
myelodysplasia are not common; however, patients with these disorders
do present in pediatric orthopaedic and neuromuscular clinics. These
disorders include the muscular dystrophies and congenital myopathies,
spinal muscular atrophy, Friedreich ataxia, hereditary motor sensory
neuropathies (HMSN), and poliomyelitis. It is important that an
accurate diagnosis be established so that an effective treatment
program can be planned and initiated. Delaying the diagnosis of these
disorders may lead to inappropriate treatment; furthermore, the mother
of an affected child might have further pregnancies and give birth to
another child with the genetic disorder (1). Accurate diagnosis requires a careful evaluation history, physical examination, and appropriate diagnostic studies (2).
delivery, and growth and development of the child involved. Questions
should be asked regarding in utero
activity, complications of delivery, birth weight, Apgar score,
problems during the neonatal period, age at achievement of
developmental motor milestones, age at onset of the current symptoms,
and information that will clarify whether the condition is static or
progressive. Systemic symptoms, such as cardiac disease, cataracts,
seizures, or other abnormalities, should also be ascertained.
these disorders, with the exception of poliomyelitis, are genetic in
origin. In order to arrive at an accurate diagnosis, family members of
the child or adolescent involved may need to be examined for subtle
expressions of the same disorder, and may also be required to undergo
hematologic or other studies.
neuromuscular disorder usually have one or more of the following: a
delay in developmental milestones, abnormal gait, foot deformity, or
spinal deformity. There is usually a history of progression. Physical
examination consists of a thorough musculoskeletal and neurologic
evaluation. Observing the child walking and performing simple tasks,
such as rising from a sitting position on the floor, can be useful.
Observation of the gait may reveal decreased arm swing, circumduction
of the legs, scissoring, or short cadence. Standing posture may reveal
increased lumbar lordosis or a wide base position for balance. Also, in
the standing position, the appearance of the feet should be observed.
Pes cavus or cavovarus deformities are common physical findings in many
of these disorders. Having the child walk on the heels and toes gives a
gross assessment of motor strength, and having the child run may reveal
an increase in muscle tone or ataxia. There is an increased incidence
of scoliosis in patients with neuromuscular disorders (3,4).
the patient with spinal muscular atrophy and congenital myotonic
dystrophy should become familiar to orthopaedic surgeons. The tongue
should be examined to detect evidence of fasciculation suggestive of
anterior horn cell diseases. Excessive drooling is common in both
cerebral palsy and congenital myotonic dystrophy. In the latter, nasal
speech may also be present. A thorough ophthalmologic examination is
necessary in order to elicit external ophthalmoplegia or retinitis
pigmentosa. In myotonic dystrophy, cataracts may develop during
adolescence.
myopathic disorders selectively affect proximal limb muscles before
affecting distal muscles. Early in the disease process, the muscles
demonstrate proportionally greater weakness than would be expected from
the degree of atrophy. The converse is true in neuropathies.
musculoskeletal examination. Sensory responses must be checked
individually and recorded. Decreased vibratory sensation may be present
in HMSNs such as Charcot-Marie-Tooth disease. In spinal muscular
atrophy, the deep-tendon reflexes may be absent, but in cerebral palsy,
they are increased. A positive Babinski sign confirms upper motor
neuron disease. Abnormalities in the Romberg test and rapid alternating
movements may indicate cerebellar involvement. Mental function
evaluation may be necessary, because organic mental deterioration may
be part of some neurologic syndromes. In many cases, the assistance of
a pediatric neurologist can be invaluable in performing a careful
neurologic and mental evaluation, because minor subtleties may offer
clues to diagnosis.
These can be divided into hematologic studies; electromyography (EMG)
with nerve conduction studies and needle electrode exam; muscle biopsy;
and nerve biopsy. Molecular diagnostic studies have become available
for many of these disorders, including Duchenne and Becker muscular
dystrophies, myotonic dystrophy, the hereditary sensory motor
neuropathies, and spinal muscular atrophy.
the most sensitive test for demonstrating abnormalities of striated
muscle function. The level of elevation parallels the rate and amount
of muscle necrosis and decreases with time as the muscle is replaced by
fat and fibrous tissue. The highest CPK levels are typically seen in
the earliest stages of Duchenne or Becker muscular dystrophy, in which
increases of 20 to 200 times the normal values may be found (6).
The level of elevation of CPK does not correlate with the severity or
rate of progression of the disorder. The highest levels are usually
found in Duchenne muscular dystrophy. Umbilical cord blood CPK levels
should be obtained in all male infants who are suspected of having this
disorder (7). Birth trauma may elevate the CPK
in umbilical cord blood, but in the healthy child this elevation
disappears promptly, whereas the enzyme level remains elevated in
muscular dystrophy. Serum CPK may be mildly or moderately elevated in
other dystrophic disorders, such as facioscapulohumeral muscular
dystrophy and Emery-Dreifuss muscular dystrophy. It is also mildly
elevated in female carriers of Duchenne muscular dystrophy, although
they are asymptomatic. In congenital myopathies and peripheral
neuropathies, the CPK levels are usually normal or only mildly
elevated. In other neuromuscular disorders that do not directly affect
striated muscle, the CPK levels are normal. Serum enzymes, such as
aldolase and serum glutamic oxaloacetic transaminase (SGOT), are also
important in the study of striated muscle function. Aldolase levels
correlate well with the CPK levels.
neuropathic process but is rarely helpful in establishing a definitive
diagnosis. Characteristics of neuropathic disorders include the
presence of fibrillation potentials, increased insertional activity,
and high-amplitude, increased-duration motor unit potentials (6). The fibrillation potential represents denervated individual muscle fibers firing spontaneously.
Myopathies rarely demonstrate EMG changes characteristic of a
neuropathy, although in an inflammatory muscle disease with significant
muscle breakdown, there may be prominent fibrillations. The use of an
experienced electromyographer is imperative in the accurate performance
of the test and interpretation of EMG data.
establishment of the diagnosis of peripheral neuropathy in children.
Nerve conduction velocities are normal in children with anterior horn
cell diseases, nerve root diseases, and myopathies. The normal value in
the child older than 5 years is 45 to 65 meters per second. In infants
and younger children, the velocity is lower because myelination is
incomplete.
Charcot-Marie-Tooth disease) before clinical deficits are present. The
nerve conduction studies can help determine whether the neuropathy
involves an isolated nerve or is a disseminated process.
test in determining the diagnosis of a neuromuscular disorder. More
recently, molecular genetic testing has become equally, if not more,
important. Muscle biopsy material is usually examined by routine
histology, special histochemical stains, and electron microscopy. The
criterion for selecting the muscle for biopsy is clinical evidence of
muscle weakness. Muscles that are involved but are still functioning
are selected in chronic diseases, such as Duchenne muscular dystrophy,
because they demonstrate the greatest diagnostic changes. A more
severely involved muscle may be chosen in an acute illness because the
process has not had sufficient time to progress to extensive
destruction. In patients who have proximal lower extremity muscle
weakness, biopsy of the vastus lateralis is performed, whereas in those
with distal weakness, a biopsy of the gastrocnemius is performed.
Biopsy of the deltoid, biceps, or triceps is performed for shoulder
girdle or proximal upper extremity weakness.
The biopsies are obtained under general anesthesia, spinal anesthesia,
regional nerve block, or a field block surrounding the area of
incision. It is important that local anesthetic is not infiltrated into
the muscle because this may alter the morphology of the muscle. The
vastus lateralis is the most common muscle chosen. A 4-cm incision is
made and the underlying fascia is incised longitudinally. The muscle is
directly visualized in order to avoid including normal fibrous septae
in the specimens. Muscle clamps are used for obtaining three specimens.
The clamps are oriented in the direction of the muscle fibers. A 2-mm
to 3-mm piece of muscle is grasped in each end of the clamp. The muscle
is cut at the outside edge of each clamp and a cylinder of muscle is
excised. The use of a muscle clamp helps keep the muscle at its resting
length and minimizes artifact. One specimen is quickly frozen in liquid
nitrogen (-160°C) to prevent loss of soluble enzymes. This specimen is
used for light microscopy with a variety of special preparations. The
other specimens are used for routine histology and electron microscopy.
The wound is subsequently closed in layers. Electrocautery may be used
during the closure. If it is used before the biopsy, it may
inadvertently damage the specimens and alter the morphology.
demyelinating disorders. Usually, the sural nerve is selected for
biopsy because of its distal location and lack of autogenous zone of
innervation. The patient notices no sensory change or only a mild
sensory diminution after excision of the 3-cm to 4-cm segment of the
nerve. Hurley et al. (8) reported a single
incision for combined muscle and sural nerve biopsy. An incision over
the posterolateral aspect of the calf allows access to the nerve and
either the soleus or the peroneal muscle. This avoids the necessity for
making two incisions. This technique was demonstrated to be useful in
disorders in which both a muscle and a nerve biopsy may be necessary
for arriving at a diagnosis.
diagnosis of a neuromuscular disorder include electrocardiogram (ECG),
pulmonary function studies, magnetic resonance imaging (MRI),
ophthalmologic evaluation, amniocentesis, and pediatric neurology
evaluation.
myotonic dystrophy demonstrate ECG abnormalities. Duchenne muscular
dystrophy is frequently associated with mitral valve prolapse secondary
to papillary muscle involvement (10,11). Arrhythmias under anesthesia have been reported with both Duchenne and Emery-Dreifuss muscular dystrophies (12,13).
respiratory muscles, but they do not establish the diagnosis. If
respiratory muscle involvement is present, the rate of deterioration
can be followed up with periodic studies. This is important if surgery
is contemplated in children or adolescents with muscular dystrophy,
spinal muscular atrophy, or Friedreich ataxia. The forced vital
capacity (FVC) is the most important study after arterial blood gas
measurements (14).
Imaging estimates of the disease severity by degree of muscle
involvement correlate well with clinical staging. MRI may also be
important in selecting appropriate muscles for biopsy.
has tremendously enhanced our understanding of the genetic aspects of
many of these disorders (16,17).
The determination of the exact location of chromosomal and gene defects
has led to the possibility of genetic engineering being used to correct
these disorders. Unfortunately, genetic testing is quite costly, and
for many disorders, such testing is not commercially available. Also, a
negative test does not necessarily exclude certain disorders. For this
reason, the decision to carry out genetic testing should be made only
by a neuromuscular specialist or geneticist. In each of the various
disorders, the current status of genetic and molecular biology research
is discussed in this chapter.
inherited disorders with a progressive degeneration and weakness of
skeletal muscle that has no apparent cause in the peripheral or the
central nervous system (CNS). These have been categorized according to
clinical distribution, severity of muscle weakness, and pattern of
genetic inheritance (Table 17.1). An accurate
diagnosis is important, both for prognosis and management of the
individual patient and for identification of genetic factors that may
be crucial in planning for subsequent children by the family involved.
Transmission is by an X-linked recessive trait. A single gene defect is
found in the short arm of the X chromosome. The disease is
characterized by its occurrence exclusively in the male sex, except for
rare cases associated with Turner syndrome. In this rare event, the XO
karyotype who carries the defective gene may demonstrate the phenotype
found in male patients with the disorder (6).
This disorder is associated with a high mutation rate, and a positive
family history is present in approximately 65% of the cases. Duchenne
muscular dystrophy occurs in approximately 1 in 3,500 live male births,
with about one third of the children involved having acquired the
disease because of a new mutation.
and less severe form of muscular dystrophy. It occurs in approximately
1 in 30,000 live male births, becomes apparent later in childhood, and
has a more protracted and variable course than Duchenne muscular
dystrophy. This disorder is discussed later but is mentioned here
because of the similar inheritance pattern and molecular biology
abnormality.
|
TABLE 17.1 CLASSIFICATION OF MUSCULAR DYSTROPHIES
|
|
|---|---|
|
evident when the child is at an age of between 3 and 6 years. Earlier
onset may also occur. The family may have observed that the child’s
ability to achieve independent ambulation was delayed or that he has
become a toe walker. Children at the age of 3 years or older may
demonstrate frequent episodes of tripping and falling, in addition to
difficulty in activities requiring reciprocal motion, such as running
or climbing stairs. Inability to hop and jump normally is commonly
present.
weakness in the proximal muscle groups that descend symmetrically in
both lower extremities, particularly the gluteus maximus, gluteus
medius, quadriceps, and tibialis anterior muscles. The abdominal
muscles are involved. Involvement of the shoulder girdle muscles (i.e.,
trapezius, deltoid, and pectoralis major muscles) and lower facial
muscles occurs later. Pseudohypertrophy of the calf muscles caused by
the accumulation of fat is common but not invariably present. Most
patients have cardiac involvement, most commonly a sinus tachycardia
and right ventricular hypertrophy. Life-threatening dysrhythmia or
heart failure ultimately develops in approximately 10% of patients.
Many also have a static encephalopathy, with mild or moderate mental
retardation (19). Death from pulmonary failure and occasionally from cardiac failure occurs during the 2nd or 3rd decades of life.
develops compensatory changes in gait and stance as weakness
progresses. Sutherland et al. (20,21)
documented disease progression by measuring the gait variables of
cadence, swing phase, ankle dorsiflexion, and anterior pelvic tilt. The
hip extensors, primarily the gluteus maximus, are the first muscle
group to be involved. Initially the patient compensates by carrying the
head and shoulders behind the pelvis, maintaining the weightline
posterior to the hip joint and center of gravity (Fig. 17.1).
This produces an anterior pelvic tilt and increases lumbar lordosis.
Cadence and swing-phase ankle dorsiflexion decrease, and the patient
develops a waddling, wide-based gait with shoulder sway to compensate
for gluteus medius weakness. Muscle weakness requires that the force
line remain behind the hip joint and in front of the knee joint
throughout single limb support (20,21,22),
and hip abductors and quadriceps muscles force the patient to
circumduct during the swing phase of gait while at the same time
shifting the weight directly over the hip joint. The generalized pelvic
weakness requires considerable forward motion to be generated by the
spine for the patient to advance. Ankle-plantar flexion becomes fixed,
and the stance phase is reduced to the forefoot, resulting in even more
difficulty with balance and cadence. Foot inversion develops as
peroneal strength diminishes. The
tibialis
posterior muscle, which is one of the last muscles to be involved, is
responsible for the inversion or varus deformity of the foot.
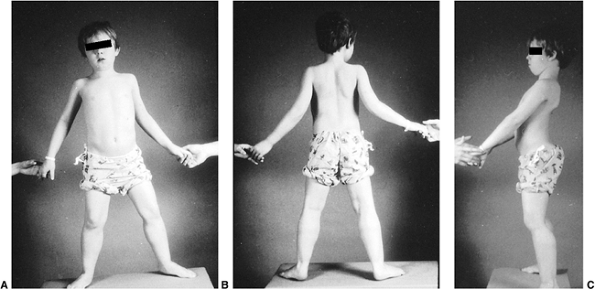 |
|
Figure 17.1 A:
A 7-year-old boy with Duchenne muscular dystrophy demonstrates precarious stance due to mild hip abduction contractures. Observe the pseudohypertrophy of the calves. B: Posterior view demonstrates mild ankle equinus in addition to the calf pseudohypertrophy. C: Side view shows an anterior tilt to the pelvis and increased lumbar lordosis, and the head and the shoulders are aligned posterior to the pelvis. This characteristic posture maintains the weight line posterior to the pelvis and center of gravity, compensates for the muscle weakness, and helps maintain balance. |
years later, precludes the use of crutches to aid in ambulation. It
also makes it difficult to lift the patient from under the arms. This
tendency for the child to slip a truncal grasp has been termed Meyeron sign.
As the weakness in the upper extremities increases, the child becomes
unable to move his or her arms. Although the hands retain strength
longer than the arms, use of the hands is limited because of weakness
of the arms.
established by physical examination, including gait and specific muscle
weakness, and by the absence of sensory deficits. The upper extremity
and knee deep-tendon reflexes are lost early in the disease, whereas
the ankle reflexes remain positive until the terminal phase. A valuable
clinical sign is the Gower sign. The
patient is placed prone or in the sitting position on the floor and
asked to rise. This is usually difficult, and the patient may require
the use of a chair for assistance. The patient is then asked to use his
or her hands to grasp the lower legs and force the knees into
extension. The patient then walks his or her hands up the extremities
to compensate for the weakness in the quadriceps and gluteus maximus.
This sign may also be found in congenital myopathies and spinal
muscular atrophy. The contracture of the iliotibial band can be
measured by the Ober test. To perform this
test, the child is placed on his or her side with both hips flexed. The
superior leg is then abducted and extended and allowed to fall into
adduction. The degree of abduction contracture can be measured by the
number of degrees the leg lacks in coming to the neutral position.
Tendo-Achilles contractures also occur. Contracture of the
tendo-Achilles and the iliotibial band are the most consistent
deformities noted during the physical examination.
continuously. A rapid deterioration may be noted after immobilization
in bed, even for short periods after respiratory infections or,
perhaps, extremity fractures. Every effort should be made to maintain a
daily ambulatory program. In the absence of treatment, children are
usually unable to ambulate effectively by the age of 10 years (5,23,24,25). The chief cause is loss of strength in the hip extensors and ankle dorsiflexors (26).
These two factors can be used as a guide to predict when ambulation
will cease. With loss of standing ability, the child becomes wheelchair
dependent. This results in a loss of the accentuated lumbar lordosis
that protected the child from kyphoscoliosis (27). As a consequence, most patients subsequently develop a progressive spinal deformity.
changes are present in more than 90% of children with Duchenne muscular
dystrophy. The average intelligence quotient of these patients has been
shown to be approximately 80 (19).
of Duchenne muscular dystrophy. This may be 200 to 300 times the normal
value, but decreases as the disease progresses and muscle mass is
reduced. CPK levels are also elevated in female carriers of the disease
(2 to 3 times the normal value for women and girls), although not to
the same extent as in affected boys. There is an 80% consistency in the
results when the CPK test is repeated at three consecutive monthly
intervals (28). Aldolase and SGOT levels may also be elevated, but the elevations are not unique to striated muscle disease.
if the clinical findings and CPK are both suggestive of a muscular
dystrophy, this test is typically not necessary. EMG shows
characteristic myopathic changes with reduced amplitude, short
duration, and polyphasic motor action potentials (6).
subsequent loss of fiber, variation in fiber size, proliferation of
connective tissue and, subsequently, of adipose tissue as well (6).
Increased cellularity is present, with occasional internal migration of
the sarcolemmal nuclei. Histochemical testing reveals loss of clear-cut
subdivisions of fiber types, especially with adenosine triphosphatase
reaction, and a tendency toward type I fiber predominance. In the past,
this was the diagnostic procedure of choice. However, the standard
today is to first obtain blood samples for DNA polymerase chain
reaction (PCR) testing for dystrophinopathies. If this is positive,
there is no need for a muscle biopsy. If PCR testing is negative, then
muscle biopsy is indicated for arriving at a definitive diagnosis.
chromosome has been identified as being responsible for both Duchenne
and Becker muscular dystrophies (16,17,29,30). The status of genetic and molecular biology in Duchenne muscular dystrophy has been summarized by Shapiro and Specht (6). The gene is located at the Xp21.2 region and spans 2 million base pairs (31,32).
It includes 65 exons (i.e., coding regions) and encodes the 400-kDa
protein dystrophin. The large size of the gene correlates with the high
rate of spontaneous mutation. Dystrophin is a component of cell
membrane cytoskeleton and represents 0.01% of skeletal muscle protein.
Its distribution within skeletal, smooth, and cardiac muscle and within
the brain correlates well with the clinical features in Duchenne and
Becker muscular dystrophies. A structural role for the dystrophin
protein is suggested by studies that demonstrate concentration of the
protein in a lattice organization in the cytoplasmic membrane of
skeletal muscle fibers (33,34). Demonstrable mutations, deletions, or duplications of dystrophin are found in 70% to 80% of the affected male patients (31,32,35,36).
The reading frame hypothesis distinguishes the mutations that correlate
with the more severe Duchenne muscular dystrophy from those that
correlate with the less severe Becker muscular dystrophy. Mutations
that disrupt the translational reading frame or the promoter (i.e., the
specific DNA sequence that signals where RNA synthesis should begin)
result in a presumably unstable protein, and this correlates with
Duchenne muscular dystrophy. In contrast, mutations that do not disrupt
the translational reading frame or the promoter have a lower molecular
weight and semifunctional dystrophin. This correlates with the less
severe Becker muscular dystrophy (31,37).
mutation analysis (by PCR or DNA Southern blot analysis), or both,
provide methods of differentiating between Duchenne and Becker muscular
dystrophies on the one hand, and other initially similar disorders
(such as dermatomyositis, limb-girdle muscular dystrophy (LGMD),
Emery-Dreifuss muscular dystrophy, and congenital muscular dystrophy)
on the other (36,38,39).
In the latter disorders, the dystrophin is normal. In patients with
Duchenne muscular dystrophy, there is a complete absence of dystrophin,
whereas in Becker muscular dystrophy, dystrophin is present but is
altered in size, decreased in amount, or both. Nicholson et al. (40)
reported a positive relation between the amount of dystrophin and the
age at loss of independent ambulation in 30 patients with Duchenne
muscular dystrophy and in 6 patients with Becker muscular dystrophy.
The researchers found that even low concentrations of dystrophin in
Duchenne muscular dystrophy may have functional significance and may
explain the variability of age at which ambulation ceases. The presence
of partially functional dystrophin protein is sufficient to minimize
the phenotypic expression, leading to the milder disorder of Becker
muscular dystrophy (31,35,38). The same tests can be used to improve detection of female carriers (36,39).
On the basis of smaller-than-normal dystrophin protein, two atypical
forms of Becker muscular dystrophy have been recognized. These are
myalgia without weakness in male patients (similar to metabolic
myopathy), and cardiomyopathy with little or no weakness in male
patients (41).
dystrophin replacement in diseased muscles. This involves the
implantation of myoblasts, or muscle precursor cells, into the muscles
of patients with Duchenne muscular dystrophy (42). This has been successful in producing dystrophin in the murine mdx model of Duchenne muscular dystrophy (43). Unfortunately, the results in human male patients have been disappointing (44,45,46,47,48).
as prednisone and deflazacort, have been shown to preserve or improve
strength, prolong ambulation, and slow the progression of scoliosis (49,50,51,52,53,54,55,56). However, the side effects—weight gain, osteoporosis with vertebral fractures, and myopathy—limit their usefulness (37,51,52,53,57). Azathioprine has also been evaluated in Duchenne muscular dystrophy but has not shown beneficial effects (58). Aminoglycoside therapy with intravenous gentamicin administration has been studied in two trials (59,60). A decrease in serum CPK levels was demonstrated, but there was no effect on muscle strength.
difficult, primarily because of the size of the viral vectors and also
because of the complications of immune reactions that may occur.
Therefore, gene therapy is still very much in the early investigational
stages. This treatment has been reviewed in detail by Chamberlain (61).
Dystrophin delivery to muscle has been attempted with four primary
vectors: adenovirus, retroviruses, adeno-associated viruses, and
plasmids. Complications of this technology included triggering of a
cellular immune response, poor integration of the vector into the host
gene, and lack of a sustained response, to name only a few (62).
Stem cell therapy may be a promising intervention for the
dystrophinopathies. In the mdx mouse, bone marrow transplantation and
injection of normal muscle-derived stem cells led to partial
restoration of dystrophin expression (63).
dystrophy include decreasing ambulatory ability, soft tissue
contractures, and spinal deformity (5,6,18,64). The goals of treatment should be to improve or maintain the functional capacity of the affected child or adolescent.
include medical therapy, physical therapy, functional testing, use of
orthoses, fracture management, surgery, use of wheelchair,
cardiopulmonary management, and genetic and psychological counseling.
preserving strength, prolonging ambulation, and slowing the progression
of scoliosis. However, this therapy is not in wide use because of the
attendant complications as described in the earlier text.
functional muscle strength, prevention or correction of contractures by
passive stretching, gait training with orthoses and transfer
techniques, ongoing assessment of muscle strength and functional
capacity, and inputs regarding wheelchair and equipment measurements.
been established and before muscle strength has deteriorated, a program
of maximum-resistance exercises should be commenced, to be performed
several times a day. This may help preserve strength and delay the
onset of soft tissue contractures. Physical therapy is more effective
in preventing or delaying contractures than in correcting them.
Contractures develop in the ambulatory patient because the progression
of muscle weakness results in the development of adaptive posturing to
maintain lower extremity joint stability. A home exercise program can
be effective in minimizing hip and ankle soft tissue contractures.
Exercises should be performed twice a day on a firm surface, and should
include stretching of the tensor fascia lata, hamstrings, knee flexors,
and ankle plantar flexors. Occasionally, serial casting may be useful
in correcting existing deformities before physical therapy.
Knee-flexion contractures of less than 30 degrees may benefit from
serial or wedge casting. This enhances the use of knee-ankle-foot
orthoses (KAFOs). Unless orthoses are used after casting and in
conjunction with physical therapy, these contractures rapidly recur.
muscle testing. Muscle strength is tested by measurement of the active
range of motion of a joint against gravity. This type of testing allows
assessment of the rate of deterioration as well as the functional
capacity of the individual.
KAFOs are used in independently ambulatory patients when gait becomes
precarious, when early soft tissue contractures of the knees and ankle
are developing, and after surgical correction of these deformities (65,66,67,68). AFOs can also be helpful in improving tendo-Achilles contractures, especially when worn both during the day and at night (69).
KAFOs are usually supplemented with a walker because of the excessive
weight on the orthoses and the risk of falling. Important prescription
components include partial ischial weight-bearing support, posterior
thigh cuff, and a spring-loaded, drop-lock knee joint with an ankle
joint set at a right angle. Ambulation may be extended for up to 3
years by the combined use of surgery and orthoses. The maintenance of a
straight lower extremity also enables the nonwalking patient to stand
with support, and thereby assists in transfers.
spinal deformities, but wheelchair-bound patients, especially those
with severe cardiopulmonary compromise and severe scoliosis, may
benefit from the use of a custom wheelchair, a thoracic suspension
orthosis, or a custom-made thoracic-lumbar spinal orthosis (TLSO). A
mobile arm-support orthosis attached to the wheelchair may help the
patient in performing personal hygiene tasks and self-feeding (70).
children with Duchenne muscular dystrophy. This is due to decreased
bone mineral density from
disuse osteoporosis, steroid induced osteoporosis, or both (71,72,73,74). Fractures can result in a permanent loss of function (71,73,74).
This occurs predominantly after ambulation has ceased and the child is
wheelchair-bound. These fractures are best treated by closed reduction
and cast immobilization. Occasionally, open reduction and internal
fixation may be needed. In children who are still ambulatory, it is
important that they be placed on a program of early mobilization to
allow weight bearing. This may require the use of an electrically
powered circle bed. Once early healing is present, the child can be
returned to the KAFO to decrease weight and enhance mobility.
weakness impair ambulation. Surgery is indicated when independent
ambulation becomes precarious and when contractures are painful or
interfere with essential daily activities. The major contractures that
are amenable to surgical intervention include equinus and equinovarus
contractures of the ankle and foot, knee-flexion contractures, and
hip-flexion and -abduction contractures. In thin individuals, these
contractures may be released by percutaneous techniques (64,75).
For ambulatory patients, orthotic measurements should be obtained
before surgery. This allows the orthoses to be applied shortly after
surgery to assist in rapid restoration of ambulation. Correction of
contractures and the use of orthoses can prolong effective ambulation
and assisted standing ability by a period of 1 to 3 years (5,18,22,65,66,67,68,75,76,77,78,79,80). Hsu and Furumasu (22)
reported a mean prolongation of walking of 3.3 years in 24 patients
with Duchenne muscular dystrophy ranging in age from 8 to 12 years at
the time of surgery. It is usually not possible to restore functional
ambulation once the patient has been unable to walk for more than 3 to
6 months (65). Each patient must be
individually assessed to determine the functional needs and the best
procedures. Common contraindications for correction of lower extremity
contractures include obesity, rapidly progressive muscle weakness, or
poor motivation (those who prefer to use a wheelchair rather than
attempt ambulation) (6).
equinovarus contractures. This is because of a combination of
tendo-Achilles contracture and muscle imbalance induced by the stronger
tibialis posterior muscle. This latter muscle retains good function
despite the progression of muscle weakness in other areas. These
equinovarus deformities can be managed by a combination of
tendo-Achilles lengthening by means of percutaneous open tenotomy (18,64,67,68,76,77) with or without resection, or by Vulpius (5) or open Z-lengthening (79), and tibialis posterior lengthening, tenotomy, or transfer through the interosseous membrane to the dorsum of the foot (5,6,18,25,64,66,67,68,76,77,81,82,83). Scher and Mubarak have also recommended toe flexor tenotomies (84).
Tibialis posterior transfer prevents recurrence of equinovarus
deformities and maintains active dorsiflexion of the foot. Some
orthopaedists, however, have questioned the necessity of a transfer,
because it is a more extensive procedure. They prefer tenotomy,
recession, or lengthening (64,66,76). Postoperative gait analysis has shown that the transferred tibialis posterior muscle is electrically silent (85). Greene (81)
has reported that tibialis posterior myotendinous junction recession in
six patients (12 feet) resulted in an increased recurrence rate when
compared with transfer in nine patients (18 feet), making the former a
less desirable procedure. Percutaneous tendo-Achilles lengthening under
local anesthesia is usually reserved for nonambulatory patients, who
typically have an equinus deformity and cannot wear shoes. The
nonambulatory patient with a moderately severe equinovarus deformity
may require open tenotomies of the tendo-Achilles, the tibialis
posterior, and long toe flexors. Severe equinovarus contractures have
been managed effectively by talectomy.
contractures and develop rapidly when the patient is wheelchair bound.
These contractures limit proper positioning in bed and may lead to the
development of hamstring spasm, causing considerable discomfort when
the patient attempts to transfer. A Yount procedure (86)
(release of the distal aspect of the tensor fascia lata and iliotibial
band) is the most common procedure used in correcting knee-flexion
contractures (18,64,66,67,68).
Hamstring tenotomies, recession or Vulpius-type lengthening, and formal
Z-lengthening may also be necessary. These procedures enhance
quadriceps power and function and also relieve symptoms.
Postoperatively, KAFOs are necessary in order to prevent recurrence.
lordosis and interfere with the ability to stand and to lie comfortably
supine. Patients with hip-flexion contractures may experience low back
pain. Correction of flexion contractures involves release of the tight
anterior muscles, including the sartorius, rectus femoris, and tensor
fascia femoris (6,18,64). Abduction contractures are improved by release of the tensor fasciae lata proximally with use of the Ober procedure (87), modified Soutter release, the Yount procedure distally (86), or by complete resection of the entire iliotibial band (251).
patients with Duchenne muscular dystrophy and found that 15 had
unilateral subluxation, 1 had bilateral subluxation, and 3 had a
unilateral dislocation. They recommended serial pelvic radiographs in
patients with this disorder. They also felt that any pelvic obliquity
should be corrected at the time of spinal stabilization.
with Duchenne muscular dystrophy, but usually do not require treatment.
These contractures
include
shoulder adduction, elbow flexion, forearm pronation, wrist flexion,
metacarpophalangeal and proximal interphalangeal joint flexion, and
others. These usually do not preclude the use of wheelchairs. Muscle
weakness is the most devastating aspect of upper extremity involvement.
Wagner et al. (89)
demonstrated wrist ulnar deviation and flexion contractures in addition
to contractures of the extrinsic and intrinsic muscles of the fingers
in adolescents with Duchenne muscular dystrophy. These contractures
produce boutonniere and swan neck deformities and hyperextension of the
distal interphalangeal joints. The treatment of upper extremity
contractures involves physical therapy with daily passive
range-of-motion exercises. When passive wrist dorsiflexion is limited
to neutral, a nighttime extension orthosis may be helpful. Surgery is
rarely indicated for these contractures.
This typically begins to occur when ambulation ceases, and it is
rapidly progressive. Approximately 25% of older ambulating patients,
however, have mild scoliosis (23,95).
Prolongation of ambulation by appropriate soft tissue releases of the
lower extremity contractures, thereby maintaining accentuated lumbar
lordosis, can delay the onset of scoliosis (78).
The curves are usually thoracolumbar, associated with kyphosis, and
lead to pelvic obliquity. Scoliosis cannot be controlled by orthoses or
wheelchair seating systems (90,96,97,98,99,100).
Although orthotic management may slow curve progression, it does not
slow the systemic manifestations of Duchenne muscular dystrophy (e.g.,
decreasing pulmonary function and cardiomyopathy). These may complicate
spinal surgery at a later time. As the scoliosis progresses, it can
result in a loss of sitting balance, produce abnormal pressure, and
occasionally cause the patient to become bedridden. Heller et al. (101)
reported improved sitting support with an orthosis in 28 patients who
either refused surgery or who were considered to be inoperable.
Fusion extends from the upper thoracic spine (T-2 or T-4) to L-5 or the
pelvis. It is important to center the patient’s head over the pelvis in
both the coronal and sagittal planes. This usually allows complete or
almost complete correction of the deformity, maintains sitting balance,
improves head control, and allows more independent hand function.
Although autogenous bone grafting is used in most patients, there
appears to be no difference in fusion rates when allograft bone is used
(106,107,108,109). Segmental spinal instrumentation techniques using Luque rod instrumentation are most commonly used (18,64,90,95,98,102,106,107,110,111,112,113,114,115).
Other segmental instrumentation systems, such as Cotrel-Dubousset,
Texas Scottish Rite Hospital (TSRH), Isola, and others, can also be
used (106,107,111,116). These allow sufficient fixation so that postoperative immobilization is not necessary (Fig. 17.2). Fixation to the pelvis is achieved using the Galveston or other techniques (103,106,110,111,112,113,114,115,116,117).
These techniques are thought to maintain better correction of pelvic
obliquity. Some authors believe that fusion to L-5 is sufficient, and
that there will be no spinopelvic deformity throughout the remainder of
the patient’s life (108,118,119,120). However, a postoperative spinopelvic deformity can occur and progress, and most authors recommend fusion to the pelvis (113,115,121). Mubarak et al. (118)
recommend fusion to L-5 if the curve is greater than 20 degrees, the
FVC is greater than 40%, and the patient is using a wheelchair full
time, except for occasional standing. If the patient’s curve is greater
than 40 degrees or if there is pelvic obliquity greater than 10
degrees, then fusion to the sacropelvis is recommended.
function studies and cardiology consultation, is mandatory because of
the associated pulmonary and cardiac abnormalities and the risk of
malignant hyperthermia (2,3,122,123,124,125,126).
Children with Duchenne muscular dystrophy have a decreased FVC,
commencing at approximately the age of 10 years, because of weakness of
the intercostal muscles and associated contractures. There is a linear
decrease over time (14,91,94,105,122). Kurz et al. (14)
observed a 4% decrease in FVC for each year of age or each 10 degrees
of scoliosis. It stabilizes at approximately 25% of normal until death.
The presence of severe scoliosis may increase the rate of decline in
the FVC. Jenkins et al. (122) reported that
when the FVC is 30% or less, there is an increased risk of
postoperative complication such as pneumonia and respiratory failure.
Smith et al. (94) found that most patients with
curves of more than 35 degrees had FVC less than 40% of predicted
normal values. They therefore recommend that spinal arthrodesis be
considered for all patients with Duchenne muscular dystrophy when they
can no longer walk. Nevertheless, successful surgery can be performed
in many patients with FVC as low as 20% of predicted normal valves (107). Marsh et al. (123)
recently reported similar results in 17 patients with FVC greater than
30% and 13 patients with FVC less than 30%. They concluded that spinal
fusion could be offered to patients in the presence of a low FVC.
longevity, although it definitely increases the quality of the
remaining life (91,107,110).
In a study of 55 patients with Duchenne muscular dystrophy, of whom 32
underwent spinal fusion and 23 did not, Galasko et al. (91)
found that FVC remained stable in the operated group for 36 months
postoperatively and then fell slightly. In the nonoperated group, it
progressively declined. The survival data showed that a significantly
higher mortality rate was seen in the nonoperated group. This study
indicated that spinal stabilization can increase survival for several
years if it is done
early,
before significant progression has occurred. Other studies, however,
have shown that posterior spinal fusion has no effect on the steady
decline in pulmonary function when compared with unoperated patients (104,107,127,128,129).
In addition to correction and stabilization of the spine, patients
experience improved quality of life, as measured by ability to
function, self-image, and cosmesis (104,110,111,130). Parents also reported improvement in their ability to provide care to their child.
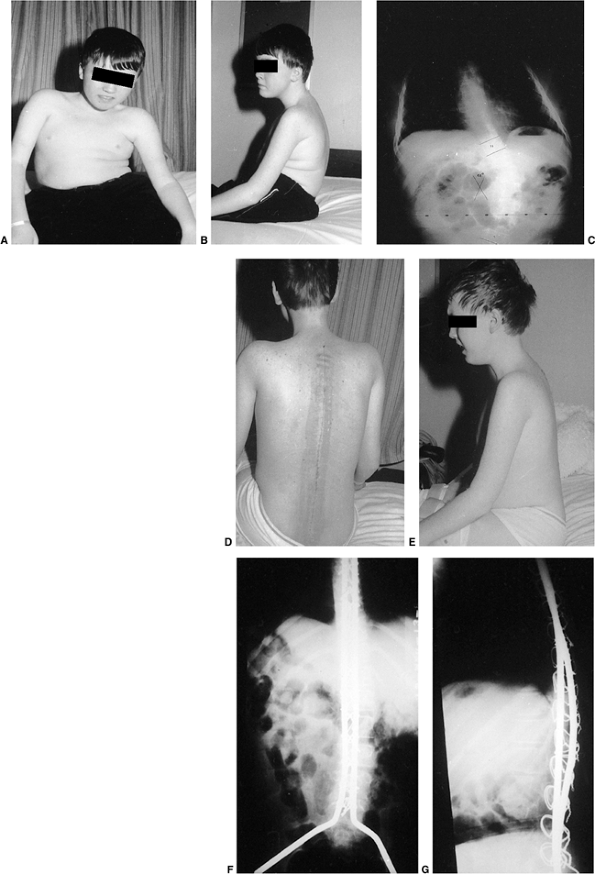 |
|
Figure 17.2 A:
An 11-year-old boy with Duchenne muscular dystrophy with a rapidly progressive right thoracolumbar scoliosis and decreasing sitting balance. He uses his hands to maintain sitting balance. B: Side view shows an associated mild kyphotic deformity. C: Preoperative sitting posteroanterior radiograph demonstrates a long, sweeping, 48-degree thoracolumbar curve between T-11 and L-5. Six months earlier, no clinical or radiographic deformity was evident. D: Postoperatively, an immediate improvement in spinal alignment and sitting balance is noted. E: Side view demonstrates correction of the associated kyphosis. F: Postoperative sitting radiograph after posterior spinal fusion and Luque rod instrumentation from T-4 to the sacrum. The Galveston technique, with insertion of the Luque rod into the wing of the ilium, was used for pelvic fixation. Almost complete correction of his spinal deformity was achieved. G: Postoperative lateral radiograph shows improved sagittal alignment. |
These include excessive intraoperative blood loss, neurologic injury,
cardiopulmonary compromise, postoperative infection, poor wound
healing, curve progression, hardware problems, and late pseudarthrosis.
Intraoperative blood loss can be minimized by early surgery and the use
of hypotensive anesthesia (108). The increased
intraoperative blood loss in patients with Duchenne muscular dystrophy
appears to result from inadequate vasocontraction caused by the lack of
dystrophin in the smooth muscle (131).
children with Duchenne muscular dystrophy is controversial. Noordeen et
al. (132) reported that a 50% decrease in amplitude was suggestive of neurologic impairment.
capable of independent ambulation. This is typically a motorized
wheelchair that allows the patient to be independent of parents or
aides, especially while attending school. The wheelchair may be fitted
with a balanced mobile arm orthosis for the purpose of facilitating
personal hygiene and self-feeding (70).
constant threat and is the most common cause of death early in the 3rd
decade of life. Kurz et al. (14) found that the
vital capacity peaks at the age when standing ceases, then declines
rapidly thereafter. The development of scoliosis compounds the problems
and leads to further diminution of the vital capacity (128).
The complication rate in spinal surgery increases when the FVC is less
than 30% of the normal value. Programs of vigorous respiratory therapy
and the use of home negative-pressure and positive-pressure ventilators
may allow patients with Duchenne muscular dystrophy to survive into the
3rd and 4th decades of life (133,134,135,136).
After initially responding to digitalis and diuretics, the involved
cardiac muscle becomes flabby, and the patient goes into congestive
heart failure. Myocardial infarction has been reported in boys as young
as 10 years. There is no correlation between the severity of pulmonary
dysfunction and cardiac function, or between age and cardiac function (137). The cardiomyopathy of Duchenne muscular dystrophy exists clinically as a separate entity.
prevent the birth of additional male infants with Duchenne muscular
dystrophy. It must be remembered that approximately 20% of families
have already conceived and delivered a second affected male infant
before the diagnosis is made in the first (68,138).
Genetic counseling with parents and family groups is important in the
management of psychological problems arising when the genetic nature of
the diagnosis becomes known.
muscular dystrophy in clinical appearance and distribution of weakness,
but it is less severe (139,140).
Onset is generally after the age of 7 years and the rate of progression
is slower. The patients usually remain ambulatory until adolescence or
the early adult years. The Gower maneuver may occur as the weakness
progresses (Fig. 17.3). Pseudohypertrophy of the calf is common, and eventually equinus and cavus foot deformities develop (Fig. 17.4).
Cardiac involvement is frequent. There may be a family history of
atypical muscular dystrophy. Pulmonary problems are less severe and the
patient’s life expectancy is greater.
associated with Becker muscular dystrophy is essentially the same as in
Duchenne muscular dystrophy. Steroid therapy (prednisone) has recently
been shown to decrease serum creatine kinase levels and improve
strength (141). Ankle and forefoot equinus occur commonly. Shapiro and Specht (6)
have reported good outcome with the Vulpius tendo-Achilles lengthening
in patients with equinus contractures. A tibialis posterior tendon
transfer is performed if
necessary.
Forefoot equinus may require a plantar release and possibly a midfoot
dorsal-wedge osteotomy for correction. The use of orthotics is also
beneficial because the rate of progression is slower and the remaining
muscle strength greater than in Duchenne muscular dystrophy. The
incidence of scoliosis is high, especially in those adolescents who
have ceased walking. These patients require careful evaluation and
periodic spinal radiographs. Posterior spinal fusion and segmental
instrumentation, usually Luque, are useful for patients in whom there
is progression (142).
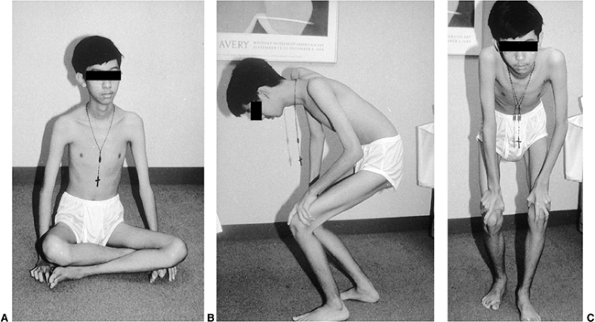 |
|
Figure 17.3 A: A 13-year-old boy with suspected Becker muscular dystrophy uses the Gower maneuver to stand from a sitting position. B: Manually assisted knee extension is necessary to achieve upright stance. C: Front view.
|
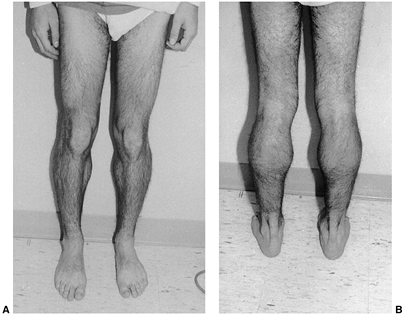 |
|
Figure 17.4 A: Pseudohypertrophy of the calves in an 18-year-old man with Becker muscular dystrophy. He is a brace-free ambulator. B: Posterior view.
|
sex-linked recessive disorder characterized by early contractures and
cardiomyopathy (12). The typical phenotype is
seen only in the male sex, although milder or partial phenotypes have
been reported in female carriers (143,144,145).
Affected boys show mild muscle weakness in the first 10 years of life
and a tendency for toe walking. The Gower maneuver may be present in
young children. The distinctive clinical criteria occur in late
childhood or early adolescence. These include tendo-Achilles
contractures, elbow-flexion contractures, neck-extension contracture,
tightness of the lumbar paravertebral muscles, and cardiac
abnormalities involving brachycardia and first-degree, and eventually
complete, heart block (145,146).
The muscle weakness is slowly progressive, but there may be some
stabilization in adulthood. Most patients are able to ambulate into the
5th and 6th decades of life. Obesity and untreated equinus contractures
can lead to the loss of ambulatory ability at an earlier age (6).
dystrophy is only mildly or moderately elevated. EMG and muscle biopsy
reveal myopathy. The diagnosis of this form of muscular dystrophy
should be considered in patients with a myopathic phenotype, after
Duchenne and Becker muscular dystrophies have been ruled out (usually
by testing for dystrophin) (6). The condition should also be distinguished from scapuloperoneal muscular dystrophy and the rigid spine syndrome (147).
Emery-Dreifuss muscular dystrophy, the X-linked recessive form, has
been localized, in linkage studies, to the long arm of the X chromosome
at Xq28 (148,149).
Rarely, an autosomal dominant form and, even less frequently, an
autosomal recessive form may be seen. The autosomal dominant and
autosomal recessive forms have an identified gene mutation on the lamin
A/C gene on chromosome 1q21 (150). The specific type of gene testing depends on the family history and sex of the affected individual.
similar to what is used in other forms of muscular dystrophy. The goals
are to prevent or correct deformities and maximize function. Treatment
modalities include physical therapy, correction of soft tissue
contractures, spinal stabilization, and cardiologic intervention.
contractures, elbow-flexion contractures, and tightness of the lumbar
paravertebral muscles. Decreased neck flexion, which is characteristic
of this disorder, can begin as early as the 1st decade of life, but is
usually not present until the 2nd decade. This is due to contracture of
the extensor muscles and the ligamentum nuchae. According to Shapiro
and Specht (6), this contracture does not
progress past neutral. Lateral bending and rotation of the neck also
become limited as the extensor contractures progress. Physical therapy
can be helpful in maintaining limited flexion of the neck.
capsulotomy, combined with anterior transfer of the tibialis posterior
tendon, can be helpful in providing long-term stabilization of the foot
and ankle (6,145).
Elbow-flexion contractures usually do not require treatment. These
contractures can be as severe as 90 degrees, although most do not
exceed 35 degrees (6). Full flexion from this
position and normal forearm pronation and supination are preserved.
Physical therapy may be helpful in slowing the progress of the
elbow-flexion contractures. Surgery has not been shown to be beneficial.
but it shows a lower incidence of progression. This has been attributed
to contractures at the lumbar and ultimately the thoracic paravertebral
muscles, which seem to prevent progression (6,145).
Patients with scoliosis need to be followed closely, but most do not
require treatment. Curves that progress beyond 40 degrees may require
surgical stabilization.
been a major cause of sudden death in these patients. Most of them do
not have cardiac symptoms preceding death. Merlini et al. (146)
reported that 30 out of 73 patients with Emery-Dreifuss muscular
dystrophy died suddenly, of whom only four were symptomatic. It is
recommended that a cardiac pacemaker be inserted shortly after
confirmation of the diagnosis (146,151).
forms of muscular dystrophy. It is a rather heterogeneous group of
disorders with various classifications proposed
for
it over the years. The age at onset and rate of progression of muscle
weakness are variable. It usually begins in the 2nd or 3rd decade of
life. It is transmitted as an autosomal recessive trait, but an
autosomal dominant pattern of inheritance has been reported in some
families (152,153,154).
muscular dystrophy, except that the facial muscles are not involved.
The initial muscle weakness involves either the pelvic or shoulder
girdle. The rate of progression is usually slow, with soft tissue
contractures and disability developing 20 years or more after the onset
of the disease. The patients remain ambulatory for many years.
Duchenne and Becker muscular dystrophies. The iliopsoas, gluteus
maximus, and quadriceps muscles are involved early in the disease
process. Usually, shoulder girdle involvement occurs at about the same
time. The serratus anterior, trapezius, rhomboid, latissimus dorsi, and
sternal portions of pectoralis major muscles are affected most often.
The disease later spreads to involve other muscles, such as the biceps
brachia and the clavicular portion of the pectoralis major. Deltoid
involvement may occur, but usually only later in the course of the
disease. In patients with severe involvement, weakness may involve the
distal muscles of the limbs, such as the wrist and finger flexors and
extensors.
and a scapulohumeral form. The latter is rare, with symptoms involving
primarily the shoulder girdle. Involvement of the pelvic girdle may not
occur for many years. In the pelvic-girdle type, there is weakness of
the hip extensors and abductors, resulting in accentuated lumbar
lordosis, gait abnormalities, and hip instability.
LGMD. The clinical characteristics are indistinguishable from those of
sporadic Becker muscular dystrophy, carriers of Duchenne or Becker
muscular dystrophies, and those of childhood acid-maltase deficiency (6). Therefore, a dystrophin assay is essential in establishing the diagnosis (152).
Becker muscular dystrophies. Significant scoliosis rarely occurs
because of the late onset of the disease process. When present, it
usually is mild and does not require treatment (142). Patients usually succumb to the disease process before the age of 40 years.
for this heterogeneous group of muscular dystrophies. The European
Neuromuscular Center workshop on LGMD adopted a nomenclature to help
categorize this complex and heterogeneous group of disorders.
Presently, five autosomal dominant and nine autosomal recessive
conditions have been identified that fit into this clinical grouping (154).
MD) is being identified more frequently. It is a severe variant of the
more common later-onset facioscapulohumeral muscular dystrophy (155,156,157). A Mobius type of facial weakness may also be present and progress asymptomatically at a relatively slow pace (158).
Although many of these infants represent sporadic cases, genetic
diagnosis is positive for many of them and is identical to that seen in
adults (159). Facial diplegia is noted in
infancy, followed by sensorineural hearing loss in childhood (mean age,
5 years). Ambulation begins at a normal age, but because of progressive
muscle weakness, most patients become wheelchair bound during the 2nd
decade of life. Weakness causes the child to walk with the hands and
forearms folded across the upper buttocks to provide support for the
weak gluteus maximus muscles (6,155,157). This marked lumbar lordosis is progressive and is almost pathognomonic for IFSH MD (Fig. 17.5).
After the patient becomes wheelchair dependent, the lordosis leads to
fixed hip flexion contractures. Equinus or equinovarus deformities and
scoliosis occur less frequently.
compromised pulmonary functions and succumb in early adolescence.
Shapiro et al. outlined the possible treatment modalities for children
with IFSH MD. Flexible equinus and equinovarus deformities respond well
to AFOs. Occasionally, a Vulpius-type tendo-Achilles lengthening may be
necessary. Hip-flexion contractures usually do not require treatment in
ambulatory patients, because treatment may decrease function. Spinal
orthoses control the lordosis but do not provide correction because the
spine remains flexible early in the course of the disorder. Because an
orthosis interferes with ambulation, it is usually not employed. When
wheelchair use is full time, a modified wheelchair with an orthosis may
be useful, or perhaps a posterior spinal fusion and segmental
instrumentation, depending on the severity of the deformity.
Scapulothoracic stabilization is not indicated because the severity of
dysfunction is so great that minimal or no improvement in shoulder
function can be achieved.
 |
|
Figure 17.5
Marked lumbar lordosis in a 15-year-old girl with infantile facioscapulohumeral muscular dystrophy. She is still ambulatory but having increasing back pain. |
The disease is characterized by muscular weakness in the face, shoulder
girdle, and upper arm. It is caused by a gene defect, FRG1, on chromosome 4q35 (160,161). There is selective sparing of the deltoid, the distal part of the pectoralis major muscle, and the erector spinae muscles (162).
This results in decreased scapulothoracic motion, with scapular winging
and a marked decrease in shoulder flexion and abduction. Glenohumeral
motion is usually preserved. The onset may occur at any age but is most
common in late childhood or early adulthood. The disease occurs in both
genders but is more common in women. Abortive (minimally affected)
cases are common. Progression is insidious and periods of apparent
arrest may occur. Cardiac and CNS involvement are absent. Life
expectancy is relatively good.
involved, but they may be affected only mildly for many years. Facial
signs, which may be present in infancy, include lack of mobility,
incomplete eye closure, pouting lips with a transverse smile, and
absence of eye and forehead wrinkles. It tends to produce a “popeye”
appearance. The shoulder girdle weakness leads to scapular winging. The
weight of the upper extremities, together with the weakness of the
trapezius, permits the clavicles to assume a more horizontal position.
It also leads to a forward-sloping appearance of the shoulders. As the
disease progresses, pelvic girdle and tibialis anterior muscle
involvement may also occur. Scoliosis is rare because of the late onset
of the disease process.
muscular dystrophy are usually normal. The diagnosis is made by
physical examination and DNA confirmation. Presently, genetic testing
is more than 95% sensitive and highly specific for FSHD (163).
flexion and abduction, is the major orthopaedic problem in
facioscapulohumeral muscular dystrophy. The deltoid, supraspinatus, and
intraspinatus muscles are usually normal, however, or minimally
involved. Posterior scapulocostal fusion or stabilization
(scapuloplexy) by a variety of techniques can be helpful in restoring
mechanical advantage to the deltoid and rotator cuff muscles (164,165,166,167,168,169,170).
This can result in increased active abduction and forward flexion of
the shoulder, and improved function as well as cosmesis. Jakab and
Gledhill (166) reported the results of a
simplified technique for scapulocostal fusion. The technique involves
wiring of the medial border of the scapula to ribs three through seven.
Internal fixation is achieved with 16-gauge wire. The wires ensure firm
fixation and eliminate the need for postoperative immobilization and
subsequent rehabilitation. The child uses a sling for 3 to 4 days
postoperatively, and then begins a physical therapy program. Jakab and
Gledhill found that shoulder flexion increased 28 degrees (range, 20 to
40 degrees) and abduction 27 degrees (range, 20 to 35 degrees) at a
mean follow-up of 2.9 years. This allowed all patients to raise their
arms above their heads, conferring a greater mechanical advantage. The
beneficial effects do not seem to deteriorate with time (164,165,169,170).
It typically begins in young adults. It is transmitted as an autosomal
dominant trait. The initial involvement is in the intrinsic muscles of
the hand. The disease process spreads proximally. In the lower
extremities, the calves and tibialis anterior are involved first. The
absence of sensory abnormalities, especially vibratory, differentiates
this from Charcot-Marie-Tooth disease.
is another rare form of muscular dystrophy. It typically begins in the
adolescent years. The extraocular muscles are affected, resulting in
diplopia and ptosis. This is followed by limitation of ocular movement (171).
The upper facial muscles are often affected. The disease is slowly
progressive and may involve the proximal upper extremities. The pelvis
may be involved late in the disease process. Most patients with this
disorder have an identifiable mitochondrial myopathy (172).
autosomal dominant pattern with complete penetrance, and begins in the
3rd decade of life. It is particularly common in French Canadians (173).
in dysphasia, which leads to repetitive regurgitation and weight loss.
This condition necessitates cricopharyngeal myotomy, a procedure that
does not alter pharyngeal function (174,175). Ptosis develops in middle life.
inability of skeletal muscle to relax after a strong contraction from
either voluntary movement or mechanical stimulation. This is best
demonstrated by the slowness with which a clenched fist relaxes in such
patients. The most common myotonias include myotonic dystrophy,
congenital myotonic dystrophy, and myotonia congenita. These are all
rare disorders that are transmitted by autosomal dominant inheritance (6,17).
by myotonia, progressive muscle weakness, gonadal atrophy, cataracts,
frontal baldness, heart disease, and dementia (176). The genetic defect is located on chromosome 19q (177,178).
The distal musculature is affected first, and the myotonia begins to
disappear as muscle weakness progresses. The onset occurs usually in
late adolescence or early adulthood. In women, the diagnosis is
frequently made only after they have given birth to a child who is more
severely involved. The disease spreads slowly proximally and involves
the quadriceps, hamstrings, and eventually the hip extensors. The lower
extremities are more involved than the upper extremities. The most
common presenting symptoms are weakness of the hands and difficulty in
walking. Patients may be unable to relax their fingers after shaking
hands and may need to palmar flex the hand to open the fingers. Muscles
of the face, mandible, eyes, neck, and distal limbs may also be
affected. The levels of serum enzymes are normal. Muscle biopsies show
type I atrophy of the muscle fibers and the presence of some internal
nuclei. These are nonspecific findings. The “dive-bomber” pattern on
EMG is diagnostic (6). DNA testing that demonstrates a cytosine-thymine-quanine (CTG) expansion affecting a protein kinase is confirmatory (178).
a fish mouth that is difficult to close. There is marked wasting of the
temporal, masseter, and sternocleidomastoid muscles. Deep-tendon
reflexes are diminished or lost. Slit-lamp examination of the eyes
reveals that most patients have lenticular opacities, cataracts, and
retinopathy. Cardiac involvement is also common and includes mitral
valve prolapse and arrhythmias (177). Organic
brain deterioration may also occur. Frontal baldness in men and
glaucoma in both sexes occur in mid-adult life. The course of the
disease is one of steady deterioration. Most patients lose the ability
to ambulate within 15 to 20 years of onset of symptoms (177).
There are no characteristic orthopaedic deformities, although a slight
tendency toward increased hindfoot varus has been observed (6). Life span is shortened, and death is usually caused by pneumonia or cardiac failure.
because the onset is usually after skeletal maturity. An AFO may be
helpful in patients with a drop foot caused by weakness of the tibialis
anterior and peroneal muscles.
expression that occurs most frequently in children whose mothers have
either a forme fruste or mild clinical involvement (179,180,181,182). Although it has autosomal dominant transmission, it is predominantly transmitted from mother to child (181).
This is an exception in autosomal dominant disorders and indicates
additional maternal factors. Approximately 40% of patients have severe
involvement or die in infancy, whereas 60% will be affected later (183).
The child may have an expressionless, long, narrow face; hypotonia;
delayed developmental milestones; facial diplegia; difficulty in
feeding because of pharyngolaryngeal palsy; respiratory failure; and
mild mental retardation. The ability to swallow improves with growth,
but the hypotonia persists. Examination shows diffuse weakness and
absent deep-tendon reflexes. The appearance is similar to spinal
muscular atrophy. Ambulation is usually delayed. If the mother is the
carrier, the child may have other organic disorders later in life.
Cataracts usually occur after the age of 14 years.
As in the adult form, there appears to be an expansion of a
highly-repeated sequence of three nucleotides: cytosine, thymine, and
guanine. The trinucleotide repeat is at the 3′ end of a protein kinase
gene on chromosome 19, which lengthens as it passes from one generation
to another. The length of the sequence correlates with the severity of
the disorder. DNA testing is readily available for this disorder and is
the diagnostic test of choice.
include congenital hip dislocation and talipes equinovarus (i.e.,
clubfeet). There is a tendency to develop soft tissue contractures of
other major joints of the lower extremities. Clubfeet may behave like
those in arthrogryposis multiplex congenita (185). Serial casting may be tried, but most require surgery, such as an extensive, complete
release. If this fails, a talectomy or Verebelyi-Ogston procedure may be useful (186). Scoliosis is also common and may require orthotic or surgical intervention (142). Spine surgery is fraught with a high incidence of complications, such as cardiac arrhythmias and postoperative infection (187).
Nevertheless, because life expectancy is at least up to the early adult
years, aggressive orthopaedic management improves the quality of life.
not become clinically apparent until after the age of 10 years. In some
cases it may present as low back pain or impaired athletic ability (188,189,190).
The severity of the myotonia varies considerably. The distribution is
widespread, although it is more marked in the lower extremities than in
the upper extremities (191). Myotonia is most
evident during the initial movement. Repetitive movement decreases the
myotonia and facilitates subsequent movements. The stiffness usually
disappears within 3 to 4 minutes, and normal activities, including
running, are possible. Some patients appear herculean (massively
muscled) because of generalized muscle hypertrophy, particularly in the
buttocks, thighs, and calves. Children with myotonia congenita have no
associated weakness and no other endocrine or systemic abnormalities.
The disease is compatible with a normal life span. A patient’s
disability is not great when the limits of the disease have been
accepted. Procainamide and diphenylhydantoin (Dilantin) have been used
with some success to decrease the myotonia, but they should be used
only in severe cases (192). There are no characteristic orthopaedic deformities (6).
The disorder, a chloride channelopathy, is caused by various mutations
in the skeletal muscle voltage-gated chloride channel gene ClCN1 (193,194). To date, four mutations of the ClCN1 gene on chromosome 7q35 have been identified with myotonia congenita (195).
cause the baby at birth or in early infancy to be “floppy” or
hypotonic. When these conditions occur in an older child, they can
present as muscle weakness. These disorders are not well understood
clinically or at the molecular level. The diagnostic categorization is
not uniform or predictive. They are defined histologically from muscle
biopsies (6,196,197). When the biopsy findings are abnormal but not dystrophic, the patient is diagnosed as having a nonspecific myopathy (6). When considerable fibrosis is present along with necrotic fibers, congenital muscular dystrophy may be diagnosed.
nemaline myopathy (rod-body myopathy), myotubular myopathy
(centronuclear), congenital fiber-type disproportion, and metabolic
myopathies. Differentiation between these types can be accomplished
through histochemical analysis and electron microscopy of muscle biopsy
specimens (6,196,197,198).
dominant congenital myopathy that frequently presents as hypotonia in
infants and as delayed motor developmental milestones in young children
(196,197,199,200).
Independent ambulation may not be achieved until the age of 4 years.
The distribution of muscle involvement is similar to that found in
Duchenne muscular dystrophy, with the trunk and lower extremities
showing more involvement than the upper extremities, and the proximal
muscles more than the distal muscle groups. The pelvic girdle shows
more involvement than the shoulder. Use of the Gower maneuver is
common. No deterioration in strength occurs with time; sensation is
normal; and the deep-tendon reflexes are either decreased or absent.
Muscle wasting is a common finding, but progression of muscle weakness
is rare. Muscle biopsies show mostly type I fibers, containing central
circular or oval regions that are devoid of oxidative enzymes,
adenosine triphosphate activity, and mitochondria. Serum CPK and nerve
conduction studies are normal, whereas EMGs show myopathic
abnormalities. Scoliosis, soft tissue contractures, neuromuscular hip
subluxation and dislocation, talipes equinovarus, pes planus, and
hypermobility of joints (especially the patella) are the most common
musculoskeletal problems, and they may require treatment (199,200,201,202). Scoliotic deformities have patterns similar to those of idiopathic scoliosis, progress rapidly, and tend to be rigid (201).
Posterior spinal fusion and segmental instrumentation yield
satisfactory results. Soft tissue contractures around the hip and knee
may need to be released. Clubfeet require extensive soft tissue
releases in order to achieve correction. Congenital dislocation of the
hip can be treated by open or closed reduction techniques, but the
recurrence rate is high and may require osseous procedures such as
pelvic or proximal femoral osteotomies (202).
Central core disease is one of the disorders in which patients are
susceptible to malignant hyperthermia. This association with malignant
hyperthermia has led researchers to link both disorders with the long
arm of chromosome 19 as the probable site of mutation (203,204).
childhood, with hypotonia affecting all skeletal muscles (6,196,197,205,206).
There is no involvement of cardiac muscle. Elongated facies, with a
high-arched palette and a nasal, high-pitched voice, are frequently
noted. Skeletal changes may resemble those seen in arachnodactyly.
Martinez and Lake, in a review of the literature relating to 99
patients, recognized these distinct forms: neonatal (severe),
congenital (moderate), and adult onset (205).
The neonatal form is characterized by severe hypotonia, with 90%
mortality in the first 3 years of life because of respiratory
insufficiency. The mean survival after birth was 16 months. The
moderate congenital form, which is the most common and prototypic, is
diagnosed during or after the neonatal period and is characterized by
mild or moderate hypotonia, weakness, and delayed developmental
milestones. Most patients begin to walk at the age of 2 to 4 years, and
the weakness is usually nonprogressive or only slowly progressive. The
mortality rate is approximately 5% in the congenital form. Death is
usually caused by severe involvement of the pharyngeal and respiratory
muscles (207,208,209).
The adult-onset form is characterized by proximal weakness that
occasionally progresses acutely. There is no correlation between the
number of rods and the phenotype in nemaline myopathy (206).
The inheritance pattern in this disorder is variable, with autosomal
recessive, autosomal dominant, and sporadic cases identified. However,
all mutations identified to date follow an autosomal recessive
inheritance pattern (210).
myopathy. The major musculoskeletal problems are scoliosis and lumbar
lordosis. Posterior spinal fusion and segmental instrumentation may be
indicated in progressive scoliotic deformities (6).
Lower extremity orthoses can be helpful in providing stability to the
joints and in aiding ambulation. Because of their diminished pulmonary
function and the heightened risk for malignant hyperthermia, patients
undergoing surgery require careful monitoring during the administration
of anesthesia (211).
Muscle biopsies demonstrate persistent myotubes that would be normal in
fetal life. There are X-linked recessive, autosomal recessive, and
autosomal dominant forms (212,213). The defect in the X-linked recessive form is at the locus Xq28. The defective gene has been identified and named as MTM1 (214). Mutation detection analysis is now available, and sensitivity of testing is up to 72% (215).
These children have varying degrees of weakness, generally noted in
infancy. Patients with X-linked recessive forms are usually severely
involved and die in infancy. The infant with the autosomal recessive
form of the disease is hypotonic at birth, but the hypotonia is not
progressive and may improve with time. Most of these children are able
to walk. They may have a myopathic facies, high-arched palate, and
proximal muscle weakness. There is an increased incidence of cavovarus
foot deformities, scoliosis, lumbar lordosis, and scapular winging. By
late adolescence or early adult life, some patients lose their ability
to ambulate.
generalized hypotonia at or shortly after birth. The histologic
findings (from muscle biopsies) that may suggest this diagnosis include
a predominance of type I fibers of reduced size and relatively large
type II fibers. It is recognized as a nonspecific pathologic change
that occurs in many patients and has a myopathic, neuropathic, or CNS
origin (216). The degree of weakness is
variable, and sequential examinations determine the prognosis. Most
patients become ambulatory. The most serious problem is the
vulnerability to life-threatening respiratory infections during the
first years of life. Proximal muscle weakness is frequently associated
with acetabular dysplasia (216). To prevent
postural contractures from developing, an appropriate lower-extremity
splint should be used until the patient achieves ambulation. Severe,
rigid scoliosis can occur. Orthoses are usually ineffective, and early
spinal arthrodesis may be necessary (6).
abnormalities that are generally clinically evident in the 1st two
decades of life (217). These include disorders
of glycolysis, lipid metabolism, mitochondrial dysfunction, and purine
nucleotide cycle defects. Myopathies caused by metabolic errors in the
first step of glycolysis, for example, myophosphorylase and
phosphofructokinase deficiencies, are clinically associated with
cramping, weakness, and exercise intolerance with anaerobic activity
(i.e., short-duration but vigorous activity). The other glycolytic
disorders, such as acid maltase or debrancher enzyme deficiencies, are
associated with progressive muscle weakness and wasting (218).
Carnitine palmitoyl transferase deficiency, which is a disorder of
lipid metabolism, presents with muscle cramping, weakness, and
myoglobinuria following prolonged exercise. Myopathies caused by
deficiencies in mitochondrial enzymes are less well defined and may be
associated with severe benign exercise intolerance and progressive
myopathic syndromes (218,219,220).
which babies are “floppy,” with generalized muscle weakness and with
the involvement of respiratory and facial muscles (221). It is a muscle disorder in which the muscle biopsy
demonstrates dystrophic features characterized by considerable
perimysial and endomysial fibrosis. It is different from Duchenne
muscular dystrophy and Becker muscular dystrophy because it affects
children of both sexes, is not associated with massively elevated
levels of CPK, does not involve abnormalities of the dystrophin gene or
protein, and is associated with a more variable prognosis (6).
There are several forms of congenital muscular dystrophy. In one, the
infant is weak at birth. Many have severe stiffness of joints, whereas
others do not. A few infants have rapid progression and do not survive
after the first year of life. Most, however, stabilize and survive into
adulthood (222). Another type is seen in Japanese infants and has been termed Fukuyama congenital muscular dystrophy. It is characterized by a marked developmental defect in the CNS (223,224).
There is progressive muscle degeneration and mental retardation. Severe
joint contractures develop, and many children with this condition die
in the first decade of life. Three disorders are associated with
congenital muscular dystrophy and CNS malformations: Fukuyama
congenital muscular dystrophy, Walker-Warburg syndrome, and
muscle-eye-brain disease. Merosin-deficient congenital muscular
dystrophy is associated with changes in the white matter of the brain
as seen on MRI and has been linked to chromosome 6q2 (225,226).
dislocation and subluxation, tendo-Achilles contractures, and talipes
equinovarus (Fig. 17.6). Because most patients
survive, aggressive orthopaedic management is warranted. This may
include physical therapy, orthoses, soft tissue releases, and perhaps
osteotomy (6,227).
Early physical therapy may be helpful in preventing soft tissue
contractures. Soft tissue releases in the treatment of congenital
dislocation of the hip are characterized by a high incidence of
recurrent dislocation (Fig. 17.7) (227).
Progressive scoliosis may be initially treated by an orthosis, although
most patients require surgical stabilization similar to the procedure
used in other forms of muscular dystrophy (113).
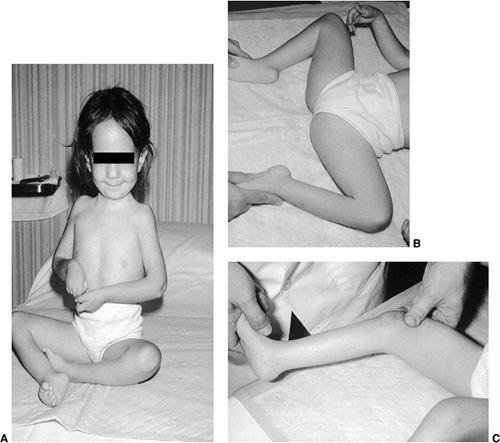 |
|
Figure 17.6 A:
Clinical photograph of a 3-year-old girl with congenital muscular dystrophy. Observe the position of the upper and lower extremities. B: The hips are flexed, abducted, and externally rotated. C: Moderate knee-flexion contractures are present. |
 |
|
Figure 17.7
Pelvic radiograph of an 11-year-old girl with congenital muscular dystrophy, 3 years after posterior spinal fusion and Luque rod instrumentation, including the Galveston technique. She is wheelchair-dependent and has developed bilateral asymptomatic hip dislocations despite extensive soft tissue releases in early childhood. |
characterized by degeneration of the anterior horn cells of the spinal
cord, and occasionally the neurons of the lower bulbar motor nuclei,
resulting in muscle weakness and atrophy (228,229,230,231,232). They are autosomal recessive disorders that occur in approximately 1 in 6000 to 10,000 individuals (233). The prevalence of carriers is estimated at 1 in 40 to 1 in 50 (234).
The loss of anterior horn cells is considered to be an acute event
without progression. The neurologic deterioration may stabilize and
remain unchanged for long periods (235). The
progression of muscle weakness is a reflection of normal growth that
exceeds muscle reserve. Respiratory function is compromised, and
atelectasis and pneumonia are the usual causes of death.
widely and are based on the age at onset and the functional capacity of
the child at the time of diagnosis. This has led to the disorder being
classified into three types. These include Type I (severe), or acute
Werdnig-Hoffman disease; Type II (intermediate), or chronic
Werdnig-Hoffman disease; and Type III (mild), or Kugelberg-Welander
disease (236). All three fall within the
spectrum of the same disorder, but each has its specific diagnostic
criteria and prognosis. There is a considerable overlap between these
three disorders, however, and most authors consider them to be a single
disorder, namely, spinal muscular atrophy (237). Generally, the earlier the onset, the worse the prognosis.
clinical onset between birth and 6 months. These children typically
have severe involvement with marked weakness and hypotonia. They
usually die from respiratory failure between the ages of 1 and 24
months. Because of their young age and severe involvement, orthopaedic
intervention is not indicated in these children. Pathologic fractures
may occur because of in utero osteoporosis secondary to decreased movement at birth, thereby suggesting the presence of osteogenesis imperfecta (238). These fractures heal rapidly with immobilization.
occurs at between the ages of 6 and 24 months. These children show less
severe involvement than those with Type I spinal muscular atrophy, but
are never able to walk. They may, however, live into the 4th and 5th
decades of life.
occurs after the age of 2 years and usually before the age of 10 years.
Walking is usually possible until late childhood or early adolescence.
These patients are usually not able to run. Their motor capacity
decreases with time, and they have difficulty rising from the floor
because of weakness of the pelvic-girdle muscles; this is known as the Gower sign.
There is atrophy of the lower limbs, with pseudohypertrophy of the
calves. Cranial nerve muscles are usually not affected. These patients
have normal intelligence and may function effectively in society. Both
the quality and quantity of life may be extended in Type II and Type
III spinal muscular atrophy by the use of nighttime or full-time
assisted ventilation (239).
placed in a sitting position but are unable to stand or walk, even with
orthotics.
homogeneity for the three types of spinal muscular atrophy occur at the
same locus on chromosome 5q (16,17,228,241). Two genes have been found to be associated with disease, the survival motor neuron gene (SMN) and the neuronal apoptosis inhibitory protein gene (NAIP) (229,242,243).
The presence of large-scale deletions involving both genes corresponds
to a more severe phenotype. Prenatal diagnosis is available with the
use of PCR amplification assays. No specific gene therapy is available.
according to the clinical classification. The clinical characteristics
common to all groups are relatively symmetric limb and trunk weakness,
and muscle atrophy that affects the lower extremities more than the
upper extremities and the proximal muscles more than the distal
muscles. Hypotonia and areflexia are present. Sensation and
intelligence are normal. In infants, gross fasciculations of the tongue
and fine tremors of the fingers are commonly present (235,244).
The only muscles not involved are the diaphragm, sternothyroid,
sternohyoid, and the involuntary muscles of the intestine, bladder,
heart, and sphincters (228,236).
muscular atrophy include laboratory studies, EMG, nerve conduction
studies, DNA testing, and muscle biopsies. Hematologic studies in
spinal muscular atrophy are not particularly useful (232).
The CPK and aldolase levels are normal to only slightly elevated. In
patients with spinal muscular atrophy, electrophysiologic studies such
as EMG show typical neuropathic changes such as increased amplitude and
duration of response (232). Denervational
changes, manifest as prominent fibrillation potentials, are a hallmark
of this disorder. Nerve conduction velocities are typically normal,
although the compound muscle action potential (CMAP) amplitude is
typically markedly diminished (245). Muscle biopsies are usually diagnostic, demonstrating muscle fiber degeneration and atrophy of fiber groups (232).
However, with the recent advent of genetic testing for this disorder,
muscle biopsy is usually not necessary. DNA testing is highly sensitive
for this disorder and is readily available. DNA PCR for spinal muscular
atrophy is now the diagnostic procedure of choice.
are useful in making the diagnosis of spinal muscular atrophy. The most
common radiographic abnormalities are nonspecific and include hip
subluxation or dislocation and progressive spinal deformity (232).
Spinal radiographs, posteroanterior and lateral, should be obtained in
the sitting position to avoid the compensations seen in the standing
and supine positions.
spinal muscular atrophy include the presence of soft tissue
contractures of the lower extremities, hip subluxation and dislocation,
and spinal deformity (231,232).
the result of progressive muscle degeneration and replacement with
fibrous tissue. Ambulation may be promoted and soft tissue contractures
delayed by the use of orthoses such as KAFOs (246).
Contractures tend to occur most frequently after the child becomes
wheelchair bound. The prolonged sitting posture enhances hip- and
knee-flexion contractures. Contractures of the soft tissues of the hip
may also result in abnormal growth of the proximal femur, predisposing
the patient to coxa valga and progressive hip subluxation. Soft tissue
contractures without an associated osseous deformity usually do not
require treatment. Even when they are released, the sitting posture of
the child promotes their recurrence.
It is important that hip dislocation be prevented in order to provide
comfort and sitting balance and to maintain pelvic alignment. A
comfortable sitting posture is important if the adolescent or young
adult is to function in society. Periodic anteroposterior radiographs
of
the
pelvis, beginning in mid- to late childhood, are important in order to
ensure early recognition of coxa valga and subluxation. Once diagnosed,
it is usually progressive because of the continued muscle weakness and
soft tissue contractures. Procedures that have been used with some
success include soft tissue releases such as adductor tenotomy,
iliopsoas recession, and medial hamstring lengthening. This restores
some balance to the proximal musculature. A varus derotation osteotomy
is frequently indicated if the hip is severely subluxated (232).
If the hip is dislocated, an open reduction with capsulorrhaphy and
pelvic osteotomy of the Chiari type may be of benefit to the patient.
The usual rotation osteotomies (e.g., Salter, Sutherland, Steel)
sacrifice posterior coverage to gain lateral (superior) and anterior
coverage. In the child who will be predominantly in a sitting position,
this lack of posterior coverage may predispose the patient to a
posterior subluxation and pain. Therefore, the pelvic osteotomy method
chosen must allow improved posterior coverage. This is usually
accomplished with the Chiari osteotomy or perhaps a shelf procedure.
Even after satisfactory alignment of the hip, resubluxation and
dislocation can occur because of the progressive degeneration of the
proximal muscles (248). These children require annual clinical and radiographic evaluation to assess the hips postoperatively. Thompson and Larsen (248)
reported four cases of recurrent hip dislocation after corrective
surgery. Two patients had 2nd operations followed by recurrent
dislocation. Therefore, these orthopaedists question the advisability
of treatment of hip dislocations in patients with spinal muscular
atrophy. Sporer and Smith recently documented that patients with a hip
dislocation had minimal pain or problems with sitting, and no
difficulty with perineal care. They suggested observation rather than
surgery for hip dislocation (247). Thus,
treatment of hip subluxation and dislocation in spinal muscular atrophy
is controversial. Each patient must be evaluated individually. The
presence of pain, rather than the radiographic appearance of the hips,
should be the main indication for treatment.
adolescence develop a progressive spinal deformity. This occurs in 100%
of the children and adolescents with Type II disease, and most of those
with Type III, especially when they lose their ability to walk (240,249,250,251,252,253,254). As in other neuromuscular disorders, the progression of the curve has an adverse effect on pulmonary function (252).
life because of severe truncal weakness. Once the deformity begins, it
is steadily progressive and can reach a high magnitude of severity
unless appropriately managed. The thoracolumbar paralytic C-shaped and
single thoracic patterns, usually curved to the right, are most common.
Approximately 30% of the children also have an associated kyphosis,
which is also progressive (250,254).
In Type II spinal muscular atrophy, the mean expected increase in
scoliosis is 8.3 degrees per year, whereas in Type III it is 2.9
degrees per year.
progression of scoliosis or kyphosis in children with spinal muscular
atrophy (231,240,250,251,254,255,256,257,258). However, it can be effective in improving sitting balance and slowing the rate of progression in young ambulatory children (258).
This has the advantage of allowing them to reach an older, more
suitable age for undergoing surgical intervention. Orthotic treatment
may help maintain overall posture, aid sitting posture, and slow the
curve progression in younger nonambulatory children with deformities
between 20 and 40 degrees. The TLSO is the most common orthosis used in
children with spinal muscular atrophy. This orthosis must be carefully
molded in order to distribute the forces over a large surface area.
This is necessary for preventing skin irritation and breakdown, which
is a major problem for children with neuromuscular diseases. Furumasu
et al. (259) found that orthoses had the effect
of decreasing the ability to function because of decreased spinal
flexibility. It is also important to ensure that the TLSO does not
further compromise the child’s limited pulmonary functions.
Occasionally, wheelchair modifications can also be effective in
controlling truncal alignment and improving sitting posture (232).
This may also be helpful in slowing the rate of curve progression.
Unfortunately, almost all children with spinal muscular atrophy
eventually require surgery for spinal deformity.
muscular atrophy include: curve magnitude greater than 40 degrees,
satisfactory flexibility on supine lateral bending as seen on
radiographs, and an FVC greater than 40% of normal (230).
When these criteria are met, a posterior spinal fusion with segmental
spinal instrumentation techniques such as Luque rod instrumentation and
sublaminar wires is used (Fig. 17.8) (102,110,112,113,231,249,250,252,253,255,256,257,259,260).
Other segmental systems, such as Cotrel-Dubousset, Texas Scottish Rite
Hospital (TSRH), and Isola, can also be utilized. However, these do not
usually distribute the forces of instrumentation throughout the spine
as efficiently as the Luque rods with sublaminar wiring do. The spine
is usually osteopenic, and there is a risk of bone failure unless the
forces produced by instrumentation are minimized by extensive
distribution. Fixation to the pelvis using the Galveston technique (116) or other techniques (117,260)
is common. In most children who are nonambulatory and have pelvic
obliquity, fusion to the pelvis provides improved spinopelvic stability
and alignment. Anterior spinal fusion and instrumentation are rarely
indicated in view of the compromised pulmonary status of these
children, which could predispose them to pulmonary complications
postoperatively (257). Anterior fusions alone are too short to adequately
stabilize the entire spine. When the procedure is performed, it is
combined with a simultaneous or staged posterior spinal fusion, usually
with Luque rod instrumentation (102).
Whatever posterior instrumentation system is used, it is important to
ensure that no postoperative immobilization is necessary; this enhances
sitting balance and pulmonary status and makes transfers easier.
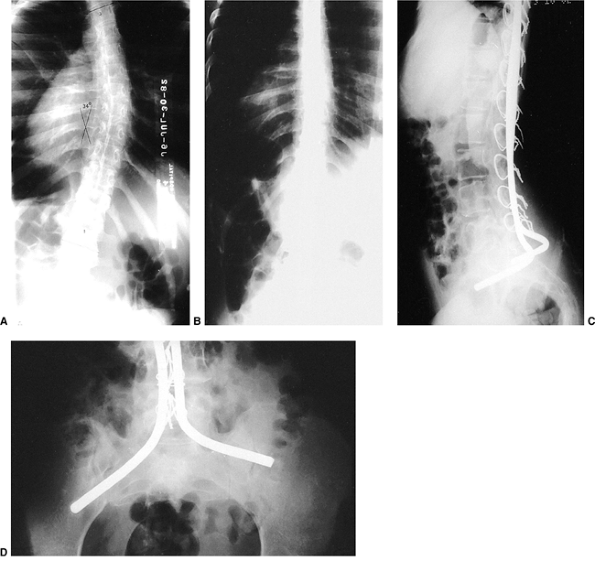 |
|
Figure 17.8 A:
Sitting posteroanterior spinal radiograph of an 18-year-old woman with spinal muscular atrophy. A slowly progressive scoliosis has affected her wheelchair sitting balance. B: Postoperative radiograph after posterior spinal fusion and Luque rod instrumentation using the Galveston technique provided almost complete correction of the spinal deformity. Thirteen years postoperatively she functions independently despite the subsequent need for a tracheostomy and ventilator support. C: Lateral view demonstrates preservation of lumbar lordosis, which is important for proper sitting balance. D: Anteroposterior view of the pelvis shows proper positioning of the Luque rods in the ilium. They should penetrate as far into the ilium as possible for maximum strength. |
Although spinal alignment and sitting balance are improved, the loss of
spinal mobility decreases the function of the upper extremities and
activities of daily living such as performing transfers and maintaining
personal hygiene. Askin et al. (261)
recommended early surgery to preserve function. They found as well that
the patient’s functional ability may not improve following surgery, but
the cosmetic results are gratifying, and the caregivers also find it
easier to carry out their tasks. Bridwell et al. (110)
reported improved function, self-image, cosmesis, and caregiver ability
in 21 patients with spinal muscular atrophy followed for a mean of 7.8
years postoperatively (range, 2 to 12.6 years). Growing rods or rods
that can be elongated periodically may be helpful in young children
with spinal muscular atrophy who have severe deformities (262). This allows definitive surgery to be delayed until an older age (262).
neuromuscular disorders. These include excessive blood loss, pulmonary
complications, neurologic injury, wound infection, loss of fixation
(caused by osteopenia), pseudarthrosis, and even death (102,132,249,251,254,255,257,260).
The use of segmental spinal instrumentation techniques and aggressive
preoperative and postoperative respiratory therapy may lead to fewer
complications. Hypotensive anesthesia
and
intraoperative spinal cord monitoring may be helpful in decreasing
intraoperative blood loss and neurologic injury. Noordeen et al.
reported that a 50% decrease in amplitude of motor action potential may
be indicative of an impending neurologic injury (132).
relatively uncommon disorders that are hereditary and progressive.
Friedreich ataxia is the most common form and has orthopaedic
implications because it is associated with a high incidence of
scoliosis. In whites, this disorder accounts for up to half of all
cases of hereditary ataxia (263). Friedreich
ataxia is characterized by slow, progressive spinocerebellar
degeneration. It occurs in approximately 1 in 50,000 live births. It is
autosomal recessive and occurs most commonly in North America in people
of French-Canadian heritage. Both sexes are affected equally.
consisting of (i) ataxia (which is usually the presenting symptom);
(ii) areflexia of the knees and ankles; and (iii) a positive plantar
response, or the Babinski sign (232,263). Geoffroy et al. (264) established strict criteria for the clinical diagnosis of typical Friedreich ataxia. This has been modified by Harding (265,266).
The primary symptoms and signs that occur in all affected patients
include: onset before the age of 25 years; progressive ataxia of limbs
and gait; absent knee and ankle deep-tendon reflexes; positive plantar
response; decreased nerve conduction velocities in the upper
extremities, with small or absent sensory action potentials; and
dysarthria. The secondary symptoms and signs that are present in more
than 90% of the cases include scoliosis, pyramidal weakness in the
lower extremities, absent reflexes in the upper extremities, loss of
position and vibratory sense in the lower extremities, and an abnormal
ECG. Supplementary symptoms and signs are present in fewer than 50% of
the cases. These include optic atrophy, nystagmus, distal weakness and
wasting, partial deafness, pes cavus, and diabetes mellitus.
demonstrated that the muscle weakness is always symmetric, initially
proximal rather than distal, more severe in the lower extremities, and
rapidly progressive when the patients become nonambulatory. The first
muscle to be involved is the hip extensor (gluteus maximus). They also
demonstrated that muscle weakness is not the primary cause of loss of
ambulatory function. Ataxia and other factors also play a role. Death
usually occurs in the 4th or 5th decade because of progressive
hypertrophic cardiomyopathy, pneumonia, or aspiration (263,265).
sensory action potentials in the digital and sural nerves. Conduction
velocity in the motor and sensory fibers of the median and tibial
nerves is moderately slowed. An EMG shows a loss of motor units and an
increase in polyphasic potentials. The ECG in adults typically shows a
progressive hypertrophic cardiomyopathy. Hematologic tests such as CPK
are normal, but there is increased incidence of clinical and chemical
diabetes mellitus.
It is now known that this condition is caused by a trinucleotide repeat
of GAA, which causes loss of expression of the frataxin protein. There
is an inverse relation between the number of trinucleotide repeats and
the age at onset of the disease (271). Various
medications such as physostigmine, tryptophan, buspirone, and
amantadine have been tried for symptomatic treatment, with generally
disappointing results (272,273,274,275). DNA testing is available and is the diagnostic test of choice.
ataxia. It is slowly progressive and tends to become rigid. When
combined with ataxia, it can result in decreased ability to stand and
walk. Orthotic management is usually ineffective in preventing the
deformity, but an AFO can be used after surgery to stabilize the foot
and ankle and to prevent recurrent deformity. Surgical procedures can
be used in ambulatory patients to improve balance and walking ability.
Procedures that have been shown to be effective include tendo-Achilles
lengthening and tibialis posterior tenotomy, lengthening, or anterior
transfer to the dorsum of the foot (232,263).
The tibialis anterior muscle may also be involved and may require
tenotomy, lengthening, or centralization to the dorsum of the foot to
prevent recurrence. In fixed, rigid deformities, a triple arthrodesis
may be necessary for achieving a plantigrade foot.
incidence of curve progression has been shown to correlate to the age at clinical onset of the disease process. Labelle et al. (278)
demonstrated that when the disease onset is before the age of 10 years
and scoliosis occurs before the age of 15 years, most scoliotic curves
progress to greater than 60 degrees and require surgical intervention.
When the disease onset is after the age of 10 years and the scoliosis
occurs after the age of 15 years, the curve progression is not as
severe; most do not reach 40 degrees by skeletal maturity, and do not
progress thereafter. There was found to be no correlation between curve
progression, degree of muscle weakness, level of ambulatory function,
and duration of the disease process. The patterns of scoliosis in
patients with Friedreich ataxia are similar to those in adolescent
idiopathic scoliosis rather than to those in neuromuscular scoliosis.
The pathogenesis of scoliosis in Friedreich ataxia appears to be not
muscle weakness but ataxia that causes a disturbance of equilibrium and
postural reflexes. Double major (i.e., thoracic and lumbar) and single
thoracic or thoracolumbar curves are the most common curve patterns (276,277,278).
Only a few patients have lumbar or long C-shaped thoracolumbar curves.
About two thirds of these patients develop an associated kyphosis
greater than 40 degrees (278). The treatment of scoliosis in Friedreich ataxia can be by either orthotic or surgical methods.
ambulatory patients having 25- to 40-degree curves. It is usually not
well tolerated, but it may slow the rate of progression although it
rarely stabilizes the curve (263,276).
In ambulatory patients, an orthosis may interfere with walking because
it prevents the compensatory truncal movement that is necessary for
balance and movement.
especially in older adolescents confined to wheelchairs, a single-stage
posterior spinal fusion stabilizes the curve and yields moderate
correction. Curves between 40 and 60 degrees can be either observed or
treated surgically, depending on the patient’s age at clinical onset,
the age when scoliosis was first recognized, and evidence of curve
progression. Posterior segmental instrumentation using Harrington rods
and sublaminar wires or Luque rod instrumentation has been demonstrated
to be effective in achieving correction and a solid arthrodesis (102,276,277,278).
Other segmental systems (e.g., Cotrel-Dubousset, Isola, and TSRH)
should also be effective. Fusions are typically from the upper thoracic
(T-2 or T-3) to lower lumbar regions. Fusion to the sacrum is usually
unnecessary, except in C-shaped thoracolumbar curves with associated
pelvic obliquity (277). Autogenous bone
supplemented with banked bone, when necessary, usually produces a solid
fusion. Anterior surgery, with or without instrumentation, usually
followed by a posterior spinal fusion and instrumentation, is limited
to rigid curves that are greater than 60 degrees and associated with
poor sitting balance. Surgery is performed only after a thorough
cardiopulmonary evaluation and under careful intraoperative and
postoperative monitoring. Postoperative immobilization should be
avoided. Vertebral osteopenia and spinal stenosis are not problems in
Friedreich ataxia.
They usually begin in the late adolescent or early adult years and
worsen with time. The spasms are characterized by a sudden onset and
short duration. The hip adductors and the knee extensors are commonly
involved. Initial treatment is usually massage, warming, and perhaps
muscle relaxants, such as diazepam and baclofen. In adults, if the
adductor or quadriceps spasms are interfering with perineal care or
sitting balance, the patient may benefit from tenotomies. However, this
is rarely necessary.
HMSN types I, II, and III are encountered predominantly in pediatric
orthopaedic and neuromuscular clinics, whereas HMSN types IV, V, VI,
and VII tend to be late-onset and occur in adults (232).
|
TABLE 17.2 CLASSIFICATION OF HEREDITARY MOTOR SENSORY NEUROPATHIES
|
||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
includes disorders referred to as peroneal atrophy, Charcot-Marie-Tooth
disease (hypertrophic form), or Roussy-Levy syndrome. It is a
demyelinating disorder that is characterized by peroneal muscle
weakness, absent deep-tendon reflexes, and slow nerve conduction
velocities. HMSN Type II is the neuronal form of Charcot-Marie-Tooth
disease with progressive axon loss. It is characterized by persistently
normal reflexes, sensory and motor nerve conduction times that are only
mildly abnormal, decreased compound motor action potentials, and
variable inheritance patterns (232). These two
types are clinically similar, although HMSN Type II often causes less
severe weakness and has a later onset than HMSN Type I. HMSN Type III
is the autosomal recessive disorder, Dejerine-Sottas disease. This
disorder begins in infancy and is characterized by more severe
alterations in nerve conduction and by sensory disturbances that are
more extensive than in HMSN Types I and II. The HMSN Types I and III
are caused by demyelinization of peripheral nerves, whereas Type II is
caused by axon loss. These are characterized by muscle weakness in the
feet and hands, absent deep-tendon reflexes, and diminution of distal
sensory capabilities, particularly light touch position and vibratory
sensation (232).
rarely seen by pediatric orthopaedists or in pediatric neuromuscular
clinics: HMSN Type IV, Refsum disease, is characterized by excessive
phytanic acid; HMSN Type V is an inherited spastic paraplegia, with
distal weakness in the limbs presenting in the 2nd decade of life, and
characterized by an awkward gait and equinus foot deformities; HMSN
Type VI is characterized by optic atrophy in association with peroneal
muscle atrophy; and HMSN Type VII is associated with retinitis
pigmentosa, distal weakness in the limbs, and muscle atrophy.
combination with EMG, nerve conduction studies, and genetic testing.
The EMG findings in HMSN show typical neuropathic changes, with
increased amplitude and duration of response. Nerve conduction studies
in patients with the demyelinating HMSN Types I and III show marked
slowing of the rate of impulse conduction in the muscles involved. A
biopsy specimen of a muscle such as the gastrocnemius demonstrates
typical neuropathic findings, including atrophy of the fiber group,
with all of the fibers in an abnormal group having uniformly small
diameter. A biopsy specimen of a peripheral nerve, usually the sural
nerve, shows typical demyelinization, confirming the diagnosis of
peripheral neuropathy.
of a portion of the short arm of chromosome 17 in the region of p11.2
to p12 (17,280,281,282). Additional studies have shown a human peripheral myelin protein-22 gene to be contained within the duplication (283,284,285).
It is thought that the abnormality in the peripheral myelin protein-22
gene, which encodes the myelin protein, has a causative role in
Charcot-Marie-Tooth disease. Either a point mutation in peripheral
myelin protein-22 or duplication of the region that contains the
peripheral myelin protein-22 gene can result in the disorder (286).
occurring either as an autosomal dominant or as an autosomal recessive
trait (287). Chromosome linkage has been identified at 1p35-36 (288), at 8p21 involving the neurofilament-light gene (289), and on 7q11-q21 (290).
HMSN Type III, previously referred to as Dejerine-Sottas disease, also
shows genetic heterogeneity, with multiple loci identified to date.
Inheritance typically follows an autosomal recessive pattern.
disturbance or foot deformities. The severity of involvement is
variable. In severe involvement, there may be proximal muscle weakness.
The major orthopaedic problems include pes cavovarus, hip dysplasia,
spinal deformity, and hand and upper extremity dysfunction.
has been the orthopaedic approach. Recently, however, there have been
promising results with the use of progesterone receptor antagonists. In
transgenic rat studies, administration of selective progesterone
receptor antagonists led to decreased overexpression of PMP22 and
improved CMT phenotype (291). Presently, human studies are underway and appear promising (292).
with HMSN and other neuromuscular disorders is becoming better
understood (293,294,295,296,297,298,299).
The components of the pes cavovarus deformity include claw toes;
plantar-flexed first metatarsal with adduction and inversion of the
remaining metatarsals; midfoot malposition of the navicular, cuboid,
and cuneiforms, leading to a high arch (cavus); and hindfoot varus
malposition between the talus and calcaneus (Fig. 17.9).
Initially, HMSNs affect the more distal muscles. The mildest cases show
involvement of the toes and forefoot, whereas the midfoot and hindfoot
are progressively affected with progression of the disease process. In
a computed tomography study of 26 patients with HMSN I, II, or III,
Price et al. (300) found that the interossei
and lumbrical muscles of the feet demonstrated earlier and more severe
involvement than the extrinsic muscles. These intrinsic muscles have
the most distal innervation. Even with minimal weakness, the invertor
muscles, such as the tibialis
anterior
and tibialis posterior muscles, are stronger than the evertors, such as
the peroneus longus; this relation favors the development of adduction
and varus deformities.
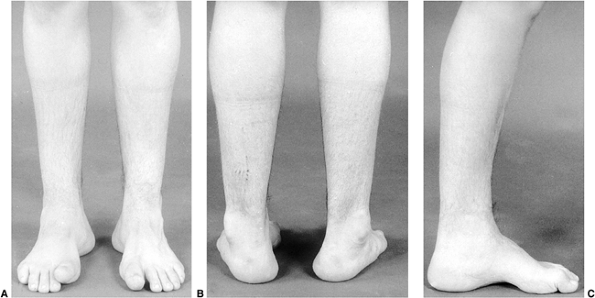 |
|
Figure 17.9 A:
Front view of the lower legs and feet of a 16-year-old boy with hereditary motor sensory neuropathy Type I (i.e., Charcot-Marie-Tooth disease). His calves are thin, and he has mildly symptomatic cavus feet. Clawing of the toes is minimal. B: Posterior view demonstrates moderate heel varus. C: The cavus foot deformity is most apparent when viewed from the medial side. A mild flexion deformity of the great toe interphalangeal joint is present. |
is variable, even among patients belonging to the same family.
Initially, the deformity is flexible but later becomes rigid. Shapiro
and Specht identify the plantar-flexed first metatarsal as the key
finding (232). As the first metatarsal becomes
increasingly plantar flexed, increasing hindfoot varus and supination
and cavus of the forefoot and midfoot follow. The block test is useful
for determining the mobility of the remainder of the foot in children
with a rigid plantar-flexed first metatarsal (296).
children with HMSN include maintenance of a straight, plantigrade, and
relatively flexible foot during growth (298,299).
This maximizes function and minimizes the development of osseous
deformities that may require more extensive surgery (such as a triple
arthrodesis) in adolescence and early adult years.
deformities include: plantar release, plantar-medial release, tendon
transfers, calcaneal osteotomy, midtarsal osteotomy, triple
arthrodesis, and correction of toe deformities (295,296,298,299).
deformity, a plantar release may be helpful in correcting the
plantar-flexed first metatarsal and providing correction of the
associated flexible deformities of the hindfoot and midfoot (301). In the radical plantar release described by Paulos et al. (296),
selective Z-lengthening of the long toe flexor tendons and the tibialis
posterior tendon are performed if there is a “bowstring” effect after
plantar release.
deformity is rigid and leading to fixed varus deformity, the plantar
release may be combined with a medial release (296).
The medial structures to be released include the ligamentous and
capsular structures between the talus and calcaneus (except the
posterior talocalcaneal ligament), and the capsule of the talonavicular
joints. The navicular is then reduced onto the head of the talus and
secured with a smooth Steinmann pin. The posterior ankle and subtalar
joint ligaments and the tendo-Achilles are not disturbed because they
are necessary for counter-resistance during postoperative serial
casting. Once the incision has healed, a series of corrective
weight-bearing casts are applied. Excellent correction of the entire
foot has been reported after this technique.
deformities in which active inversion is associated with relative
weakness of the evertor muscles, a transfer of the tibialis anterior
tendon to the dorsum of the midtarsal region in line with the 3rd
metatarsal may be helpful (302). The transfer is designed to balance strength,
but the foot must be aligned initially by a plantar release and perhaps the plantar-medial release.
the individual needs of the patient. These may include tendo-Achilles
lengthening, anterior transfer at the tibialis posterior tendon, long
toe extensors to the metatarsals or midfoot, and flexor-to-extensor
tendon transfers for claw toes (296,302).
Tendo-Achilles lengthening is rarely necessary, as the equinus is due
to the plantar-flexed first metatarsal and forefoot. The hindfoot is
typically in a calcaneus position.
mild but fixed deformity, a calcaneal osteotomy may be helpful in
correcting the varus deformity of the hindfoot (232).
This osteotomy does not interfere with growth because it is not made
through a cartilaginous growth area. To allow lateral translation, the
osteotomy is cut slightly obliquely, passing from a superior position
on the lateral surface to a more inferior position on the medial
surface. It is possible to translate the distal fragment by as much as
one third of its transverse diameter, thereby allowing conversion of
weight bearing from varus to mild valgus. In patients who are older
than 10 years or who are more severely affected, a lateral
closing-wedge calcaneal osteotomy, with lateral translation of the
distal and posterior fragments, is performed (Fig. 17.10) (232). In both procedures, the osteotomy is stabilized with staples or Steinmann pins.
of a dorsal and slightly laterally based wedge, with the proximal
osteotomy cut through the acicular and cuboids, and the distal cut
through the cuboids and three cuneiforms. Moderate deformities can be
corrected satisfactorily with this procedure, especially if it is
augmented with a plantar release, calcaneal osteotomy, and perhaps an
anterior transfer of the tibialis anterior tendon. Equinus deformities
of the midfoot and varus deformities of the forefoot can be corrected
with appropriate wedge resections. Growth retardation and limitation of
mobility are minimal when compared with the situation after a triple
arthrodesis. Recently, the use of the Ilizarov external fixator and a
V-osteotomy have been shown to be effective in achieving a painless
plantigrade foot (303). This approach can obviate the need for a triple arthrodesis in selected patients.
who have a severe deformity, walk with difficulty, and cannot run, a
triple arthrodesis may be performed. Every attempt should be made to
avoid this procedure because of the associated complications of
undercorrection, overcorrection, pseudoarthrosis of the talonavicular
joint, and degenerative changes in the ankle and midfoot joints (304,305,306,307).
reported unsatisfactory results in 23 of 30 feet (16 patients) at a
mean follow-up at 21 years. The progressive muscle imbalance resulted
in recurrent pes cavovarus deformities. There was also an increased
incidence of degenerative osteoarthritis of the ankle as a consequence
of the deformity and the loss of subtalar joint motion. These surgeons
were of the opinion that triple arthrodesis should be limited to
patients with severe, rigid deformities. Saltzman et al. (308)
reported similar results in 67 feet in 57 patients, including 6 feet in
patients with Charcot-Marie-Tooth disease, at 25 and 44 years of mean
follow-up. However, 95% of the patients were satisfied with the
clinical results.
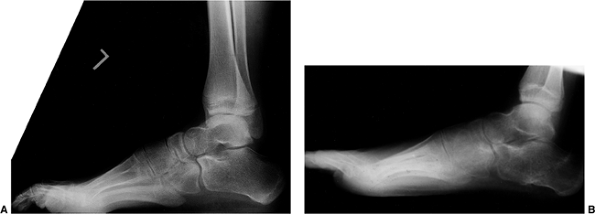 |
|
Figure 17.10 A:
Moderate cavovarus deformity of the left foot in a 14-year-old boy with Charcot-Marie-Tooth disease. His condition was managed with a closing-wedge valgus osteotomy at the calcaneus, an opening-wedge, plantar-based osteotomy of the medial cuneiform, and soft tissue balancing. B: Postoperatively, the cavovarus deformity has been improved. He is a brace-free ambulator because of restoration of muscle balance. |
surfaces of the talocalcaneal, talonavicular, and calcaneal cuboids
joints are removed, along with appropriate-sized wedges to correct the
various components of the hindfoot and midfoot deformities (Fig. 17.11).
In patients who have marked equinus of the midfoot and forefoot in
relation to a relatively well-positioned hindfoot, the Lambrinudi
triple arthrodesis may be performed (309). Once
an arthrodesis has been performed to straighten the foot, tendon
transfers to balance muscle power are of great importance.
have undergone a triple arthrodesis may be corrected by proximal and
distal interphalangeal fusion or flexor-to-extensor tendon transfer.
The great toe may require an interphalangeal joint fusion and transfer
of the extensor hallucis longus from the proximal phalanx to the neck
of the first metatarsal (Jones procedure). The latter then serves as a
foot dorsiflexor.
Occasionally, hips may be dislocatable at birth, although the
neuropathy does not become apparent for several years. It is more
likely to occur in HMSN Type I than in HMSN Type II because of the more
severe neurologic involvement in the former. Walker et al. (311)
proposed that the slight muscle weakness about the hip in growing
children with HMSN may be sufficient to distort growth and development,
leading to dysplasia. Usually, hip dysplasia is diagnosed between the ages of 5 and 15 years following mild discomfort (310,311,312,313). However, dysplasia may be present in asymptomatic patients (Fig. 17.12).
Annual anteroposterior radiographs of the pelvis have been recommended
to allow early diagnosis and treatment. Typical radiographic findings
include acetabular dysplasia, coxa valga, and subluxation. The
treatment of HMSN hip dysplasia includes soft tissue releases to
correct contractures and restore muscle balance, and pelvic or proximal
femoral varus derotation osteotomies, or both, to stabilize and
adequately realign the hip (310,312,313,314,315).
The type of pelvic osteotomy is determined by the patient’s age and the
severity of the dysplasia. Rotational osteotomies (Salter, Steel) are
useful in many children with mild dysplasia, whereas periacetabular
osteotomies are useful in adolescents and young adults (315), and the Chiari osteotomy (314) is used when there is severe dysplasia.
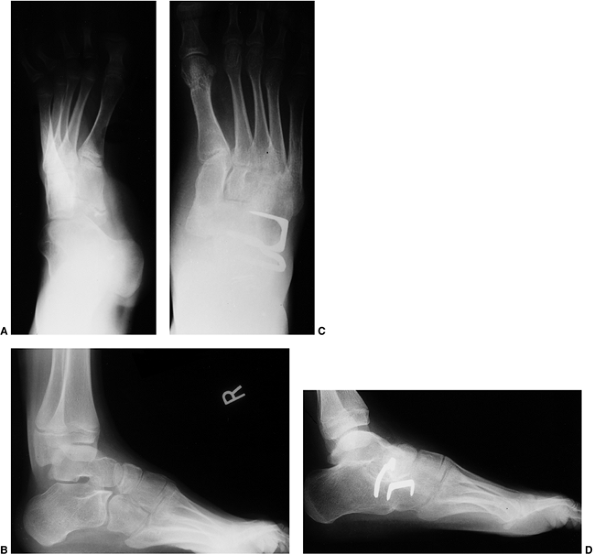 |
|
Figure 17.11 A:
Anteroposterior radiograph of severe cavovarus deformity of the right foot in a 14-year-old boy with Charcot-Marie-Tooth disease, in standing posture. B: Lateral radiograph demonstrates a varus hindfoot and midfoot, and a plantar flexed first metatarsal. C: Postoperative anteroposterior radiograph, taken in standing posture, following a Ryerson triple arthrodesis, soft tissue balancing, and correction of his claw toe deformities. D: Lateral radiograph showing markedly improved alignment. |
These children were usually ambulatory, with age of onset of spinal
deformity of approximately 10 years. A study by Walker et al. (317)
found a 37% incidence of scoliosis or kyphoscoliosis in children with
HMSN. The incidence increases to 50% in those who were skeletally
mature. Spinal deformity is more common in girls and in HMSN Type I.
Curve progression requiring orthoses or surgery is uncommon. The curve
patterns and management are similar to those in idiopathic adolescent
scoliosis, except for an increased incidence of kyphosis. As a
consequence, orthotic management can be effective in arresting
progression of the deformity. If progression reaches 45 to 50 degrees,
a posterior spinal fusion and segmental spinal instrumentation can
effectively stabilize and partially correct the deformity (316). Intraoperative spinal cord monitoring with somatosensory cortical-evoked potentials may show no signal transmission (318).
This is because of the demyelinization of the peripheral nerves and
perhaps the degeneration of the dorsal root ganglion and dorsal column
of the spinal cord. A wake-up test may need to be performed.
The involvement tends to be milder, however, and does not appear until
a later age. Intrinsic muscle weakness with decreased stability is a
relatively common finding. In a study of 68 patients with
Charcot-Marie-Tooth disease, the mean age at onset of symptoms in the
hands and upper extremities was 19 years. Intrinsic muscle function was
initially impaired, and patients became aware of motor weakness and a
lack of dexterity. Sensory changes such as numbness are usually present
concomitantly. Physical and occupational therapy may be helpful. In
some patients, operative intervention, such as transfer of the flexor
digitorum sublimis to restore opposition, nerve compression releases,
soft tissue contracture releases, and joint arthrodeses, may be
effective in improving function. Preoperative EMG has been shown to aid
in selecting optimal forearm muscles for tendon transfers to the hand (321).
infection, with localization in the anterior horn cells of the spinal
cord and certain brain stem motor nuclei. It is caused by one of three
poliomyelitis viruses known as Brunhilde (type 1),
Lansing (type 2), and Leon
(type 3). Humans are the natural host for poliomyelitis virus,
transmitting the disease by the oropharyngeal route. The poliomyelitis
viruses have varying virulence. Most poliomyelitis virus infections
have an abortive course, with only mild gastrointestinal symptoms.
Fewer than 1% of infections develop into the paralytic form of the
disease. The development of prophylactic vaccines has greatly reduced
the incidence of polio, although the disease remains a major health
problem in developing countries. Fewer than 10 cases occur in the
United States annually, and these most commonly result from
administering the active oral polio vaccine (322,323).
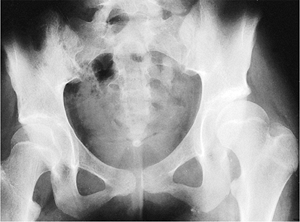 |
|
Figure 17.12
Anteroposterior pelvic radiograph of a 15-year-old girl with Charcot-Marie-Tooth disease. Asymptomatic acetabular dysplasia of the left hip is visible. The medial joint is slightly widened. The Shenton line is disrupted, and the center-edge angle is 16 degrees. This condition was first observed 6 years earlier and did not progress. |
oropharyngeal route and multiplies in the gastrointestinal tract lymph
nodes before spreading to the CNS by the hematogenous route. The
incubation period ranges from 6 to 20 days. Motor neurons in the
anterior horn cells of the spinal cord and brain stem are acutely
attacked. In the spinal cord, the lumbar and cervical regions are
particularly involved. The medulla, cerebellum, and midbrain may also
be involved. Except for the motor areas, the white matter of the spinal
cord and the cerebral cortex are uninvolved.
by viral multiplication and toxic by-products of the virus, or
indirectly from ischemia, edema, and hemorrhage in the glial tissues
surrounding the anterior horn cells. In addition to acute inflammatory
cellular reaction, edema with perivascular mononuclear cuffing occurs.
necrotic ganglion cells are surrounded and partially dissolved by
macrophages and neutrophils. After 4 months, the spinal cord is left
with residual areas of gliosis and lymphocytic cell collections
occupying the area of the destroyed motor cells. Evidence of continuous
disease activity has been found in spinal cord segments examined two
decades after the onset of the disease. Histopathologic sections
demonstrate a loss or atrophy of motor neurons, severe reaction
gliosis, and mild-to-moderate perivascular interparenchymal
inflammation, with sparing of corticospinal tracts. The skeletal muscle
demonstrates gross atrophy and histologic tests show that this lost
muscle has been replaced with fat and connective tissue. The percentage
of motor units destroyed in an individual muscle varies markedly, and
the resultant clinical weakness is proportionate to the number of lost
motor units. Sharrard reported that clinically detectable weakness is
present only when more than 60% of the motor nerve cells supplying the
muscle have been destroyed (324). The muscles
involved may range from those of just one extremity to those of all
four extremities, the trunk, and the bulbar musculature.
are the ones most frequently involved. However, involvement occurs
twice as frequently in the lower extremity as in the upper extremity
muscles. Sharrard (325) combined clinical and
histologic studies that demonstrated that muscles with short motor
nerve cell columns are often severely paralyzed, whereas those with
long motor cell columns are more frequently left paretic or weak. The
quadriceps, tibialis anterior, medial hamstrings, and hip flexors are
the lumbar innervated muscles most frequently involved. The deltoid,
triceps, and pectoralis major are most frequently affected in the upper
extremities. The sacral nerve roots are usually spared, resulting in
the characteristic preservation of the intrinsic muscles of the foot (326).
function of the anterior horn cells that have been damaged but not
destroyed. Clinical recovery begins during the 1st month after the
acute illness and is nearly complete by the 6th month, although there
is limited potential for additional recovery through the second year.
Sharrard has stated that the mean final grade of a muscle is two grades
above its assessment at 1 month and one grade above it at 6 months (324).
stage of the disease process. The stages are designated as acute,
convalescent, or chronic. Because the acute and convalescent stages are
rarely encountered in this country, orthopaedic management is usually
confined to the chronic stage. Every year, most pediatric orthopaedic
programs see several children with poliomyelitis in the chronic stage.
These children have usually been adopted from nonindustrialized nations
or from parents who have immigrated from such countries.
malaise to generalized encephalomyelitis with widespread paralysis.
Diagnosis is based on clinical findings, because there are no
diagnostic laboratory tests. This phase generally lasts 7 to 10 days.
The return to normal temperature for 48 hours and the absence of
progressive muscle involvement indicates the end of the acute phase.
This phase is usually managed by pediatricians because there may be
medical problems, especially respiratory, that may be life-threatening.
signs of acute poliomyelitis. Meningismus is reflected in the
characteristic flexor posturing of the upper and lower extremities. The
muscles involved are tender, even to gentle palpation. Clinical
examination can be difficult because of pain during the acute stage.
prevention of deformity and ensuring comfort. This approach consists of
physical therapy with gentle, passive range- of-motion exercises and
splinting. Muscle spasms, which can lead to shortening and
contractures, may respond to the application of warm, moist heat. This
can relieve muscle sensitivity and discomfort. Sharrard (324) emphasized that rapid loss of elasticity, coupled with shortening of tendons, fascia, and ligaments, leads to contractures.
after the temperature returns to normal and progression of the
paralytic disease ceases. The phase continues for 2 years, during which
spontaneous improvement of muscle power occurs. The assessment of the
rate of recovery in poliomyelitis is made by serial examination of the
muscle strength. Muscle assessment should be performed once every month
for 6 months and then at 3-month intervals during the remainder of the
convalescent stage.
strength at 3 months after the acute phase should be considered to be
permanently paralyzed. Muscles showing evidence of more than 80% return
of strength require no specific therapy. Muscles that fall between
these two parameters retain the potential for useful function, and
therapy should be directed toward recreating hypertrophy of the
remaining muscle fibers.
prevent contractures and deformity, restoration and maintenance of
normal range of motion of the joints, and help for individual muscles
to achieve maximum possible recovery. Physical therapy and orthotics
are the main treatment modalities. Physical therapy is directed toward
having individual muscles assume maximum capability within their
pattern of normal motor activity and not permitting adaptive or
substitute patterns of associated muscles to persist. Hydrotherapy can
also be helpful in achieving these goals. Orthoses, both ambulatory and
nighttime, are necessary for supporting the extremity during this phase.
and it is during this stage that the orthopaedist assumes
responsibility for the long-term management resulting from muscle
imbalance (327).
achieve maximal functional capacity. This is accomplished by restoring
muscle balance, preventing or correcting soft tissue contractures,
correcting osseous deformities, and directing allied personnel, such as
physical therapists, occupational therapists, and orthotists. Using
this approach, Arora and Tandon (328) have shown that ambulation can be restored in patients who could only crawl earlier (328).
Therefore, each patient requires a careful evaluation to determine what
procedures may be effective in restoring ambulation, if possible, and
maximizing function.
contribute to soft tissue contractures and fixed deformities in
poliomyelitis. Contractures result from the increased mechanical
advantage of the stronger muscles that continue the attenuation of
their weaker antagonists. The greater the disparity in muscle balance,
the sooner a contracture may develop.
deformity, except in cases where it is allowed to persist over a period
of years in a growing child. Static instability can be controlled
readily and indefinitely by orthoses. Dynamic instability of a joint
readily produces a fixed deformity, and orthotic control is difficult.
Deformities are initially confined to soft tissues, but later, bone
growth and joint alignment may also be affected.
osseous growth potential of young children makes them more vulnerable
to secondary osseous deformities. The worst deformities occur in young
children and those with severe muscle imbalance. Release of soft tissue
contractures and appropriate tendon transfers performed in a young
child are crucial for preventing structural changes.
instability effectively halts progression of paralytic deformity.
Tendon transfers are performed when dynamic muscle imbalance is
sufficient to produce deformity, and when orthotic protection is
required. Transfers should be delayed until the paralyzed muscle has
been given adequate postural treatment to ensure that it has regained
maximum strength and that the proposed tendon transfer is really
required. The objectives of tendon transfer are to provide active motor
power to replace function of a paralyzed muscle or muscles, to
eliminate the deformity caused by a muscle when its antagonist is
paralyzed, and to produce stability through better muscle balance.
fair before transfer, and must have adequate strength to actively
perform the desired function. On an average, one grade of motor power
is lost after muscle transfer. The length and range of motion of the
transferred muscle and that of the muscle being replaced must be
similar. Loss of original function resulting from tendon transfers must
be balanced against potential gains. Free passive range of motion is
essential in the absence of deformity at the joint to be moved by the
tendon transfer. A transfer as an adjunct to bony stabilization cannot
be expected to overcome a fixed deformity. The smooth gliding channel
for the tendon transfer is essential. Atraumatic handling of the muscle
tissue can prevent injury to its neurovascular supply and prevent
adhesions. The tendon should be rooted in a straight line between its
origin and new insertion. Attachment of the tendon transfer should be
under sufficient tension to correspond to normal physiologic conditions
and should allow the transferred muscle to achieve a maximum range of
contraction.
joints, and thereby impair the alignment of the extremities, mostly the
lower extremities, and limit their ability to function.
Osteotomies
can be helpful in restoring alignment and improving function. Because
of possible recurrence during subsequent growth, these procedures are
usually postponed, if possible, until late childhood or early
adolescence.
the foot where a subtalar, triple, or pantalar arthrodesis may be
useful in stabilization and realignment.
These guidelines include restoring ambulation, correcting the factors
that cause deformities with growth, correcting factors that reduce
dependency on orthoses, correcting upper extremity problems, and
treating spinal deformities. Understandably, these guidelines allow the
child or adolescent to achieve the maximum possible functional level.
The specific methods of achieving each guideline are multiple,
sometimes complex, and based on careful evaluation of the patient.
Because children with previous poliomyelitis are infrequently
encountered, specific details on the various procedures are not
presented. Such information can be obtained from the references in the
various sections.
each child on the basis of a thorough musculoskeletal examination—in
particular, range of motion of the joints, existing deformities, and
manual testing of the individual muscles of the extremities and trunk.
The latter should be individually recorded on a worksheet that can be
available for future reference. It is important to remember that a
muscle normally loses one grade of power when transferred. To be
functionally useful, a muscle grade of at least 4 is necessary,
although a grade 3 muscle, when transferred, may be an effective
tenodesis in preventing deformity by balancing an opposing muscle.
be less severe than that of the lower extremities. A stable upper
extremity, especially the shoulder, is necessary for supporting body
weight when using a walker or crutches. It is also necessary for
transfers or for shifting the trunk if the patient is wheelchair bound.
A functional elbow, wrist, and hand are necessary for optimum
independent functioning.
activities. Satisfactory levels of functioning of the hand, forearm,
and elbow are a prerequisite for any reconstructive surgery on the
shoulder. The major problems affecting the shoulder are paralysis of
the deltoid, pectoralis major, subscapularis, supraspinatus, and
infraspinatus muscles. Rarely are all these muscles involved because
they are innervated at different levels. Tendon transfers can
occasionally be effective in restoring shoulder stability. When there
is extensive weakness, shoulder arthrodesis may be helpful. Arthrodesis
may also be indicated where there is a painful subluxation or
dislocation. A strong trapezius serratus anterior muscle is necessary
for allowing improved functioning after fusion. El-Gammal et al. (330)
recently demonstrated that, after a shoulder fusion and a
free-functioning gracilis muscle transplantation, there was improvement
in upper extremity function in children and adolescents with a flail
shoulder and elbow caused by poliomyelitis (330).
The muscle was reinnervated by the spinal accessory or phrenic nerve.
All transplanted muscles gained at least grade 3 power. The best
results occurred with the reinnervation by the spinal accessory nerve.
flexion. When the biceps and brachialis are paralyzed, a tendon
transfer may be helpful in restoring useful elbow flexion. Possible
procedures include a Steindler flexorplasty, which transfers the origin
of the wrist flexors to the anterior aspect of the distal humerus (331).
The best functional results occur in patients whose elbow flexors are
only partially paralyzed and whose fingers and wrist flexors are
normal. Transfer of the sternal head of the pectoralis major may also
be considered. Other possible procedures include transfer of the
sternocleidomastoid and latissimus dorsi, and anterior transfer of the
triceps brachii. Paralysis of the triceps brachii muscle may occur in
poliomyelitis, but it seldom interferes with elbow function because
gravity passively extends the elbow. The triceps brachii muscles need
to function, however, for activities in which the body weight is
shifted to the hands (such as in transferring from bed to wheelchair)
or in crutch walking.
functional disabilities in children and adolescents with poliomyelitis.
Pronation contractures are the most common disability. Functioning can
be improved with release of the pronator teres and transfer of the
flexor carpi ulnaris muscle.
functioning of the hand can be considered in selected cases. The number
of possible transfers is large, and each patient requires a careful
evaluation in order to ensure maximum functional improvement. Carpal
tunnel syndrome has also been reported as one of the long-term sequelae
of poliomyelitis (332) and is associated with prolonged use of crutches or a cane.
poliomyelitis. They can have a significant impact on functional
ability, especially ambulation.
neurologic involvement. If the discrepancy is greater than 2 cm, it can
produce a great many disturbances. An appropriately timed contralateral
epiphysiodesis is the usual procedure of choice (see Chapter 28). Greater discrepancy may be treated orthotically. Lengthening is rarely considered as an option. However, D’Souze and Shah (333)
recently demonstrated that circumferential periosteal sleeve resection
of the distal femur and/or distal tibia can produce a transient growth
stimulation that can be helpful in mild discrepancies, usually 2 to 3
cm.
soft tissue contractures, internal or medial femoral torsion, coxa
valga, and hip subluxation and dislocation. Periodic anteroposterior
radiographs of the pelvis are necessary for assessing growth and the
relation between the femoral head and the acetabulum. Functioning can
be improved, and subluxation-dislocation prevented, with appropriate
soft tissue releases, tendon transfers, proximal femoral varus
derotation osteotomy, and pelvic osteotomy (Fig. 17.13) (334).
It is important that the procedures be coordinated in order to provide
as balanced a musculature as possible so that hip stability can be
maintained. Lau et al. (334) reported good or
satisfactory results in 70% of patients with paralytic hip instability
caused by poliomyelitis. The key parameters for successful management
are muscle balance, the femoral neck shaft and ante-version angles, and
the acetabular geometry.
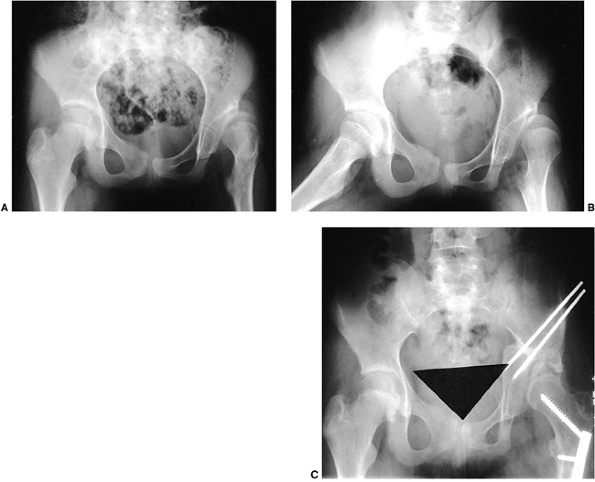 |
|
Figure 17.13 A:
Anteroposterior radiograph of the pelvis of a 13-year-old Korean girl who had poliomyelitis. She has a painful subluxation of her left hip. The acetabulum is dysplastic, the center-edge angle is 6 degrees, and a coxa valga deformity of the proximal femur is present. B: Frog-leg or Lauenstein lateral. C: Two years after a proximal femoral varus derotation osteotomy and Chiari pelvic osteotomy, there is markedly improved alignment of the left hip, and she is asymptomatic. |
valgum, and external rotation of the tibia are the common knee
deformities in poliomyelitis that can produce an adverse effect on
functional ambulation. Hamstring release, distal femoral extension
osteotomy, proximal femoral extension osteotomy, and rotational tibial
osteotomies are common procedures (335,336,337,338,339). One of the most common
soft tissue procedures is that described by Yount, in which the distal
iliotibial band, including the intermuscular septum, is released (86). This may be combined with an Ober release proximally if hip-flexion contractures are also present (87). Shahcheraghi et al. (340)
recently reported that anterior hamstring tendon transfer significantly
improved active knee extension and function in patients with paralysis
of the quadriceps femoris muscle following poliomyelitis.
has discussed possible procedures for correcting the deformities and
improving muscle balance. This is achieved by a combination of
procedures: correction of soft tissue contractures, tendon transfers,
and bone-stabilizing procedures such as calcaneal osteotomy, subtalar
arthrodesis, triple arthrodesis, and pantalar arthrodesis (342,343,344,345,346,347,348).
Recently, the use of the Ilizarov external fixator has been shown to be
helpful in correction of complex foot deformities following
poliomyelitis (349). A careful evaluation of
the patient is required for determining the appropriate procedures.
Arthrodeses produce good long-term results with a low incidence of
ankle-degenerative arthritis, because patients with poliomyelitis place
lower functional demands and stresses on the ankle (342,345,346).
poliomyelitis. The type and severity of the curvature depends on the
extent of paralysis and residual muscle power of the trunk muscles and
pelvic obliquity. The most common curve patterns are the double major
thoracic and lumbar curves, followed by the long paralytic C-shaped
thoracolumbar curve (350). Pelvic obliquity
occurs in approximately 50% of the patients with spinal deformity.
Because of severe rotation, kyphosis in the lumbar spine and lordosis
in the thoracic spine are also common.
vertical torso over a level pelvis. This permits stable sitting and
leaves the hands free for activities. It also helps prevent decubiti
and paralytic hip dislocation. In young children with curves of between
20 and 40 degrees, orthotic management with a TLSO can be tried. It
rarely provides complete stability, but can be effective in slowing the
rate of progression and allowing the child to reach a more suitable age
for surgery. In severe cases in young children, segmental spinal
instrumentation without fusion may be considered. Eberle (351),
however, reported failure of segmental spinal instrumentation in 15 of
16 children with poliomyelitis between the ages of 5 and 12 years.
Therefore, children who undergo instrumentation without fusion should
be treated with TLSO and subsequently undergo a fusion procedure as
soon as possible in order to prevent late complications. For
adolescents with a supple spine and a curve of less than 60 degrees, a
posterior spinal fusion with segmental instrumentation, usually Luque
rod instrumentation, provides stability and a low pseudoarthrosis rate (102,350).
Other segmental systems (e.g., TSRH, Cotrel-Dubousset, and Isola) are
also effective. In severe curves of 60 to 100 degrees, a combined
anterior and posterior spinal fusion is usually necessary (352).
Anterior spinal instrumentation with a Dwyer or Zielke system may be
used in thoracolumbar and lumbar curves. Anterior discectomy and fusion
is preferred for thoracic curves. The posterior spinal fusion and
instrumentation may be performed the same day, or performed 1 or 2
weeks later. Leong et al. (353) and others (352,354)
have demonstrated that combined anterior and posterior spinal fusions
provide excellent correction for postpoliomyelitis spinal deformity,
including the associated pelvic obliquity (Fig. 17.14).
Rarely is preoperative traction, or traction between staged anterior
and posterior procedures, necessary for additional correction. Fusion
to the pelvis or sacrum is usually necessary in patients with severe
pelvic obliquity (355,356).
adults, and is a sequela to poliomyelitis. Reactivation of the
poliomyelitis virus has been mistaken for amyotrophic lateral
sclerosis. Postpoliomyelitis syndrome is thought to be an overuse
syndrome (357). Diagnosis is based on five criteria and is essentially a diagnosis of exclusion. The criteria include:
-
A confirmed history of previous poliomyelitis.
-
Partial to fairly complete neurologic and functional recovery.
-
A period of neurologic and functional stability of at least 15 years’ duration.
-
Onset of two or more of the following
health problems since achieving a period of stability: unaccustomed
fatigue, muscle and joint pain or both, new weakness in muscles
previously affected or unaffected, functional loss, intolerance to
cold, and new atrophy. -
No other medical diagnosis to explain the aforementioned health problems.
those with onset later than the age of 10 years, because older children
are more likely to have severe poliomyelitis. Management of these
patients is conservative and consists of muscle strengthening,
decreasing the duration of effort, and orthotics (357). Reconstructive surgery is rarely indicated or necessary.
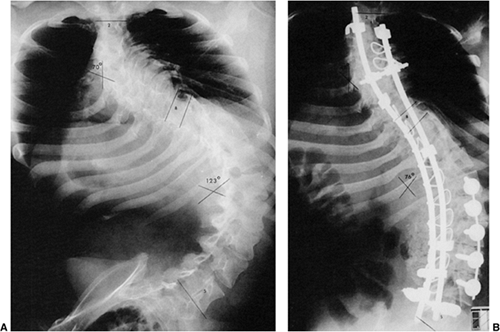 |
|
Figure 17.14 A:
Anteroposterior spinal radiograph, taken in the seated position, of a 17-year-old girl from the Middle East who has a severe paralytic scoliosis. There is a 123-degree left thoracolumbar scoliosis and a 70-degree right thoracic scoliosis. She contracted poliomyelitis at the age of 2 years, which left her with flail lower extremities and essentially normal upper extremities. She is wheelchair-dependent and has pain from rib-pelvis impingement. B: Postoperative radiograph after staged anterior spinal fusion and Zielke instrumentation and posterior spinal fusion using Isola instrumentation from T-3 to the sacrum. Pain relief was complete and sitting balance improved. The left thoracolumbar curve has been reduced to 70 degrees and the right thoracic curve to 47 degrees. |
F, Bresnan MJ. Current concepts review. Orthopaedic management of
childhood neuromuscular disease. Part III: diseases of muscle. J Bone Joint Surg Am 1982;64:1102.
F, Specht L. Current concepts review. The diagnosis and orthopaedic
treatment of inherited muscular diseases of childhood. J Bone Joint Surg Am 1993;75:439.
ME, Davids JR, Mubarak SJ. Single-incision combination biopsy (muscle
and nerve) in the diagnosis of neuromuscular disease in children. J Pediatr Orthop 1994;14:740.
SK, Leung RK, Tierney RC, et al. Mitral valve prolapse syndrome in
children with Duchenne’s progressive muscular dystrophy. Pediatrics 1979;63:116.
MH, Fenichel GM, Griggs RC, et al. Duchenne muscular dystrophy.
Patterns of clinical progression and effects of supportive therapy. Neurology 1989;39:475.
JP, De Groot IJ, Beelen A, et al. Predictive factors of cessation of
ambulation in patients with Duchenne muscular dystrophy. Am J Phys Med Rehabil 2002;81:906.
M, Araki S, Miike T, et al. Localization and characterization of
dystrophin in muscle biopsy specimens for Duchenne muscular dystrophy
and various neuromuscular disorders. Muscle Nerve 1989;12:1009.
AH, Hoffman EP, Snyder JR, et al. Exploring the molecular basis for
variability among patients with Becker muscular dystrophy. Dystrophin
gene and protein studies. Am J Hum Genet 1991;49:54.
EP, Fischbeck KH, Brown RH, et al. Characterization of dystrophin in
muscle-biopsy specimens from patients with Duchenne’s or Becker’s
muscular dystrophy. N Engl J Med 1988;318:1363.
LVB, Johnson MA, Bushby KMD, et al. Functional significance of
dystrophin positive fibres in Duchenne muscular dystrophy. Arch Dis Child 1993;68:632.
SM Jr, Lazaro RP, Lava NS, et al. Familial X-linked myalgia and cramps.
A nonprogressive myopathy associated with a deletion in the dystrophin
gene. Neurology 1989;39:1277.
TA, Morgan JE, Coulton GR, et al. Conversion of mdx myofibres from
dystrophin-negative to -positive by injection of normal myoblasts. Nature 1989;337:176.
E, Pavlath GK, Lanctot AM, et al. Normal dystrophin transcripts
detected in Duchenne muscular dystrophy patients after myoblast
transplantation. Nature 1992;356:435.
PK, Goodwin TG, Fang Q, et al. Feasibility, safety and efficacy of
myoblast transfer therapy on Duchenne muscular dystrophy boys. Cell Transplant 1992;l:235.
JR, Moxley RT, Griggs RC, et al. Randomized double-blind six-month
trial of prednisone in Duchenne’s muscular dystrophy. N Engl J Med 1989;320:1592.
AM, Schierbecker J, Renna R, et al. High dose weekly oral prednisone
improves strength in boys with Duchenne muscular dystrophy. Neuromuscul Disord 2002;12:917.
RC, Moxley RT, Mendell JR, et al. Duchenne dystrophy: randomized,
controlled trial of prednisone (18 months) and azathioprine (12
months). Neurology 1993;43:520.
JZ, Dubowitz V, Hyde SA, et al. Prolongation of walking in Duchenne
muscular dystrophy with lightweight orthoses; review of 57 cases. Dev Med Child Neurol 1985;27:149.
PJ, Wagner MB, Karlinchak B, et al. Evaluation of a program for
long-term treatment of Duchenne muscular dystrophy. Experience at the
University Hospitals of Cleveland. J Bone Joint Surg Am 1966;78:1844.
SA, Filytrup I, Glent S, et al. A randomized comparative study of two
methods for controlling tendoAchilles contracture in Duchenne muscular
dystrophy. Neuromuscul Disord 2000; 10:257.
SE, Green NE, Cole RJ, et al. Prolongation of ambulation in children
with Duchenne muscular dystrophy by subcutaneous lower limb tenotomy. J Pediatr Orthop 1993;13:331.
EB, Fernandez-Bermejo E, Heckmatt JZ, et al. Prevention of rapidly
progressive scoliosis in Duchenne muscular dystrophy by prolongation of
walking with orthoses. J Child Neurol 1988;3:269.
GM, Hsu JD, Hoffer MM, et al. Posterior tibial tendon transfer. A
review of the literature and analysis of 74 procedures. J Pediatr Orthop 1982;2:33.
AP, Craig C. Scoliosis management in Duchenne muscular dystrophy.
Prospective study of modified Jewett hyperextension brace. Arch Phys Med Rehabil 1987;68:302.
O, Lonstein JE, Winter RB, et al. Management of neuromuscular spinal
deformities with Luque segmental instrumentation. J Bone Joint Surg Am 1989;71:548.
D, Arlet V, Stricker U, et al. Modification of the original Luque
technique in the treatment of Duchenne’s neuromuscular scoliosis. J Pediatr Orthop 1997;17:743.
KH, O’Brien MF, Lenke LG, et al. Posterior spinal fusion supplemented
with only allograft bone in paralytic scoliosis. Does it work? Spine 1994;19:2658.
HJ, Thomas CH, Thompson AG. Spinal instrumentation for Duchenne’s
muscular dystrophy: experience of hypotensive anesthesia to minimize
blood loss. J Pediatr Orthop 1997;17:750.
KH, Baldus C, Iffrig TM, et al. Process measures and patient/parent
evaluation of surgical management of spinal deformities in patients
with progressive flaccid neuromuscular scoliosis (Duchenne’s muscular
dystrophy and spinal muscular atrophy). Spine 1999;24:1300.
G, Haddad F, Bull TM, et al. The treatment of scoliosis in muscular
dystrophy using modified Luque and Harrington-Luque instrumentation. J Bone Joint Surg Br 2001;83:22.
M, Asher MA, Hardacker JW. The safety and efficacy of Isola-Galveston
instrumentation and arthrodesis in the treatment of neuromuscular
spinal deformities. J Bone Joint Surg 2000;82:524.
KD, Wirtz DC, Siebert CH, et al. Spinal stabilization in Duchenne
muscular dystrophy: principle of treatment and record of 31 operative
treated cases. J Pediatr Orthop B 2001;10:18.
DK, Mehdian SH, McConnell JR, et al. Pelvic or lumbar fixation for the
surgical management of scoliosis in Duchenne muscular dystrophy. Spine 2002;27:2072.
JG, Bohn D, Edmonds JF, et al. Evaluation of pulmonary function in
muscular dystrophy patients requiring spinal surgery. Crit Care Med 1982;10:645.
MA, Johnson EW, Petty J, et al. Mechanical ventilation of patients with
late stage Duchenne muscular dystrophy. Management in the home. Arch Phys Med Rehabil 1979;60:289.
JR, O’Brien J, Krolenberg R, et al. Muscular dystrophy. Management of
end stage respiratory failure in Duchenne muscular dystrophy. Muscle Nerve 1987;10:177.
FJ. Night ventilation by body respirators for patients in chronic
respiratory failure due to late stage Duchenne muscular dystrophy. Arch Phys Med Rehabil 1981;62:270.
CA, Gilgoff I, Baydur A, et al. Gated radionuclide ventriculography in
the evolution of cardiac function in Duchenne’s muscular dystrophy. Chest 1988;94:1245.
YH, Lonstein JE, Winter RB, et al. Spinal deformities in patients with
muscular dystrophy other than Duchenne. A review of 11 patients having
surgical treatment. Spine 1985;10:614.
L, Granata C, Dominici P, et al. Emery-Dreifuss muscular dystrophy.
Report of five cases in a family and review of the literature. Muscle Nerve 1986;9:481.
GG, Thomas NST, Stayton CL, et al. Assignment of Emery-Dreifuss
muscular dystrophy to the distal region of Xq28. The results of a
collaborative study. Am J Hum Genet 1991;48:468.
N, Williams H, Elsas LJ, et al. Localization of the gene for
Emery-Dreifuss muscular dystrophy to the distal long arm of the
X-chromosome. J Med Genet 1986;23:596.
G, Di Barletta MR, Varnous S, et al. Mutations in the gene encoding
lamin A/C cause autosomal dominant Emery Dreifuss muscular dystrophy. Nat Genet 1999;21:285.
LC, Jackson JA, Elsas LJ. Emery-Dreifuss humeroperoneal muscular
dystrophy. An X-linked myopathy with unusual contractures and
bradycardia. Ann Neurol 1981;10:230.
E, Hoffman EP, Kaido M, et al. The frequency of patients with
dystrophin abnormalities in a limb-girdle patient population. Neurology 1991;41:1491.
BR, Bresnan MJ, Shapiro F, et al. Fascioscapulohumeral dystrophy
presenting in infancy with facial diplegia and sensorineural deafness. Ann Neurol 1985;17:513.
F, Specht L, Korf BR. Locomotor problems in infantile
facioscapulohumeral muscular dystrophy. Retrospective study of 9
patients. Acta Orthop Scand 1991;62:367.
WH, Siegal IM. Scapulothoracic arthrodesis in facioscapulohumeral
muscular dystrophy. Review of seventeen procedures with three to
twenty-one year follow-up. J Bone Joint Surg Am 1993;75:372.
E, Fardeau M, Lytle JO, et al. Scapulothoracic arthrodesis for patients
who have fascioscapulohumeral muscular dystrophy. J Bone Joint Surg Am 1990;72:78.
D, Coerwinkel-Driessen M, van Dalen I, et al. Definition of
subchromosomal intervals around the myotonic dystrophy gene region at
19q. Genomics 1989;4:384.
MC, Pericak-Vance MA, Yamaoka L, et al. Presymptomatic and prenatal
diagnosis in myotonic dystrophy by genetic linkage studies. Neurology 1990;40:671.
C, Schmidt-Rose T, Fontaine B, et al. ClC-1 chloride channel mutations
in myotonia congenita: variable penetrance of mutations shifting the
voltage dependence. Hum Mol Genet 1998;7:1753.
E, Gervais A, Eymard B, et al. Novel muscle chloride channel (ClCN1)
mutations in myotonia congenita with various modes of inheritance
including incomplete dominance and penetrance. Neurology 1998;50:1176.
K. Congenital non-progressive myopathy, associated with scoliosis:
clinical, histological, histochemical and electron microscopic studies
of seven cases. Nippon Seikeigeka Gakkai Zasshi 1980;54:381.
C, Nonaka I. Nemaline myopathy: comparative muscle histochemistry in
the severe neonatal, moderate congenital and adult-onset forms. Pediatr Neurol 1989;5:25.
C, Clarke A, Samson F, et al. The myotubular myopathies: differential
diagnosis of the X-linked recessive, autosomal dominant and autosomal
recessive forms and present state of DNA studies. J Med Genet 1995;32:673.
J, Hu LJ, Kretz C, et al. A gene mutated in X-linked myotubular
myopathy defines a new putative tyrosine phosphatase family conserved
in yeast. Nat Genet 1996;13:175.
NPC, Lake BD, McMeniman P. Congenital fibre type disproportion
myopathy. A histological diagnosis with an uncertain clinical outlook. Arch Dis Child 1979;54:735.
F, Mastaglia FL, Serena M, et al. Mitochondrial myopathies. A
clinico-pathological study of cases with and without extra-ocular
muscle involvement. Aust NZ J Med 1986;16:185.
Y, Osawa M, Suzuki H. Congenital progressive muscular dystrophy of the
Fukuyama type—clinical, genetic and pathological considerations. Brain Dev 1981;3:1.
D, Leclerc A, Faure S, et al. Localization of merosin negative
congenital muscular dystrophy to chromosome 6q2 by homozygosity
mapping. Hum Mol Genet 1994;3:1657.
J, Sewry C, Pennock J, et al. Clinical phenotype in congenital muscular
dystrophy: correlation with expression of merosin in skeletal muscle. Neuromuscul Disord 1995;5:301.
R, Kahn R, Hughes S, et al. Congenital muscular dystrophy. The
importance of early diagnosis and orthopaedic management in the
long-term prognosis. J Bone Joint Surg Br 1979; 61:13.
F, Bresnan MJ. Current concepts review. Orthopaedic management of
childhood neuromuscular disease. Part I. Spinal muscular atrophy. J Bone Joint Surg Am 1982;64:785.
F, Specht L. Current concepts review. The diagnosis and orthopaedic
treatment of childhood spinal muscular atrophy, peripheral neuropathy,
Friedreich ataxia and arthrogryposis. J Bone Joint Surg Am 1993;75:1699.
PE, Parsons DW, Simard LR, et al. Identification of proximal spinal
muscular atrophy carriers and patients by analysis of SMNt and SMNc
gene copy number. AM J Hum Genet 1997;60:1411.
BS, lannaccone ST, Buncher CR, et al. Spinal muscular atrophy. New
thoughts on the pathogenesis and classification schema. J Child Neurol 1992;7:347.
LM, Lehner T, Castilla LH, et al. Genetic mapping of chronic
childhood-onset spinal muscular atrophy to chromosome 5q 11.2. Nature 1990;334:540.
C, Cornelio F, Bonfiglioli S, et al. Promotion of ambulation of
patients with spinal muscular atrophy by early fitting of
knee-ankle-foot orthoses. Dev Med Child Neurol 1987;29:221.
RN, MacEwen GD. Spinal deformity associated with heritable neurological
conditions. Spinal muscular atrophy. Friedreich’s ataxia, familial
dysautonomia, and Charcot-Marie-Tooth disease. J Bone Joint Surg Am 1976;58:13.
GN, Hallett R, Hare N, et al. The outcome of scoliosis surgery in the
severely physically handicapped child. An objective and subjective
assessment. Spine 1997;22:44.
LC, Scoles PV, Thompson GH, et al. Submuscular Isola rods with or
without limited apical fusion in the management of severe spinal
deformities in young children: preliminary report. Spine 2001;26:2044.
F, Bresnan MJ. Current concepts review. Orthopaedic management of
childhood neuromuscular disease. Part II: peripheral neuropathies,
Friedreich’s ataxia and arthrogryposis multiplex congenita. J Bone Joint Surg Am 1982;64:949.
G, Barbeau A, Breton G, et al. Clinical description and roentgenologic
evaluation of patients with Friedreich’s ataxia. Can J Neurol Sci 1976;3:279.
AE. Friedreich’s ataxia. A clinical and genetic study of 90 families
with an analysis of early diagnostic criteria and intrafamilial
clustering of clinical features. Brain 1981;104:589.
A, DeMichele G, Caruso G, et al. Genetic data and natural history of
Friedreich’s disease. A study of 80 Italian patients. J Neurol 1990;237:345.
H, Beauchomp M, LaPierre Duhaime M, et al. Pattern of muscle weakness
and its relation to loss of ambulatory function in Friedreich’s ataxia.
J Pediatr Orthop 1987;7:496.
R, Hanauer A, Vincent A, et al. Physical mapping of two loci (D9S5 and
D9S15) tightly linked to Friedreich ataxia locus (FRDA) and
identification of nearby CpG Islands by pulsefield gel electrophoresis.
Genomics 1991;10:915.
V, Montermini L, Motto MD, et al. Friedreich’s ataxia: autosomal
recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996;271:1423.
K, Hermsdorfer J, Deger K, et al. Double-blind crossover study with
levorotatory form of hydroxytryptophan in patients with degenerative
cerebellar diseases. Arch Neurol 1995;52:451.
P, Serratrice G, Laplane D, et al. Levorotatory form of
5-hydroxytryptophan in Friedreich ataxia. Results of a double-blind
drug-placebo cooperative study. Arch Neurol 1995;52:456.
PI, Roa BB, Welcher AA, et al. The gene for the peripheral myelin
protein pmp-22 is a candidate for Charcot-Marie-Tooth disease type IA. Nat Genet 1992;1:159.
V, Nelis E, Van Hut W, et al. The peripheral myelin protein gene pmp-22
is contained within the Charcot-Marie-Tooth disease type IA
duplication. Nat Genet 1992;1:171.
LJ, Bolhuis PA, Zorn I, et al. The peripheral myelin gene PMP-22/GAS-3
is duplicated in Charcot-Marie-Tooth disease type IA. Nat Genet 1992;1:166.
BB, Garcia CA, Suter U, et al. Charcot-Marie-Tooth disease type IA.
Association with a spontaneous point mutation in the PMP22 gene. N Engl J Med 1993;329:96.
ENMC International Workshop on Classification and Diagnostic Guidelines
for Charcot-Marie-Tooth Type 2 and Distal Hereditary Motor Neuropathy,
26-28 September 1997, Naarden, The Netherlands. Neuromuscul Disord 1998;8:426.
Othmane K, Middleton LT, Loprest LJ, et al. Localization of a gene
(CMT2a) for autosomal dominant Charcot-Marie-Tooth disease type 2 to
chromosome 1p and evidence of genetic heterogeneity. Genomics 1993;17:370.
IV, Perepelov AV, Polyakov AV, et al. A new variant of
Charcot-Marie-Tooth disease type 2 is probably the result of a mutation
in the neurofilament-light gene. Am J Hum Genet 2000;67:37.
SM, Fedotov VP, Dadali EL, et al. A new locus for autosomal dominant
Charcot-Marie-Tooth disease type 2 (CMT2F) maps to chromosome 7q11-q21.
Eur J Hum Genet 2001;9:650.
MW, Meyer zuHorste G, Sater U, et al. Therapeutic administration of
progesterone antagonist in a model of Charcot-Marie-Tooth disease. Nat Med 2003;9:1457.
M, Lyttle D. Pathogenesis of Charcot-Marie-Tooth disease. Gait analysis
and electrophysiologic, genetic, histopathologic, and enzyme studies in
a kinship. Clin Orthop 1984;184:223.
FC, Schottstaedt ER, Larsen LJ. Plantar dissection. An operation to
release the soft tissues in recurrent or recalcitrant talipes
equinovarus. J Bone Joint Surg Am 1960;42:151.
DC, Hsu JD. Triple arthrodesis in the treatment of fixed cavovarus
deformity in adolescent patients with Charcot-Marie-Tooth disease. Foot Ankle 1992;13:1.
CL, Fehrle MJ, Cooper RR, et al. Triple arthrodesis: twenty-five and
forty-four-year average follow-up of the same patients. J Bone Joint Surg Am 1999;81:1391.
SJ, Marks HG, Bowen JR, et al. Hip dysplasia associated with
Charcot-Marie-Tooth disease in the older child and adolescent. J Pediatr Orthop 1985;5:511.
WR, Lester EL, Watson P. Dynamics of hip joint remodeling after Chiari
osteotomy. 10 patients with neuromuscular disease followed for 8 years.
Acta Orthop Scand 1997;68:128.
YH, Lonstein JE, Winter RB, et al. Spinal deformities in patients with
Charcot-Marie-Tooth disease. A review of 12 patients. Clin Orthop 1986;202:219.
GA, Gordon MJ, Neville HE, et al. Restoring hand function in patients
with severe polyneuropathy: the role of electromyography before tendon
transfer surgery. J Hand Surg Am 1999;24:732.
PM, Sutter RW, Cochi SL, et al. Epidemiology of poliomyelitis in the
United States one decade after the last reported case of indigenous
wild virus-associated disease. Clin Infect Dis 1992;14:568.
HG. Management of common third world orthopaedic problems. Paralytic
poliomyelitis, tuberculosis of bones and joints, Hansen’s disease
(leprosy), and chronic osteomyelitis. Instr Course Lect 1992;41:471.
TA, El-Sayed A, Kotb MM. Shoulder fusion and free-functioning gracilis
transplantation in patients with elbow and shoulder paralysis caused by
poliomyelitis. Microsurgery 2002;22:199.
GW, Dingeman RD, Gausewitz SH. The results of tenodesis of the tendo
Achilles to the fibular for paralytic pes calcaneus. J Bone Joint Surg Am 1988;70:320.
JAC, Marti RK, Besselaar PP, et al. The influence of subtalar and
triple arthrodesis on the tibiotalar joint. A long-term follow-up
study. J Bone Joint Surg Br 1997;79:644.
PJ, Edwards JW, Dove J, et al. Post-poliomyelitis paralytic scoliosis.
A review of curve patterns and results of surgical treatment in 118
consecutive patients. Spine 1981;6:573.
CF. Failure of fixation after segmental spinal instrumentation without
arthrodesis in the management of paralytic scoliosis. J Bone Joint Surg Am 1988;70:696.
JP, Yau ACMC, Gertzbien S, et al. Combined staged anterior and
posterior correction of the spine in scoliosis following postmyelitis. Clin Orthop 1975;110:81.
YL, Lonstein JE, Winter RB, et al. Luque-Galveston procedure for
correction and stabilization of neuromuscular scoliosis and pelvic
obliquity. A review of 68 patients. J Spinal Disord 1991;4:399.