetics: >90% of cases associated with t(12:16)(q13;p11) leading to the TLS/CHOP fusion protein; t(12;22) has been described less frequently
P.299
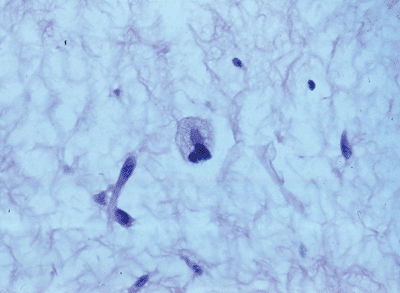 |
|
Figure 12-1
Well differentiated liposarcoma; relatively mature adipocytic tissue with varying cell size admixed with hyperchromatic and multinucleated stromal cells. |
Epidemiology
-
Frequency: accounts for about one third of all liposarcomas
-
Age: incidence peaks during the fourth and fifth decades of life
-
Site: most commonly arises in the deep tissues of the thigh
Classification
-
Grade is often equated to the size of the
round cell component, usually expressed as a percentage (>5% round
cells = high grade).
Diagnosis
-
Myxoid component is characterized
histologically by mixture of lipoblasts and uniform nonlipogenic
mesenchymal cells in prominent myxoid stroma associated with a fine
capillary network (Fig. 12-2). -
Round cell component is characterized histologically by more cellular regions of round cells (see Fig. 12-2).
 |
|
Figure 12-2 (A)
Myxoid liposarcoma. Mixture of lipoblasts and uniform nonlipogenic mesenchymal cells in prominent myxoid stroma associated with a fine capillary network. (B) High-grade myxoid liposarcoma with a predominant round cell component. |
Fibroblastic/Myofibroblastic Tumors
Hemangiopericytoma
-
Historically evolving term used to refer
to neoplasms with a thin-walled branching vascular pattern; its current
status as a discrete entity is controversial, as the pattern appears
within numerous other types of tumors -
Synonym: extrapleural solitary fibrous tumor
Pathogenesis
-
Genetics: breakpoints in chromosomes 12 and 19 reported but sporadic
-
May cause hypoglycemia due to secretion of insulin-like growth factor
Epidemiology
-
Age: occurs in middle-aged adults, most often in the pelvic retroperitoneum
Diagnosis
-
Characterized histologically by
monomorphic, evenly distributed cellularity arranged around thin-walled
vessels with a staghorn pattern (Fig. 12-3) -
Immunohistochemistry: positive for CD34
Adult Fibrosarcoma
Pathogenesis
-
Genetics: inconsistent aberrations
Epidemiology
-
Frequency: accounts for 1% to 3% of all soft tissue sarcomas
-
Age: occurs in middle-aged and older adults
-
Site: most commonly affects the trunk, head, and neck
P.300
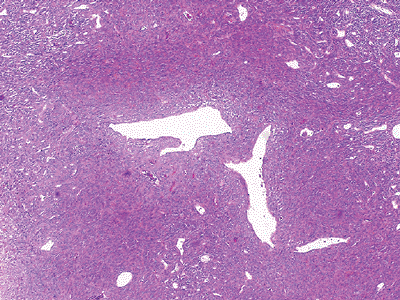 |
|
Figure 12-3 Hemangiopericytoma. Monomorphic evenly distributed cellularity arranged around thin-walled vessels with a staghorn pattern.
|
Classification
-
Termed “infantile fibrosarcoma” when it occurs in children
Diagnosis
-
Fibroblasts with variable collagen
production arranged in long intersecting fascicles classically with a
herringbone architecture (Fig. 12-4)
Myxofibrosarcoma
-
Synonym: myxoid malignant fibrous histiocytoma
Pathogenesis
-
Local recurrence is notoriously high at >50%, independent of grade.
-
Low-grade tumors may acquire a higher grade in recurrence, thereby increasing their metastatic potential.
-
Genetics: inconsistent aberrations
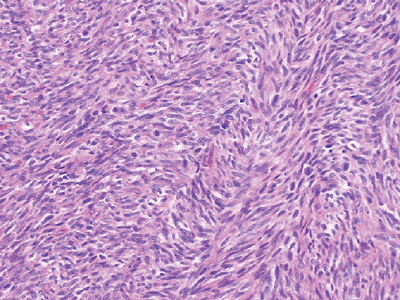 |
|
Figure 12-4
Adult fibrosarcoma. Fibroblasts with variable collagen production arranged in long intersecting fascicles with a classic herringbone architecture. |
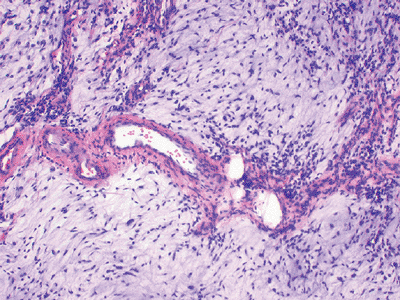 |
|
Figure 12-5
Low-grade myxofibrosarcoma. Nodular growth pattern of spindle cells in a myxoid stroma composed of hyaluronic acid with characteristic elongated, curvilinear blood vessels surrounded by a condensation of tumor cells. |
Epidemiology
-
One of the more common soft tissue sarcomas of adulthood
-
Frequency: incidence peaks in sixth to eighth decades of life
-
Site: two thirds occur in the subcutaneous tissue, usually in the limbs
Diagnosis
-
Characterized histologically by nodular
growth pattern of spindle cells in a myxoid stroma composed of
hyaluronic acid with characteristic elongated, curvilinear blood
vessels surrounded by a condensation of tumor cells (Fig. 12-5)
Fibrohistiocytic Tumors
Undifferentiated High-Grade Pleomorphic Sarcoma, Not Otherwise Specified
-
Formerly termed pleomorphic malignant fibrous histiocytoma (MFH)
-
Definition: Once considered the most
common soft tissue sarcoma of adulthood, MFH has been declassified as a
distinct diagnostic entity; it is now thought to represent a variety of
poorly differentiated or dedifferentiated neoplasms and has been
renamed “undifferentiated pleomorphic sarcoma.”
Pathogenesis
-
Genetics: extensive heterogeneity
Etiology
-
2% to 3% arise in a previously radiated field (radiation-induced sarcoma).
P.301
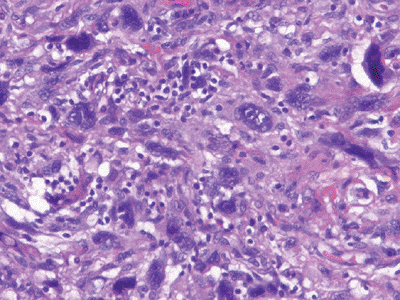 |
|
Figure 12-6
Undifferentiated pleomorphic sarcoma. Marked cellularity and nuclear pleomorphism with bizarre tumor giant cells admixed with spindle and histiocyte-type cells. |
Epidemiology
-
Frequency and age: The pleomorphic
sarcomas are the most common soft tissue sarcomas in patients >40
years of age, with an increasing incidence with increasing age, peaking
during the sixth and seventh decades of life. -
Site: most commonly arise in the deep tissues of the lower limbs
Diagnosis
-
Considered a diagnosis of exclusion
-
Characterized histologically by marked
cellularity and nuclear pleomorphism with a storiform pattern and often
bizarre tumor giant cells admixed with spindle cells and
histiocyte-type cells (Fig. 12-6)
Smooth Muscle Tumors
Leiomyosarcoma
Pathogenesis
-
Genetics: heterogeneous, though losses on chromosomes 3, 8, and 13 are common; the latter is associated with an RB1 mutation
Epidemiology
-
Frequency: accounts for 10% to 15% of limb sarcomas
-
Age: may occur in any age, though more common in middle-aged adults
-
Site: commonly arise primarily in the pelvis from the uterus and large blood vessels (inferior vena cava)
Diagnosis
-
Characterized histologically by elongated cigar-shaped spindle cells with blunt-ended nuclei interspersed with myxoid changes (Fig. 12-7)
-
Immunohistochemistry: positive for smooth muscle antigen (SMA) and desmin
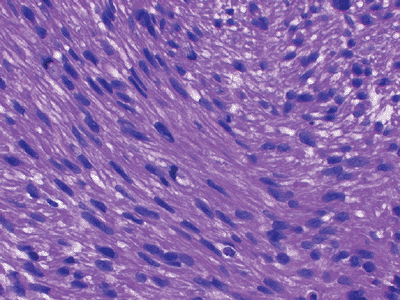 |
|
Figure 12-7 Leiomyosarcoma. Spindle cells with blunt-ended nuclei interspersed with myxoid changes.
|
Skeletal Muscle Tumors
Embryonal Rhabdomyosarcoma
-
Synonyms: myosarcoma, rhabdomyosarcoma, rhabdosarcoma
Pathogenesis
-
Genetics: losses on chromosome 11
Epidemiology
-
Frequency: most common soft tissue sarcoma in children and adolescents, with 4.6 cases per million persons <15 years of age
-
Site: Most arise in the head and neck, followed by the genitourinary tract; limb and trunk involvement is less common (<10%).
-
Spindle cell variant most commonly arises in the scrotum.
-
Botryoid variant arises beneath mucosal epithelial surfaces (e.g., bladder, biliary tract, pharynx).
-
-
Age: ~45% occur in children <5 years of age.
Classification
-
Embryonal is the most common subtype of rhabdomyosarcoma and encompasses the spindle cell, botryoid, and anaplastic variants.
Diagnosis
-
Immunohistochemistry: desmin and actin positivity variable; MyoD1 and myogenin highly specific
-
Histologically characterized by a
constellation of rhabdomyoblasts in various stages of differentiation,
with the more primitive cells possessing oval nuclei and more
differentiated cells demonstrating elongated nuclei (“tadpole” cells)
with eosinophilic cytoplasm (Fig. 12-8)
P.302
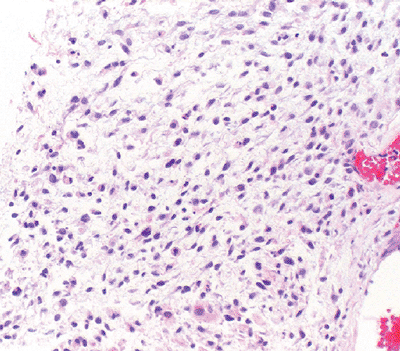 |
|
Figure 12-8
Embryonal rhabdomyosarcoma. A constellation of rhabdomyoblasts in various stages of differentiation. The more primitive cells possess oval nuclei, while more differentiated cells demonstrate elongated nuclei (“tadpole” cells) with eosinophilic cytoplasm. |
Alveolar Rhabdomyosarcoma
-
Synonyms: rhabdomyoblastoma, rhabdomyopoetic sarcoma, monomorphous round cell rhabdomyosarcoma
Pathogenesis
-
Genetics: t(2;13)(q35;q14) leading to the PAX3/FKHR fusion protein; t(1;13) in rare cases
Epidemiology
-
Frequency: less common than embryonal rhabdomyosarcoma
-
Age: occurs most commonly in adolescents and young adults
-
Site: most commonly arises in the extremities, followed by paraspinal, perineal, and paranasal locations
Diagnosis
-
Histologically resembles lymphoma and
other small blue round cell tumors but characterized by alveolar
pattern similar to that in the lung, with cellular areas separated by
fibrovascular septa (Fig. 12-9) -
Immunohistochemistry: positive for desmin, actin, myogenin, and MyoD
Vascular Tumors
Epithelioid Hemangioendothelioma
-
Synonyms: angioglomoid tumor, myxoid angioblastomatosis
Pathogenesis
-
Genetics: t(1;3) has been reported, though consistency is unknown.
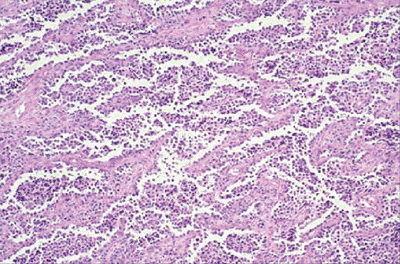 |
|
Figure 12-9
Alveolar rhabdomyosarcoma. Nest of small blue round cells separated by fibrovascular septa to produce a morphologic pattern similar to alveoli in lung. |
Epidemiology
-
Age: affects all ages
-
Site: usually arises in the extremity, often originating from a small vein
-
Often presents with a multifocal distribution in an entire limb bud involving both soft tissue and bone
Diagnosis
-
Clinical presentation: unlike other soft tissue sarcomas, typically presents as a painful mass
-
Characterized histologically by short
strands of eosinophilic epithelioid endothelial cells embedded in a
blue to pink acidic matrix (Fig. 12-10) -
Immunohistochemistry: positive for CD31, CD34, and FLI1
Angiosarcoma
-
Synonyms: lymphangiosarcoma, malignant hemangioendothelioma, hemangiosarcoma, hemangioblastoma
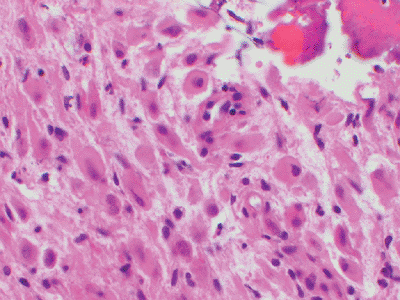 |
|
Figure 12-10 Epithelioid hemangioendothelioma. Short strands of bland eosinophilic endothelial cells in a deep pink hyaline matrix.
|
P.303
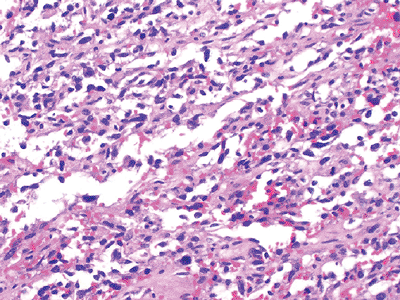 |
|
Figure 12-11
Angiosarcoma. Spindle and epithelial cells arranged morphologically in loose, irregular vascular channels. Areas of hemorrhage are common. |
Pathogenesis
-
Genetics: inconsistent chromosomal aberrations
Etiology
-
One third of tumors are associated with a
pre-existing condition such as chronic lymphedema, neurofibromatosis-1,
vascular implants, Klippel-Trenaunay syndrome, Maffucci syndrome, and
previous radiation.
Epidemiology
-
Age: Incidence peaks in the seventh decade of life, though may occur at any age.
-
Site: Most occur in the subcutaneous tissue; the majority of deep angiosarcomas occur in the thigh.
Diagnosis
-
Histologically characterized by nodular
hemorrhagic masses composed of epithelioid and spindle cells attempting
to form interconnecting rudimentary vascular channels (Fig. 12-11) -
Immunohistochemistry: positive for von Willebrand factor, CD31, and CD34
Chondro-Osseous Tumors
Extraskeletal Osteogenic Sarcoma
-
Synonym: soft tissue osteosarcoma
Pathogenesis
-
~10% associated with previous radiation (radiation- induced sarcoma)
-
Genetics: inconsistent aberrations
Epidemiology
-
Frequency: accounts for <2% of all soft tissue sarcomas and 2% to 4% of all osteogenic sarcomas
-
Age: Incidence peaks during fifth to seventh decades of life.
-
Site: most commonly arises in the deep soft tissue of the lower extremity
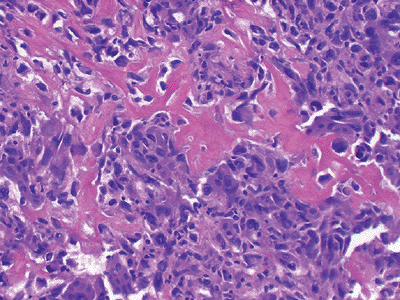 |
|
Figure 12-12
Extraskeletal osteogenic sarcoma. Neoplastic bone in a lacy, sheet-like pattern interspersed with atypical, mitotically active tumor cells. |
Diagnosis
-
Histologically characterized by same features as skeletal osteosarcoma; lace-like osteoid produced by pleomorphic cells (Fig. 12-12)
-
Immunohistochemistry: variable positivity for many antigens, most specifically osteocalcin
-
Prognosis is worse than in skeletal osteogenic sarcoma (likely due to lack of chemotherapy response).
Tumors of Peripheral Nerves
Malignant Peripheral Nerve Sheath Tumor (MPNST)
-
Synonym: neurofibrosarcoma (generally abandoned)
Pathogenesis
-
Genetics: alterations at the NF-1 locus on chromosome 17; p53 mutations
Epidemiology
-
50% of cases occur in the setting of neurofibromatosis-1.
-
Two thirds arise from neurofibromas.
-
More common in large nerves
-
Sciatic nerve most commonly affected
-
Diagnosis
-
Histologically characterized by elongated, tapered nuclei in a fibrosarcoma-type background (Fig. 12-13)
-
Herringbone pattern suggestive of fibrosarcoma arising within a major nerve is highly suggestive of MPNST.
-
Immunohistochemistry: positive for S-100
-
Triton tumor: MPNST with rhabdomyosarcomatous differentiation; associated with a poor prognosis (5-year survival ~12%)
P.304
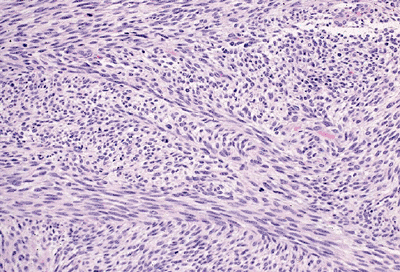 |
|
Figure 12-13 Malignant peripheral nerve sheath tumor. Elongated, tapered nuclei in a background of fibrosarcoma-like spindle cells.
|
Tumors of Uncertain Differentiation
Epithelioid Sarcoma
Pathogenesis
-
Genetics: inconsistent aberrations
Epidemiology
-
Age: occurs in young adults in the second to fourth decades of life
-
Site: commonly arises on the flexor surfaces of fingers, hands, wrist, and forearm
Diagnosis
-
Clinically, often associated with superficial ulceration
-
Histologically challenging: may be misdiagnosed as a benign granulomatous process (Fig. 12-14)
-
Immunohistochemistry: positive for cytokeratins and epithelial membrane antigen (EMA)
Alveolar Soft Part Sarcoma
Pathogenesis
-
Genetics: t(X;17)(p11;q25) resulting in the ASPL/TFE2 fusion protein
-
Associated with very high metastatic potential; brain and lung metastases are common
Epidemiology
-
Frequency: very rare, accounting for <1% of all soft tissue sarcomas
-
Age: Incidence peaks during the second to fourth decades of life.
-
Site: most commonly arises in deep tissues of the thigh
-
In children, most commonly arises in the head and neck
-
 |
|
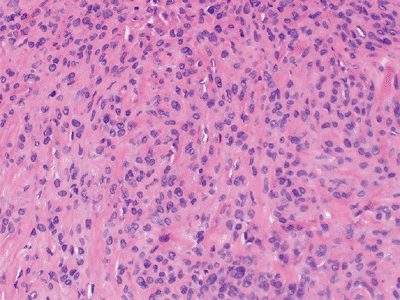
Figure 12-14 Epithelioid sarcoma. Nodular mixture of eosinophilic epithelial cells and spindle cells.
|
Diagnosis
-
Histologically characterized by nests of
tumor cells separated by sinusoidal vascular partitions, resulting in a
pseudoalveolar architecture (Fig. 12-15)
Clear Cell Sarcoma
-
Synonym: malignant melanoma of soft parts
Pathogenesis
-
Genetics: t(12;22)(q13;q12) resulting in the EWS/ATF1 fusion protein
-
Associated with high metastatic potential, especially to lymph nodes
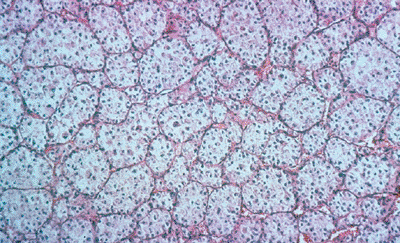 |
|
Figure 12-15
Alveolar soft part sarcoma. Nests of tumor cells separated by sinusoidal vascular partitions resulting in a pseudoalveolar architecture. |
P.305
Epidemiology
-
Age: Incidence peaks during the third and fourth decades of life.
-
Site: >90% arise in the extremities, with almost half of those cases involving the foot and ankle.
-
Often attached to tendons and aponeuroses
-
Diagnosis
-
Histologically characterized by large cells with clear cytoplasm
-
Immunohistochemistry: positive for melanoma antigens and S100
Extraskeletal Myxoid Chondrosarcoma
Pathogenesis
-
Genetics: t(9:22) (q22;q12) resulting in the EWS/NR4A3 fusion protein is seen in ~50% of cases.
-
Despite the name, there is no evidence of cartilaginous differentiation.
-
Late disease recurrence is common.
Epidemiology
-
Frequency: accounts for <3% of soft tissue sarcomas
-
Age: Incidence peaks during sixth decade of life.
-
Site: most commonly arises in the deep tissues of the proximal limb girdles (thigh > arm)
Diagnosis
-
Histologically characterized by uniform round and oval cells in a blue chondromyxoid hypovascular stroma (Fig. 12-16)
-
No characteristic immunohistochemistry findings, although variable S100, cytokeratin, EMA
Synovial Sarcoma
-
Synonyms: malignant synovioma, tendosynovial sarcoma, synovial cell sarcoma (but all of these terms have been abandoned)
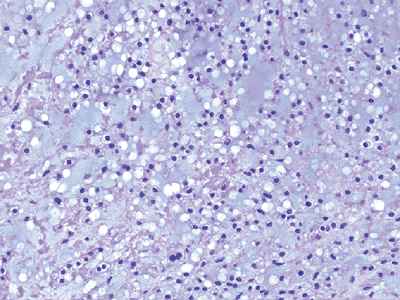 |
|
Figure 12-16 Extraskeletal myxoid chondrosarcoma. Uniform round and oval cells in a blue chondromyxoid hypovascular stroma.
|
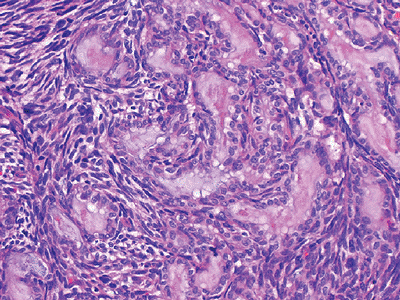 |
|
Figure 12-17
Biphasic synovial sarcoma. Mixture of both spindle cells and epithelial cells. The epithelial cells display ovoid nuclei and form mucin-containing glands. |
Pathogenesis
-
Can be deceptively slow-growing; not unusual to present as mass present for years without change
-
Genetics: t(x;18)(p11;q11) results in the SYT/SSX1, SYT/SSX2, or SYT/SSX4 fusion protein.
-
Monophasic synovial sarcoma with the SSX2 gene variant is associated with a better prognosis.
Epidemiology
-
Frequency: accounts for 5% to 10% of all soft tissue sarcomas
-
Age: may occur at any age but peaks during second to fourth decades of life
-
Site: 80% arise in the extremity.
-
~5% arise intra-articular.
-
Diagnosis
-
Histologically characterized by both mesenchymal background cells with nests of epithelial cells (biphasic) (Fig. 12-17)
or simply mesenchymal or epithelial tissue (monophasic), the latter
similar in appearance to the herringbone pattern of fibrosarcoma -
Immunohistochemistry: for tumors with an epithelial component, positive for cytokeratins and EMA
Suggested Reading
Adjuvant
chemotherapy for localised resectable soft-tissue sarcoma of adults:
meta-analysis of individual data. Sarcoma Meta-analysis Collaboration. Lancet 1997;350(9092):1647–1654.
chemotherapy for localised resectable soft-tissue sarcoma of adults:
meta-analysis of individual data. Sarcoma Meta-analysis Collaboration. Lancet 1997;350(9092):1647–1654.
AJCC Cancer Staging Handbook, 6th ed. New York: Springer, 2002.
Alektiar KM, Leung D, Zelefsky MJ, et al. Adjuvant brachytherapy for primary high-grade soft tissue sarcoma of the extremity. Ann Surg Oncol 2002;9(1):48–56.
Borden EC, Baker LH, Bell RS, et al. Soft tissue sarcomas of adults: state of the translational science. Clin Cancer Res 2003;9(6):1941–1956.
Eilber FC, Brennan MF, Eilber FR, et al. Validation of the postoperative nomogram for 12-year sarcoma-specific mortality. Cancer 2004;101(10):2270–2275.
Fong
Y, Coit DG, Woodruff JM, Brennan MF. Lymph node metastasis from soft
tissue sarcoma in adults. Analysis of data from a prospective database
of 1772 sarcoma patients. Ann Surg 1993;217(1):72–77.
Y, Coit DG, Woodruff JM, Brennan MF. Lymph node metastasis from soft
tissue sarcoma in adults. Analysis of data from a prospective database
of 1772 sarcoma patients. Ann Surg 1993;217(1):72–77.
McCarter MD, Jaques DP, Brennan MF. Randomized clinical trials in soft tissue sarcoma. Surg Oncol Clin North Am 2002;11(1):11–22.
O’Sullivan
B, Davis AM, Turcotte R, et al. Preoperative versus postoperative
radiotherapy in soft-tissue sarcoma of the limbs: a randomised trial. Lancet 2002;359(9325):2235–2241.
B, Davis AM, Turcotte R, et al. Preoperative versus postoperative
radiotherapy in soft-tissue sarcoma of the limbs: a randomised trial. Lancet 2002;359(9325):2235–2241.
Pathology and Genetics of Tumours Soft Tissue and Bone. Lyon: International Agency for Research on Cancer, 2002.
Pisters
PW, Leung DH, Woodruff J, et al. Analysis of prognostic factors in
1,041 patients with localized soft tissue sarcomas of the extremities. J Clin Oncol 1996;14(5):1679–1689.
PW, Leung DH, Woodruff J, et al. Analysis of prognostic factors in
1,041 patients with localized soft tissue sarcomas of the extremities. J Clin Oncol 1996;14(5):1679–1689.
Pollack
A, Zagars GK, Goswitz MS, et al. Preoperative vs. postoperative
radiotherapy in the treatment of soft tissue sarcomas: a matter of
presentation. Int J Radiat Oncol Biol Phys 1998;42(3):563–572.
A, Zagars GK, Goswitz MS, et al. Preoperative vs. postoperative
radiotherapy in the treatment of soft tissue sarcomas: a matter of
presentation. Int J Radiat Oncol Biol Phys 1998;42(3):563–572.
Weiss SW. Enzinger and Weiss’ Soft Tissue Tumors, 4th ed. St. Louis: Mosby, 2001.
Weitz
J, Antonescu CR, Brennan MF. Localized extremity soft tissue sarcoma:
improved knowledge with unchanged survival over time. J Clin Oncol 2003;21(14):2719–2725.
J, Antonescu CR, Brennan MF. Localized extremity soft tissue sarcoma:
improved knowledge with unchanged survival over time. J Clin Oncol 2003;21(14):2719–2725.
Williard
WC, Hajdu SI, Casper ES, et al. Comparison of amputation with
limb-sparing operations for adult soft tissue sarcoma of the extremity.
Ann Surg 1992;215(3):269–275.
WC, Hajdu SI, Casper ES, et al. Comparison of amputation with
limb-sparing operations for adult soft tissue sarcoma of the extremity.
Ann Surg 1992;215(3):269–275.