
but signs such as delayed motor development, fatigability, muscle
cramps during activity, muscle wasting, and orthopaedic conditions such
as cavus feet, claw hands or toes, a long C-shaped scoliosis, and
dynamic scapular winging are often seen (111).
Most muscle and nerve disorders can be diagnosed by a careful history
and physical examination, and by specific laboratory tests,
electromyography, nerve conduction studies, muscle and nerve biopsies,
and genetic evaluation.
determine its onset, duration, exacerbating or relieving factors, and
response to any treatment. The history can provide important clues to
help in the diagnosis. Was the weakness present at birth or of recent
onset and is it progressive? Weakness present at birth but not
progressive may describe a child with a congenital myopathy, whereas
onset of weakness in a young boy with gradual worsening is typical of
muscular dystrophy. Detecting muscle weakness, usually by observation
or muscle testing, is a major component of the clinical examination.
Generalized muscle weakness results in hypotonia (floppiness), ptosis,
a tent shaped mouth (bouche de tapir) and delayed motor development.
Localized muscle atrophy is often observed at the shoulder girdle and
the quadriceps muscle.
as walking, dressing or undressing, and by testing individual muscles.
Grading of activity-related muscle strength is a good screening method,
especially in young, uncooperative patients. The activities are
considered by regions: the hips, legs, shoulders, arms, and bulbar area
(ie, respiratory function). Weakness can result in delayed development
of the motor milestones (e.g., head control, sitting, crawling,
standing, walking, running), Meryon’s sign (i.e., reduced muscle
resistance of the shoulder against the examiner’s hand when lifted
under the arms), Gowers’ sign (i.e., use of the hands to “climb up the
legs” to a standing position when rising from a sitting position on the
floor), and difficulty in climbing steps or rising from a chair (117,137,229).
A 5-year-old boy with Duchenne muscular dystrophy may have a normal
walk but when asked to run, the pelvic girdle and quadriceps weakness
is quickly unmasked.
localizing the distribution of weakness, but it requires patient
cooperation and can be difficult if there are associated fixed
deformities. Agonist muscles (ie, prime movers) and antagonist muscles
(ie, stabilizers) are graded for strength through the range of joint
mobility. For example, the muscles controlling the foot may be tested
for strength in dorsiflexion, plantar flexion, inversion, and eversion.
tedious, and almost impossible in young, uncooperative children, it is
essential as a baseline study for patients with suspected muscle or
nerve disease. Muscle testing often is better performed in a special
therapy session in which adequate time can be allotted. The Medical
Research Council scale is generally accepted and grades muscle power as
follows: 0, no contraction; 1, flicker or trace of contraction; 2,
active motion with gravity eliminated; 3, active motion against
gravity; 4, active motion against gravity and resistance; and 5, normal
power (226). Myometric (dynamometric) methods
are also useful in quantitating muscle strength, especially in
evaluating therapeutic techniques (97).
ankle should be tested, along with the superficial reflexes of the
abdomen and the great toe plantar response. The quality of reflex is
judged by the briskness of muscle contracture and is best graded as
absent, hypoactive, normal, or hyperactive. Children with spinal
muscular atrophy and peripheral neuropathies typically have absent
reflexes, whereas myopathic disorders such as muscular dystrophy have
reflexes until later in the course of the disease.
light touch, deep touch, two-point tactile, vibration, and temperature.
Self-mutilation and Charcot joint changes are almost always
manifestations of sensory loss. In neuropathies, multiple modalities
may be affected, producing a “glove” or “stocking” distribution of
loss, paresthesia, “pins and needles” sensation, and dysesthesia.
Bulbar involvement is evaluated by cranial nerve testing. Cerebellar
testing, particularly the Romberg sign for ataxia, is important when
the differential diagnosis includes Friedreich’s ataxia.
or hands, is common in neuropathic disorders such as spinal muscular
atrophy.
the aminotransferases (transaminases), aldolase, lactate dehydrogenase,
and creatine phosphokinase (CPK). The serum CPK level is a sensitive
and valuable screening test
to demonstrate disease of striated muscle (343).
Skeletal muscle, heart muscle, and brain tissue contain CPK, which
catalyzes the release of phosphate from creatine phosphate. The high
CPK level seen in Duchenne’s muscular dystrophy (50 to 100 times
normal) and other muscle disorders represents leakage from the muscle
cell during necrosis. Aldolase and serum glutamic-oxaloacetic
transaminase levels also may be elevated in muscle disease but are not
as sensitive as the CPK level (352) and are also elevated by hepatic dysfunction.
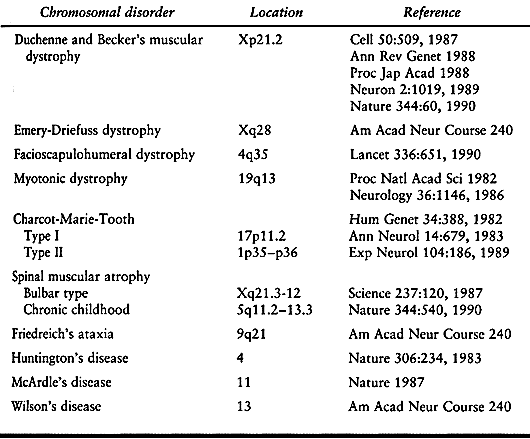
enhanced our knowledge of the gene abnormalities causing neuromuscular
diseases (Table 178.1). This information should eventually result in more effective management of these disorders.
 |
|
Table 178.1. Gene Location of Some Neuromuscular Disorders
|
other rare neuromuscular disorders in children. Metabolic myopathies,
which are caused by abnormalities of glycogen, glucose, or lipid
metabolism, have been well characterized. Mitochondrial DNA defects or
mutations cause a broad spectrum of mitochondrial encephalomyopathies,
which feature variable weakness with encephalopathic, cardiac, and
visceral manifestations.
children with neuromuscular disease. Careful evaluation allows optimal
daily management and an assessment of preoperative risks.
Patients with muscular dystrophy may develop cardiomyopathy or mitral
valve prolapse secondary to papillary muscle weakness. Children with
Friedreich’s ataxia, Emery-Dreifuss dystrophy, and infantile myasthenia
gravis may have arrhythmias (99,113,166,288,295,378).
can be assessed by questions about shortness of breath, frequency of
pulmonary infections, and more objectively, by pulmonary function or
sleep studies.
neuropathy from a myopathy but are seldom specific. A normal muscle is
silent at rest and produces an interference pattern at maximal
activity. In neuropathies, fibrillations and fasciculations occur at
rest and the interference pattern is reduced at maximal activity. In
myopathies, the muscle is silent at rest and has polyphasic individual
potentials
of
low amplitude and short duration during activity. Myotonia frequently
presents as a classic pattern of spontaneous bursts of potentials that
wax and wane and give an acoustic pattern resembling a dive bomber.
Muscle evaluation should include the areas of the body involved in the
weakness, and examination of four muscles is usually sufficient. The
deltoid and vastus lateralis are good muscles to study in children,
because they are frequently involved in neuromuscular diseases of
childhood. In myopathies, there may be no correlation between the
severity of the muscle weakness and the electromyography.
myelination and the diameter of the neuron. The median, ulnar,
peroneal, and posterior tibial nerves most commonly are studied, and
normal adult values are 45 to 65 m/sec. In infants, the velocity is
about half that of the adult level, which is reached by 3 to 5 years of
age (235). Motor conduction velocity typically
is delayed in demyelinating neuropathies but is normal in anterior horn
cell disease, root disease, or myopathies. Repetitive stimulation of
the motor nerve can reveal pathologic fatigability, as in myasthenia
gravis (34). Sensory conduction velocities are
delayed and occasionally are helpful in diagnosing the mixed
neuropathies, such as peroneal muscle atrophy or Friedreich’s ataxia.
One in about 15,000 people in the general population develops malignant
hyperthermia during general anesthesia. Although it has been reported
to be associated with many disorders including the congenital
myopathies, muscular dystrophy, osteogenesis imperfecta,
myelomeningocele, and King’s syndrome (ie, short stature, scoliosis,
cryptorchidism, pectus carinatum, characteristic facial features) (82,174,175,189,225,328,336,360),
it is mainly associated with central core myopathy, with which it is
closely linked. There is an abnormality of the ryanodine receptor gene
at the 19q13.1 locus (121).
triggered by the administration of depolarizing muscle relaxants (e.g.,
succinylcholine chloride) or inhalational anaesthetics (e.g.,
halothane). If possible, these medications should be avoided in
patients at known risk. Patients with previous episodes or a positive
family history (transmitted as an autosomal dominant trait) of
malignant hyperthermia may be treated prophylactically with sodium
dantrolene. Preoperative assessment of risk is difficult (315).
In vitro muscle contraction tests to various triggering agents are
available but require a muscle biopsy. There are presently no
commercially available means of establishing the presence of the
malignant hyperthermia gene.
at the earliest signs of tachycardia, tachypnea, or a rigid masseter
muscle, because survival is unlikely after significant hyperthermia.
Treatment consists of termination of all anesthetic agents, ventilation
with 100% oxygen, cooling (e.g., ice packs, iced intravenous fluids,
gastric irrigation, cooling blankets), intravenous sodium bicarbonate
for metabolic acidosis, and administration of sodium dantrolene (1
mg/kg/min, up to 10 mg/kg total dose) (187,193).
Treatment is continued for as long as 6 hours after an attack. In
patients at high risk for malignant hyperthermia, the recommended local
anesthesia is procaine, and for general anesthesia, narcotics,
barbiturates, or neuroleptic drugs, and prophylactic dantrolene sodium
are used (239).
Three issues are important: selection of the muscle, technique of the
biopsy, and specimen care. Muscles with mild involvement should be
selected in chronic disease, but severely involved muscle should be
chosen in acute disease. The histology of severely involved muscle in
chronic disease may show only secondary changes and not be diagnostic.
Commonly selected muscles are the vastus lateralis, rectus femoris,
deltoid, gastrocnemius, and biceps brachii. Obtain an adequate specimen
from the belly of the muscle. Avoid areas of musculotendinous junction,
scar from previous surgery, immunization sites, and electrode insertion
sites for EMG (103).
to the technologist or have it transported quickly to the laboratory.
Traditional techniques of maintaining muscle length are not needed for
routine muscle biopsy. A moderate volume (250 mg) is needed to assay
for enzyme systems to characterize metabolic myopathies. Part of the
specimen is sent for genetic and protein analysis, and part of the
specimen is frozen rapidly in liquid nitrogen to preserve the enzymes
and prevent. Histochemical evaluation includes staining with
hematoxylin and eosin, Verhoeff–van Gieson, periodic acid–Schiff (PAS)
stain for glycogen, Gomori trichrome, oil red O for lipids, and
methylene green–pyronine for RNA. Staining for adenosine triphosphatase
(ATPase) at selected pH determines fiber types. In skeletal muscle, the
ratio of muscle fiber types I and II is 1 to 2. Type I fibers have low
ATPase activity and glycogen and high oxidative activity, and type II
fibers have the opposite relative amounts. At present, fibers are
subtyped based on ATPase activity (88).
fibers, but it has the disadvantage of unsightly scars. Needle biopsy
is cosmetically better but has the disadvantage of producing a small
sample with disoriented fibers (63,96). Coordinate the method of specimen handling and biopsy technique with the pathologist to ensure adequate results.
anesthesia (1% Xylocaine without epinephrine) and sedation. The muscle
must not be infiltrated with Xylocaine. Make a 2.5 cm incision,
preferably following the skin lines, over the belly of the selected
muscle. Expose a 2 × 0.5 cm cylinder of muscle (the long axis parallel
to the muscle fibers), and excise the specimen with a scalpel.
Electrocautery should not be used before removing the specimen. Sutures
at either end of the specimen tied over a tongue blade or muscle biopsy
forceps can be used to maintain specimen length. The procedure is
usually performed on an outpatient basis, and complications are
uncommon.
of needle biopsy. The Bergstrom needle, consisting of a cannula and
sliding trocar with cutting blade, typically obtains a specimen of
about 200 fibers. After administration of local anesthesia (1%
Xylocaine without epinephrine), make a stab incision over the belly of
the muscle. Insert the needle into the muscle, and activate the cutting
blade to obtain the specimen. Suction can be applied to the needle hub
to improve the biopsy size (239). Several
repeat specimens may be obtained through the same skin incision by
changing the direction of the needle. Close the skin by a single stitch
or adhesive strip and apply pressure over the muscle for several
minutes to reduce the risk of hematoma formation.
neuropathy but is rarely required. The sural nerve, which is entirely
sensory, usually is selected because it innervates only a small area of
the skin over the dorsolateral aspect of the foot, and the sensory loss
is not usually a functional problem. Light and electron microscopy, the
latter of which requires glutaraldehyde fixation are used for specimen
evaluation. Preparation of the specimen needs to be coordinated with
the pathologist before the biopsy (327).
incision over the posterolateral aspect of the leg parallel to the
interval between the tendo Achilles and the peroneus brevis muscle. The
nerve courses beside the lesser saphenous vein. Isolate 2.5 cm of the
nerve in the interval and cut it sharply (not with scissors). If less
than 1 cm of the nerve is taken, the ends can be reapproximated with
microvascular sutures, but this repair is very time consuming for such
a mild sensory loss and not usually done. If the nerve is not
resutured, secure the proximal end in the deep layer of the
subcutaneous fat, which helps to protect against painful neuroma
formation.
and constitute a diverse group of conditions that include structural
congenital myopathies, diseases that typically present as a floppy
infant, with muscle biopsy demonstrating structural abnormalities
within the muscle cell; dystrophies, conditions in which the muscle
initially develops and functions normally but then progressively
degenerates and atrophies; myotonias, syndromes characterized by the
delayed relaxation of muscle; and metabolic conditions, considered to
include diseases with specific metabolic abnormalities and acquired
myopathies, such as those secondary to infections, autoimmune
disorders, and conditions related to toxins (62).
illnesses that present with hypotonia and weakness from birth; muscle
biopsy demonstrates structural abnormalities in the muscle cell. Most
myopathies have an autosomal dominant transmission, are nonprogressive,
and are characterized by symmetric proximal muscle weakness. Serum
enzyme levels are normal or mildly elevated, and an EMG may show
myopathic changes. The types of myopathies are differentiated by
genetic studies, histochemical, or electron microscopic evaluation of
the muscle biopsy specimen and include nemaline myopathy (rod body),
central core disease, myotubular myopathy (central nuclear), congenital
fiber-type disproportion, minicore disease, and nonspecific congenital
myopathies (4,15,36,50,70,88,101,102,104,134,241,303,321).
Myotubular myopathy has a gene abnormality at the xq28 locus, central
core disease at 19q13.1, and nemaline myopathy at 1q21 (2-tropomyosin
gene).
children with congenital myopathies. They are usually easily reducible
in early infancy but require prolonged treatment to achieve stability.
Any lax-jointed, low-toned infant or any older child who presents
before walking with an easily reducible dislocated hip without
contractures should be suspected of having a myopathy. The hips are
treated in the early stages similar to those in infants with typical
congenitally dislocated hips, except that the total time of treatment
is often prolonged and stability of the hip is difficult to achieve.
reduction in newborns and young infants. The hips reduce initially in
flexion of about 110° and mild abduction of 45°. Instruct caregivers
not to dislocate a hip inadvertently by positioning it in adduction.
These hips tend to redislocate easily, requiring frequent (initially,
almost daily) adjustment of the Pavlik harness. Prone positioning in
the Pavlik harness is helpful. It is important not to allow the hip to
remain persistently posteriorly dislocated in the Pavlik harness,
because this creates a severe treatment complication. After the hip is
stable, maintain the harness in about 90° of flexion and 45° of
abduction until adequate bony and cartilaginous support develops. There
is no time-honored rule for the length of treatment, but the total
course should be long enough to allow joint stability and formation of
a normal cartilaginous acetabulum.
the hip or whose hip does not easily reduce initially in skin traction
until the femoral head approaches the area of the acetabulum. With the
patient under general anesthesia, gently reduce the hip and apply a
cast to maintain stability. Cast treatment may be necessary for as long
as 6 months. Hip dysplasia after treatment for dislocation from
hypotonia and joint laxity may improve with abduction bracing. Use an
abduction brace and a standing frame with the legs in abduction for
these children, in whom the development of walking skills is usually
delayed. After ambulation is achieved, an abduction brace is helpful,
but most children have difficulty walking in the brace. If dysplasia
persists despite brace therapy in an ambulating child, perform a varus
derotation proximal femoral osteotomy and, if needed, an acetabular
redirectional osteotomy.
progressive, often has a long C-shaped or double-curve pattern, and is
most difficult to treat in a hypotonic patient with respiratory
compromise. The orthopaedic dilemmas include poor tolerance from
respiratory compromise in a thoracolumbosacral orthosis or spinal
fusion with subsequent inhibition of spinal growth in a young child,
resulting in a short trunk. As soon as scoliosis is recognized,
institute treatment by linearly posturing the spine on pads when supine
and, if respiratory capacity permits, by a thoracolumbosacral orthosis
with abdominal relief. Pulmonary function studies of the patient in and
out of the orthosis are necessary to determine the safety of orthotic
treatment. If the orthosis cannot be used, a wheelchair can be modified
with asymmetric lateral supports, a slightly reclined back, and a firm
seat.
posterior spinal fusion is necessary, unless cardiopulmonary incapacity
is life threatening, even though this fusion inhibits longitudinal
growth of the spine if performed before skeletal maturity. Internal
fixation is essential, preferably with segmental instrumentation. The
fusion extends from the high thoracic area to the sacrum. The Luque
technique has been performed without fusion to allow spinal growth in
the young child, but this remains an unproven technique because of
certain obvious risks, such as wire breakage, rod breakage, and growth
over the upper ends of the rods, with subsequent kyphoscoliosis. We
recommend a posterior spinal fusion with the unit rod, a modification
of the Luque technique, from the first or second thoracic vertebra to
the sacrum, and we discourage the use of anterior spinal procedures in
patients with respiratory compromise. The unit rod is preferred because
it prevents cephalad-caudad movement of the rods, spinal rotation, and
pelvic obliquity without the use of rod interconnecting devices. In
nonambulatory patients with progressive scoliosis, we recommend
stabilization to the sacrum.
-
Place the patient in the prone position on a four-poster scoliosis operative frame so that the abdomen is free of pressure (6,25,37,77,110,213,214,218,231,247).
-
Make a dorsal longitudinal skin incision
over the spinous processes of the vertebrae to be fused, and forcibly
retract the skin margins with Weitlander retractors to reduce the
bleeding. -
Incise the subcutaneous tissue to the dorsolumbar fascia, exposing the tips of the spinous processes from T1 to the sacrum.
-
Slit the cartilaginous caps of the
spinous processes longitudinally, and interconnect them by splitting
the supraspinous ligament. -
With a Cobb elevator, retract the
cartilaginous caps laterally, perform a subperiosteal dissection down
each side of the lamina, and pack gauze between the bone and
paraspinous muscle to maintain hemostasis. -
Then expose each vertebra subperiosteally
laterally from inferiorly to superiorly to the tips of the transverse
processes in the thoracic area and to the base of the transverse
processes in the lumbar area. -
Place the self-retaining Weitlander
retractors progressively deeper in the wound, and spread them widely
against the paraspinous muscles to minimize bleeding. -
Promptly electrocauterize soft-tissue bleeding and control bone bleeding with small quantities of bone wax.
-
Expose the posterior superior iliac spines and adjacent iliac crest by elevating the erector spinae off the sacrum
-
Expose the outer table of the ilium
subperiosteally down to the greater sciatic notch. With the drill guide
developed for the unit rod, drill holes in the ilium from the bottom of
the posterior superior spines to pass 1 to 2 cm above the sciatic notch
(Fig. 178.1). Be very careful to stay within the intraosseous area, and probe the hole to confirm this.![]() Figure 178.1.
Figure 178.1.
The holes in the ilium are drilled by using a drill guide that hooks
into the sciatic notch. The hole enters at the posterior superior iliac
spine and is drilled 2 cm past the sciatic notch. (From Dias RC, Miller
F, Dabney K, et al. Surgical Correction of Spinal Deformity Using a
Unit Rod in Children with Cerebral Palsy. J Pediatr Orthop 1996;16:734, with permission.) -
Carefully remove the ligamentum flavum from the midline for sublaminar wire passage.
-
Pass dual stainless steel wire strands
under each lamina, except at the top of the fusion and at L5, where two
dual strands are used for strength. Be extremely cautious not to cause
neural damage while passing the wires. -
With a rongeur, osteotome or power burr,
decorticate and perform facetectomies of the vertebrae in the area to
be fused. Corticocancellous allograft is typically used in these
children (150 to 250 g). -
If instrumentation with the unit rod is required, select the appropriate length of rod.
-
Place a flexible measuring rod along the
lamina on the concave side of the scoliosis. The unit rods are prebent
to the contour of the normal spine, which corresponds to the desired
postoperative spinal posture. The unit rod is available in 1/4- and
3/8-inch sizes, the smaller being used in children weighing less 30 to
40 pounds or in very osteopenic children. -
Secure the unit rod to the pelvis in a manner similar to the Galveston technique for Luque segmental spinal fusion (6,77).
-
Cross the pelvic legs of the rod, and
insert them into the pelvis, being careful to be in line with the
drilled holes. Rod holders can be used to guide the legs during gradual
alternate side impaction (Fig. 178.2).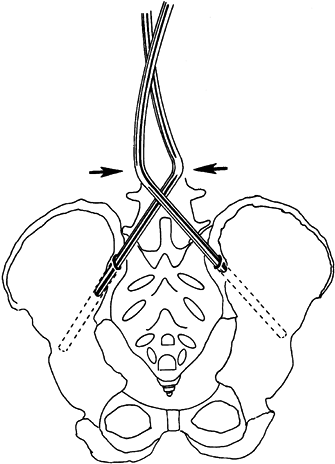 Figure 178.2.
Figure 178.2.
The rod is inserted into the pelvis by crossing the pelvic legs,
keeping them aligned with the orientation of the drilled holes. (From
Dias RC, Miller F, Dabney K, et al. Surgical Correction of Spinal
Deformity Using a Unit rod in Children with Cerebral Palsy. J Pediatr Orthop 1996;16:734, with permission.) -
Then tighten the sublaminar wires by
twisting at each level starting at L5 and moving proximally. Push the
rod to the spine at each level, resulting in gradual correction of the
deformity. -
Close the fascia with interrupted and overlying continuous suture with no drains. Perform subcutaneous and skin closure.
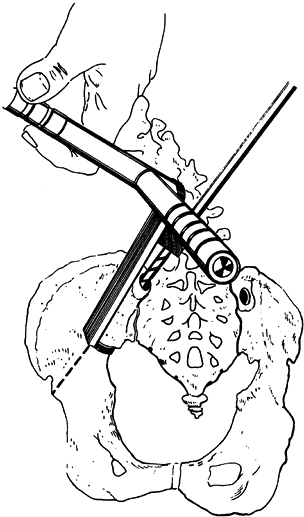
Luque rods, one for the concave side of the scoliosis and one for the
convex side. The Luque rods are available in the same sizes as the unit
rods, and the suggestions for use are outlined above. Before the
operative procedure, obtain lateral bending radiographs to determine
the flexibility of the scoliosis, and bend the Luque rods to achieve no
more than 10° additional correction beyond the preoperative bending
radiographs. Bend the superior end of the convex rod and the inferior
end of the concave rod into the shape of an L. Apply the rod on the
concave side of the scoliosis first, and place the L portion between
spinous processes or through a drill hole in the spinous process to
prevent migration of the rod. If stabilization to the pelvis is
required, bend the inferior end of the Luque rods as described for the
Galveston technique (6). As described for the
unit rod instrumentation, secure the rod to the lamina using the
sublaminar wires. Apply the convex wires in similar manner, and cut and
carefully tighten all
wires.
The two rods are connected inferiorly and superiorly to provide
additional stability. We prefer to interconnect the rods securely to
prevent cephalad-caudad shifting, spinal rotation and loss of pelvic
obliquity correction. Rod connectors can prevent shifting of the rods.
The fusion technique, bone grafting, and wound closure are identical to
that described earlier for the unit rod.
muscle disorders characterized by progressive muscle weakness due to
primary degeneration of muscle fibers. It has become apparent that
these disorders are caused by specific gene abnormalities (301,340).
groups of muscles first affected, genetic transmission, and areas of
body with progressive weakness (301).
boys and usually has an early childhood onset, leading to loss of
ability to walk and eventual death (128,229).
It is transmitted genetically as a sex-linked recessive trait and is
due to a mutation or deletion of DNA at a locus (Xp21) on the short arm
of the X chromosome (106,120,163,164,200,312).
About two thirds of the boys inherit the gene abnormality from the
mother, and one third are thought to be due to new mutations. The onset
is initially insidious, often with delayed motor milestones, with
weakness clinically apparent by 3 years age (385).
The weakness first involves the pelvic-girdle musculature, followed by
the shoulder girdle musculature, and then distal musculature of the
upper and lower extremities (2,387).
and has difficulty climbing steps and running. Pseudohypertrophy of the
gastrocsoleus (Fig. 178.3), deltoid, and
serratus anterior muscles is secondary to the dystrophic process and
accumulation of fat within the muscles and fibrous tissue. The weakness
results in a wide-based waddling gait associated with increased lumbar
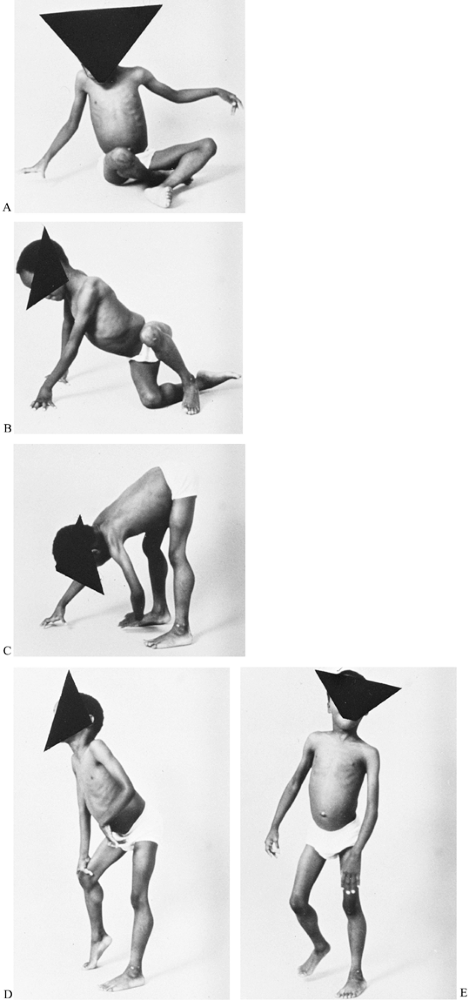
lordosis (Fig. 178.4). Gowers’ sign (Fig. 178.5) is a characteristic way for a child with this type muscular dystrophy to rise from the floor to a standing position (137).
This maneuver may be demonstrated by placing the boy prone on the
floor. First, he crawls into the knee-elbow position; then, with hands
and feet on the floor, he raises his hands consecutively
to
the knees and pushes to an upright posture. The knee reflexes are
diminished and sensation is normal. Progressive weakness is
unremitting, and ambulation becomes more difficult, until between ages
9 and 12 years, the boy loses the ability to walk. Scoliosis and
increasing contractures of the lower extremities develop. The weakness
progresses until total care is required and severe cardiopulmonary
compromise occurs between ages 17 and 22 years (5,14,44,49,173,201,314,319,387).
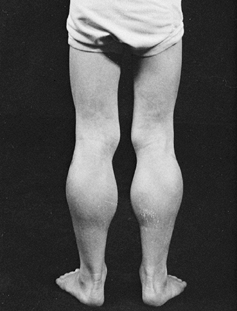 |
|
Figure 178.3. Pseudohypertrophy of the calf muscles in a boy with Duchenne muscular dystrophy.
|
 |
|
Figure 178.4. A boy with Duchenne muscular dystrophy who stands with lordotic spinal posture and has pseudohypertrophy of the calf muscles.
|
 |
|
Figure 178.5. Gowers’ sign is a characteristic way for a child with Duchenne muscular dystrophy to rise from the floor (A to C) by first using the hands to crawl into the knee-elbow position (D to E)
and then to push on the knees to achieve an upright position. Gowers’ maneuver demonstrates weakness in the shoulder and pelvic muscles. |
history, clinical presentation, and an elevation of serum creatinine
phosphokinase (often 100 times normal in young children).
Traditionally, muscle biopsy has been used to confirm the diagnosis but
modern techniques of DNA analysis using peripheral blood can provide a
definitive diagnosis, help to identify carriers, and allow prenatal
diagnosis for 70% to 80% of the children (298).
The molecular basis of Duchenne and Becker’s muscular dystrophy is the
absence or abnormality of dystrophin (a subsarcolemmal protein) and
dystrophin-associated glycoproteins (found in the sarcolemma or muscle
cell membrane) (106,120,163,164,200,312). These proteins are found in skeletal, smooth and cardiac muscle, and
brain. In the 20% of children who do not have a diagnosis by DNA
analysis, muscle biopsy can be diagnostic. The biopsy typically from
the vastus lateralis shows muscle fiber degeneration, regeneration,
fibrosis, fatty infiltration, central nuclear migration and
hypertrophic muscle cells (86).
Absence of dystrophin is diagnostic. EMG, rarely required, shows
myopathic changes with muscle action potentials of reduced amplitude
and brief duration. The nerve conduction velocities are normal.
strength and walking ability for as long as practical and prevention of
deformities (144,179,309,353,356,367,381,382).
The single most important factor in maintaining strength is prevention
of prolonged immobilization. If a boy with Duchenne muscular dystrophy
is immobilized by any method, functional losses tend to be permanent.
Therefore, make every reasonable effort to maintain strength by
resistive muscle exercises. The patient should perform stretching
exercises, especially of the muscles most subject to contractures
(e.g., tensor fasciae latae, hip and knee flexors, and ankle plantar
flexors) several times each day.
successfully in these children from about 5 years of age to improve
muscle strength (142). Complications from this
treatment include weight gain, hypertension, behavior disturbances,
increased appetite, cushingoid features, and osteopenia. Take into
account chronic steroid useage when administering general anesthesia.
muscular dystrophy to falls, and relative inactivity results in
osteopenic bone (162). Fractures of long bones
occur in 20% of children with Duchenne muscular dystrophy, typically
occurring with falls during daily activity or physical therapy (26,162,305,320).
These fractures often herald the end of the ambulatory stage. Treat
nondisplaced fractures of the femur and tibia in lightweight long-leg
casts or splints. Encourage weight bearing on the noninvolved leg
immediately and within days on the fractured leg. Bed rest and traction
are contraindicated. Displaced fractures of the lower limbs require
prompt surgical stabilization.
Apply
a lightweight orthosis to the leg over the area of internal fixation
and begin ambulation. The family should be aware that fracture
complications are higher with early mobilization but that the added
risk is necessary to avoid even more serious problems.
controlling the severity greatly enhances the quality of life. Toe
walking, caused by contractures of the tendo Achilles, can sometimes be
detected in patients as young as 3 years of age, and it responds to
stretching therapy or serial plaster casts. Tendon lengthening in young
ambulatory patients is discouraged because of resulting weakness. For
the ambulatory patient, a nighttime ankle-foot orthosis helps eliminate
the typical equinus posturing of the foot during sleep, and muscle
stretching therapy delays progression of contractures (320).
Despite aggressive therapy, the ability to walk becomes threatened
between 8 and 12 years of age from quadriceps muscle weakness (331);
contractures of the hip flexors (Thomas test), hamstring muscles, and
iliotibial tract (Ober test); and equinovarus deformities of the feet.
As these children lose the ability to walk independently, surgical
releases of lower extremity contractures and long-leg bracing can
prolong standing and ambulation for several years (9,26,29,64,73,156,265,272,279,310,316,320,354,355,367).
When surgical releases are delayed until the children have almost
stopped walking, the severity of weakness usually means that standing
or walking is possible only with knee-ankle-foot orthoses. Earlier
surgery followed by physical therapy and limited bracing (ankle-foot
orthoses) can be just as effective (13,108,179,266).
muscular dystrophy include Yount fasciotomy of the iliotibial tract,
Ober release of the iliotibial band, distal hamstring lengthening,
transfer of the posterior tibialis tendon to the dorsum of the foot,
percutaneous lengthening of the tendo Achilles, and open lengthening of
the tendo Achilles.
The late weakness of the posterior tibialis muscle often causes a varus
deformity of the foot, which is sometimes treated by a posterior
tibialis tendon transfer through the interosseous membrane of the tibia
and fibula to the dorsum of the foot, which maintains a plantigrade
foot. If, however, the varus is not severe and the posterior tibialis
muscle is weak, a tenotomy can be performed easily just posterior to
the medial malleolus (308). The tenotomy is often the treatment of choice, because the child must wear an orthosis in any case.
 |
|
Figure 178.6.
A boy with Duchenne muscular dystrophy who has undergone operative release of the iliotibial tract, distal hamstring muscle release, and percutaneous tendo Achilles lengthening. He now ambulates with long-leg braces. |
-
Expose the iliotibial tract through a 2 cm lateral longitudinal incision, located proximal to the lateral femoral condyle.
-
Incise the iliotibial tract, fascia lata,
and intramuscular septum transversely at a level 2.5 cm proximal to the
patella. Protect the biceps tendon and the common peroneal nerve
posteriorly. -
A segment of the iliotibial tract and septum may be removed in patients with severe contractures to prevent recurrence.
-
With the patient in a lateral decubitus
position, make an incision from 3 cm posterosuperior to the greater
trochanter of the femur obliquely to 2 cm inferior to the
anterosuperior iliac spine. -
Incise the iliotibial band from the anterior portion of the gluteus maximus muscle anteriorly to the anterosuperior spine.
-
Incise the fascia surrounding the tensor fasciae latae transversely.
-
Through a 2 cm medial longitudinal
incision centered over the talonavicular joint, free the posterior
tibialis tendon from its distal insertions. -
Make a second 2 cm incision
midlongitudinally at the musculotendinous junction of the posterior
tibialis tendon just posteromedial to the tibia, and isolate the
posterior tibialis tendon. -
Place a blunt, smooth elevator beneath
the tendon and lift it medially, drawing the tendon into the proximal
wound. Elevate the muscle origin from the tibia and interosseous
membrane for several centimeters proximally. -
Make a third incision of 1 cm anterolaterally between the tibia and the fibula, 2.5 cm above the ankle joint.
-
Direct a long, curved tendon passer from
the second incision posterior to the tibia, through the interosseous
membrane, to the third incision. -
Open the jaws of the tendon passer to
create an opening in the interosseous membrane. Some surgeons fashion a
2-cm long window in the membrane using a scalpel; this reduces the
chance of adhesions between the tendon and membrane (although this is
not a major consideration in patients with Duchenne muscular dystrophy). -
Place a heavy, nonabsorbable suture in
the distal end of the tibialis posterior tendon. Using a suture, draw
the posterior tibial tendon forward through the interosseous membrane
to the third incision. -
Make a fourth 2 cm incision over the dorsal surface of the third cuneiform or the third metatarsal.
-
Retract the extensor tendons and incise the periosteum in a cruciate fashion.
-
Drill a 0.6 cm hole plantarly through the bone.
-
Direct the tendon passer subcutaneously
from the fourth incision to the third incision, and deliver the tendon
subcutaneously to the fourth incision. During passing, be careful to
allow no twisting or kinking of the tendon. Some surgeons prefer to
pass the tendon beneath the extensor retinaculum, but in our
experience, this procedure is unnecessary, and it may become the site
of tendon adhesion that restricts motion. -
Pass the sutures attached to the tendon
end through the plantar surface of the foot with long, straight
needles. The needles and sutures should exit the plantar surface of the
foot in a non-weight-bearing area. -
Direct the tendon into the drill hole,
hold the foot in a neutral position, and anchor the sutures snugly over
a heavily padded button. The tendon can also be fixed to the midfoot
using one of the available anchor systems. -
Further secure the tendon to the drill hole site by interrupted sutures to the periosteum.
-
Close the incision and apply a
well-padded long-leg cast with the foot in a slightly dorsiflexed
position. Particular attention should be directed toward padding the
proximal fibula, where the peroneal nerve is most cutaneous.
and apply a short-leg walking cast for an additional 4 weeks. After
cast removal, remove the button, place the extremity in a brace, and
begin active exercises.
-
Perform a percutaneous tenotomy with the patient in a prone position.
-
Dorsiflex the foot to maintain the tendo Achilles in a taut position.
-
Palpitate the posterior tibial artery in
the neurovascular bundle, and protect it during the procedure. The
tendo Achilles rotates 90° on the longitudinal axis between its origin
and insertion, and the medial fibers proximally are posterior at the
insertion. -
Make a longitudinal 3 mm stab wound
medial to the tendo Achilles and 1 cm superior to the calcaneus, and
divide the anterior two thirds of the tendon fibers. -
Make a second stab wound incision
dorsally 2 cm below the palpable musculotendinous junction and divide
the medial two thirds of the tendon fibers. -
Dorsiflex the foot and the tendo Achilles lengthens.
-
Close the stab wounds, and apply a well-padded cast at 5° dorsiflexion for 4 weeks.
-
With the patient in a prone position,
make a 5 cm longitudinal incision from the superomedial aspect of the
calcaneus proximally along the medial border of the tendo Achilles. -
Divide the subcutaneous tissue and tendon sheath, and evacuate the rotation of the tendo Achilles.
-
Incise the midposterior area of the
tendon longitudinally by a stab wound. Place a clamp in the stab wound
incision, and open it so that the tendon splits in the longitudinal
direction of the fibers. -
Cut one part of the split tendon transversely distally and the other proximally, creating a Z-plasty tenotomy.
-
Dorsiflex the foot to 5° above neutral
and suture the tendon in the lengthened position. Apply a well-padded
cast for 6 weeks, followed by a brace.
procedure, and lightweight long-leg casts are applied with the knees in
extension and the feet neutral.
return to walking is encouraged. We incorporate a strip of polyurethane
foam (7.5 × 1.5 cm) for padding dorsally under the standard long-leg
cast. Ten days later, the casts are removed, long-leg orthoses are
measured, and casts are reapplied until the orthoses are fabricated.
The knee-ankle-foot orthosis prescription should include lightweight
materials (usually plastics), contouring proximally to allow the
buttocks to rest on the brace, drop-lock hinges
at the knee, solid ankles in the neutral position, tarsal straps, and extension beyond the metatarsal heads (377).
to a wheelchair. At this stage, contractures of the knees and hips are
inevitable. Surgical releases usually are not required; instead, a
program to maintain motion is indicated to prevent the progression of
contracture that would hinder a good sitting position in the
wheelchair. Occasionally, a patient, especially one who earlier refused
surgery to prolong walking or refused orthotics, develops a severe,
rigid equinovarus deformity of the foot that causes pain on the
anterolateral aspect of the foot or the inability to wear shoes.
Multiple tenotomies (tendo Achilles, tibialis posterior, and flexor
digitorum longus) and postoperative casting can achieve a satisfactory
foot position. Severe, stiff longstanding deformity can be corrected
operatively with a tarsal medullostomy, but postoperative foot edema
may persist for at least 6 months.
-
Place the patient in the supine position, and control hemostasis during the procedure with a pneumatic tourniquet.
-
Make a 3 mm stab wound skin incision obliquely over the sinus tarsi.
-
Introduce a 3 mm oval curette into the
sinus tarsi. Curette the talonavicular, calcaneocuboid, and subtalar
joints, leaving the outer cortical margins intact. Carefully avoid
injuring the neurovascular structures. -
Close the stab wound incision with a
single stitch, and place the leg in a well-padded long-leg cast with
the knee flexed and the foot in neutral position.
long-leg cast for 1 month, and a short-leg cast for an additional 2
months (Fig. 178.7).
 |
|
Figure 178.7. A: A boy with Duchenne muscular dystrophy and a severe equinovarus foot deformity. B: The deformity was corrected by a tarsal medullostomy. C: After surgery, the foot has maintained a neutral position.
|
a boy with Duchenne muscular dystrophy is able to walk, spinal lordosis
develops to compensate for weakness of the trunk and pelvic muscles. By
the time the child must use a wheelchair, a functional kyphosis
typically develops. Wilkins hypothesized that the kyphosis unlocks the
posterior facet joints, causing an unstable spin and progressive
scoliosis (269,366).
For whatever reason, scoliosis typically becomes apparent during the
last months of walking or soon after confinement to a wheelchair (41,78,230,307).
The peak rate of progression occurs between 13 and 15 years, with up to
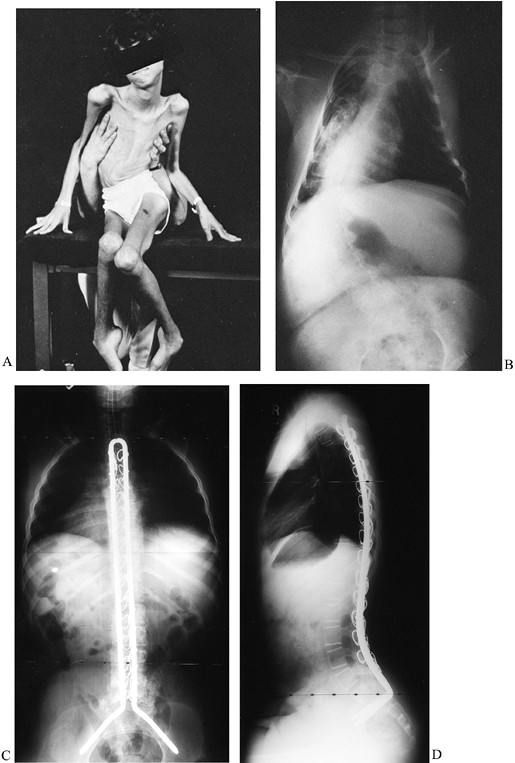
3° of progression per month. The scoliosis is typically a long C-shaped
thoracolumbar curve that progresses steadily until a severe deformity
results (Fig. 178.8A, Fig. 178.8B).
The scoliosis eventually involves the pelvis, leading to severe pelvic
obliquity and difficulty in sitting, back pain, skin breakdown,
pulmonary compromise, and loss of hand function because of obligatory
use of the hands for support of the trunk (313).
A few patients do not develop the spinal kyphosis but persist with the
lordotic posture after ambulation ceases. They develop a less
progressive scoliosis, but by 6 to 8 years after ambulation ceases,
severe scoliosis usually develops. Various spinal orthoses and
wheelchair adaptations have been used to control the scoliosis, but
none has been totally successful, and at best, they only delayed the
progression of curvature.
 |
|
Figure 178.8. A:
After becoming nonambulatory from Duchenne muscular dystrophy, this boy developed a severe scoliosis, which led to pelvic obliquity and difficulty sitting. B: The radiograph demonstrates a long C-shaped pattern of scoliosis. AP (C) and lateral radiographs obtained after a posterior spinal fusion with Unit rod instrumentation (D). |
for progressive scoliosis of 30° or greater. Preoperative cardiac and
pulmonary evaluation must be done to determine the anesthetic risk. A
vital capacity of less than 35% normal indicates that pulmonary
complications are more likely (178,232,270).
fusion with the unit rod or Luque instrumentation with Galveston pelvic
fixation from T2 to the sacrum (37,115,284,329,330,332,362).
The unit rod technique is described earlier in this chapter. The
patients are ventilated for the first 24 hours in the intensive care
unit and then quickly mobilized to return muscle strength and to avoid
pulmonary and gastrointestinal complications. No postoperative bracing
is required.
Careful intraoperative hemostasis, use of allograft, and speed will
reduce blood loss. Preoperative autologous blood donation,
intraoperative cell-saver techniques, and reinfusion of postoperative
drainage will reduce the need for homologous transfusion.
a clinical pattern similar to that of the Duchenne type, but it is
milder and with slower progression (22,23,31).
It results from a deletion in the same gene that causes the Duchenne
type. Dystrophin is present but in reduced amounts, typically above 20%
(164,199). Proximal
girdle muscle weakness and pseudohypertrophy are apparent by 7 years of
age, with maintenance of walking ability until age 16 and death
occurring in middle adult life after cardiopulmonary failure (100).
Clinically, the patient with Becker’s muscular dystrophy resembles a
“strong” patient with Duchenne muscular dystrophy in the juvenile
years. The CPK level is markedly elevated, with levels similar to those
seen in Duchenne muscular dystrophy, and the results of muscle biopsy
resemble those in Duchenne muscular dystrophy. Orthopaedic problems
include equinus, cavus, and scoliosis (186).
The early major orthopaedic problem is progressive contracture of the
tendo Achilles, which may be controlled by muscle-stretching therapy
and by a nighttime ankle-foot orthosis to prevent the typical equinus
posturing of the foot during sleep. Periodic serial stretching casts
usually control the mild contractures, and tendo Achilles lengthening
has occasionally been necessary.
gait and exaggerated spinal lordosis. In the teenage years, ambulation
can be facilitated by canes for balance and a knee-ankle-foot orthosis.
Scoliosis can occur and is managed as for the Duchenne type (186).
group of patients with muscle weakness in a girdle distribution and
autosomal inheritance. Leyden (209) described a type with predominantely pelvic girdle weakness, and Erb (105)
described a shoulder girdle type. Typical findings include elevation of
the CPK level, myopathic changes on EMG, and dystrophic changes on
muscle biopsy. Recent advances in molecular genetics have established
several syndromes with different gene abnormalities that had been
classified as limb girdle dystrophies (87).
There is a wide range of clinical severity. For example, severe
autosomal recessive muscular dystrophy of childhood is characterized by
absence of a sarcoglycan called adhalin (87) and can present with a clinical course similar to that of Duchenne’s muscular dystrophy or with a later onset, milder type.
types of muscular dystrophies, and the principles of management are the
same.
has an onset usually in adolescence, involves the facial and then the
shoulder girdle muscles, and has a slow progression, with an expected
average to long life span (87,148,204,349). The disease is uncommon, with a prevalence of about 1 in 20,000 (249,250).
The early weakness results in a lack of facial mobility, sloping of the
shoulders, and difficulty in raising the arms above the head. The upper
arm and scapular muscles are involved earlier than the deltoid and
forearm muscles. In longstanding, severe cases, the wrist extensor
muscles are more involved than the flexors, producing a wrist drop
called the praying mantis posture. The CPK level is normal to slightly
elevated, and muscle biopsy shows only slight changes, such as isolated
atrophic fibers and variation in fiber size. The gene abnormality has
been localized to 4Q35, but no gene product has been identified (114,365).
variations: typical, as described; late exacerbation type, which may
consist of only mild facial weakness for years and then rapid
deterioration in midlife; infantile type, which has an onset before age
1 year and is severely crippling, requiring a wheelchair by age 9 years
(38); and scapuloperoneal syndrome, in which the peroneal and tibialis anterior muscles are involved early in the illness (16,109).
several diseases that must be differentiated, including myotubular
myopathy, nemaline central cord disease. and mitochondrial myopathy (34).
scapular instability, and back pain. The dropped wrist is treated by a
splint and the dropped foot by an ankle-foot orthosis with a rigid
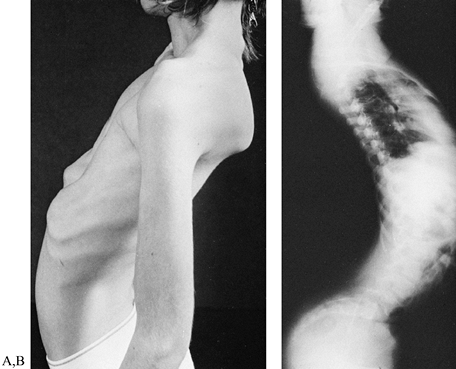
ankle in neutral position. Lordosis (Fig. 178.9A, Fig. 178.9B),
which is initially flexible, usually develops in the lumbar spine but
may become so severe and rigid that the sacrum becomes horizontal. The
lordosis produces severe back pain that may be treated by a lumbosacral
orthosis. In ambulatory patients, the orthosis should support the back
but not necessarily correct the lordosis, which may be necessary for
walking. Surgery for back pain is almost never indicated.
 |
|
Figure 178.9. Photograph (A) and radiograph of a boy with facioscapulohumeral dystrophy and severe spinal lordosis (B). The lordosis frequently results in disabling back pain.
|
and latissimus dorsi muscles limits elevation of the shoulder by
allowing scapular winging and rotation (Fig. 178.10A, Fig. 178.10B).
If the deltoid muscle is strong and scapular winging inhibits function
so that the arm cannot be raised above the horizontal level, a
scapulothoracic arthrodesis may be effective in improving shoulder
flexion and abduction (42,43,61,177,188,207,321) (Fig. 178.10C). The operation initially should be performed on one side; if helpful, it can be considered for the contralateral side.
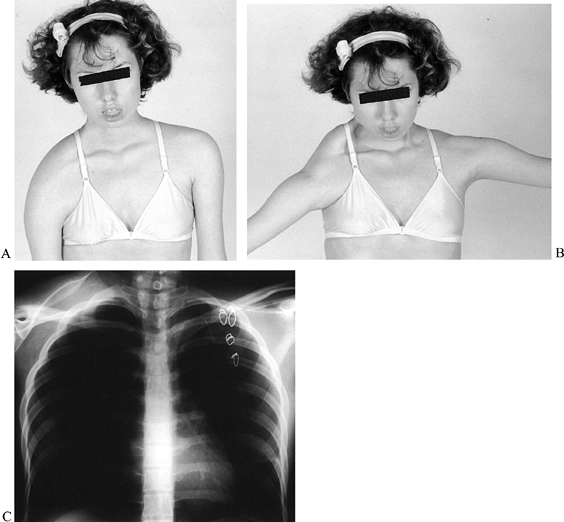 |
|
Figure 178.10. Facioscapulohumeral dystrophy in a young woman with shoulders in neutral (A) and abduction (B). Note the improved abduction and appearance of left shoulder after scapulothoracic arthrodesis. Radiograph (C) demonstrates the wiring technique and preoperative position of the right scapula.
|
-
Place the patient prone on bolsters. Free
drape the shoulder and arm to allow shoulder motion and access to the
brachial and radial pulses. -
Make a 6 cm incision along the medial border of the scapula, with the scapula in a reduced position.
-
Divide the insertion of the rhomboids
into the vertebral border of the scapula to provide access to the
thoracic surface of the scapula. -
Elevate subperiosteally the medial 2 cm
of the subscapularis and the supra and infraspinatus. The medial origin
of subscapularis needs to be resected to allow scapulothoracic contact. -
Position the vertebral end of the
scapular spine at rib 3 or 4. Expose ribs 3,4,5, and 6 subperiosteally
(avoid damaging the parietal pleura). -
Drill two sets of holes at the scapular
spine and one set at the vertebral border of the scapula over each of
the lower three ribs. -
Harvest a corticocancellous iliac graft,
and fashion three 2 cm cortical pieces, and space two drill holes 1 cm
apart in each piece. -
Place two 18-gauge stainless steel wires
under rib 3 and one under the lower ribs, and thread them through the
scapula and the pieces of autograft. Position the scapula so there is a
20° angle between the medial border of the scapula and the spine. -
Decorticate the ribs and undersurface of the scapula, apply cancellous autograft, and tighten the wires.
-
Check for normal pulses in the arm and
for desired shoulder range of motion. Use a shoulder immobilizer for
postoperative immobilization.
describe an autosomal recessive group of disorders that present with
muscle weakness at birth or shortly thereafter and a dystrophic pattern
on muscle biopsy. Neonatal hypotonia is typical, but some children
present with joint contractures (which may be an arthrogrypotic
picture). The condition tends to remain static, but there can be slow
progression or, alternatively, functional improvement and achievement
of walking ability. Respiratory and swallowing problems depend on the
severity of the weakness. The CPK may be slightly elevated, EMG reveals
a myopathic pattern, and the muscle biopsy indicates severe dystrophic
changes (87,224,384).
in association with central nervous system involvement have been
described: Fukuyama-type congenital muscular dystrophy,
muscle-eye-brain disease, and the Walker-Warburg syndrome (87,124,125,190).
Children with congenital muscular dystrophy have severe mental
retardation and guarded prognoses. A subgroup of children with
classical congenital muscular dystrophy with white matter changes in
the brain lack laminin M (merosin), an extracellular protein (87,345).
Mild contractures respond to therapy and splinting but tend to recur.
Rigid and recurrent deformities are difficult and are treated as
described in the section on arthrogryposis.
delayed relaxation of skeletal muscle after cessation of voluntary
contracture or mechanical stimulation. It does not occur spontaneously
but is initiated by voluntary contracture or stimulation and is usually
accentuated by cold or periods of rest (74).
Myotonia is seen in a number of illnesses, such as myotonic dystrophy,
myotonia congenita, paramyotonia congenita, Schwartz-Jampel syndrome,
drug-induced myotonia, and muscle contracture induced by exercise (80,227,344,372). Significant orthopaedic deformities occur in myotonic dystrophy and in Schwartz-Jampel syndrome, which is rare.
contracture after a blow to the muscle belly by a reflex hammer or by a
hand-grasp test, in which the patient grasps the observer’s hand, tries
to release, but immediate relaxation fails and an unwinding motion of
the fingers must occur to unclasp the hand (34).
In infants, myotonia may be manifest by delayed opening of the eyes
after closure with crying. EMG demonstrates a characteristic pattern
and confirms the clinical myotonia. The frequency of discharge is
initially increased, followed by a gradual decrease and cessation,
which produces an acoustic pattern that sounds like a dive bomber.
dominant disorder characterized by myotonia, a progressive dystrophic
process of muscle leading to weakness and atrophy and various endocrine
and systemic abnormalities (48,60,75,374).
The molecular abnormality is an unstable expansion of DNA with a
variable number of trinucleotide repeats in the myotonia protein kinase
gene on chromosome 19 (12,35,45,123,151,152,157,216,291,298).
Myotonic mothers usually have premature onset of labor, postpartum
hemorrhage, and poor uterine tone, which predisposes the infant to
cerebral damage and static encephalopathy (ie, cerebral palsy). The
pregnancy is frequently complicated by poor fetal movements and
hydramnios. In children with severe encephalopathy, an underlying
myotonia can be overlooked. The child appears to have only a complex
form of cerebral palsy, but evaluation of the mother demonstrates the
characteristics of myotonia, and the diagnosis in the child can be
suspected. In the neonatal period, the main characteristic features are
hypotonia with difficulty in sucking and respiratory distress (27).
The neonate frequently has facial paresis and a triangular, fish-shaped
mouth, with the upper lip forming an inverted V. Mental retardation is
common, and the mean IQ is approximately 66 (51).
There is marked wasting of the sternocleidomastoid and trapezium
muscles, and in most patients, muscle function improves in the first
decade of life. Most of these children walk by age 4 or 5 years.
Cataracts usually do not occur until about 14 years of age. Conduction
abnormalities and myocardial dysfunction are common, and these children
routinely require electrocardiograms and echocardiograms routinely (118).
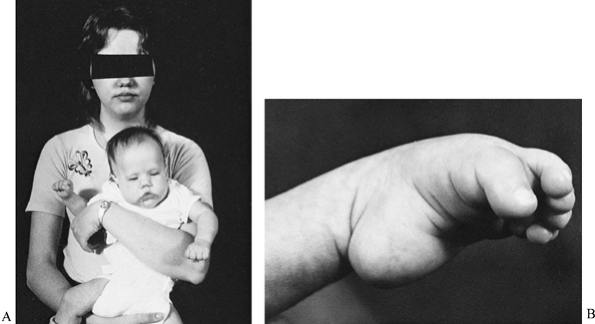 |
|
Figure 178.11. A: A mother with the adult type of myotonic dystrophy and her daughter with the congenital type of myotonic dystrophy. B:
A foot with talipes equinovarus from congenital myotonic dystrophy. The equinus is severe, the forefoot is plantar flexed on the hindfoot, and the first toe is more flexed than the other toes. |
have orthopaedic problems, including talipes equinovarus, congenital
dislocation of the hip, severe truncal weakness, and contractures of
the extremities (167). Children with congenital
myotonic dystrophy should be treated aggressively orthopaedically
because their conditions improve for several years after birth (24,51,153,154,246,351,386).
The talipes equinovarus is not a typical clubfoot. The equinus is the
most dramatic component of the deformity. The forefoot is usually
plantarflexed on the hind foot, and the
first
toe is usually more plantarflexed than the other toes. There is usually
a wide space between the first and second toes. In the newborn, the
clubfoot is best treated by serial casting, gradually bringing the foot
to the neutral position. There is considerable variation in the
stiffness of the foot, from a rigid foot to a hypotonic positional
deformity. In most patients, the foot can be casted to a neutral
position. Careful attention should be given to the toe plate on the
cast to maintain a neutral position; otherwise, the toes curl around
the end of the cast, resulting in an increasingly severe toe deformity.
The most resistant component of the clubfoot is usually the equinus
deformity; therefore, attention should be directed toward molding the
cast at the longitudinal arch of the foot, so that a breach of the arch
will not occur. After the foot is casted to a neutral position, it must
be maintained until ambulation begins. Frequently, these patients are
severely hypotonic, and walking may be delayed until they are 3 to 4
years old, making prolonged maintenance a difficult orthopaedic
problem. Bivalved casts are most useful, although they have a tendency
to slip distally on the foot, and orthoses require frequent
modifications to accommodate growth.
that cannot be corrected with serial casting and thus require surgical
correction. We recommend that surgical procedures not be performed
before the child is 1 year of age, because the children are exceedingly
hypotonic, and the risks of anesthesia and subsequent surgical
complications are high. A posteromedial lateral release usually
corrects the deformity (52,223,311,347,348).
Overzealous correction should not be performed, because a severe valgus
deformity of the foot may occur. The foot must be maintained in a
neutral position after surgery, which may require an orthosis for
several years.
An orthosis typically prevents progression in the childhood years until
the pubertal growth spurt, when progression occurs. Spinal fusion and
instrumentation is effective, but mesenteric artery syndrome has been
reported and experienced by the authors (67).
noticeable in late adolescence or early adult life, although myotonia
may be present in childhood. Muscle cramps, nasal voice character, and
progressive weakness are observed first. The facial muscle atrophy of
the temple, jaw, and neck muscles produces a lower lip droop called an
inverted smile and a characteristic “hatchet-face” or “swan-neck”
facies. The
muscle
weakness is more prominent distally, especially in the calf muscles and
forearms, but the intrinsic muscles of the hands and feet frequently
are spared until later in the disease. Smooth muscle may be involved,
resulting in dysphasia, recurrent pulmonary infections, and lower
gastrointestinal tract dysfunction (135).
Cataracts, frontal baldness, mild mental retardation, and cardiac
abnormalities are seen. The endocrine abnormalities result in abnormal
glucose tolerance and insulin release, with frank diabetes mellitus,
hypothyroidism, and gonadal dysfunction (171).
Deep tendon reflexes are usually absent or diminished in the distal
muscles, and the disease is progressive until death in the fifth to
sixth decade of life from cardiorespiratory compromise.
level, hypogammaglobulinemia with low IgA, and a glucose tolerance test
of the diabetic type (143). The EMG demonstrates typical myotonia. The muscle biopsy shows changes that include internal nuclei and type I fiber atrophy.
neck muscles, pain, or mild subluxation that usually responds
symptomatically to a soft cervical collar. Equinus deformity of the
foot may be controlled by an ankle-foot orthosis. Occasionally, a pes
cavovarus or pes planus deformity develops, and orthotic inserts
typically resolve painful callosities. If a rigid foot deformity causes
pain and instability, a triple arthrodesis to realign the hind foot and
midfoot is helpful. Toe deformities consist of a flexion and a valgus
deformity of the hallux, which may curl under the second toe. The
second to fifth toes develop flexion contractures of the distal
interphalangeal joints with dorsal callus formation. If these
deformities are severe, the hallux is treated by an osteotomy of the
middle phalanx with realignment and Steinmann pin fixation, and the
second through fifth toes are treated by partial phalangectomy of the
distal aspect of the proximal phalanx and internal fixation with
Kirschner wires. These surgical techniques are described later in the
section on peripheral neuropathies.
may have motor signs manifested as weakness or decreased deep tendon
reflexes; sensory abnormalities, which are usually more severe distally
than proximally; or autonomic changes, resulting in impaired sweating,
atrophy of the skin, or loss of hair.
which is limited to a nerve, nerve root, or plexus (e.g., entrapment
syndrome, brachial plexus injury, Bell’s palsy); mononeuropathy
multiplex, which involves multiple peripheral nerves (e.g.,
polyarteritis nodosa); and acquired polyneuropathies (e.g., diabetes,
vitamin deficiencies, alcoholic neuropathy, drug-induced neuropathy,
collagen vascular disease, genetic polyneuropathies such as
Charcot-Marie-Tooth disease). Peripheral neuropathy, demyelinating
polyneuropathy, myositis, encephalopathy, and recurrent infection are
associated with acquired immunodeficiency syndrome (AIDS) (17,71,208).
The incidence of neuromuscular disease in AIDS patients is unknown, but
the diagnosis must be considered in inflammatory polyneuropathy or
myopathy. Complex orthopaedic problems occur in many of the
neuropathies.
determined diseases with nerve fiber degeneration or segmental
demyelinization. The pathology of nerve fiber degeneration involves a
“dying-back” phenomenon that usually begins peripherally in the longest
nerve fibers. Segmental demyelinating diseases involve inadequate
peripheral myelinization by the Schwann cells and result in slowed
nerve conduction velocities.
motor and sensory neuropathies (e.g., Charcot-Marie-Tooth disease),
hereditary sensory neuropathy (e.g., congenital absence of pain),
spinocerebellar degeneration (e.g., Friedreich’s ataxia), and metabolic
defects with neuropathy. The hereditary metabolic neuropathies are rare
and include metachromatic leukodystrophy (ie, sulfatide lipidosis),
Bassen-Kornzweig syndrome (ie, beta-lipoproteinemia), infantile and
juvenile amaurotic idiocy, globoid cell leukodystrophy (ie, Krabbe’s
disease), angiokeratoma corporis diffusum (ie, Fabry’s disease),
Chédiak-Steinbrinck-Higashi syndrome, Tangier disease (ie,
alpha-lipoproteinemia), and Cockayne’s syndrome.
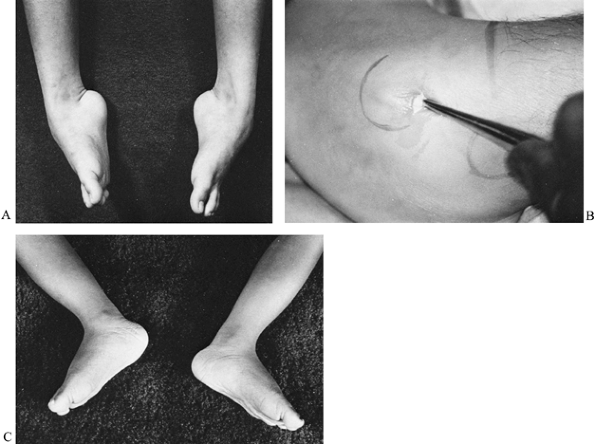
inherited group of diseases of peripheral nerves with sensory and motor
involvement. The general clinical characteristics include distal
symmetric muscle weakness, manifested by equinus feet, steppage gait,
cavus feet, and loss of fine motor function in the hands; there is a
mild sensory abnormality, usually causing poor balance and clumsiness
and decreased deep tendon reflexes. Evaluate any patient with distal
muscle weakness and cavus deformity of the feet for hereditary
sensory-motor neuropathy. The disorders have been classified according
to clinical manifestations, pathology, heredity, and electrophysiologic
changes (72,92,94,95,98,130,141,149,185,263,264,277,278,324) (Table 178.2).
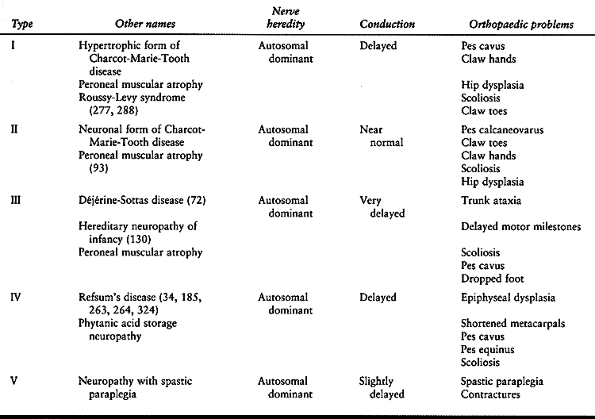 |
|
Table 178.2. Classification of Hereditary Sensory-Motor Neuropathies
|
Charcot-Marie-Tooth disease, which is also called peroneal muscular atrophy (54,346).
The hypertrophic form of Charcot-Marie-Tooth disease (type I) is
associated with segmental demyelinization, reduced nerve conduction
velocity, and an enlarged palpable superficial nerve with an insidious
onset in the first or second decade of life. There is a broad range of
clinical severity. The progression is slow, although a wheelchair is
often required in middle to late adult life. Type IA is usually caused
by a duplication of a gene on chromosome 17 for peripheral myelin
protein 22 (PMP-22), a glycoprotein expressed in the myelin sheath.
Type IB, which has a similar presentation to type IA, is caused by a
mutation of the Po gene on chromosome 1n41. The neuronal form of
Charcot-Marie-Tooth disease (type II) is clinically similar to the
hypertrophic form, except that the pathogenesis involves axonal
degeneration and not demyelination, nerve conduction velocities are
near normal, the nerves are not enlarged, and leg atrophy is severe,
producing a stork-leg appearance. Type II has been linked to chromosome
1p35-p36. An X-linked type has an abnormality in the connexin gene at
xq13. The orthopaedic problems of Charcot-Marie-Tooth disease include
pes cavus, claw toes, drop feet, hip dysplasia, and scoliosis (69,358).
are type III, or Déjérine-Sottas disease, which is similar to type I
but more severe; and type IV, or Refsum’s disease (92). Of interest to the orthopaedic surgeon is hereditary neuropathy with a liability to pressure palsies (HNPP) (371).
The onset is in adolescence, and nerve palsies can occur with minor
trauma to the peripheral nerves. Extreme care must be taken during
surgery to avoid traction on peripheral nerves and, if possible, to
avoid the use of a tourniquet.
commonly seen in peripheral neuropathies, spina bifida, poliomyelitis,
Friedreich’s ataxia, spinal cord lesions, and several less common
neurologic illnesses. The pathogenesis involves muscle imbalance,
but the specific mechanism is unknown (32,33,252,283,287).
A cavus foot has a pathologic elevation of the longitudinal arch of
less than 150° on the lateral radiograph at the intersection of the
axis of the first metatarsal and the calcaneus (169).
There are three forms of pes cavus, as determined by the orientation of
the os calcis during stances: pes cavovarus if the os calcis is
inverted; pes calcaneocavus if the os calcis pitch (long axis of the os
calcis to the floor) is greater than 30° or the long axis of the tibia
to the long axis of the os calcis is greater than 130°, and pes
equinocavus if the os calcis pitch is less than 20° (287).
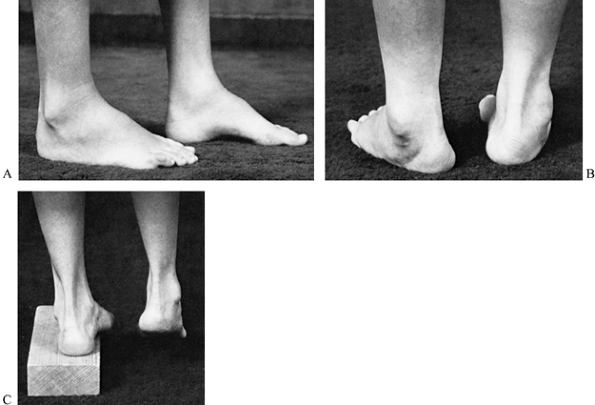 |
|
Figure 178.12. A: A cavus foot in a patient with Charcot-Marie-Tooth disease. B: A cavus foot, demonstrating heel varus during stance. The depression of the first metatarsal forces the foot into varus. C:
The Coleman block test, demonstrating the flexibility of the hindfoot. When the depressed first metatarsal is allowed to hang off the block, the heel is no longer forced into varus during stance. |
deformity combined with decreased proprioception results in difficulty
in walking, lack of balance, and painful callosities. The peripheral
neuropathy initially causes weakness of the intrinsic muscles of the
foot and peroneal muscles, resulting in a forefoot drop, relative
shortening of the long toe extensors because of the forefoot drop, and
hyperextension at the metatarsophalangeal joints. Hyperextension leads
to a secondary tightening of the long toe flexor tendons and to flexion
of interphalangeal joint and a claw toe deformity. The posterior
tibialis muscle initially remains strong, and the first metatarsal
droops more than the remainder of the forefoot. During stance, the
plantarflexed first metatarsal forces the foot into supination, and
contracture of the relatively strong posterior tibialis tendon holds
the heel in varus (Fig. 178.12B). Initially,
the foot is flexible, but the plantar fascia also shortens,
contributing to the depression of the first metatarsal and a fixed
varus deformity of the heel. The rigidity of the heel varus is
determined by the Coleman lateral block test (Fig. 178.12C),
in which a plantarflexed first metatarsal is allowed to hang over a 2.5
cm block, eliminating the forced forefoot pronation (negating the
tripod effect); if the heel returns to a neutral position with this
maneuver, the hindfoot deformity is not fixed (58,59,252). Therefore, attention may be directed toward the midfoot and forefoot.
the cavovarus deformity becomes more rigid, painful calluses develop
over the heel, the base of the fifth metatarsal, and the head of the
first metatarsal (tripod foot). Subsequent bony adaptations result in a
rigid equinocavovarus foot deformity (Fig. 178.13).
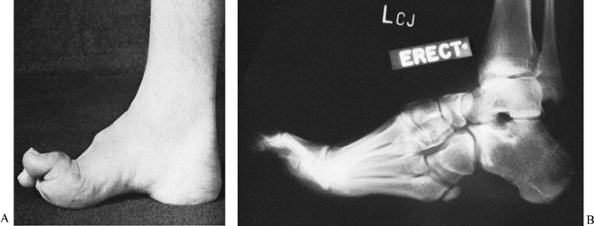 |
|
Figure 178.13. A: A rigid cavus foot caused by Charcot-Marie-Tooth disease in a skeletally mature patient. B:
Radiograph of a rigid cavus foot, demonstrates the elevated longitudinal arch, heel varus, hypertrophy of the fifth metatarsal, and claw toes. |
age, flexibility of the foot, bony deformity, and muscle imbalance. In
the early stages, the whole foot may be slightly supinated, the arch
moderately elevated, and the great toe slightly cocked upward. As soon
as the diagnosis is confirmed, begin daily manipulation of the foot to
resist the depression of the first metatarsal, stretching of the
plantar fascia and tendo Achilles, and extension of the toes. A
nighttime ankle-foot orthosis in a neutral ankle position is
recommended to prevent the foot from dangling into the equinus posture
during sleep and to delay the onset of a fixed deformity. A supple foot
can be treated nonoperatively by manipulation followed by serial
stretching in a short walking cast, followed by an ankle-foot orthosis
with a rigid ankle in neutral position and a lateral heel extension to
resist varus.
becomes necessary to maintain a plantigrade foot. The goals of surgery
are to correct deformity, restore muscle balance, and if necessary,
stabilize the foot. Rigid deformities, usually consisting of a heel
varus and a plantarflexed first metatarsal, must be corrected before
tendon transfers are performed.
Other procedures that can be performed include a plantar medial release
to reduce the midfoot contracture; extension osteotomy of the first
metatarsal to correct a rigid plantarflexed first metatarsal; transfer
of the extensor hallucis longus tendon to the neck of the first
metatarsal and interphalangeal fusion (Jones procedure); transfer of
the extensor digitorum longus tendons to the third cuneiform (Hibbs
procedure); transfer of the posterior tibialis tendon through the
tibiofibular interosseous membrane to the dorsum of the foot to remove
a deforming force and achieve dorsiflexion of the ankle; Dwyer or
medial translation calcaneal osteotomy to correct heel varus if the
heel does not correct with the Coleman block test; and a metatarsal
osteotomy to correct the forefoot (90,91,161,181,273,297,302,333,359,361).
In children younger than 8 to 10 years of age, the heel and first ray
are usually flexible and will correct with soft-tissue releases and
serial casting. Hindfoot equinus and tendo Achilles contracture are
often present. Tendo Achilles lengthening should not be performed
simultaneously with the plantar medial release because it is important
to have a stable hindfoot to allow correction of the longitudinal arch
with serial casting. In the older child with greater weakness, a rigid
hindfoot, and fixed plantar flexion of the first metatarsal, the
plantar medial release is combined with an extension osteotomy of the
first metatarsal (and adjacent metatarsals if required), calcaneal
osteotomy, and a posterior tibialis tendon transfer to the dorsum of
the foot (Fig. 178.14A, Fig. 178.14B). In the skeletally immature, avoid damage to the first metatarsal physis when performing proximal osteotomies.
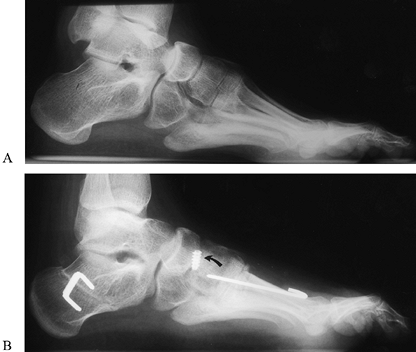 |
|
Figure 178.14. Preoperative (A) and postoperative radiographs of a cavovarus foot (B).
Surgery included a plantarmedial release; extension osteotomy of the first metatarsal, Dwyer calcaneal osteotomy, and anterior transfer of the tibialis posterior tendon (note method of fixation of tendon at arrow). |
procedures include radical plantar fascial release (Steindler plantar
flexor tenotomy) to relieve midfoot cavus; transfer of the posterior
tibialis tendon through the tibiofibular interosseous membrane to the
dorsum of the foot to achieve dorsiflexion of the ankle; transfer of
the extensor hallucis longus tendon to the neck of the first metatarsal
and
interphalangeal fusion (Jones procedure); correction of claw toes by
interphalangeal joint arthrodesis; and extensor tendon release or
transfer, calcaneal and metatarsal osteotomies to correct a rigid cavus
foot; or as a salvage procedure for severe deformity, triple
arthrodesis to correct severe heel varus and severe midfoot deformity
and achieve hind foot stability (7,10,57,90,91,176,181,210,281,325,361,376) (Fig. 178.15A, Fig. 178.15B). Tendon transfer is rarely indicated in the adult foot without an associated bone realignment.
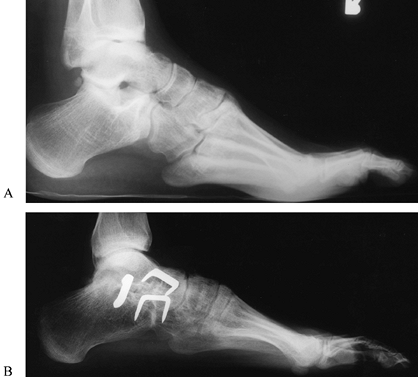 |
|
Figure 178.15. Pre- (A) and postoperative radiographs of a cavovarus foot treated by a triple arthrodesis (B).
|
indicates satisfactory results, but there is a high incidence of
radiologic ankle and midfoot arthritis (289,364).
A study of triple arthrodesis using force plate analysis demonstrates
increased midfoot load bearing and load concentration under the
metatarsal heads (318). The incidence of
pseudarthrosis, typically in the talonavicular joint, is reported to
occur up to 25% of the time. This can be symptomatic and require
revision surgery (289).
-
Make a slightly curved, longitudinal incision on the dorsum of the common extensor tendons, centered over the third cuneiform (126).
-
Incise the subcutaneous tissue by sharp dissection, and protect any neurovascular structures.
-
Divide the common extensor tendons as far distally as possible.
-
Drill a 6.3 mm diameter hole in the third cuneiform from dorsal to plantar.
-
With a pullout suture, draw the proximal
ends of the common extensor tendons into the drill hole, and secure the
pullout suture on the plantar surface over a padded button. -
Close the wound, and apply a well-padded short-leg cast for 6 weeks.
-
Make a dorsolongitudinal skin incision
over the extensor tendons to the third and fourth toes, beginning just
proximal to the midtarsal joints and extending distally through the
middle of the metatarsals (57). -
Separate the extensor tendons to the third and fourth toes, and excise the periosteum overlying the tarsal bones.
-
Identify the tarsal bones radiographically.
-
Remove the dorsal wedge, and elevate the
forefoot to close the wedge osteotomy. Be sure to correct for rotation
deformities (e.g., pronation, supination) of the forefoot at this time. -
Close the periosteum with interrupted
sutures, and approximate the skin. Apply a short-leg plaster cast from
the toes to the knee. Adequate healing is usually obtained within 4
weeks.
-
Expose the lateral area of the calcaneus through an incision parallel, posterior, and inferior to the peroneus longus tendon (89).
-
Strip the periosteum of the lateral area of the calcaneus superiorly and inferiorly.
-
Remove a laterally based wedge of bone
from the calcaneus just posterior, inferior, and parallel to the
peroneus longus tendon. The medial edge of the wedge should not
penetrate the medial cortex of the calcaneus. -
Correct the varus deformity of the
calcaneus by closing the osteotomy and fracturing its medial cortex.
Dorsiflex the forefoot against the pull of the tendo Achilles to
stabilize the osteotomy. The varus deformity should be corrected with
the heel in neutral or slight valgus position. -
Close the wound and immobilize the foot in a short-leg cast until the osteotomy is healed in about 6 weeks.
-
A Jones procedure is transfer of the
extensor hallucis longus tendon to the neck of the first metatarsal and
fusion of the interphalangeal joint. -
Make a dorsolongitudinal 1.5 cm incision over the interphalangeal joint of the hallux (181).
-
Expose the extensor hallucis longus tendon, and cut it transversely 1 cm proximal to the interphalangeal joint.
-
Cut the dorsal capsule of the
interphalangeal joint transversely, and excise the cartilaginous
surfaces of the interphalangeal joint. -
Insert a Kirschner wire from distal to
proximal across the interphalangeal joint; cut the wire approximately 3
mm from the cutaneous margin, and bend it to prevent migration. -
Close the skin by interrupted sutures.
-
Expose the neck of the first metatarsal
through a dorsomedial 2.5 cm incision, dissect the extensor hallucis
longus tendon, and protect the short extensor tendon. -
Pass the extensor hallucis longus tendon through the hole, and suture it on itself with interrupted sutures.
-
Close the wounds, and apply a well-padded short-leg cast, with a plantar extension to protect the toes.
-
Make a longitudinal 4 cm incision along the medial side of the foot from the calcaneal tuberosity distally (325).
Separate the plantar aponeurosis from the plantar foot muscles, and
incise the plantar fascia transversely at the plantar surface of the
calcaneus. -
With a blunt periosteal elevator, lift
the origins of the abductor hallucis muscle, the flexor digitorum
brevis muscle, and the abductor digitiminimi muscle from the periosteum
of the calcaneus. Avoid removal of cortical bone or periosteum with the
fascia and muscle attachments. -
Carry the dissection to the
calcaneocuboid joint, releasing the quadratus plantae and the long
plantar ligament. The entire dissection should be carried out near the
periosteum of the calcaneus, avoiding neurovascular structures. -
Then dorsiflex the foot to the corrected position and close the skin.
-
Apply a well-padded short-leg cast with
particular attention to adequate padding over the metatarsal heads and
dorsum of the foot to prevent pressure necrosis of the skin.
serial stretching casts on a weekly basis to obtain adequate
correction, if desired, beginning about 1 week after surgery.
-
Make a straight 5 cm incision obliquely over the sinus tarsi, extending from the peroneus brevis tendon to the extensor tendons.
-
Dissect the subcutaneous tissue sharply, and distally reflect the tendinous origin of the extensor brevis tendon.
-
Incise the capsule of the calcaneocuboid joint, and remove the articular surfaces with an osteotome.
-
Then incise the lateral capsule of the
talonavicular joint, and remove the articular surfaces of this joint
with an osteotome. A small incision over the medial aspect of the
talonavicular joint allows better visualization for cartilage removal. -
Incise the capsule of the subtalar joint.
With an osteotome, excise the surfaces of the anterior, medial, and
posterior articular facets of the subtalar joint. Carefully remove the
medial borders of the subtalar joint so that the neurovascular bundle
and tendons are not damaged. -
Remove wedges of bone as necessary to
correct any deformity, and position the foot in the neutral position.
Maintain the foot in the neutral position, and suture the reflected
flap of the extensor digitorum brevis in the sinus tarsi. Fixation can
be achieved with staples, Steinman pins, or cannulated screws. -
Close the wound and overlay it with a
gauze dressing. Apply a well-padded long-leg cast as the foot is
maintained in neutral position.
12 weeks to heal. In patients in whom continued ambulation and standing
are necessary for maintenance of function, walking may begin as soon as
tolerable, but periodic radiographs are necessary to verify proper foot
position.
is caused by intrinsic muscle weakness that allows the long toe flexor
muscles to continue to flex the interphalangeal joint and the long toe
extensor muscles to extend the metatarsophalangeal joint without
stability provided by the weak intrinsic muscles. Callosities develop
over the heads of the proximal phalanges from contact in the shoes and
under the metatarsal heads. Initially, clawing is flexible and should
receive daily manipulative therapy. Fixed clawing requires operative
treatment, which is usually performed simultaneously with other
procedures for the cavus foot. Our choice of treatment of the second to
fifth toes includes a tenotomy of the long extensor tendons, dorsal
capsulotomy of the metatarsophalangeal joint, and arthrodesis of the
interphalangeal joint if the toes cannot come down into 20° of passive
flexion.
 |
|
Figure 178.16. A: Preoperative photograph of flexible claw toes in a patient with Charcot-Marie-Tooth disease. B: The toes corrected. C: Preoperative photograph of rigid claw toes in a patient with Charcot-Marie-tooth disease. D: Postoperative radiograph demonstrates straightening of the toes by an interphalangeal fusion with Kirschner wires.
|
-
Stabilize the interphalangeal arthrodesis with intramedullary Kirschner wire fixation.
-
Transfer the extensor tendons to the
middorsal area of the foot to assist in active dorsiflexion of the
foot, if desired (Hibbs procedure). -
For clawing of the hallux, transplant the
long extensor tendon to the neck of the first metatarsal (Jones
procedure), and perform interphalangeal joint fusion. Take care not to
damage the extensor brevis tendon, or the hallux will droop.
-
Make a 1 cm dorsolongitudinal incision over the proximal interphalangeal joint of the toe.
-
By sharp dissection, identify the common extensor tendon, and incise it transversely.
-
With an osteotome, excise the articular surfaces of the interphalangeal joint, and then close the incision.
-
Extend the toe to a neutral position, and
place a smooth Kirschner wire from the tip of the distal phalanx
through the distal phalangeal joint and proximal interphalangeal joint
to the proximal phalanx. -
Cut the Kirschner wire 0.5 cm from the tip of the phalanx and bend it at least 45° to prevent proximal migration.
short-leg cast with the plantar surface extending well past the
phalanges, or have the patient wear a hard-sole wooden shoe until
interphalangeal arthrodesis has occurred in about 4 weeks.
The other types of hereditary sensory-motor neuropathy also have
associated scoliosis, but the incidence is unknown. Progressive curves
between 20° and 40° in immature patients are treated by a
thoracolumbosacral orthosis until maturity. The amount of progression
after treatment is unknown. Progressive curves greater than 40° to 50°
in immature patients are treated by a posterior spinal fusion and
instrumentation. The patterns are similar to those seen in idiopathic
scoliosis. The fusion should include the measured curve (i.e., Cobb
method) and additional vertebrae as necessary to achieve adequate
truncal balance because of the associated truncal weakness. No patients
in our series have required fusion to the sacrum or to the
cervical
area. Because there is a high incidence of failure of
somatosensory-evoked potential monitoring, preparation must be made for
a wake-up test (195).
interferes with fine motor coordination, as in writing, and wrist
weakness makes sports and carrying heavy objects difficult. Most
patients tolerate the weakness by adjusting their lifestyle or by using
simple adaptive equipment. In some patients with type I hereditary
sensory-motor neuropathy and moderate weakness, opponensplasty with
intrinsic muscle reconstruction is helpful (234,375). Kling and Drennan (192) reported on a small group of patients with type II
hereditary sensory-motor neuropathy who developed severe upper
extremity atrophy, which leads to a functionless hand. An orthosis may
help to maintain a neutral posture.
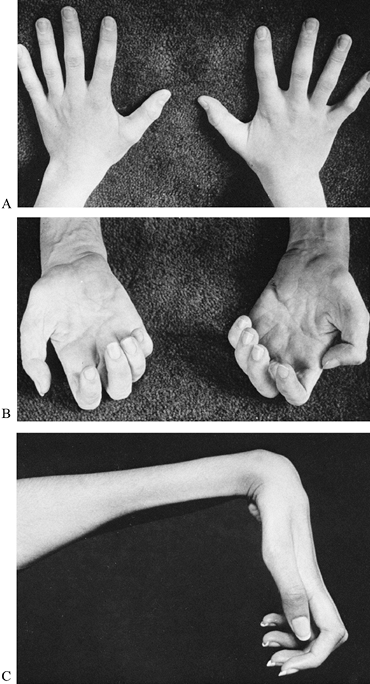 |
|
Figure 178.17. A: Hands with intrinsic muscle weakness caused by Charcot-Marie-Tooth disease. B: Claw hands caused by muscle weakness and contractures from Charcot-Marie-Tooth disease. C: A functionless hand with severely atrophic muscles from Charcot-Marie-Tooth disease.
|
The dysplasia can be asymptomatic or minimally symptomatic and may
remain undetected until early adolescence. Children with
Charcot-Marie-Tooth disease need to be evaluated radiographically to
detect early dysplasia. Typically, the femur has mild coxa valga and
anteversion, and the superolateral corner of the acetabulum is
deficient, which allows lateral subluxation during ambulation (Fig. 178.18).
The acetabular dysplasia is treated by redirection of the acetabulum by
single, double, or triple innominate osteotomies or shelf arthroplasty,
depending on the severity of the deformity (286,323).
The Chiari procedure may be necessary if severe changes in the
acetabulum prevent the femoral head from being fully reduced into the
true acetabulum.
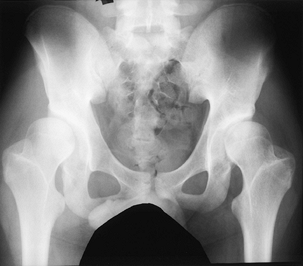 |
|
Figure 178.18. Hip dysplasia in a 16-year-old boy with CMT IA.
|
decreased specific sensory perceptions, painless ulcerations, and
Charcot joints. There are four major hereditary sensory neuropathies:
autosomal dominant hereditary sensory neuropathy, autosomal recessive
hereditary sensory neuropathy, familial dysautonomia (Riley-Day
syndrome), and familial sensory neuropathy with anhidrosis (248,259).
neuropathy have marked loss of sensation to pin prick and temperature
in the lower extremities that can result in severe trophic changes. The
upper extremities are less involved. A nerve biopsy shows a decreased
number of myelinated sensory fibers. In autosomal recessive hereditary
sensory neuropathy, the sensation of touch is more severely affected
than those of pain and temperature, but the upper and lower extremities
are severely affected, and nerve biopsy shows a total absence of
myelinated sensory nerve fibers.
nervous system secondary to a gene defect at the 9q31-33 locus, is
characterized by a reduction in small and large myelinated nerve
fibers. Clinical manifestations include a labile blood pressure,
insensitivity to pain, abnormal gastrointestinal mobility, lack of
fungiform papillae on the tongue, ataxia, areflexia and kyphoscoliosis.
It occurs in Ashkenazi Jews with a prevalence of 1/3600. The children
develop a progressive kyphoscoliosis, and death usually occurs in
infancy or childhood from chronic pulmonary insufficiency and
aspiration (267,268,379).
Familial sensory neuropathy with anhidrosis is characterized by
decreased temperature perception, intact touch sensation, absent axon
reflex to histamine, below-normal intelligence, and anhidrosis.
neuropathy include Charcot joints, joint dislocations, fractures,
chronic osteomyelitis, and severe kyphoscoliosis (215,256).
Treatment of spinal deformity is difficult because of the typical high
rigid kyphosis, osteopenia, labile autonomic nervous system, and
insensitivity to pain. Spinal fusion and instrumentation have a high
incidence of complications but is beneficial (280).
arthropathy in which the synovium is hypertrophic, ligamentous laxity
causing joint instability, and eventual joint surface destruction. The
lack of protective sensation allows unrestricted repetitive trauma, and
large weight-bearing joints are most frequently and severely involved.
Recurrent microtrauma causes synovial inflammation, hemarthrosis,
periosteal elevation with subsequent cortical bone thickening, physeal
widening, osteonecrosis, and osteochondritis dissecans. Minor trauma
can lead to undetected dislocations and fractures (1,180,194).
trauma by patient training, adaptive tools to reduce traumatic
exposure, and protective orthoses. Joint instability usually can be
treated by an orthosis, or if it is severe, a fusion can be attempted.
Fusions are difficult to achieve and require prolonged immobilization;
because delayed union and pseudarthrosis are common, augmentation with
a bone graft is often required. A fusion also tends
to
transfer stress to adjacent joints, which can become deformed. Periodic
radiographic spinal evaluation should be performed to detect early
Charcot changes of vertebrae and to allow treatment by a spinal fusion
to prevent neurologic damage. Osteomyelitis by hematogenous or
contiguous spread occurs frequently in Charcot joints. Cutaneous ulcers
should be protected to eliminate repetitive trauma and should be
treated aggressively with local care to obtain healing. After a joint
is infected, the prognosis is poor. Antibiotic therapy, protection of
the extremity, and excision of infected or necrotic bone may contain
the infection, but uncontrolled infection may require amputation (136).
anesthetized area. In children, the hands may be mutilated by
repetitive biting or other trauma, causing ulceration, osteomyelitis,
and autoamputation of the phalanges. In childhood, ulcerations of the
feet are most common (Fig. 178.19). The only effective therapy is to teach the patient protective techniques and to use protective orthoses.
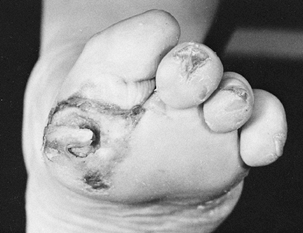 |
|
Figure 178.19. Neuropathic ulcer in a patient with hereditary sensory neuropathy.
|
ataxia) is the most common spinocerebellar degenerative disease. It
usually is autosomal recessive, although it may be autosomal dominant
or sex linked (20). The gene abnormality is on chromosome 9 (55).
The onset is at the end of the first decade and is characterized by
progressive ataxia of the limbs and of gait, the presence of Romberg’s
sign, absent knee and ankle reflexes, extensor plantar responses,
dysarthria, scoliosis, pes cavus, weakness, loss of position and
vibration sense in the legs, cardiomyopathy, and diabetes (30,131,202).
ataxic; by 20 years of age (average, 15.8 years), most are confined to
a wheelchair; and by 40 years of age (average, 36 years), death occurs
from cardiopulmonary failure (47,160,338).
Sural nerve biopsies show axonal degeneration; the central nervous
system exhibits changes in the posterolateral columns of the spinal
cord and cell loss in the deep cerebellar nuclei. Muscle biopsy reveals
fiber group atrophy characteristic of denervation (88).
treatment of the orthopaedic deformities of pes cavus and scoliosis
substantially enhances the quality of life, especially in the less
severely involved patient.
equinus foot, pes cavovarus, tripod stance, and claw toes. As in
Charcot-Marie-Tooth disease, the intrinsic muscles are weak, with the
foot everters and dorsiflexors being weaker than the foot inverters and
plantar flexors, unlike the usual Charcot-Marie-Tooth disease, the
severe ataxia causes a markedly unstable foot.
plantigrade foot, balancing muscle function, and correcting deformity.
In the immature foot, which is almost always flexible, an ankle-foot
orthosis and stretching exercises are usually sufficient. More rigid
foot deformities occur during the adolescent years. Our preference for
treatment of the cavus foot and instability is a triple arthrodesis;
for the muscle imbalance, a transfer of the posterior tibialis tendon
through the interosseous membrane to the dorsum of the foot is
indicated; and for claw toes, an interphalangeal joint arthrodesis is
preferred (217). These surgical techniques were previously described under peripheral neuropathies.
presenting between ages 9 and 21 years and is relentlessly progressive,
even after skeletal maturity (47,66,112,157,160,203).
Scoliosis in this disorder is not a collapsing spine disorder, such as
that seen in poliomyelitis. It is very similar to idiopathic scoliosis
in its behavior. A thoracolumbar orthosis may be prescribed for a curve
between 20° and 40°, but there is insufficient evidence to support the
efficacy of this treatment, and at best, it only slows progression (47,66).
The orthosis is poorly tolerated in ataxic, unstable ambulatory
patients, because it eliminates trunk motion used to correct imbalance
during gait. Nighttime bracing often is an acceptable compromise. For
progressive scoliosis greater than 40° to 60°, extension of the fusion
to the sacrum is not always required. A posterior spinal fusion from
the high thoracic area is indicated. Segmental instrumentation should
be used, which allows for rapid postoperative mobilization without a
brace. A
thorough cardiopulmonary evaluation and ongoing monitoring are mandatory because virtually all patients have cardiomyopathy.
illnesses with proximal muscle weakness caused by degeneration of the
anterior horn cells of the spinal cord and bulbar motor nuclei (337).
The hypotonia is symmetric, the lower extremities are weaker than the
upper extremities, there is no sensory loss or upper motor neuron
signs, and fasciculations of the tongue and tremors of the hands are
common (46,350).
Cardiac function is not impaired. The prognosis for life expectancy
depends on respiratory capacity. The spinal muscular atrophies are
divided into several syndromes according to the severity of weakness,
age at onset of symptoms, distribution of muscle weakness, and pattern
of heredity (334) (Table 178.3). The clinical presentations of the syndromes vary and may overlap in the spectrum of syndromes (133). The most common forms are types I, II, and III (184,253,254).
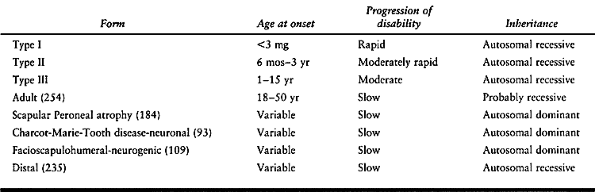 |
|
Table 178.3. Types of Spinal Muscular Atrophy
|
disease) is the most severe illness. The onset occurs within the first
3 months of life, and the patient presents with progressive weakness
and hypotonia with spontaneous movement confined to the toes, fingers,
and hands. Infants with this disease are never able to develop head
control or roll over, bulbar weakness results in difficulty in sucking,
tendon reflexes are absent, and death usually occurs within the first
year of life, before significant orthopaedic problems develop (40,165,363). Types I, II and III are caused by abnormalities of the survival motorneuron gene (SMN) on chromosome 5.
to severe spinal muscular atrophy) the infant develops normally for the
first 6 months of life and learns to sit, but symmetric proximal muscle
weakness ensues (155). The children never stand
or walk. Fasciculations of the tongue and a fine motor tremor of the
hand are helpful diagnostic signs. The weakness is moderate, with
survival until late adolescence or early adult life. The patients must
use a wheelchair, and they develop postural contractures of hip
flexion, knee flexion, and foot equinus. Scoliosis is common,
progressing rapidly and markedly reducing respiratory ability.
Prognosis is related to the progressive muscle weakness and spinal
deformity, which eventually lead to respiratory failure.
Kugelberg-Welander disease, mild spinal muscular atrophy) has an
insidious onset in children from 2 to 12 years of age with episodes of
weakness. Patients have a waddling gait, with difficulty in climbing
steps and getting up from the floor. The prognosis is good, and there
is only a minimal respiratory deficit. Patients tend to have flat feet
(eversion of the foot), pelvic girdle weakness, a mild hand tremor, and
scoliosis (129,196).
patients with spinal muscular atrophy according to function. Group I
children are unable to sit independently and usually die in infancy of
pulmonary failure. Group II children develop head control, can sit
independently, and usually survive until young adult life. Group III
patients pull to stand and walk, and may live into the fourth decade.
Group IV patients can walk, run and climb steps, but usually require
wheelchairs by the third decade and have a long life expectancy (107,383). The major orthopaedic problems are spinal deformities, contractures, and hip instability.
learn to walk and run. The most common deformity is a long C-shaped
scoliosis, and severe pelvic obliquity develops as the curve
progresses. A kyphosis is usually associated with the scoliosis, and a
progressive spinal deformity decreases pulmonary capacity and
jeopardizes sitting posture (66,81,150,157,228,294).
In a series of patients followed at the Alfred I. duPont Hospital for
Children (Wilmington, DE), the average age at onset of scoliosis was
7.6 years (range, infancy to 13 years) (8). In
a mild deformity, a soft thoracolumbosacral orthosis with a large
abdominal relief provides trunk support and may delay progression of
the curve, but the orthosis must not compress the chest and further
compromise respiratory function (228).
Pulmonary function evaluations before and after application of the
orthosis are used to determine safe parameters, because often an
orthosis decreases the tidal volume by as much as 20% (8).
In severely compromised patients, the posterior half of an orthosis may
be used alone, but this is less effective. Typically, scoliosis is
progressive, and when the curve reaches 40° to 45°, a posterior spinal
fusion from the high thoracic area to the sacrum with internal fixation
is recommended. In a child younger than 10 years old with a progressive
scoliosis greater than 60°, surgery is indicated.
evaluation must be performed. Severe postoperative respiratory problems
are seen with a vital capacity less than 25% normal (228).
An intense respiratory rehabilitation program, emphasizing inhalation
strength, coughing, and maintenance of good pulmonary hygiene, can
temporarily improve respiratory ability by about 15% to help during the
perioperative period. After surgery, the pulmonary function usually
returns to the preoperative level and gradually declines, but one study
suggests an improvement (271).
Galveston pelvic fixation or the unit rod in this group of patients
because they allow rapid postoperative mobilization, which helps in the
general care and especially inpulmonary care (39,258).
Patients with spinal muscular atrophy typically have severely
osteopenic bone, and supplementary bone grafts are necessary to achieve
an adequate fusion mass. Loss of pelvic fixation due to the rods
cutting out of the ilium anteriorly requires revision because colonic
perforation can occur (228). In young children
with a large deformity and osteopenic bone, do not attempt an extensive
correction, and use the smaller ¼ or 3/16 inch rods. Rod
connectors when using Luque rods prevent rod shift and increasing
deformity. For 3 to 4 months postoperatively, a thoracolumbosacral
orthosis can be used when the patient is sitting or standing to give
additional support and to prevent the wires of the Luque rod or unit
rod from pulling loose from the osteopenic bone. In patients with
severe respiratory compromise with a large curve, the usual methods of
internal fixation may be impossible, and halo-dependent traction
followed by spinal fusion and limited instrumentation may be the only
reasonable alternative. Anterior spinal fusions generally are not
recommended in patients with spinal muscular atrophy because
postoperative respiratory compromise is severe (8,228).
Postoperative ventilation for 24 to 48 hours in an intensive care unit
is often required. For all patients, the period of immobilization
should be only a few days, and sitting or ambulation should be started
as soon as tolerable, except for the rare problem requiring the
halo-dependent technique. Even in this situation, the patient can be up
in a wheelchair in overhead traction during the day.
and are more frequently seen in the severely weak children. Flexion
contractures of the elbow and adduction contractures of the shoulder
are common but rarely a functional problem. Bedridden infants assume a
frog-leg position, and lower extremity contractures occur with the hip
in flexion abduction, the knee flexed, and the foot in equinus.
Wheelchair-dependent patients develop flexion contractures of the hip
and knee and equinovarus deformity of the foot (107).
Ambulatory patients have only mild contractures, which seldom interfere
with walking. A maintenance motion-therapy program with intermittent
splinting can control most contractures, and surgery is seldom
required. Soft-tissue procedures are effective in reducing contractures
but are indicated only if a contracture inhibits some functional
ability or becomes painful. Surgical releases are used to get these
children into long-leg braces or a standing frame. Hip flexion
contractures require release of the fascia lata, sartorius, rectus
femoris, and iliopsoas. Knee flexion contractures require tenotomy of
the medial and lateral hamstrings and iliotibial band. Foot deformities
seldom require surgery, but a severe equinovarus contracture
occasionally causes a skin ulceration on the foot. Tenotomy of the
tendo Achilles and posterior tibialis tendon with manipulation and
casting for 6 weeks with lightweight plastic material are usually
adequate to correct the foot posture.
Hip dislocation was seen in 50% of the functional group I children, 39%
of group II, 37% of group III, but it did not occur in group IV,
according to an unpublished series. The highest incidence of hip pain
occurred in the group II children. In children who are limited
ambulators or confined to a wheelchair and have a mobile painless
dislocated hip, nonoperative management is recommended.
brain stem motor nuclei. New cases have been rare since prophylactic vaccines became available in the 1960s (282,285).
The virus is transmitted by the oropharyngeal-fecal route, and humans
are the reservoir. The incubation period is 6 to 20 days, and an
estimated 1% to 2% of infected people develop neural symptoms.
Coxsackieviruses and echoviruses produce a similar illness and should
be considered in sporadic cases.
convalescent, and chronic. The acute stage begins with gastrointestinal
symptoms (e.g., nausea, vomiting, sore throat), a febrile illness
(usually less than 103°F), followed by meningeal symptoms (e.g.,
headache, nuchal rigidity, back pain, pain on straight-leg raising),
and severe muscle pain. An asymmetric paralysis occurs within 2 days of
the meningeal symptoms and usually reaches its maximum within 48 hours.
The legs are usually weaker than the arms, but weakness may occur in
any muscle group, including the bulbar muscles. The most commonly
involved muscles are the gluteal muscles, hip flexors, quadriceps,
tibialis anterior, medial hamstrings, deltoid, triceps, and pectoralis
major (299). There is no sensory loss, and the
cerebrospinal fluid has a high protein level in the active stage. The
EMG and muscle biopsy results show denervation. During the acute phase,
patients should minimize activities (usually by bed rest), apply hot
packs to painful muscles, position extremities in the anatomic position
to prevent contractures, and perform general passive range of motion on
all joints to the limits of tolerance (293). The acute stage ends when the temperature is normal for 48 hours and there is absence of progressive muscle involvement.
temperature has returned to normal and continues for as long as 2
years, during which time muscle strength improves spontaneously. The
major recovery occurs in the first month, with the exception of the
triceps surae and deltoid muscles, which improve much more slowly.
Sharrard (300) reports recovery of muscle
strength to average two grades above the level at 1 month and one grade
above the level at 6 months. Orthopaedic treatment consists of
restoring a full range of motion of joints, correcting contractures,
and maximizing muscle strength. Overactivity of muscles in the early
convalescent stage can inhibit functional return, and contractures of
antagonistic muscles must be stretched before weaker muscles are
exercised. Braces and orthoses may be used at night to prevent
deformities and during therapy sessions to assist function. Equinus
deformity of the foot requires a short-leg brace, and quadriceps
weakness of grade IV or less requires a long-leg brace with knee-hinge
locks.
weakness, and no further muscle strength recovery is anticipated.
Orthopaedic treatment consists of managing the chronic consequences of
paralysis, muscle imbalance, and growth. Joint imbalance is classified
into two types: flaccid joints (i.e., negative, static) and active
joints with muscle imbalance (i.e., dynamic). Flaccid joints deform
only if allowed to be postured in an abnormal position over prolonged
periods (139). Deformity can be prevented by
maintaining the joint in a neutral position by an orthosis and an
exercise program. Active joints with muscle imbalance develop fixed
deformities that begin as soft-tissue contractures and evolve into bony
deformities that worsen with growth.
Orthoses can permanently stabilize a flaccid joint or augment function
in a dynamically imbalanced joint. Spring-loaded orthoses are useful to
counterbalance unopposed muscle tensions. Muscle-balancing procedures
consist of tendon transfers, which produce joint stability and active
motor power and eliminate deforming forces (292).
The principles of tendon transfer demand that muscle of normal or good
strength must be used because typically one grade of strength is lost
after a transfer, and loss of function of the transferred muscle must
be balanced or an iatrogenic deformity will occur (158,221,236,244).
The joint to be moved by the transferred tendon must be free of
deformity and have an acceptable range of motion, or the transferred
tendon cannot overcome the deformity. The excursion and strength of the
transferred muscle must be similar to those of the muscle being
replaced, and good surgical technique is essential. The tendon must be
routed straight from its origin to insertion, have a smooth gliding
channel, and be placed under tension. Postoperative management of a
transferred tendon usually includes immobilization for 4 weeks with the
joint in a slightly overcorrected position, followed by guarded motion
for an additional 3 to 4 weeks. Postoperative muscle training is
important to obtain optimal function of transferred tendons.
 |
|
Table 178.4. Selected Procedures for the Treatment of the Involved Hip in Poliomyelitis
|
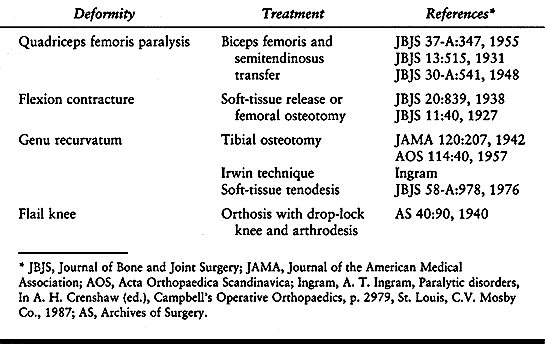 |
|
Table 178.5. Selected Procedures for the Treatment of the Involved Knee in Poliomyelitis
|
 |
|
Table 178.6. Selected Procedures for the Treatment of the Involved Shoulder in Poliomyelitis
|
 |
|
Table 178.7. Selected Procedures for the Treatment of the Involved Elbow in Poliomyelitis
|
 |
|
Table 178.8. Selected Procedures for the Treatment of the Involved Foot and Ankle in Poliomyelitis
|
Residual growth and loss of joint function must be considered before
performing a bony procedure. Ideally, bony procedures are not performed
until the patient reaches about 12 years of age, when adequate growth
has been achieved.
disorder associated with neurogenic and myopathic disease. It is
characterized by rigid or dislocated joints, muscle atrophy or aplasia,
fusiform or cylindrical extremities with thin subcutaneous tissue and
lack of skin creases, and normal sensation and intelligence (3,19,79,119,255,275,276,326). The pathogenesis involves severe weakness during early intrauterine life that restricts fetal mobility,
with subsequent loss of muscle mass and contractures of joints. All
extremities are involved in 46% of patients with arthrogryposis, only
lower extremities in 43%, and only upper extremities in 11% (335).
Serum CPK, chromosomal analysis, EMG, and nerve conduction studies are
rarely helpful in diagnosis, but a muscle biopsy may show neuropathic
or myopathic changes. More than 90% of these patients have neurogenic
illnesses, and 7% have myogenic disorders (18,342).
with congenital contractures, amyoplasia congenita or arthrogryposis
multiplex congenita accounts for the classic form of arthrogryposis
that is characterized by symmetric contractures; internally rotated
shoulders, extended elbows, markedly flexed hands and wrists, flexed or
extended knees, severe talipes equinovarus, frequently dislocated hips,
and a port-wine stain over the forehead area (122,146).
Even though these patients are severely disabled, many develop
ambulatory potentials; 46.6% have independent ambulation, 33.3% walk
with braces, and 20% use wheelchairs (322).
Another infrequent form of arthrogryposis is distal arthrogryposis,
which has autosomal dominant transmission with variable penetrance. The
hands and feet are primarily involved, but knees and hips occasionally
are affected (147).
arthrogrypotic patients produces severe disabilities, and the goals of
orthopaedic treatment are to obtain the maximum range of motion of
joints and maintain the extremities in a functional position.
Twenty-five percent of infants with multiple congenital contractures
sustain birth fractures (usually of the femoral shaft) or epiphyseal
separation, which are attributed to difficult deliveries. If they are
not displaced, these fractures heal rapidly with splint immobilization (76).
Passive stretching exercises are performed at least four times daily
and are guided by the patient’s parents, who are trained and supervised
by physical therapists.
Avoid
rigid cast fixation for most patients. Many parents tend to exercise
the extremity as a unit, and this should be discouraged; specific
attention should be directed to passive stretching of independent
joints. After each joint is passively stretched, apply thermoplastic
splints between sessions to maintain position. The thermoplastic
splints require frequent modifications as the patient grows and the
joint mobility increases. In the knee and elbow, the contractures may
be so severe that radiographs are occasionally necessary to determine
the proper plane for passive flexion and extension exercises.
children with arthrogryposis are to provide an extremity that can be
brought to the mouth and stabilized for feeding and to provide for
toilet care or pulling up from sitting (370).
successful to obtain motion, but the wrist, shoulder, and fingers are
the most resistant. The elbow achieves the most significant benefit
from the stretching therapy, and mild changes substantially improve the
ability to dress, self-feed, and care for personal hygiene. Defer most
surgery until the patient is old enough to demonstrate functional
achievements (211).
useful if the hand is in a nonfunctional position from shoulder
rotation. Perform the derotation osteotomy at the proximal third of the
humerus, and immobilize the site in a shoulder spica cast until healing
occurs. Tricepsplasty
(i.e., lengthening of the triceps tendon and posterior capsulotomy of the elbow joint) is recommended by Williams (369) to restore motion to the extended elbow (235).
Unfortunately, some motion is lost with growth. Flexor muscle power to
the elbow can be facilitated by triceps tendon transfer, but this is
done at the expense of active extension. In our experience, the
pectoralis transfer by fascia lata extension through to the ulna has
been most desirable. The maintenance of the triceps function is very
helpful for the mobility of the patient. Transfer of function can be
successful only if an adequate range of motion has been obtained by
conservative or, if necessary, surgical methods.
row carpectomy or wrist fusion toward the end of growth. Surgery of
small joints of the hand has not been successful in achieving motion or
improving function (21).
-
Make a posterolongitudinal incision from the distal third of the triceps tendon to 2 cm inferior to the olecranon.
-
Expose the triceps tendon, and incise it as an inverted V just below the musculotendinous junction.
-
Reflect the insertion of the triceps
tendon distally, and incise the capsule between the humerus and
olecranon. Gently flex the elbow. Tight collateral ligaments and
occasionally the radiohumeral capsule may require division to obtain
adequate flexion. -
Protect the ulnar nerve by removing it
from its sheath and allowing it to prolapse forward if any tension
develops. The elbow should flex to at least 90°. -
Repair the triceps tendon in the lengthened position by the V-Y technique, and close the wound.
-
Postoperative immobilization is necessary
for 10 days, after which physical therapy is instituted to maintain the
increased range of motion.
elbow flexion is indicated if there are active, strong forearm pronator
and flexor muscles (e.g., pronator teres, flexor carpi radialis,
palmaris longus, flexor digitorum sublimis, flexor carpi ulnaris), the
elbow has adequate motion, the triceps muscle extends the elbow, and
the elbow flexor muscles are paralyzed.
-
Make an approximately 9 cm curved
longitudinal incision over the medial side of the elbow, beginning
about 7 cm proximal to the medial epicondyle, extending over the
posterior aspect of the medial epicondyle, and ending distally on the
volar surface of the forearm along the pronator teres muscle. Identify
the ulnar nerve. -
Remove the common attachment of the flexors to the medial epicondyle with an osteotome.
-
Free the muscles distally for about 4 cm.
-
Flex the elbow, and reattach the excised
medial epicondyle with its muscles to the humerus with a screw or
strong sutures 5 cm proximally at the intermuscular septum between the
brachialis and triceps. -
Close the wound, and apply a cast with the elbow flexed and the forearm supinated.
-
Place the patient in a supine position, with the arm held in mild abduction and the elbow extended.
-
Use two incisions. Make the first
incision from the inferior half of the axillary crease distally to the
level of the middle third of the humerus. Expose the insertion of the
pectoralis major muscle, and free it from the humerus. -
Make a transverse incision at the
anterior aspect of the elbow. Carry dissection down bluntly to the
anterior surface of the ulna. -
Using the tip of the olecranon as a guide, expose the ulna anteriorly about 2 cm distal to this point.
-
Incise the periosteum and drill a hole in the ulna from anterior to posterior. Leave the drill in place.
-
Make an incision along the lateral aspect of the thigh from the greater trochanter to just above the knee.
-
Dissect free a strip of fascia lata as long and wide as possible, and remove it.
-
Close the wound in a routine manner, with no attempt to close the defect in the fascia lata.
-
Sew the fascia lata into a tube with
interrupted stitches. Then pass the rolled-up fascia lata
subcutaneously between the wounds on the forearm. -
Crisscross a strong absorbable suture
along the lower end of the fascia lata, and thread it onto two straight
needles that are placed through the hole in the ulna after the drill
point has been removed. Pull the end of the fascia lata down in to the
ulna, and tie the free ends of the suture over a sponge and button. -
Flex the elbow 70° to 90°, and drape the
other end of the fascia lata around and suture it under slight tension
to the pectoralis major. -
Close the wounds, and apply a splint to hold the elbow at 90°.
dial lock brace to allow flexion and block extension just below a right
angle. Gradually increase the extension over the next month, and have
the patient exercize the elbow into flexion several times daily.
-
Dissect the triceps tendon off the olecranon with a strip of periosteum from the upper shaft of the ulna.
-
Mobilize the triceps off the humerus to the midhumeral level.
-
Flex the elbow 90°, and separate the interval between the brachioradialis and pronator teres muscles.
-
Roll the periosteal elongation of the
triceps and tendon into a tube, pass it around the lateral side of the
arm superficial to the radial nerve, and attach it to the bicipital
tubercle of the radius through drill holes. -
Secure the tendon under tension with the elbow almost fully flexed.
-
Close the wound.
-
Expose the carpal bones through a transverse posterior incision.
-
Retract the extensor tendons medially and laterally to expose the dorsal area of the proximal row of carpal bones.
-
Excise the proximal row of carpal bone,
and dorsiflex the wrist to test the position. If inadequate
dorsiflexion is achieved, all carpal bones except the pisiform may be
excised to obtain adequate dorsiflexion. Dorsiflex the wrist to about
20°, and hold the forearm in a neutral position.
by a short-arm cast for an additional 2 to 3 weeks. After removal of
the cast, have the patient wear a wrist splint part time, usually at
night, to prevent recurrence of the flexion deformity.
-
Make a 6-cm incision dorsally, extending
midway from the base of the second and third metacarpals, across
Lister’s tubercle proximally, and up the dorsal radial surface. -
Dissect the interval between the extensor pollicis longus and the extensor digitorum communis.
-
Denude the dorsal surface of the radius,
carpal bones, and proximal area of the second and third metacarpals of
fibrous or granulation tissue to expose the distal radius, dorsal
carpal bone, and proximal area of the second and third metacarpals. -
Cut a 1 cm wide bony slot in the dorsal
radius across the carpal bones (i.e., lunate, scaphoid, capitate,
trapezoid) into the bases of the second and third metacarpals. -
Place a corticocancellous iliac bone
graft in the slot, and close the wound. A Steinman pin placed down the
third metacarpal into the radius provides some fixation.
long-arm cast. This cast is worn until the arthrodesis is firm, usually
10 to 12 weeks.
most patients with arthrogryposis can walk or stand if the knee is in a
suitable position (317). Motion from at least
15° to 45° of flexion is desirable but may not be obtained in rigid
extremities. Rigid knees that have 35° to 40° of flexion are stable for
standing and sitting; full extension makes sitting awkward, and
excessive flexion makes standing impossible. Knee flexion contractures
(65% of patients) are more common than extension contractures (6%).
Daily passive stretching with thermoplastic maintenance splinting is
the most effective treatment to increase motion in infants and young
children. Forty percent of patients can be managed by nonoperative
treatment programs (339).
juvenile and adolescent patients, recalcitrant contractures, often with
posterior subluxation of the tibia, respond to two-pin traction. Place
the proximal pin in the proximal metaphysis of the tibia, so that the
anterior traction reduces the knee subluxation, and place the distal
pin in the distal metaphysis of the tibia, so that longitudinal
traction can reduce the flexion contracture of the knee. The two-pin
traction method requires prolonged hospitalization and may be combined
with hamstring lengthening and posterior capsulotomy. Recurrence of the
flexion deformities may occur in growing children because the joint
capsule and rigid periarticular soft tissues do not stretch adequately
during growth.
joint deformities, making joint motion impossible. An osteotomy,
usually of the distal femur, is used to reposition the knee to 35° of
flexion, which allows adequate sitting and standing. The angulation of
the distal femur causes a mild iatrogenic cosmetic deformity.
resistant to therapy. The patella may be dislocated laterally, and the
tibia may be dislocated anteriorly. A radiograph of the knee may be
necessary for orientation before initiating to stretching therapy. For
the dislocated knee in the infant, apply longitudinal skin traction to
the tibial area of the legs, initially at 1 lb (0.45 kg) and
progressing to a maximum of 5 lb (2.25 kg). Continue traction until the
tibial plateau is beneath the femoral condyle, and then initiate
flexion therapy. As soon as the knee can be flexed to about 30° in the
reduced position, discontinue traction and apply splints to maintain
reduction. If reduction cannot be achieved by traction, open reduction,
consisting of
a
quadriceps lengthening, release of the lateral patellar retinaculum,
and occasionally, knee ligament lengthening is necessary (65). After flexion is acquired, prolonged orthotic management is necessary to maintain motion.
The absolute treatment goal for hip contractures is to obtain a
functional position of motion in the flexion–extension plane, which is
a major benefit. At birth, the hips are frequently in a nonfunctional
position of abduction, flexion, and external rotation (i.e.,
Buddha-like position) and should be passively stretched and sprinted.
In the newborn, a cloth band can be wrapped around the proximal part of
the leg to reduce the abduction contracture. In patients between 1 and
3 years of age, casts are applied to the legs, with a bar incorporated
between the casts to control abduction and rotation, and the hips are
passively stretched, placing the patient in a prone position. A hip
flexion contracture of 30° is acceptable, because the lumbar spine can
provide adequate compensatory motion. If adequate correction has not
been obtained in the patient by 2 years of age, a soft-tissue release
is helpful, or a varus extension intertrochanteric osteotomy may allow
the desired position without producing additional muscle weakness. If a
rigid contracture prevents ambulation, trochanteric osteotomies can
reposition the extremity at about 35° of hip flexion and bring the hip
out of the abducted position to allow adequate sitting and standing (290).
bilateral or unilateral. Occasionally, there is a reasonably mobile,
nonteratogenic dislocated hip that can be treated in a manner similar
to a typical congenitally dislocated hip if treatment is initiated
before 3 months of age. Bilateral hip dislocations are best left
untreated and attention directed toward obtaining adequate motion (172).
A teratogenic unilateral hip dislocation was formerly thought to cause
severe contractures, pelvic obliquity, and secondary scoliosis, but
this is not always the case in arthrogryposis (84).
An ipsilateral knee flexion contracture should be corrected before
reduction is attempted, because knee therapy can redislocate the hip.
Skin traction (i.e., a home traction program) and passive stretching
therapy are used to obtain as much hip motion as possible (183).
Occasionally, the femoral head reduces to the level of the acetabulum,
and a closed reduction can be performed similar to that done for a
typical congenitally dislocated hip, but postoperative casting should
not allow the hip to contract in a nonfunctional position.
unilateral dislocations of the hip require open reduction through an
anterior approach, extensive soft-tissue release, and possibly femoral
shortening to obtain an adequate reduction. After surgery, a maximum of
6 weeks of casting from the thorax to the toes, followed by prolonged
orthotic control, is necessary for adequate joint remodeling.
Inadequate reduction with subsequent redislocation or subluxation does
occur. if the hip cannot be reduced adequately, the pelvic obliquity is
treated by an intertrochanteric femoral osteotomy to position the leg
functionally, and a shoe lift is used to accommodate the limb length
discrepancy until a properly timed ipsilateral epiphysiodesis is
performed (28,257). If
there is any question about obtaining good motion, it is better to
leave the unilateral hip dislocation unreduced. A stiff hip produces a
more significant problem than the other possible related conditions.
is talipes equinovarus. Convex pes valgus, calcaneovalgus, and
cavovarus deformities are seen occasionally. These severely deformed
feet are often extremely stiff, and the goal of treatment is to obtain
a pain-free plantigrade foot at maturity.
taping and passive stretching therapy. The passive stretching is
performed four times each day, and the taping is changed every other
day to reflect changes in position. Thirty-one of the 80 patients in
our series have had successful nonsurgical treatment, even though a
perfect result was not obtained (251). In
patients with nonplantigrade feet but reasonable motion, a
posteromediolateral release was satisfactory in 15 of 20 feet. After a
posteromediolateral release, prolonged orthotic care and extensive
passive stretching are necessary.
best treated by a triple arthrodesis. In the rigid foot, a talectomy
performed when the child is between 1 and 2 years old gives the most
satisfactory plantigrade foot (84,138,145,170,191). Recurrence after the age of 10 years is best treated by a triple arthrodesis.
-
Make a skin incision from just
distolateral to the head of the talus, extending obliquely
inferoposteriorly to 1 inch (2.5 cm) inferior to the lateral malleolus
(Ollier approach). -
Retract the peroneus longus and brevis tendons inferiorly, and incise the talocalcaneal portion of the bifurcate ligament.
-
Turn the foot medially to expose the talar neck. The talus may be excised as one piece or in fragments.
-
Strip the ligaments from both the medial
and lateral aspects of the malleolus, and displace the foot posteriorly
on the tibia so that the medial malleolus is in contact with the
navicular and the lateral malleolus is at the calcaneocuboid joint. -
Align the foot to the desired rotation (i.e., long axis of the foot is perpendicular of the anterior tibia), and
P.4544
eshape the malleoli to fit the calcaneus in the new position. Any tight
tendons or ligaments may be lengthened to allow proper orientation of
the foot. The foot should align without tension. -
Close the wound, and apply a long-leg
cast with the knee flexed and the foot in the corrected position. If
the knee motion is inadequate to achieve cast support, place a
Steinmann pin through the tibia and incorporate it into a short leg
cast.
a short-leg cast for an additional 10 weeks. Allow weight bearing about
1 month after surgery. After the cast is removed, use a solid
ankle-foot orthosis to maintain position.
Noncongenital scoliosis may be present at birth or may develop in early
childhood; it is progressive, becomes rigid, and is associated with
pelvic obliquity (83,116,304).
Congenital scoliosis is far less common. A thoracolumbosacral orthosis
is preferred in curves between 20° and 40°, and a posterior spinal
fusion and instrumentation is preferred in progressive curves of more
than about 40°. Because the scoliosis tends to be rigid, the fusion
should not be delayed in progressive curves, or a severe deformity will
develop. Arthrodesis is difficult to achieve, and a high incidence of
pseudarthrosis is reported.
release followed by a posterior spinal fusion to the sacrum with
instrumentation enhances correction of the curve and pelvic obliquity (233).
scheme: *, classic article; #, review article; !, basic research
article; and +, clinical results/outcome study.
MA, Johnson EW, Petty J, Stauch D. Mechanical Ventilation of Patients
with Late Stage Duchenne Muscular Dystrophy: Management in the Home. Arch Phys Med Rehabil 1979;60:289.
RM, Koenigsberger R, Mellinger J, Lovelace RE. Central Core Disease
with Congenital Hip Dislocation: Study of Two Families. Neurology 1971;21:369.
JR, McKeon J. Orthopedic Surgery and Rehabilitation for the
Prolongation of Brace-free Ambulation of Patients with Duchenne
Muscular Dystrophy. Am J Phys Med Rehabil 1991;70:323.
JR, O’Brien J, Krolenberg R, Alba AS. Muscular Dystrophy. A Management
of End Stage Respiratory Failure in Duchenne Muscular Dystrophy. Muscle Nerve 1987;10:177.
DF, Moseley CF, Koreska J. Unit Rod Segmental Spinal Instrumentation in
the Management of Patients with Progressive Neuromuscular Spinal
Deformity. Spine 1989;14:1301.
JD, McCurrach ME, Harley HG, et al. Molecular Basis of Myotonic
Dystrophy: Expansion of a Trinucleotide (CTG) Repeat at the 3′ End of a
Transcript Encoding a Protein Kinase Family Member. Cell 1992;68:799.
Proceedings of the Second International Congress on Muscle Diseases.
Part 2, Perth, Australia, 1971. Amsterdam: Excerpta Medica, 1973.
J, Shelbourne P, Davies J, et al. Detection of an Unstable Fragment of
DNA Specific to Individuals with Myotonic Dystrophy. Nature 1992;355:547.
J, Marie P. Sur une Forme Particuliere d’Atrophic Musculaire
Progressive, Souvent Familiale, Debutant par les Pieds et les Jambes et
Atteignant Plus Tard les Mains. Rev Med 1886;6:97.
YH, Lonstein JE, Winter RB, Bradford DS: Spinal Deformities in Patients
with Friedreich’s Ataxia: A Review of 19 Patients. J Pediatr Orthop 1985;5:553.
Y, Lonstein J, Winter R, Bradford DS. Spinal Deformities in Patients
with Muscular Dystrophy Other Than Duchenne—A Review of 11 Patients
Having Surgical Treatment. Spine 1985;10:614.
YH, Lonstein JE, Winter RB, Bradford DS. Spinal Deformities in Patients
with Charcot-Marie-Tooth Disease. A Review of 12 Patients. Clin Orthop 1986;202:219.
DS, Klutzow FW. Congenital Rod Disease. Further Evidence of
Innervational Abnormalities as the Basis for the Clinicopathologic
Features. J Neurol Sci 1974;23:371.
PJ. Inherited Neuronal Degeneration and Atrophy Affecting Peripheral
Motor Sensory and Autonomic Neurons. In: Dyck PJ, Thomas PK, Lambert
EH, eds. Peripheral Neuropathy. Philadelphia: W.B. Saunders, 1975:825.
PJ, Chance PF, Lebo RV, Carney JA. Hereditary Motor and Sensory
Neuropathies. In: Dyck PJ, Thomas PJ, Griffin JW, et al, eds. Peripheral Neuropathy, 3rd ed. Philadelphia: WB Saunders, 1993:1094.
PJ, Lambert EH. Lower Motor and Primary Sensory Neuron Diseases with
Peroneal Muscular Atrophy. Part 1: Neurologic, Genetic and
Electrophysiologic Findings in Hereditary Polyneuropathies. Arch Neurol 1968;18:603.
PJ, Lambert EH. Lower Motor and Primary Sensory Neuron Diseases with
Peroneal Muscular Atrophy. Part II: Neurologic, Genetic, and
Electrophysiologic Findings in Various Neuronal Degenerations. Arch Neurol 1968;18:619.
L, Try K, Stokke O, et al. Dietary Effects on Serum Phytanic-Acid
Levels and on Clinical Manifestations in Heredopathia Atactica
Polyneuritiformis. Lancet 1966;1:691.
GD, Epstein BS, Engel WK, et al. Malignant Hyperthermia and Central
Core Disease in a Child with Congenital Dislocating Hips. Arch Neurol 1978;35:189.
JM, Ohlendieck K, Kahl SD, et al. Deficiency of a Glycoprotein
Component of the Dystrophin Complex in Dystrophic Muscle. Nature 1990;345:315.
A, DeMichele G, Caruso G, et al. Genetic Data and Natural History of
Friedreich’s Disease: A Study of 80 Italian Patients. J Neurol 1990;237:345.
Y, Osawa M, Suzuki H. Congenital Progressive Muscular Dystrophy of the
Fukuyama Type—Clinical, Genetic, and Pathological Considerations. Brain Dev 1981;3:1.
H. Congenital Myopathies. In: Adachi M, Scher J, eds. Current Trends in
Neurosciences—Neuromuscular Disease. New York: Igaku-Shoin, 1990:197.
RC, Moxley RT, Mendell JR, et al. Prednisone in Duchenne Muscular
Dystrophy—A Randomized, Controlled Trial Defining the Time Course and
Dose Response. Arch Neurol 1991;48:383.
J, Dubowitz V, Hyde S, et al. Prolongation of Walking in Duchenne
Muscular Dystrophy with Lightweight Orthoses: Review of 57 Cases. Dev Med Child Neurol 1985;27:149.
RN, MacEwen GD. Spinal Deformity Associated with Heritable Neurological
Conditions: Spinal Muscular Atrophy, Friedreich’s Ataxia, Familial
Dysautonomia, and Charcot-Marie-Tooth Disease. J Bone Joint Surg 1976;58-A:13.
LC, Jackson JA, Elsas LJ. Emery-Dreifuss Humeroperoneal Muscular
Dystrophy: An X-Linked Myopathy with Unusual Contractures and
Bradycardia. Ann Neurol 1981;10:230.
R. A Case of Congenital Defect of the Muscular System (Dystrophia
Muscularis Congenita) and Its Association with Congenital Talipes
Equinovarus. Proc R Soc Med 1908;1:157.
JE, Carr CR. Progressive Muscular Atrophy of the Peroneal Type
(Charcot-Marie-Tooth Disease), Orthopaedic Management and End Result
Study. J Bone Joint Surg 1950;32-A:27.
JG, Bohn D, Edmonds JF, et al. Evaluation of Pulmonary Function in
Muscular Dystrophy Patients Requiring Spinal Surgery. Crit Care Med 1982;10:645.
R, Khan R, Hughes S, Dubowitz V. Congenital Muscular Dystrophy, The
Importance of Early Diagnosis and Orthopaedic Management in the
Long-term Prognosis. J Bone Joint Surg 1979;61-B:13.
W, Wagener H. Conversion of H3-Phytol to Phytanic Acid and Its
Incorporation into Plasma Lipid Fractions in Heredopathia Atactica
Polyneuritiformis. Metabolism 1966;15:687.
M, Iwasaki Y, Wada F, Segawa M. A Case of Congenital Polymyositis—A
Possible Pathogenesis of “Fukuyama Type Congenital Muscular Dystrophy.”
Rinsho Shinkeigaku 1980;20:911.
C, Gluer S, Thermann H, et al. Non-union of the Ulna in a 10-month-old
Child Who Had Type IV Hereditary Sensory Neuropathy. J Bone Joint Surg 1997;79A:1232.
K: Congenital Non-progressive Myopathy, Associated with
Scoliosis—Clinical, Histological, Histochemical and Electron
Microscopic Studies of Seven Cases. J Jpn Orthop Assoc 1980;54:381.
SJ, Marks HG, Bowen JR, MacEwen GD. Hip Dysplasia Associated with
Charcot-Marie-Tooth Disease in the Older Child and Adolescent. J Pediatr Orthop 1985;5:511.
L, Dejerine J. De la Myopathie Atrophique Progressive (Myopathic
Hereditaire), Debutant, Dans I’Enfance, par le Face, sans Alteration du
Systeme Nerveux. Coll R Acad Sci Paris 1884;98:53.
PK, Bertorini TE, Goodwin TG, et al. Dystrophin Production Induced by
Myoblast Transfer Therapy in Duchenne Muscular Dystrophy. [Letter.] Lancet 1990;14:114.
E, Fardeau M, Lytle JO, et al. Scapulothoracic Arthrodesis for Patients
Who Have Facioscapulohumeral Muscular Dystrophy. J Bone Joint Surg 1990;72-A:78.
RM, Bredesen DE, Rosenblum ML. Neurological Manifestations of the
Acquired Immunodeficiency Syndrome (AIDS): Experience at UCSF and
Review of the Literature. J Neurosurg 1985;62:475.
PR, Sanchez JJ. Anterior Transplantation of the Posterior Tibial Tendon
for Persistent Palsy of the Common Peroneal Nerve. J Bone Joint Surg 1961;43-A:60.
M, Tsilfidis C, Saboourin L, et al. Myotonic Dystrophy Mutation: An
Unstable CTG Repeat in the 3′ Untranslated Region of the Gene. Science 1992;255:1253.
WJ, Rinsky LA, Gamble JG: Simultaneous Correction of Pelvic Obliquity,
Frontal Plane and Sagittal Plane Deformities in Neuromuscular Scoliosis
Using a Unit Rod with Segmental Sublaminar Wires: A Preliminary Report.
J Pediatr Orthop 1990;10:742.
(Shriners Hospital for Crippled Children, University of California)
Presented at SICOT 90, XVIII, World Congress, Montreal, 1990.
EB, Fernandez-Bermejo E, Heckmatt JZ, Dubowitz V. Prevention of Rapidly
Progressive Scoliosis in Duchenne Muscular Dystrophy by Prolongation of
Walking with Orthoses. J Child Neurol 1988;3 0 9.
G, Levy G. Sept Cas d’une Maladie Familiale Particulaire: Troubles de
la Marche, Pieds, Bets et Areflexie Tendineuse Generalisee, avec
Accessoirement, Legere Maladresse des Mains. Rev Neurol 1926;54:427.
RB, Dubos JP. The First Fifteen Years’ Personal Experience with
Innominate Osteotomy in the Treatment of Congenital Dislocation and
Subluxation of the Hip. Clin Orthop 1974;98:72.
SK, Leung RK, Tierney RC, et al. Mitral Valve Prolapse Syndrome in
Children with Duchenne’s Progressive Muscular Dystrophy. Pediatrics 1979;63:116.
D, Coerwrankel-Driessen M, vanDalen I, et al. Definition of
Subchromosomal Intervals Around the Myotonic Dystrophy Gene Region at
19q. Genomics 1989;4:384.
F, Specht L. Current Concepts Review: The Diagnosis and Orthopaedic
Treatment of Childhood Spinal Muscular Atrophy, Peripheral Neuropathy,
Friedreich Ataxia and Arthrogryposis. J Bone Joint Surg 1993;75(A):1699.
F, Specht L: Current Concepts Review. The Diagnosis and Orthopaedic
Treatment of Inherited Muscular Diseases of Childhood. J Bone Joint Surgery 1993;75(A):439.
SE, Green NE, Cole RJ, et al. Prolongation of Ambulation in Children
with Duchenne Muscular Dystrophy by Subcutaneous Lower Limb Tenotomy. J Pediatr Orthop 1993;13:336.
JA, Conner SB. Comparison of Harrington Instrumentation and Segmental
Spinal Instrumentation in the Management of Neuromuscular Spinal
Deformity. Spine 1982;7:299.
NS, Williams H, Eisas LJ, et al. Localization of the Gene for
Emery-Dreifuss Muscular Dystrophy to the Distal Long Arm of the
X-Chromosome. J Med Genet 1986;23:596.
VJ. Surgical Correction of the Resistant Clubfoot: One-Stage
Posteromedial Release with Internal Fixation. A Preliminary Report. J Bone Joint Surg 1971;53-A:466.
VJ. Resistant Congenital Clubfoot—One-Stage Posteromedial Release with
Internal Fixation. A Follow-up Report of a 15-Year Experience. J Bone Joint Surg 1979;61-A:805.
FH, Stephens FE. Studies in Disorders of Muscle. II: Clinical
Manifestations and Inheritance of Facioscapulohumeral Dystrophy in a
Large Family. Ann Intern Med 1950;32:640.
PJ Jr, Wagner MB, Kaplan JS, Spencer GE. Predicting the Success of
Reambulation in Patients with Duchenne Muscular Dystrophy. J Bone Joint Surg 1983;65-A:719.
PJ, Wagner MB, Karlinchak B, Katirji B. Evaluation of a Program for
Long-Term Treatment of Duchenne Muscular Dystrophy. Experience at the
University Hospitals of Cleveland. J Bone Joint Surg 1996;78A:1844.
G. Zwen fruhintantile hereditare Falle von progressiver Muskelatrophie
unter dem Bilde der Dystrophic, aber auf Neurotischer. Grundlage Arch Psychiatr Nervenkrankh 1891;22:437.
AH, Sonnenblick EH, Engel WK. Stokes-Adams Syndrome and Atrial
Arrythmias as the Presenting Symptoms of Myotonic Dystrophy, with
Response to Electrocardioversion. Ann Intern Med 1966;65:1260.
K, Rudnik-Schoneborn S, Forrest E, et al. A Collaborative Study on the
Natural History of Childhood and Juvenile Onset Proximal Spinal
Muscular Atrophy. J Neurosci 1997;146:67.