
Editors: Tornetta, Paul; Einhorn, Thomas A.; Damron, Timothy A.
Title: Oncology and Basic Science, 7th Edition
Copyright ©2008 Lippincott Williams & Wilkins
> Table of Contents > Section
II – Specific Bone Neoplasms and Simulators > 6 – Bone Sarcomas >
6.2 – Ewing Sarcoma and Primitive Neuroectodermal Tumor of Bone
II – Specific Bone Neoplasms and Simulators > 6 – Bone Sarcomas >
6.2 – Ewing Sarcoma and Primitive Neuroectodermal Tumor of Bone
6.2
Ewing Sarcoma and Primitive Neuroectodermal Tumor of Bone
Bruce Rougraff
Ewing sarcoma was a feared cancer of childhood, with
very few survivors, prior to the use of chemotherapy, surgical
resection, and radiation. The disease manifests as chronic increasing
pain in the area of a lytic, destructive bone lesion of flat bones and
the diaphysis of long bones. The initial presentation is frequently
confused with osteomyelitis, and it can be mistakenly treated as that
for a period of time before the diagnosis is established. Ewing sarcoma
and a less virulent related disease, primitive neuroectodermal tumor of
bone (PNET), metastasize to lung,
very few survivors, prior to the use of chemotherapy, surgical
resection, and radiation. The disease manifests as chronic increasing
pain in the area of a lytic, destructive bone lesion of flat bones and
the diaphysis of long bones. The initial presentation is frequently
confused with osteomyelitis, and it can be mistakenly treated as that
for a period of time before the diagnosis is established. Ewing sarcoma
and a less virulent related disease, primitive neuroectodermal tumor of
bone (PNET), metastasize to lung,
P.188
bone,
and bone marrow most frequently. Current management includes biopsy,
staging, neoadjuvant chemotherapy, surgical resection and/or radiation,
and further chemotherapy. The 5-year survival rate is 60% to 65% for
nonmetastatic disease and 25% to 30% for metastatic disease.
Pathogenesis
Etiology
-
Unknown; associated with reciprocal
translocation of chromosomes 11 and 22 (90% of cases), which involves
bands q24 and q12 of both chromosomes respectively -
This results in a new chimeric EWS/FLI-1
fusion product, which produces the EWS/FLI-1 or MIC2 protein, stained
for by the CD99 immunohistochemistry marker.
Epidemiology
-
Third most common primary bone sarcoma (after osteosarcoma and chondrosarcoma, respectively)
-
Three times less common than osteosarcoma
-
Rare in African-Americans (0.5% of Ewing cases); peak incidence in the second decade of life
-
Male:female ratio 1.3:1
Pathophysiology
-
Sheets of monotonous, small, round blue cells with indistinct cytoplasm (Fig. 6.2-1)
-
Glycogen granules in the cytoplasm can be seen after periodic acid Schiff (PAS) staining or with electron microscopy.
-
PAS-positive granules sensitive to digestion with diastase
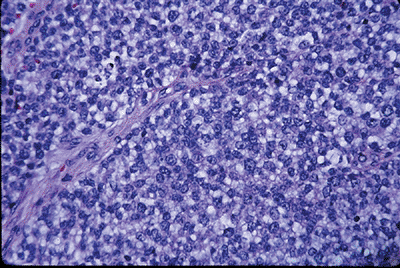 Figure 6.2-1
Figure 6.2-1
This hematoxylin-and-eosin (H&E) staining shows typical Ewing
sarcoma features of monotonous sheets of small, round blue cells with
indistinct cytoplasm.![]() Figure 6.2-2
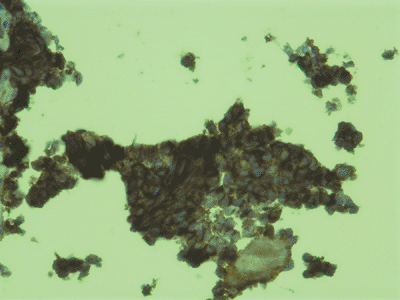
Figure 6.2-2
This CD-99 immunohistochemical staining shows positivity that is
consistent with Ewing sarcoma. Cytogenetics is used to confirm
Ewing/PNET (t11:22). -
The nuclear chromatin is finely granular, with one to three small nucleoli per nuclei.
-
CD99 immunohistochemistry marker stains for EWS/FLI1 fusion or MIC2 protein, which is present in 90% of cases. (Fig. 6.2-2)
Classification
-
Ewing sarcoma: most common, least differentiated, worst prognosis
-
PNET: more neural differentiation, better prognosis
-
Askin’s tumor: primary in thoracopulmonary region, best prognosis
Diagnosis
Clinical Features
-
Typical age at diagnosis: 5 to 30 years
-
Rare in patients <5 years old; this distinguishes it from metastatic neuroblastoma
-
Usually presents as a painful mass
-
May be accompanied by fever and weight loss, which are poor prognostic signs
Radiographic Features
-
Location
-
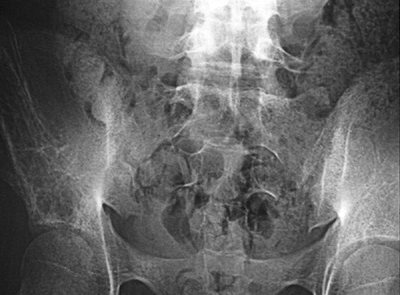
Mostly, lytic destructive lesion with “onion-skinning” periosteal reaction (Figs. 6.2-3, 6.2-4, 6.2-5, 6.2-6, 6.2-7, and 6.2-8)
-
90% of patients have soft tissue mass.
-
A destructive, lytic, diaphyseal lesion in a child is two times more likely to be a Ewing sarcoma than an osteosarcoma.
-
A destructive, lytic, metaphyseal lesion in a child is 12 times more likely to be an osteosarcoma than a Ewing sarcoma.
Treatment
Surgical Indications/Contraindications
-
Traditionally surgery for Ewing sarcoma was reserved for expendable bones.
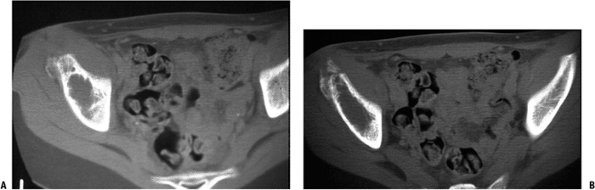
![]() Figure 6.2-4 Computed tomographic scans demonstrating the lytic destructive changes of the ilium.
Figure 6.2-4 Computed tomographic scans demonstrating the lytic destructive changes of the ilium. Figure 6.2-5 Anteroposterior radiograph of a 19-year-old with chronic hip pain. Note the cortical destruction and periosteal changes.P.190
Figure 6.2-5 Anteroposterior radiograph of a 19-year-old with chronic hip pain. Note the cortical destruction and periosteal changes.P.190![]() Figure 6.2-6
Figure 6.2-6
Magnetic resonance imaging shows large area of soft tissue involvement
without a stress fracture. This was diagnosed as a Ewing sarcoma of
bone and was treated with chemotherapy, wide resection, and bone
grafting. -
Because of improved local control with
surgery compared to radiation alone, most Ewing sarcoma patients have
surgical resection if adequate margins are attainable and the defect is
reconstructable (Fig. 6.2-9). -
Spine and acetabulum are sites that pose difficulties with resection and reconstruction.
-
Most resections are reconstructed with bone-grafting procedures.
-
Frequent diaphyseal location lends itself to intercalary allograft reconstruction.
-
Young age (small skeletal size) often requires expandable prostheses if growth plate has to be resected.
-
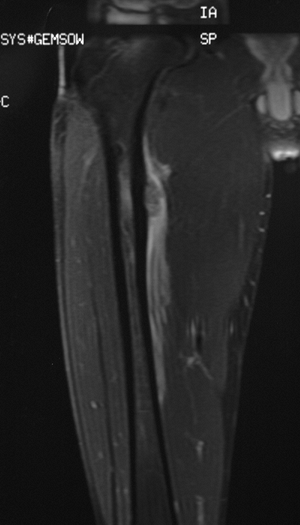
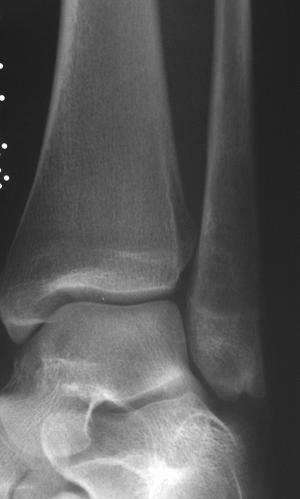 |
|
Figure 6.2-7 Subtle distal fibula lytic lesion without periosteal signs.
|
Results and Outcome
-
Local relapse rate with radiation alone is 25%; with surgery and radiation it is 8%.
-
It is unclear whether this difference affects survival.
-
Five-year disease-free survival rate for non-metastatic Ewing sarcoma is 60% to 65%.
-
Recurrent disease after 5 years for patients with nonmetastatic disease is very unusual.
-
-
Five-year survival rate for patients with metastatic disease at the time of diagnosis is 25% to 30%.
Postoperative Management
-
Early postoperative pain control is very important.
-
Patient-controlled analgesia
-
Regional and epidural pain management
-
-
Early rehabilitation emphasizing range of motion is critical.P.191
![]() Figure 6.2-8 Magnetic resonance imaging shows marrow replacement with tumor. This was diagnosed as a Ewing sarcoma.
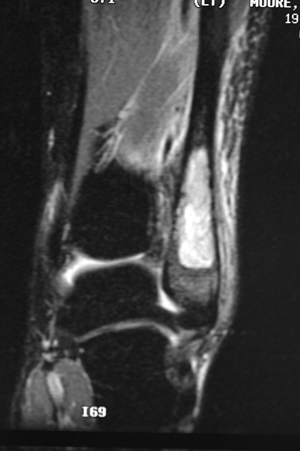
Figure 6.2-8 Magnetic resonance imaging shows marrow replacement with tumor. This was diagnosed as a Ewing sarcoma.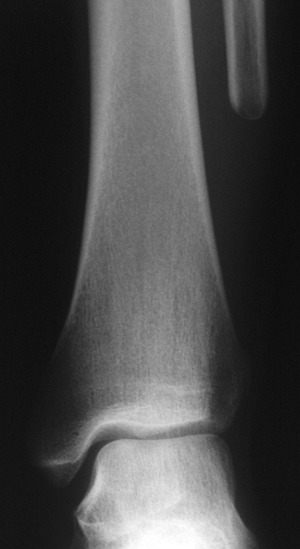 Figure 6.2-9
Figure 6.2-9
The patient was treated with wide resection and chemotherapy. No
reconstruction was performed, and no instability of the ankle was noted
on follow-up examinations. -
Avoid weight bearing until after bony union.
-
Long-term antibiotics for segmental allografts
Suggested Reading
Bacci
G, Ferrari S, Longhi A, et al. Local and systemic control in Ewing’s
sarcoma of the femur treated with chemotherapy, and locally by
radiotherapy and or surgery. J Bone Joint Surg [Br] 2003;85:107–114.
G, Ferrari S, Longhi A, et al. Local and systemic control in Ewing’s
sarcoma of the femur treated with chemotherapy, and locally by
radiotherapy and or surgery. J Bone Joint Surg [Br] 2003;85:107–114.
Nesbit
ME, Gehan EA, Burgert EO, et al. Multimodal therapy for the management
of primary, nonmetastatic Ewing sarcoma of bone: A long-term follow-up
of the first intergroup study. J Clin Oncol 1990;8:1664–1674.
ME, Gehan EA, Burgert EO, et al. Multimodal therapy for the management
of primary, nonmetastatic Ewing sarcoma of bone: A long-term follow-up
of the first intergroup study. J Clin Oncol 1990;8:1664–1674.
Picci
P, Rougraff BT, Bacci G, et al. Prognostic significance of
histopathologic response to chemotherapy in nonmetastatic Ewing’s
sarcoma of the extremities. J Clin Oncol 1993;11:1763–1769.
P, Rougraff BT, Bacci G, et al. Prognostic significance of
histopathologic response to chemotherapy in nonmetastatic Ewing’s
sarcoma of the extremities. J Clin Oncol 1993;11:1763–1769.
Sluga M, Windhager R, Lang S, et al. The role of surgery and resection margins in the treatment of Ewing’s sarcoma. Clin Orthop 2001;392:394–399.
Sucato DJ, Rougraff BT, McGrath BE, et al. Ewing’s sarcoma of the pelvis. Clin Orthop 2000;373:193–201.
Toni A, Neff JR, Sudanese A, et al. The role of surgical therapy in patients with nonmetastatic Ewing’s sarcoma of the limbs. Clin Orthop 1993;286:225–240.
Vlasak R, Sim FH. Ewing’s sarcoma. Pediatr Orthop Oncol 1996;27:591–603.
Wilkins RM, Pritchard DJ, Burgert O, Unni KK. Ewing’s sarcoma of bone. Cancer 1986;58:2551–2555.
Yang RS, Eckhardt JJ, Eilber FR, et al. Surgical indications for Ewing’s sarcoma of the pelvis. Cancer 1995;76:1388–1397.