
malformation of the central nervous system (CNS) with long-term
implications for function and morbidity. Its effect on the child and
the parents is devastating, and significant costs are borne by the
medical community and society at large. In 1989, in the United States
alone, the cost of care
for
affected individuals with myelodysplasia was estimated at 200 million
dollars and is likely higher today because of newer technologies (1).
The eventual orthopaedic care of these patients is strongly influenced
by abnormalities in the neurologic and urologic systems, societal
pressures, education, and the availability of medical resources. The
prognosis for the children who are affected has changed and improved
remarkably over the last three decades because of neurosurgical and
urologic advances.
treatment be withheld in patients with thoracic-level deformities of
hydrocephalus, kyphosis, or other congenital anomalies (2).
At that time, the prognosis for these patients was poor, and most
children born with myelomeningocele died in early infancy because of
CNS infections or hydrocephalus. Urologic problems were expected even
in those patients who did survive neurologic complications during the
first years of life. Many of these children could expect their survival
to be threatened by lower urinary tract dysfunction, which would
predictably lead to infection, reflux and upper urinary tract
dysfunction, and consequent secondary renal failure. In 1972,
neurologic renal dysfunction was the number one cause of death in
children older than 2 years with myelomeningocele.
because of early antibiotic therapy, myelomeningocele sac closure, and
insertion of a ventriculoperitoneal (VP) shunt, to prevent uncontrolled
hydrocephalus. Neurosurgical treatment is also needed in order to
prevent further motor disability by detecting and treating syrinx,
Arnold-Chiari malformation, and tethered spinal cord. Today the
condition takes a new and different course, and the whole medical team
is cognizant of the importance of detecting uncorrected or progressive
CNS abnormalities such as retethering of the spinal cord, shunt
malfunction and hydrocephalus, and development of syrinx.
urologic management of children with myelodysplasia; the grim prognosis
of eventual renal failure and death is no longer a reality.
Prophylactic management of bladder dysfunction with the development of
clean, intermittent catheterization and the use of anticholinergic
agents has done a great deal to decrease the rates of secondary
problems of the bladder, and consequent renal dysfunction (3,4).
children have survived the neurosurgical and urologic problems,
orthopaedic care has also evolved over the last 30 years. For instance,
today we are more selective in the use of hip stabilization procedures;
we have improved orthotics; we have made significant advances in spinal
surgical techniques and instrumentation; and we have a greater
understanding of the relative results of different treatments for the
myelodysplastic foot. In addition, coordinated improvement of care is
made possible through interdisciplinary treatment programs, with teams
consisting of a neurosurgeon, an orthopaedist, a urologist, a social
worker, physical and occupational therapists, educators, a
pediatrician, and a nurse-specialist. Although “failures” of these
programs do exist for families in lower socioeconomic classes, these
integrated programs have evolved with time and offer improvements
unimagined 30 years ago (5).
bifida and associated neurologic abnormalities from a pathologic
viewpoint. Neural tube defects (NTD) are grouped together under the
generic terms myelodysplasia, spinal dysraphism, and spina bifida aperta. These are not to be confused with spina bifida occulta. In the orthopaedic nomenclature, spina bifida occulta
is a term that refers to a laminar or spinous process defect that is
commonly seen on plain radiographs. In the neurosurgical literature,
the term spina bifida occulta is used to
describe various spinal cord defects (e.g., lipomyelomeningocele, split
cord malformation, etc.) that occur with intact skin. These always need
operative intervention and can be recognized through the presence of
skin anomalies (hypertrichosis, hemangioma, dimple, etc.), or
orthopedic deformities (high arched foot, hammer toes, etc.), and
urologic problems.
within the defect; this lipoma is intimately involved with the sacral
nerves and is covered with skin and soft tissues. These children may
not have hydrocephaly or other CNS abnormalities. Neurologic function,
which is almost normal at birth, may become impaired with growth. This
abnormality is similar to other abnormalities of the spine, including
dermoid sinus, dermoid cyst, and diastematomyelia (split cord).
Neurologic deficit rarely extends above the lumbosacral area; however,
progressive deterioration of the functioning neurologic levels from
tethering due to growth is a major concern in these children.
include meningocele. This is a cyst involving only the meninges but not
any neural elements; these are closed by a neurosurgeon; morbidity is
minimal, and further treatment is not usually needed. A true neural
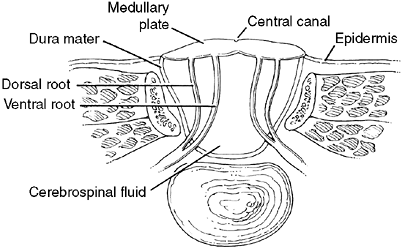
tube defect, or spina bifida, is myelomeningocele. This consists of a
spinal cord that has failed to fuse, thereby resulting in an open
defect with no dura, bone, muscle, or skin covering (Fig. 16.1).
Myelomeningoceles occur mostly in the low thoracic and lumbosacral
regions, and may present rarely in a cervical location. Cervical
dysraphia are usually skin covered and have more normal neurologic
function (6,7). Initial
management includes exploration and detethering by a neurosurgeon.
However, these patients may develop retethering with symptoms including
increased spasticity in the legs and decreased hand function (8). In the lower myelomeningocele the neural elements are abnormal, and
pronounced, peripheral neurologic deficits are common. The roof of the
myelomeningocele is composed of the spinal cord. The central canal of
the cord opens posteriorly through the dorsal columns, thereby allowing
the cerebral spinal fluid to flow to the outside from the fourth
ventricle. Unlike those with spina bifida occulta lesions, most
children who are born with a myelomeningocele have multiple brain
anomalies including hydrocephalus and Arnold-Chiari malformations.
 |
|
Figure 16.1 Cross section of myelomeningocele. The abnormal cord is part of the sac and is elevated out of the canal.
|
of the defect from further trauma and early closure of the sac within
the first 24 to 72 hours. Careful closure of the defect in layers will
prevent inclusion of epithelial elements and thereby avoid the later
occurrence of epidermal inclusion cysts.
myelomeningocele sac, growth may cause tethering to recur later in
life. Other intraspinal abnormalities such as diastematomyelia (9)
or epidermal cysts may also increase tethering. If any dermal elements
are left attached to the spinal cord, dermoid cysts may develop and
eventually cause a decrease of function in the lumbosacral roots by
tethering or direct pressure on the nerve elements.
Tethering is more often detected in patients with myelomeningocele in
the low lumbar or sacral level as they begin to lose strength and motor
function. The average age at which tethering is detected has been
reported as being between 6 and 11 years of age (10,13).
Signs of tethering include loss of motor function; increased
spasticity; change in bladder function; progressive foot deformities;
back, buttock, or leg pain; and rapid increase in magnitude of lordosis
or scoliosis. It is extremely important that the orthopaedist carry out
a detailed neurologic evaluation including upper and lower extremities
at each clinic visit, and any change in motor or sensory function
requires a referral to the neurosurgeon for appropriate management.
Tethered cord syndrome is diagnosed on clinical evaluation; a magnetic
resonance imaging (MRI) study with gadolinium will further clarify the
nature of the tether and confirm the diagnosis. An MRI is important for
ruling out a dermoid cyst, which is not infrequently seen in
association with a tethered cord (10).
before major loss of function occurs. Releasing the tethered cord in
the lumbosacral region (14) may temporarily arrest the progression of spinal deformity if it is less than 40 degrees to 45 degrees in magnitude (15,16). However, curve progression has been noted with time after the detethering (15,17).
Detethering is very reliable in reducing back pain and may also be
effective in maintaining or restoring motor function that may have been
lost (15).
fluid in the ventricles of the brain. This can be documented with
ultrasound, MRI, and computerized tomography (CT) scan. In utero,
the ventricles communicate to the persistently open central canal of
the cord (which in turn communicates with the myelomeningocele sac),
thereby relieving buildup of cerebrospinal fluid (CSF) and protecting
against massive cortical damage. Open communication between the central
canal and the fourth ventricle permits outflow of the CSF, decompresses
the ventricular pressure, and relieves the hydrocephalus. However, at
the time of operative closure, the fluid flow from the central canal is
stopped and hydrocephalus returns. If hydrocephalus is not shunted, the
fluid pressure increases in the brain and the spinal cord, causing
brain atrophy, hydromyelia and, eventually, syringomyelia.
shunting as a means of avoiding further damage to the CNS. The
potential for shunt malfunction and acute hydrocephalus should be
appreciated by the entire multidisciplinary team, and the integrity of
the shunt function should be monitored by the neurosurgeon. In the
young child, shunt failure may be due to mechanical malfunction
following intraabdominal urologic procedures (18).
Typical symptoms of acute hydrocephalus in small children include
bulging fontanelle, altered mental status, nausea, vomiting, and severe
headache (19). Symptoms of progressive
brainstem dysfunction may also occur and include stridor, swallowing
difficulty, and vocal cord paralysis. In older individuals, gradual
shunt malfunction may be marked by symptoms such as progressive spinal
deformity, increased weakness in the lower extremity, back pain,
headache, vision changes, or nausea and vomiting first thing in the
morning. Prompt referral to a neurosurgeon is necessary if any of these
symptoms is present.
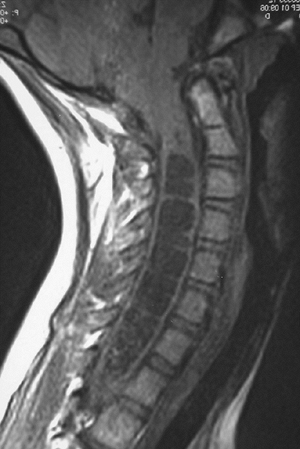
fourth ventricle with the persistent central canal of the cord (Fig. 16.2).
The dilatation of the spinal cord with CSF may be marked by increasing
scoliosis, paralysis of the lower extremities, increased spasticity,
and back pain. Cervical syrinx will also present with spasticity and
weakness of the hands and upper extremities in older teens.
 |
|
Figure 16.2
Representative MRI scan of affected individual with Arnold-Chiari malformation and associated hydromyelia (syrinx) of the cervical spine. |
shunt care. For instance, prior to orthopaedic surgery, broad-spectrum
prophylactic antibiotics that penetrate the blood-brain barrier are
needed in order to prevent secondary shunt infections. It is critical
to ensure that the shunt is functioning if cordotomy is needed to
correct spinal deformity; acute shunt malfunction may be life
threatening.
displacement of the posterior fossa structures, including the brain
stem, through the foramen magnum. This is known as Arnold-Chiari (also termed Chiari II) malformation (Fig. 16.3).
Some infants have symptomatic Arnold-Chiari deformity, and this is the
leading cause of death in infants with myelomeningocele (20).
include periodic apnea, stridor, nystagmus, weak or absent cry, and
upper-extremity spasm and weakness. Placement of a VP shunt to control
hydrocephalus may be enough to resolve brain stem symptoms, and
therefore surgical decompression of the Arnold-Chiari malformation may
not be needed. In a few infants and children, the brain stem
compressive symptoms persist or progress; in such patients, the
posterior fossa and upper spinal region need to be decompressed
surgically. Symptomatic Arnold-Chiari deformities in older children
also include spastic weakness of the lower extremities, difficulties in
swallowing, and depressed or absent cough reflex. Such individuals may
also benefit from posterior fossa decompression.
 |
|
Figure 16.3
Arnold-Chiari type II malformation of the brain stem. This is the most common type of malformation seen in myelomeningocele. The medulla oblongata is displaced distally through the foramen magnum into the cervical neural canal. The ventricle communicates with the still-open central canal of the cord. |
Approximately 6000 infants in the United States are born each year with
myelomeningocele and the overall incidence is 1 in 2000 life births
(0.15% among whites and 0.04% among African-Americans) (22). The overall incidence would likely be higher, as it is estimated that 23% of pregnancies with myelodysplasia are terminated (23).
the original hypothesis contends that the condition is caused by a
defect in neurulation (24). The formation of
the myelomeningocele occurs early in fetal life, probably between the
third and fourth weeks of gestation and before limb bud development.
This implies how important it is for the mother to take good prenatal
nutritional supplements and avoid known teratogens such as valproic
acid in the very early stages of embryogenesis. Most NTD (anencephaly,
myelomeningocele) occur as isolated malformations because of
multifactorial variables that may be either inherited or acquired. For
parents who have one affected
child,
the risk of having a second affected child is higher. Some NTDs are
associated with chromosomal abnormalities and others are caused by
single-gene mutations. Also, environmental factors such as exposure to
valproic acid may cause NTDs independently of any genetic variables.
Another important environmental factor is lack of folate in diets of
mothers who may have an unrecognized disorder of folate metabolism.
Folic-acid supplementation before conception and within the first month
of pregnancy can decrease the rate of myelomeningocele by 70%; in the
rest of the women, such supplementation may not have any effect (25).
that all women of childbearing age receive 0.4 mg folate before
conception and during early pregnancy. The Centers for Disease Control
and Prevention recommends that women who fall in the high-risk group
(prior affected child, or having a first-degree relative with a neural
tube defect) should receive a larger dose of folate, 4.0 mg per day (26).
Yates et al. have shown an association between susceptibility of
offspring with NTDs and depressed red cell folate levels that cannot be
attributed entirely to low dietary intake of folate (27).
They postulate that a factor that predisposes a person to have
offspring with NTD is an inherent disorder of folate metabolism and not
just a dietary deficiency. Unfortunately, 50% of woman of childbearing
age do not take folate supplements and many pregnancies today are
considered unplanned (21).
of gestation in all pregnant women, to look for elevated levels of
serum α-fetoprotein, this test can detect 75% to 80% of affected
pregnancies (28,29). If
serum α-fetoprotein levels are elevated, special diagnostic procedures
such as detailed ultrasound at 16 to 22 weeks, ultra-fast MRI scan (30),
and amniocentesis for α-fetoprotein and acetylcholinesterase are
indicated. Women who have had an infant with a neural tube defect carry
enough risk of having another affected child to justify undergoing
amniocentesis. Ultrasound examination of the fetal spine provides
important information about the presence and location of neural tube
defect, and is therefore a sensitive and efficient test in pregnancies
at high risk for neural tube defect. If no abnormalities are found on
detailed ultrasound examination, amniocentesis is recommended for
evaluation for α-fetoprotein and acetylcholinesterase.
family the option to either terminate or continue the pregnancy. This
is clearly an enormous decision for the family that depends on accurate
information about prognosis for the fetus, presented in in an unbiased
manner. For parents, the decision is affected by personal, religious,
and moral beliefs. If the family decides to continue the pregnancy, the
affected children are delivered at centers that are prepared to provide
optimal care for the child. These centers plan for elective cesarean
section (31,32) in
order to prevent further damage to the exposed sac during vaginal
delivery. In addition, they have pediatric neurosurgeons available for
closing the neural defect and placing a VP shunt.
implies that the disorder will be treated in the postnatal period;
however, the innovative option of antenatal treatment can be considered
with in utero surgery to correct the defect. In utero
intervention between 19 and 25 weeks of gestation has been used before
for tackling severe life-threatening problems such as tumors,
diaphragmatic hernia, obstructive uropathy, and hemolytic anemias.
Thankfully myelodysplasia is no longer considered a life-threatening
condition, yet in utero surgery is being attempted in this disorder also, in hopes of decreasing the eventual morbidity from the defect.
The rationale behind this approach is the fact that fetuses with
myelomeningocele in whom normal limb movements have been documented
later demonstrate paralysis (38,39,40,41). This implies that injury to the neural elements may not occur at the time of abnormal neurulation, but later in utero
and perhaps during birth. Further support for this theory is based on
the relatively lower incidence of neural damage in cervical dysraphism
and lipomeningocele, in which the neural elements are relatively better
protected by skin and fatty tissue. On the basis of this observation,
the goal of in utero correction is to prevent later chemical or mechanical damage from meconium, membrane rupture, or endometrium (7).
Contraindications are chromosomal abnormalities, ultrasound
confirmation of multiple congenital anomalies including clubfeet, and
absence of movement in the legs of the fetus (7).
In preliminary studies, these surgical procedures have been successful
in closing the defect, leading to a decrease in the incidence of
hydrocephalus and Arnold-Chiari malformation, and a reduced need for VP
shunting (34,35,36,37,42). Unfortunately, no consistent improvements have been found in lower extremity motor function or bladder function (34,35,43,44).
becomes routinely accepted by the public and the medical community. For
instance, the risks to the infant and mother need to be more clearly
delineated and the indications for the different levels of severity of
myelodysplasia need to be clarified. Once these are known, we need a
concrete understanding of the cost-benefit ratio for each family.
Surveys
have
been performed that reveal that families expect these procedures to be
extremely reliable in providing fairly normal ambulatory potential,
urinary continence, and independence from VP shunting (45). These lofty goals may not be obtainable with in utero
surgery, and the indications would therefore be questionable;
especially, high complication rates [preterm labor and prematurity,
lethal pulmonary hypoplasia (46), uterine rupture and small bowel obstruction] are present (47,48). In addition, in utero
closure has been associated with dermoid inclusion cysts and cord
tethering; will the rates of occurrence of these problems be higher
than in patients who have more standard postnatal closures? Clearly
long-term as well as immediate follow-up are needed.
Medicine has revealed that 56% of members are of the opinion that the
method has not been validated (49). Because of
these concerns, a National Institutes of Health (NIH)–funded
prospective study is currently under way at two centers to determine
the efficacy of in utero repairs and the
morbidity associated with such procedures. In the meantime, a
moratorium has been imposed on these surgeries being performed at other
centers so as to concentrate all the experience at the participating
centers.
community, ethical questions will undoubtedly arise. For instance, who
is the patient (mother or fetus)? If a definite improvement in the
outcome of the fetus can be reliably achieved, is the mother obligated
to undergo surgery with the attendant risks in order to improve the
life of the viable fetus (47,50)?
If so, does that imply that the fetus should be considered to be a
child (with rights), and how does that relate to constitutional issues
of abortion and abortion rights? Does the mother have the right to
subject the fetus to the risks of surgery for a currently
non–life-threatening condition? Finally, if proven successful, will
this treatment be equally available to affluent and poor mothers?
functioning and control of the genitourinary and gastrointestinal
systems. In patients with myelodysplasia, the normal milestones of
bowel and bladder control are typically delayed or absent. The need for
diaper use, self-catheterization, and reproductive dysfunction in male
patients with myelodysplasia further widens the perceived gap between
their own identity and that of “normal” individuals. In many respects,
advances in the treatment of urinary and fecal incontinence have done
more to improve patients’ self-esteem than advances in the management
of other organ systems (51). It is important to
realize that families and patients give priority to the importance of
appropriate urologic and gastrointestinal care above many other aspects
of management. The psychosocial impact of incontinence is extremely
important, and the development of the child’s self-esteem and sexual
identity are paramount (51). More importantly,
medical and surgical methods for achieving low-pressure storage and
timely evacuation of urine have decreased the morbidity of renal
dysfunction that was so pervasive in the 1970s and early 1980s.
sphincter dysfunction) is noted in 90% of patients with myelodysplasia.
Over time the significance of these abnormalities has changed. In the
early 1970s, when little attention was paid to intravesical storage
pressures, renal failure and urosepsis were the number one cause of
death in children older than 2 years (51). As
mentioned earlier, lower urinary tract dysfunction would predictably
lead to infection, reflux, and upper urinary tract dysfunction, and
secondary renal failure. Over the last several decades, there has been
tremendous improvement in the urologic management of children with
myelodysplasia, and such a grim prognosis is no longer expected.
Prophylactic management of bladder dysfunction with the development of
clean, intermittent catheterization and the use of anticholinergic
agents has done a great deal to decrease the rates of secondary
problems of bladder and renal dysfunction (3,4,51).
Contemporary goals of treatment have evolved from typical incontinent
urinary diversion to the goal of diurnal continent urinary diversions
and decreased rates of urinary tract infections. Obtaining these goals
should stave off upper urinary tract problems and renal failure.
directly responsible for both bladder contractility and urinary
sphincter control. It is therefore most convenient to consider each of
these components of neuropathic lower urinary tract dysfunction
independently and then consider the resultant scenarios that result
when combining the two. Bladder dysfunction can take the form of
hypercontractility (i.e., uninhibited bladder contractions and/or high
resting tone) or atony with the former a result of upper motor neuron
injury and the latter a result of a lower motor neuron lesion.
Sphincter dysfunction can take the form of either failure to relax/open
while the bladder is contracting (so-called detrussor-sphincter dysynergia, DSD)
or sphincteric incompetency. Any combination of bladder and sphincteric
dysfunction can exist. The combination that is of most concern in the
context of bladder and kidney prognosis is that of a high-pressure
bladder with failure of the sphincter to relax. This combination of
so-called hostile bladder dynamics results
in (a) an increased incidence of vesico-ureteral reflux, (b) stagnation
of urine leading to urinary tract infection, and (c) high-pressure
urine storage with backpressure on the kidneys and resultant renal
injury. Furthermore, increased bladder pressures lead to progressive
thickening of the bladder itself. These changes
decrease compliance and increase pressure, which sets up a vicious cycle of even further bladder thickening.
months after sac closure. Renal ultrasounds and urodynamic evaluations
assess the resting pressure of the bladder. For patients with
pathologically high intravesical pressures (i.e., higher than 40 cm H2O)
or indirect signs of increased bladder pressure such as hydronephrosis,
an attempt is made to improve bladder emptying with clean, intermittent
catheterization, and decrease the bladder tone with anticholinergic
medications. If pressures cannot be adequately reduced and
hydronephrosis persists, the child may require the temporary creation
of a cutaneous vesicostomy to protect the upper urinary tracts.
toward the two problems that face patients with myelodysplasia. These
measures are typically not undertaken before the age at which the
patient’s peers have achieved a state of voluntary bladder control
(i.e., not before age 4). Decreased compliance of the bladder is
treated with augmentation procedures to increase the volume of the
bladder with a consequent decrease in the tone and pressure.
Augmentation cystoplasty can be performed with multiple organs,
including ureter, stomach, ileum, and sigmoid colon. Today, most
augmentation cystoplasty is performed with the small bowel. Thankfully,
because of improved prophylactic care outlined in the preceding text,
bladder augmentations are not required as much today (51).
can be treated medically with α-sympathomimetics, which are used for
increasing the tone of the external sphincter. However, generally
speaking, these medical measures are by themselves inadequate to
achieve a state of bladder control. Surgical methods (bladder neck
tubulization, urethral lengthening, and bladder neck suspension) are
occasionally needed in order to increase the resistance in the bladder
neck (52,53,54).
Other methods to increase the tone of the extrinsic bladder sphincter
include the use of mechanical prosthetic devices, such as the
artificial urinary sphincter (55,56).
Additional methods for increasing resistance of the extrinsic urethral
sphincter include injection of the periurethral area with bulking
agents such as collagen and dextranomer in hyaluronic acid (57,58).
and developing a catheterizable conduit from the abdominal wall can be
performed. Catheterizable conduits have been developed from the ureter,
small bowel, and appendix. In the latter procedure, the appendix is
sutured to the abdominal wall with a continent valve construction and
then sutured to the bladder. The development of continent urinary
diversion methods has made a significant improvement in the care of
patients with spina bifida. These devices provide improved facility for
catheterization and continence, especially in patients who have a
posterior spinal fusion (59,60,61,62,63).
Finally, in patients with fecal incontinence, a Malone antegrade
continence enema (MACE) procedure can be developed for maintaining
continence of feces (64). In this procedure the
appendix is used in making a conduit for antegrade continence enemas to
control constipation and achieve fecal continence.
bifida has seen as many improvements as the neurosurgical field has.
The orthopaedist needs to be cognizant of the importance of maintaining
bowel and bladder functions in these patients and must, wherever
possible, coordinate with the urologist and perform surgical procedures
without compromising the bowel and bladder functions of patients.
Examples of such orthopaedic surgical procedures would include spine
fusion to try to improve upper renal drainage, bladder volume, and
access to vesicostomy portals. In this scenario, any potential benefit
of spine fusion on urinary function would be negated if there were
damage (65) to functioning neural elements relating to the extrinsic bladder sphincter.
disorder, several important health concerns that impinge on the quality
of life of affected individuals attain importance. These concerns
include psychosocial issues, latex allergies, and growth and
development.
have demonstrated that affected individuals tend to be socially
immature, less likely to have social interactions out of school, more
dependent on adults, less accomplished academically and athletically,
and more likely to exhibit difficulty in concentrating or attention
disorders (66). These findings show that the
affected individuals are plagued with multiple deviations from normal
life experiences because of their physical and mental disabilities and,
in some instances, decrease in cognitive functioning. Treatment of the
physical problems is often invasive. Patients may require frequent
hospitalization for shunt malfunctions or for the treatment of
orthopaedic deformities involving the use of casts, splints, and
wheelchairs. Hospitalization and treatment programs must be planned to
interfere as little as possible with the normal developmental sequence,
particularly in infancy. Development of independence is a complex issue
between the parents and their handicapped child. Currently 15% of
affected individuals require some form of custodial care (17,67).
The parents are often overprotective of the child. This protective
attitude is also present in school and in society at large, especially
if the child is confined to a wheelchair.
disease entity rather than as a person. This prevents the child from
gaining a sense of individuality and identity. In the clinic, the
children are frequently undressed to their diapers, then paraded before
members of the clinic without regard for their embarrassment. The
tendency is to treat them as asexual beings. Development of a positive
self-image and adult sexuality is a difficult task for these patients.
Because of the physical impairment of children with myelomeningocele,
personal interactions among peers are severely restricted from
childhood into adolescence. The child with myelomeningocele looks
different because of braces, orthopaedic shoes, wheelchairs, and
deformities; these are barriers to peer acceptance. These feelings of
being different may never be erased, even if the cause of these
feelings is eliminated.
the treatment of children with myelomeningocele during the last
decades, there are still considerable societal barriers to these
children. For example, access to schools, playgrounds, amusement parks,
sporting events, movie theaters, and private homes are limited to some
extent. Also, acceptance of handicapped people in the workforce and job
market is restricted. The choice of a career is also difficult. High
school counselors are not trained to advise disadvantaged children,
especially those with perceptible motor abnormalities of the hands.
Government programs are not available until the patient is 18 years old
or has graduated from high school. Employment opportunities also are
limited by the affected person’s lack of ability to get health
insurance.
has been noted in three groups; those who work in latex production
units, health care workers, and children with myelomeningocele (68,69,70,71,72,73,74,75,76,77,78,79,80,81,82). The incidence in affected children is as high as 3.8% to 6.6% (76,83).
This is because of multiple medical and surgical procedures in infancy,
and consequent extensive exposure to latex in gloves, urologic
catheters, and other durable medical goods. All patients should be
questioned about a history of latex allergy, which would be noted with
symptoms of swelling or itching of the lips when exposed to latex
products in everyday life. Such reactions may occur from blowing up
balloons, after dental examinations, and swelling or itching of the
skin after contact with any rubber products. Other information that may
suggest increased risk of latex allergy includes eczema on the hands
and itching in the mouth after eating bananas, chestnuts, or avocados.
Preoperative skin testing or radioallergosorbent testing (RAST) testing
can be performed with latex extract, but the tests may not be sensitive
enough to identify all persons who may be at risk for latex contact
allergy (78,79,80,84,85,86,87).
myelomeningocele, regardless of history, should have all procedures
performed in a latex-free environment (83).
Items that are known to provoke sensitivity reactions need to be
avoided in the hospital environment. When planning for surgery, it must
be assumed that the child is sensitive to all latex material. A
latex-free environment is one in which no latex gloves are used by any
personnel in the operating room, and there should be no latex
accessories (e.g., catheters, adhesives, tourniquets, anesthesia
equipment) that come into direct contact with the patient. A persistent
rate of latex allergy (1.2%) exists despite discontinuing the use of
latex medical devices (83). For a child with a
known latex allergy who is to undergo invasive tests or surgery, some
physicians recommend the prophylactic use of antihistamines and
intravenous steroids in addition to the usual precautions relating to
the use of latex (74,75).
Stature and bodily dimensions are altered by skeletal deformities and
nutritional factors that may lead to obesity. Growth may be diminished
by alterations in the hypothalamic-pituitary axis. These abnormalities
result in precocious puberty and growth hormone deficiency (12).
There have been several studies on the effects of growth hormone
replacement in such patients, and they report significant increases in
strength, stature, and mobility (12,89,90).
The use of growth hormone will have positive effects such as increased
strength, decreased obesity, and improved mobility; yet the
myelodysplasia team should recognize the increased risk of worsening
any spinal deformity that may be present.
patients according to muscle function corresponding to differing levels
of paralysis (91,92,93,94).
The levels of paralysis can be considered flaccid or spastic, and there
may be associated spasticity of the upper extremities. Patients with
spasticity in the lower extremities tend to require more surgical
procedures than those in whom the lower extremities are flaccid.
Patients with upper-extremity spasticity also tend to have more
problems with activities of daily living (95).
All patients with new-onset spasticity should also be evaluated for
concurrent intraspinal pathology such as cord tethering, hydromyelia,
and shunt function (15).
are grouped differently according to different classification systems.
It is important to remember that these classification systems are
subject to intra- and interobserver variability (96).
It is likely that these issues account for the reported discrepancies
in the results of treatment for hip displacement and abnormal gait (97,98).
thoracic, upper lumbar, lower lumbar, and sacral levels of function.
Thoracic-level patients have no active hip flexion and no voluntary
muscle control in the lower extremities. Upper lumbar-level patients
have variable power with hip flexion and adduction (L-1 and L-2) and
quadriceps function (L-3). Lower lumbar-level patients have active knee
flexion against gravity (hamstring power), anterior tibialis function
(L-4), and extensor hallicus longus function (L-5). Sacral-level
functioning patients will have weakness of the peroneals and intrinsic
muscles of the foot, but will have active toe flexor function as well
as good hip extensor and abductor power. Some investigators have split
sacral-level function into upper and lower sacral function on the basis
of the quality of strength of the triceps and gluteus maximus. Lower
sacral function in patients is associated with more normal long-term
function, and such patients are less likely to require orthotics or
surgical intervention than those in the upper sacral levels (99).
of paralysis. Those with upper lumbar levels of paralysis have
Trendelenburg gait patterns marked by lateral and posterior shift of
the trunk over the stance-phase extremity (100,101).
The child “hikes” over the stance extremity, thereby allowing the
contralateral hemi pelvis and lower extremity to internally rotate and
“swing” through. The magnitude of these shifts is directly related to
the degree of hip abductor and extensor weakness (102).
Concurrent hip flexion is because of increased pelvic tilt from
weakness of hip extensors and as an accommodation to hip and
knee-flexion contractures. The latter is the result of gastrocsoleus
weakness and absent knee-extensor moment (100,101).
Many of these quantitative aberrations in gait, and consequent knee
pain, can be minimized with the use of an appropriate orthotic and
forearm crutch such as that described in the following text (103).
Community ambulators can walk in the course of most of their
activities. Household ambulators walk only indoors. Nonfunctional
ambulators walk only during therapy, and nonambulators are completely
wheelchair dependent. The predominant predictor of gait and function in
children with myelomeningocele is the motor level of paralysis (21,91,92,98,104,105,106,107,108,109,110,111,112,113,114,115,116,117).
Within each motor level the type of paralysis may also be predictive,
since patients with spastic paralysis seem to show decreases in
ambulatory potential (95,118), and if spasticity is new, it should be considered a sign of concurrent tethering of the spinal cord.
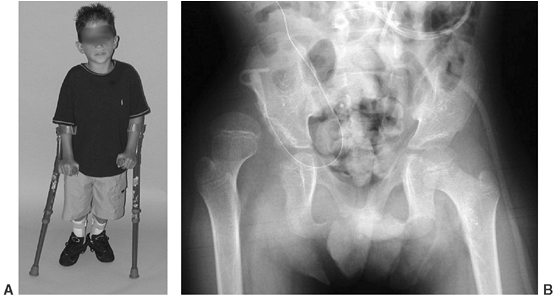
and L-4 level of motor function have a reasonably good chance for
functional walking because of the presence of power in the medial
hamstrings, which can act as hip extensors (91,106,111,112) (Fig. 16.4).
Most individuals (over 80%) who maintain L-5 and sacral function have a
good potential to maintain community ambulation throughout their lives (2,91,92,95,98,99,100,101,102,103,104,105,106,107,108,109,110,111,117).
Other reported predictors with variable effects on gait and standing
programs include obesity, gender, hydrocephalus, Arnold-Chiari
malformation, intelligence, motivation, spasticity, scoliosis,
kyphosis, pelvic obliquity, fixed-hip contractures, upper-extremity
weakness, parental compliance, and vigilance with physiotherapy (91,92,94,98,108,110,112,115,117,121,122,123,124,125,126).
Increases in age and weight are also known to be associated with
decreased ambulation; affected adolescents with marginal ambulation
skills may opt for wheelchair mobility as they
will feel the need to keep up with peers who are moving faster (110).
Decreased gait and function over time can be seen in low lumbar- and
sacral-level patients who have worsening of motor function, balance,
and spasticity and increases in neuropathic sores and infections (99,107).
In these patients a decrease in motor function and ambulation may be
associated with an increase in spinal deformity, both of which may be a
sign of tethering of the spinal cord or need for shunt revisions (99,107,118).
 |
|
Figure 16.4
Community-ambulatory child with L-3–L-4 level of function. He uses Lofstrand crutches and bilateral ankle-foot orthoses. He is able to ambulate with a swing-through gait despite a unilaterally dislocated hip. (Courtesy of Charles T. Price, MD) |
by multiple medical problems that require treatment by other medical
specialists (neurosurgeons, urologists, developmental pediatricians,
and therapists). The mental development of the child, and the formation
of parent-child bonding, may take precedence over the correction of
skeletal deformity, especially during early infancy. There are very few
orthopedic deformities that cannot have treatment delayed until the
child is 1 year old. By that time, most of the problems of infancy,
such as shunt malfunction, feeding difficulties, and respiratory
problems, have been addressed. Orthopaedic care should be coordinated
with the overall treatment plan for the child and also considered in
light of other orthopedic deformities of the spine, hips, knees, and
feet.
more procedures on aging populations of patients with myelodysplasia
are performed. Few adult orthopaedists have expertise in this area and
therefore “pediatric” orthopaedists become the primary orthopaedic
specialists for adults also. In general one can extrapolate therapeutic
plans from children to adults. However, we must recognize that
complication rates go up as patients age. These individuals are less
likely to tolerate complicated procedures, and these should perhaps be
staged in adults. In addition, one should exercise increased vigilance
for adult complications such as deep vein thrombosis, although the
incidence of such problems is low (127).
myelodysplasia, it is difficult to extrapolate the results of
orthopaedic treatment rendered in the 1970s and 1980s to contemporary
patients. Are previously reported failures of treatment potentially
more successful with the current understanding of myelodysplasia? For
instance, would procedures used in the 1980s to stabilize hip position
have been more effective if contemporary treatment of tethered cord and
hydromyelia had been available? Would ambulatory potential be better
with a reduced hip with state-of-the-art orthotics and appropriate
treatment for endocrine-induced obesity? Despite these challenges, I
have tried to integrate historical and contemporary information to
serve as a guide to the management of orthopaedic problems inherent to
myelodysplasia.
from early standing programs in patients with thoracic and upper-lumbar
function (128,129,130).
Some would argue that most patients with high levels of paralysis
should be placed in wheelchairs rather than be subjected to aggressive
standing programs. This is based on the conclusion that such
individuals are likely to be wheelchair-dependent by adulthood. Liptak
et al. compared children from Seattle who were put into wheelchairs
with those from Rochester who were treated with parapodiums. In this
study, there were shown to be minimal benefits from a standing program
with parapodiums in comparison to immediate wheelchair use (131).
The complications were different between the two groups (knee sores in
parapodium use and decubitus ulcers in wheelchair use); however, these
complications were equivalent in severity (hip dislocation in
parapodium use and knee contracture from wheelchair use). The fact that
other differences did not exist may be due to regional variations in
climate, obesity, and activities such as television watching between
these two areas of the United States. In a similar comparative study,
Mazur et al. (128) observed that the major
advantage of early walking and standing is greater overall independence
and increased ability to transfer as adolescents, and lower incidence
of fractures and pressure sores (128). It is interesting to note that results of standing programs from Rochester (131) and Australia (128)
are different when compared with immediate wheelchair use in children
from Seattle. These differences as well are likely due to regional and
international differences in socioeconomic status and functioning
between these children. A balance between therapeutic standing programs
and the practical use of a wheelchair for activities of daily living is
probably optimal. Over the lifetime of the patient, the importance of
standing programs recedes because of more significant contractures at
the hips and knees, and therefore the time and effort required to get
adolescent and young adult patients standing outweigh the benefits (105,107,124).
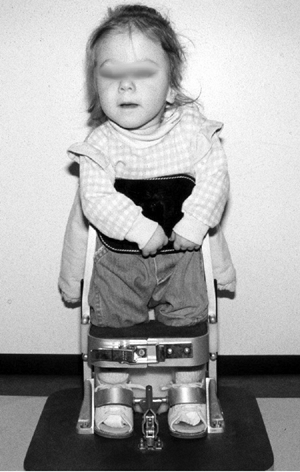
Psychological benefits include promotion of later functional
independence as well as the parents’ perception that a child who stands
is closer to being “normal” (131). Because of
these benefits, all families with affected children are given the
opportunity to participate in standing and walking programs (Fig. 16.5). However, the families are made aware of the need for surgeries and
hospitalization to maintain the conditions of the joints and to facilitate standing (128).
 |
|
Figure 16.5 Two-year-old child with thoracic-level myelomeningocele is started in her parapodium. (Courtesy of Stuart Weinstein, MD)
|
multidisciplinary teams of orthopaedic surgeons, therapists, and
orthotists. As previously described, the most important factor that
portends the ability to walk or stand is the motor level of function.
Within each functional motor level, abilities differ depending on the
patient’s weight, age, motivation, and the presence of muscle or joint
contractures, which can prevent the ability to stand upright. Surgical
treatment may be of benefit for reducing contractures and allowing
standing and walking in an upright position with appropriate orthoses.
Several different orthoses may be utilized for patients with differing
levels of function at different stages of development, and some
combination of orthoses should provide most children an ability to
ambulate to some degree (124,135,136,137).
sitting balance may be started on the program by being made to stand in
prone standers, which allow vertical positioning by 12 months of age (138).
Parapodiums may also be used; they function by holding the feet, knees,
and hips at a neutral position, and swivel bases may be added to
facilitate some propulsion in older children (137,139,140).
Individuals with thoracic level of function may ambulate with these
orthoses by swinging through with forearm crutches or a walker. With
time, thoracic- or upper-lumbar patients are eventually fitted with a
hip-knee-ankle-foot orthosis (HKAFO) or reciprocating-gait orthosis
(RGO) in order to improve ambulation. Contraindications to these
include marginal vision and balance; poor patient and family
motivation; scoliosis and poor trunk control; and weak upper
extremities (141,142).
and relies on the patient generating active hip flexion of one limb
that leads to contralateral hip extension through a cable system (138,141,142,144). Thoracic-level patients (without hip flexor power) may also benefit from the RGO (145),
but ambulation is promoted through an opposite mechanism: by leaning
back at the stance extremity, this extensor moment drives the
contralateral leg forward (141). Although
designed for reciprocal motion, a swing-through gait is also possible
with the obligate use of forearm crutches or walker.
pelvic bands, and hip and knee hinges may be utilized in
upper-lumbar-level patients during ambulation (91,136,146).
These will be of benefit in patients with weak quadriceps function and
severe hip extensor weakness. Use of forearm-based crutches or a walker
will be needed which will definitely provide stability, speed, and
power for forward motion. These patients ambulate with predominant
swing-through or swivel type of motion and are dependent on strong
upper extremities.
who utilize the RGO believe that the resulting reciprocal gait is an
improvement over the standard swing-through gait seen with statically
locked RGOs or HKAFOs (138,142,145,147,148).
Other research has demonstrated that the HKAFO is similar in regard to
oxygen consumption but is associated with increased velocity and
efficiency (141,149).
More functional use of the RGO is likely in smaller and lighter
individuals and at centers that actively promote and support this
orthosis (141,142). If there is decreased function as the patient grows, he or she may change from the RGO to the HKAFO (143). Eventually, both orthoses are abandoned as the children grow and the ratio of power to weight decreases (143).
Adolescents with upper-lumbar or thoracic level of function utilize a
wheelchair because of ease of mobility or because their obesity and hip
and knee contractures prevent adequate orthosis fitting.
more functional motor levels. These orthoses should provide stability
to the foot and ankle and should be lightweight and cosmetically
acceptable. For a variety of reasons, static splinting with an
ankle-foot orthosis (AFO) is uniformly prescribed in almost all
individuals with myelomeningocele (135,150,151).
In patients with very distal levels of motor function (S-1–S-2), AFOs
may not be needed and in fact may result in increased transverse plane
stresses (152).
There is little indication for hinged AFOs in myelomeningocele. In
low-lumbar-level patients, AFOs will prevent equinus contracture caused
by foot drop and will improve foot clearance during swing phase for
ambulatory patients. In these patients, the AFOs will provide a stable
base for ambulation and may prevent the development of calcaneus
deformity that is caused by unopposed ankle dorsiflexion in the patient
with L-4 or L-5 level of motor function. In these patients, the AFOs
can be attached to a pelvic band with twister cables that may be
effective in controlling internal rotation deformities until definitive
derotational surgery can be performed.
 |
|
Figure 16.6
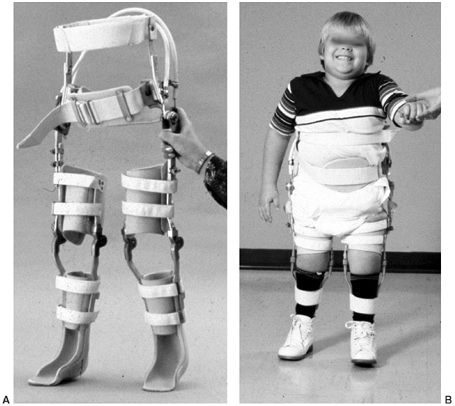
Six-year-old boy with upper-lumbar-level myelomeningocele ambulates with a reciprocal gait orthosis (RGO). (Courtesy of Stuart Weinstein, MD) |
anterior panel (anterior floor reaction, AFO) that will generate a knee
extensor moment in terminal stance phase, thereby compensating for weak
gastrocsoleus power (150,152,153,154) (Fig. 16.7).
This is beneficial in preventing knee contractures and crouching that
may lead to anterior knee pain in adults because of the increased
quadriceps power needed to accommodate triceps surae weakness (152,154).
Knee-ankle-foot orthoses (KAFO) are useful in lumbar-level patients who
have weak quadriceps function or who have excessive genu valgum that is
difficult to control in stance phase. These orthoses may have free
hinges to accommodate angular irregularities or drop-lock hinges for
patients with weak quadriceps function. The long-term benefit of the
KAFO in preventing degenerative changes in the knee is unknown (155,156,157) and may not provide any better trunk stability than is provided by an anterior floor reaction AFO (102).
deformities of the lower extremity that affect gait and make it
difficult to fit appropriate orthoses for efficient use. When the limb
is advanced, there may be an abnormal foot progression angle because of
muscle contracture, imbalance, and bony deformities. Internal and
external rotational deformities may be present at the femur and tibial
levels. Operative treatment of these rotational deformities has a high
degree of success in improving the gait (158).
dislocation of the hip or from muscle imbalances and contracture of the
external rotators (158,159). Conversely, an internally rotated limb may be dynamic because of muscle
imbalance from strong medial hamstrings compared to paralytic lateral
hamstrings. Fixed internal rotation is caused by persistent femoral
anteversion that has not remodeled with time. Fixed rotational
differences are managed with femoral derotational osteotomy, which can
be performed either proximally or distally. In milder internal
rotational deformities, lateral transfer of the medial hamstrings to
the fibula is an alternative procedure for providing dynamic external
rotation moment (158,159,160).
 |
|
Figure 16.7 This Charcot degeneration occurred in a 16-year-old, L-5 paraplegic girl who refused to wear her ankle-foot orthosis.
|
children with low lumbar levels of function. This deformity can limit
ambulation because of feet collisions during swing phase. In the young
child this can be managed with physiotherapy, fitting of a KAFO, or
using twister cables attached to a pelvic band. These modalities may be
continued until 3 to 4 years of age, by which time any natural
derotation would have occurred. At this time tibial derotational
osteotomy can be done to provide more permanent correction (160).
Tibial osteotomy alone is sufficient at the supramalleolar level for
deformities of 20 degrees or less, but concurrent fibular osteotomy is
usually needed if greater correction is called for. Care should be
taken to avoid incidental damage to the growth plate; fixation is done
with crossed wires or screws. Older children may be more appropriately
treated with plate fixation and cast immobilization; in these cases the
physis is still protected, and the plate is placed anteriorly so that
no pressure sores result from AFO use.
valgus is a common deformity seen in older ambulatory children with
myelomeningocele (158,161,162,163). An externally rotated foot progression angle greater than 20 degrees may lead to increased knee valgus (155) and decreased knee extension moment in stance phase (164).
Surgery may be required in patients in whom the deformity is greater
than 20 degrees and associated crouch is noted (lever arm dysfunction) (164).
The deformity in the tibia and fibula can be managed by supramalleolar
osteotomy to correct rotation and valgus of the ankle when it is
greater than 10 degrees (160,163,165).
Such procedures will likely decrease the knee stresses seen from
internal knee varus moment and increase the knee extension in stance
phase (166).
in that the neural arch is incomplete over the region of the defect.
The pedicles and transverse processes are splayed and externally
rotated. As a result, the absent lamina and the rotation of the
remaining posterior elements decrease the host bone that is available
to obtain a posterior fusion. Spinal deformity in myelomeningocele may
be categorized according to the predominant features in three main
varieties: paralytic scoliosis, kyphosis, and deformities associated
with congenital anomalies. Combinations of these different deformities
may exist.
prevent further progression of deformity and improve sitting balance
with a stable spine centered over the pelvis, thereby freeing the upper
extremities from having to play a supportive role in balancing the
trunk. While attempting to attain this goal, it is important to
maintain optimal mobility in the lumbosacral spine, thereby decreasing
the potential incidence of decubitus ulcers.
Such
ulcers are likely to form over insensate skin and from an increase in
shear forces after a spinal arthrodesis to the pelvis. Secondary goals
include improvement of posture, self-esteem, and transfer ability,
relief of back pain, and bowel and bladder care. Unfortunately,
obtaining these goals is associated with the highest rates of
complications encountered in the surgical management of patients with
neuromuscular scoliosis.
the reported incidence depends on the definition of scoliosis, and
whether kyphosis and congenital anomalies are included in that
definition. The incidence of scoliosis is related to the motor level of
paralysis and the last intact laminar arch (167,169,173). Eighty-three to one hundred percent of patients with thoracic level paralysis will have some degree of scoliosis (169,174,175);
approximately 60% of patients with an L-4 level of paralysis will have
spinal deformity. Patients with scoliosis usually have an accompanying
lordosis in the lumbar spine because of concurrent hip-flexion
contractures. Scoliosis is usually a long C-shaped paralytic curve, but
can occasionally manifest as a balanced curve, similar to that seen in
patients with adolescent idiopathic scoliosis (Fig. 16.8).
and tends to worsen with time and age irrespective of the magnitude of
the curve. A rapid increase in the magnitude of the scoliotic curve in
patients with myelomeningocele may be due to tethering of the cord,
undiagnosed syrinx, Arnold-Chiari malformation, or progressive
hydrocephaly (176,177).
and spinal fusion. Observation is generally recommended in patients
with a balanced spine or in curves less than 30 degrees. Boston bracing
has been used with some initial success in patients who had a curve
less than 45 degrees (178). Wherever the
fitting of orthoses are indicated, bivalved orthoses are preferred to
standard thoracolumbar-sacral orthoses (TLSOs) used in adolescent
idiopathic scoliosis. However, orthotic treatment of myelomeningocele
is generally considered “unsuccessful” because it does not result in a
cure of the deformities (180). Isolated use of
an orthosis may be beneficial in affected children who are less than 7
years of age, with a flexible curve for which the orthopaedist wishes
to delay
surgical
treatment, or in individuals with poor trunk control. Problems inherent
to bracing in myelomeningocele include rib deformities and pressure
sores (178).
In addition, circumferential bracing will increase the pressure over
the abdominal contents and can be a factor leading to incontinence, as
well as poor nutrition and failure to thrive.
 |
|
Figure 16.8
Fourteen-year-old girl with low-lumbar-level function, with a progressive scoliosis that is relatively well balanced and has a curve pattern similar to that seen in idiopathic scoliosis. In such patients, posterior spine fusion produces a well-balanced curve into the lumbar spine. (Courtesy of Stuart Weinstein, MD) |
greater than 50 degrees and in patients with progressive deformities or
spinal imbalance. Prior to surgical treatment, patients are evaluated
for the incidence of pelvic obliquity and concurrent hip-flexion
contractures. Severe hip-flexion contractures should be released prior
to spine fusion, in order to reduce the concurrent lordosis associated
with scoliosis. This can improve sitting balance and help maintain
ambulation in those patients who propel themselves forward with
movement of the pelvis in the sagittal plane. Preoperative radiographic
evaluation should include anteroposterior and lateral scoliosis films,
with the patient in a sitting posture, in order to assess the
deformity. MRI should be obtained prior to surgery to rule out tethered
cord, syrinx, and Arnold-Chiari malformation. CT scans can also be
obtained to assess the bony anatomy in patients with concurrent
congenital anomalies. Neurosurgical procedures for spina dysraphism can
be done at the same time or prior to definitive orthopaedic correction
of deformity. This needs to be done because the correction of skeletal
deformity generally results in lengthening of the spine, and would
therefore increase tethering. Cordotomy is occasionally needed to
increase exposure in patients with associated and severe congenital
kyphosis.
myelodysplasia is more challenging than in patients with adolescent
idiopathic scoliosis or in patients with other neurologic disorders. A
recent institutional review of contemporary spine techniques for
neuromuscular deformity documented that most of the complications
occurred in patients with myelodysplasia (181).
Within the last decades, the rates of serious complications have
decreased, and results of spinal arthrodesis are generally good, with
improvements in posture and pulmonary function (182,183,184), and corrections of deformity approaching 40% to 60% (181,185,186,187,188,189).
Because little spontaneous correction of compensatory curves can be
expected, it is wise to fuse the entire segment of the spinal
deformity, including the primary and compensatory curves (186).
Fusion should not end in the middle of the spinal defect. Historically,
spinal fusion has been extended to the pelvis if there is fixed pelvic
obliquity greater than 15 degrees, or if the lumbosacral scoliosis is
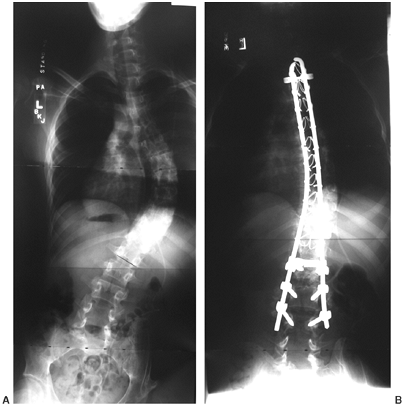
greater than 20 degrees (Fig. 16.9). Sacral pelvic fixation can be obtained with rods placed in the first sacral foramen or with iliosacral screw fixation (190) into the pelvis by the Galveston methodology or over the ala of the sacrum (Dunn-McCarthy Technique) (191,192).
 |
|
Figure 16.9
Thirteen-year-old boy with thoracic level myelomeningocele and progressive curve and with pelvic obliquity. Posterior spine fusion to the pelvis stabilizes the curve and provides a level sitting platform. (Courtesy of Stuart Weinstein, MD) |
posterior fusion and instrumentation; however, pseudoarthrosis and
hardware failure rates varied from 16% to 46% (186,187,193,194,195,196,197,198,199,200).
Failure can result from: osteoporosis; deficiency in posterior bony
elements available for fusion; limited hip motion; and increased stress
at the lumbosacral articulation. Today, anterior fusion is performed in
addition to posterior fusion in most patients with myelodysplasia in
order to avoid the pseudoarthrosis that is likely to occur after
posterior-only surgery (186,187,196,200,201,202,203,204,205,206).
Anterior fusion prevents the crankshaft phenomenon. Recent reports
indicate that the concurrent use of anterior and posterior approaches
has decreased the rate of pseudoarthrosis to less than 16% (186,200,202,207,208).
In general, anterior fusion is combined with posterior instrumentation
consisting of ¼-inch rods, hooks and pedicle screws, wires, and cables
that can be placed around the rotated pedicles and lamina (200,204,205,209). Anterior and posterior spine fusions may be done on the same day with decreased hospital and ICU time and costs (210).
However, some surgeons stage these procedures at least a week apart.
This is advisable in especially complicated cases or when both anterior
and posterior instrumentation are to be used (203).
with moderate deformity when surgery consists of anterior interbody
fusions and posterior fusion with newer generations of screws, cables,
hooks, and rods (200,204,205). In these patients improvements in sitting balance can be expected (196).
Anterior and posterior instrumentation may be needed in patients with
severe deformity, those with thoracic levels of paralysis, and those in
whom it would be ill advised to attempt fusion to the sacrum (185,187,188,189,203,208,209).
Anterior and posterior instrumentation and fusion, or posterior fusion
with contemporary segmental instrumentation (pedicle screws), may
spontaneously correct the pelvic obliquity, obviate fusion to the
sacrum, and maintain lumbosacral mobility (204,205,208).
Lumbosacral mobility has an important role in ambulation, sitting, and
activities of daily living. Marginal ambulators who have had fusion to
the pelvis may experience a decrease in ambulation because of loss of
lumbar-sacral movement that is needed for forward motion (196,211). Spinal-pelvic fixation will additionally decrease the ability of girls to perform self-catheterization of the urethra (212).
indicated as a sole method of correction of scoliosis in the rare cases
of isolated lumbar or thoracolumbar curves. Some surgeons have proposed
this as a good option with potential advantages, including high fusion
rates (206), less extensive surgical dissection
and fusion (and consequently better lumbar-sacral mechanics), and also
potentially decreasing rates of infection. Risks associated with this
approach include pseudoarthrosis and extension of the curve proximally
into the noninstrumented curve (196,206,213)
and loss of motor function. This method should be restricted to
patients who have isolated lumbar and thoracolumbar curves of less than
75 degrees, without kyphosis or history of syrinx (213).
If a concurrent tethered cord is present, neurosurgical release should
be performed prior to the spine fusion; patients with intact quadriceps
function should have intraoperative monitoring of that function.
historical rates ranging from 7% to 33% after surgical correction of
spinal deformity in myelodysplasia (186,189,193,196,203,207,208).
Infection rates are higher than those following surgical treatment of
idiopathic scoliosis, and are likely a result of ubiquitous bladder
colonization and the relatively poor condition of the surrounding skin
and muscle around the meningocele repair site (175,186,187,198,201,214,215,216).
In general, if posterior fusion is done in such a manner as to avoid
poor skin, inverted Y incisions around the previous scarring may be
helpful for avoiding concurrent infection (208).
Antibiotics are used prior to and after surgery to prevent shunt
infection and spine infection. When infections occur they may be
polymicrobial infections with enteric, gram-negative or gram-positive
organisms (216). These complications are
treated with surgical debridement of soft tissues and long-term
antibiotic therapy. Attempts are made to retain implants unless repeat
drainage is necessary after initial debridement.
Kyphosis may be noted in the perinatal period at the time of closure of
the myelomeningocele. Up to 20% of individuals with myelodysplasia will
have kyphosis that may exceed 80 degrees at presentation (219,220,221,222).
These deformities may be gradual over many levels, be sharply angled,
or involve congenital anomalies of the vertebral bodies (170,214,219,220,223,224,225). Thoracolumbar kyphosis will continue to progress with time (226,227) because of the altered anatomy of the posterior spine elements and the paravertebral muscles (197,202,220,221,228,229).
The spinous process, lamina, and intraspinal ligaments are absent and
the paravertebral spinal extensors are rotated laterally and anterior
to the axis of deformity (230). Further
progression of deformity is expected because of unopposed flexion of
the crura of the diaphragm, psoas, and quadratus lumborum muscles (224,228). These deformities may progress at a rate of 6 to 12 degrees per year (218,220,221,226,231,232).
Younger individuals and those with more sharply angulated kyphosis or
with congenital anomalies have faster progression of deformity than
those with gradual and paralytic deformity (219,220,226). This may be because of compression of the physis and retardation of growth in the vertebral endplates (226).
caudal displacement of an Arnold-Chiari malformation (217,218).
Patients who are affected at the thoracic level have difficulty with
independence because the arms are required as props while sitting. In
addition, cephalad displacement of the abdominal contents and loss of
pulmonary capacity will be factors that contribute to the child’s
inability to grow and thrive (218). Difficulties in sitting and skin breakdown over the kyphosis may eventually result (205).
The development of thoracic lordosis and severe kyphosis of the lumbar
spine may also have an untoward effect on the ability of the ureters to
drain efficiently. Further compromise in urologic function includes
difficulties in placing and accessing urethrostomy or vesicostomy (233).
treatment of thoracolumbar kyphosis is predominantly surgical. Bracing
has no long-term efficacy and may cause further skin breakdown (218,234).
Bracing may have a role only on the rare occasions when a patient has
an extremely small curve with intact sensation. In the light of this,
intermittent use of an orthosis in patients with poor trunk control may
be of benefit while waiting to perform definitive surgical correction.
Such use will promote development of activities of daily living through
occupational therapy, using the upper extremities.
deformity, skin breakdown, poor positioning, respiratory compromise,
and possible pain due to costal pelvic impingement (227).
The method of surgical correction depends upon when the deformity is
diagnosed. Many neurosurgeons can decrease the rate of subsequent
kyphosis by initially resecting kyphotic vertebra at the time of sac
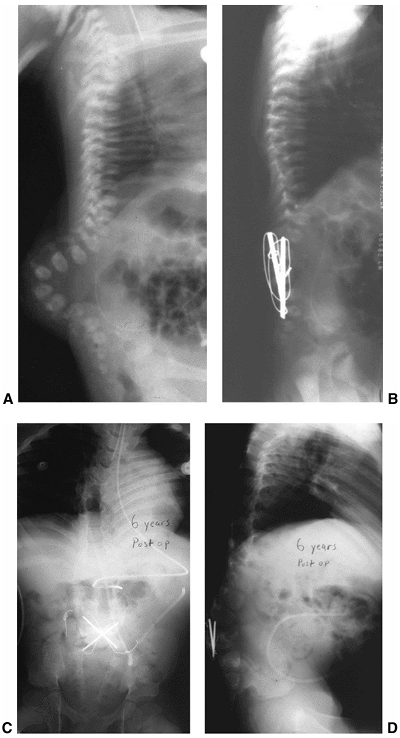
closure (222,235). Excision of apical vertebral bodies or tension band fixation at the time of closure will reduce the deformity (Figs. 16.10 A, B).
At the same time, dorsal reapproximation of the paravertebral muscles
will help cover the defect and, additionally, prevent further
progression of the deformity.
categories: a collapsing “C” shaped deformity and a more rigid “S”
shaped deformity (232) (Fig. 16.11).
The former is more likely associated with distal neurologic function,
whereas the latter is associated with the thoracic level of spinal cord
function. This classification system is helpful in understanding the
deformity and planning surgical correction. In general, C-shaped
kyphosis is treated with eggshell decancellation methods at vertebrae
above and below the apex of deformity. S-shaped deformities require
much more extensive osteotomy and excision of vertebral segments above
the apex of deformity.
kyphosis in the patient who has no associated complications is at
approximately 3 years of age (227,237).
Surgical treatment of thoracolumbar kyphosis in children older than 2
or 3 years of age is performed with spinal instrumentation, and with
the goal of completely restoring the sagittal balance. The procedure is
done in such a manner as to maintain growth (Fig. 16.12).
Many different methods of kyphosis correction and fixation have been
described over the years for treatment of C- and S-shaped deformities (197,202,221,222,225,231,232,234,235,238,239,240,241,242,243,244,245,246,247).
C-shaped spinal deformity can be reduced by either anterior release of
the spine through the disc space or posterior removal of spine
segments. In the latter method, at least one vertebral body above and
below the apex of deformity is removed. This is done posteriorly
through the pedicle where decancellation and decortication of the
posterior
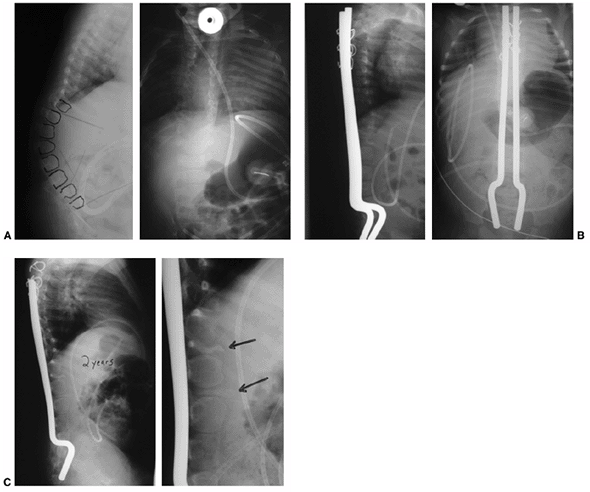
aspect of the vertebral bodies will allow for correction of the kyphosis (222,227). One can expect improvement of 45 degrees after removal of one vertebral body (248). Instrumentation to the pelvis is obtained by placing the ¼-inch rods into or over the sacral ala (192),
or into a lumbosacral foramen. Proximal fixation to the thoracic spine
is obtained with sublaminar wires or cables placed in extraperiosteal
fashion, allowing for further growth of the spine. Fixation in the
thoracic spine should extend proximally into normal spine anatomy and
across associated thoracic lordosis. Usually four fixation points
(sublaminar wires) are sufficient for each rod. Patients are usually
immobilized in a body shell for 12 months until complete healing is
obtained.
 |
|
Figure 16.10 A–D:
Newborn infant with open myelomeningocele and severe kyphosis. Closure and reduction of the kyphosis was performed simultaneously with resection of vertebral bodies and tension band fixation. B: At 6-year follow-up, the child has only mild residual kyphosis that is not progressive and does not require any further treatment. (Courtesy of Charles T. Price, MD) |
 |
|
Figure 16.11 Two types of lumbar kyphosis: the C-shaped collapsing curve (top) and the S-shaped curve (bottom). (From Loder RT, Shapiro P, Towbin R, et al. Aortic anatomy in children with myelomeningocele and congenital lumbar kyphosis. J Pediatr Orthop 1991;11:31,32,33,34,35, with permission.)
|
delayed until patients are older in order to gain as much growth as
possible. Easier instrumentation and decreased chances of proximal
deterioration are associated with surgery in older patients (249). Prior to surgery, it is critical to
determine the presence of hip-extension contractures that would
severely limit hip flexion and sitting balance once the deformity is
corrected. Rigid kyphosis is usually managed with posterior osteotomy
or excision of vertebral bodies from the apex of the deformity and then
proximally into the area of thoracic lordosis (222,232).
Stabilization is best done with segmental fixation placed around
nonfunctioning neural elements. Postoperatively, patients are usually
immobilized in a polypropylene TLSO for at least 12 to 18 months, until
healing is ensured (232,250).
 |
|
Figure 16.12 A: Anteroposterior and lateral radiographs of an 8-month-old infant with thoracic level of paralysis and C-shaped kyphosis. B:
Anteroposterior and lateral radiographs after decancellation of two vertebral bodies above and below the apex with posterior instrumentation. Rods are placed into the S1 foramen and fixed proximally with sublaminar wires in an extraperiosteal fashion. The rods are left long to allow for continued spine growth. C: Follow-up radiographs at 2 years demonstrate reduction of kyphosis and growth of the spine away from the rods proximally. Consolidation of the egg-shelled vertebrae are noted (arrows). |
recognize that the kidneys may be present in the concavity of the
deformity and therefore be susceptible to injury from inadvertent
penetration (251). Surgical exposure may be enhanced when the sac is transected (cordotomy) and the nerve roots sacrificed (243,244,249). Other benefits of cordotomy include a concurrent decrease in upper-extremity spasticity (233).
However, it is important to ensure that the shunt is functioning
preoperatively in order to avoid acute hydrocephalus following planned
cordotomy (252). The effects of cordotomy and kyphectomy on bladder function in affected patients show individual variations (233,242).
When sagittal balance is restored, improvements in bladder function are
likely to ensue because of the increase in volume and compliance of the
bladder following kyphectomy. On the other hand, flaccid incontinence
from loss of tone in the external bladder sphincter is a rare
complication due to injury of a functioning nerve root (65,233).
When considering cordotomy, preoperative urodynamic studies may
demonstrate intact innervation to the external sphincter. In surgery,
it is clearly important to transect nonfunctioning neural elements and
to preserve the functioning cord and nerve roots (65,233).
In these cases in may be wise to use distal fixation over the ala of
the sacrum (Dunn method) as opposed to placement into the sacral
foramen (65).
sagittal deformity is attained by decancellation techniques, osteotomy,
or excision of apical deformity, and is stabilized with fixation from
the pelvis to the nondysraphic thoracic spine. Better short- and
long-term results are obtained when segmental fixation such as Luque
instrumentation, with or without hooks and screws, is utilized (250).
Pelvic fixation is low profile and quite strong when the distal ends of
a rod construct are placed anterior to the sacrum or within the sacral
foramen (231,250). Consistent reduction of kyphosis from 60 to 100 degrees is recorded in the literature (227,231,232,243,244,245,250). Continued proximal spine growth of at least 1 to 3 cm is also obtained (232,238), and is preserved when the proximal fixation is placed extraperiosteally.
rates are initially high for kyphosis surgery. They include spine
infection, meningitis, skin loss and exposure of instrumentation,
failed instrumentation and pseudoarthrosis, fractures, loss of
correction, acute hydrocephalus, vascular injury, and even death (222,231,234,241,249). Vascular injury could occur because the aorta and vena cava bridge the kyphosis (236,253)
and are short relative to the spine. These structures may undergo
increased tension from the correction of the spine kyphosis. It is
obviously important to recognize altered vascular anatomy prior to
correction of kyphosis, because reports of death from aortic rupture
during surgery have been recorded (236). When
correcting the deformity it is prudent to monitor the blood flow to the
feet intraoperatively with pulse oximetry and Doppler evaluation; a
decrease in blood flow to a value below preoperative baseline status
may herald impending problems. Routine aortography is not needed except
in cases of increased risk on account of previous periaortic procedures
and potential aortic scarring (236,251).
anomalies that can occur anywhere along the spinal axis. The incidence
of congenital malformations in spinal deformity has been reported as
ranging from 15% to 38% (169,170,171). These deformities consist of fused ribs, sacral agenesis, hemivertebrae, unsegmented bars, or spondylolisthesis (254).
Spondylolisthesis may be considered to be a developmental or a
congenital condition; isthmic spondylolisthesis probably results from
increased lumbar lordosis and increased mechanical stresses caused by
spina bifida. The rate of spondylolisthesis is higher in patients with
L-5–S-1 levels of function. Spondylolisthesis in ambulatory patients
should be followed for continued progression, even though the deformity
is usually asymptomatic.
growth, and progression may be related to mechanical imbalance, hip
instability, spasticity, or diminished neurologic function. In general,
treatment of these deformities is taken in the context of whether
paralytic scoliosis or kyphosis is concurrently present. If the
congenital deformity is thought to be a contributing factor to
progression, this portion of the deformity is treated with an anterior
and posterior spine fusion. Preoperative assessment includes evaluation
with a CT scan and an MRI to rule out the high incidence of spinal cord
abnormalities. Standard operative methodology for congenital scoliosis
or kyphosis is considered if the congenital spinal deformity does not
appear to have a paralytic component. These surgical methods can
include anterior/posterior spine fusion, anterior convex
hemiepiphysiodesis, and convex posterior fusion. In rare cases a
lumbar-sacral hemivertebra can be removed if it is causing severe
pelvic obliquity.
from children born with dislocated hips to those born with located
hips, but who nevertheless develop progressive hip displacement because
of altered muscle forces across the hips. In the former, the dislocated
hip is considered teratologic and rarely presents with neonatal hip
instability.
Progressive hip instability is a result of altered forces about the hip
secondary to selective paralysis of the hip extensors and hip
abductors. Functional deformities of the hip include muscle and joint
contracture that may prevent functional standing (108).
all dysplastic hips with muscle transfers to rebalance the forces and
to prevent further hip displacement. Many of these transfers were
carried out by surgeons who had experience in muscle transfers in
patients with poliomyelitis and resultant paralysis (255,256,257,258,259,260).
In the last two decades, it has become clear that failures and
complications are associated with this procedure. It has been further
determined that an anatomically located hip does not necessarily
portend an improved level of functioning. A reassessment of priorities (261)
has led orthopaedists to refocus their attention away from hip location
and toward effective improvement of standing and gait, and prevention
of later hip arthrosis and pain. The latter problem appears to be of
minimal significance, because studies with long-term follow-up have
shown that the prevalence of pain in dislocated hips is low (109,110,117,262). Additionally, the presence of hip dislocation does not appear to affect the incidence of decubitus ulcers (109); this is more likely due to lower level of paralysis and the protective presence of sensation.
have hip dislocation or progressive instability; Carroll and Sharrard
et al. (263) found that the hips in half of the patients with myelomeningocele were either dislocated or subluxed at birth (263,264,265).
This is somewhat higher than what was reported by Samuelsson and Eklof,
who found that only 10% of hips were dislocated in neonates with spina
bifida (266). The reported incidences of hip
dislocation are directly related to how dislocation is defined, when
children are assessed for hip displacement, and the motor level of
involvement.
The incidence varies according to age, and continued hip subluxation in
thoracic-level patients may occur over time. Hip displacement in
thoracic level patients is a result of global weakness, and hips
displace because of a lack of tone. Increased muscle spasticity may
also be a contributing factor as noted in thoracic-level and
upper-lumbar-level patients (266). Hip
dislocation is seen less in patients with thoracic-level involvement
than in those with lumbar-level paralysis. The latter patients have
more profound muscle imbalance, which promotes displacement of the hips
(266).
Patients with upper-lumbar-level function have actively functioning hip
flexors (psoas and sartorius) and variable adductor power. Patients
with lower-lumbar-level involvement will have poor hip abductor and hip
extensor power, and therefore the resulting muscle imbalance may
contribute to progressive hip displacement and dysplasia. Individuals
with sacral level of function (L-5 and S-1) have abductor power and
variable hip extensor, which promotes more stable positioning;
incidences of hip displacement in this group range from 0 to 20% (266,267,268).
patient and are based upon the motor level of function of the patient,
whether one or both hips are affected, the degree of hip pathology
(contracture, dislocation, or progressive subluxation), and the
personal preference of the treating surgeon. This can be a challenging
decision in the infant or young toddler who may someday develop a
decrease in motor function and ambulatory potential because of
neurologic deterioration from tethering of the spinal cord or
syringomyelia (268,269).
Clearly such an occurrence would negate the potential benefits of
aggressive surgical methods to restore and stabilize the hip position.
However, functional goals for the hip in myelodysplasia include
maintaining enough motion to allow standing programs and effective
sitting. Historically, there has been a controversy regarding the
importance of reducing established hip dislocations and preventing
progressive hip displacement in order to benefit gait through improved
hip mechanics. In order to discern any benefit of hip location, it is
important to document the natural history of hip function in
myelomeningocele.
abduction, external rotation, and flexion. Postnatal flexion
contractures of the hip will decrease in many patients by 27 months of
age (134). Most of the flexion contractures are
seen in patients with thoracic or upper lumbar level of function and
are caused by unopposed hip flexion. Broughton et al. (271)
found that the average hip flexion contracture in children between 9
and 11 years of age was 22 to 33 degrees for thoracic- and
upper-lumbar-level patients in contrast to less than 10 degrees in
low-lumbar- and sacral-level patients (271).
Rapid increase in contractures may imply decrease in neurologic
function because of spinal cord tethering, hydrocephalus, or shunt
malfunction (108).
Controversy exists as to whether a subluxated or dislocated hip, as
shown by radiography, has any effect on gait. Some orthopaedists
suggest that radiographically located hips are essential for improving
the mechanics of gait (266,267,268,269,272). These
potential benefits should be weighed against complications (infection,
stiffness and pressure sores, fracture) and failure rates [from 30% to
50% (106,263,273,274)] that are seen from extensive surgical management (263,275).
On the other hand, others have shown that function is not directly
related to location of the hip, and many have noted that several
myelodysplastic children with dislocated hips can ambulate (104,108,109,111,117,276,277).
It is difficult to critically evaluate the existing literature to
determine the effects of hip reduction surgery on the ability of
patients to ambulate. This is caused, in part, by differences in age;
the inability to reliably and reproducibly determine neurosegmental
levels; whether hips are unilaterally or bilaterally affected; and
whether hips are reduced, subluxated, or dislocated. Much of the older
literature is unreliable because it does not take these issues into
account. However, some studies have attempted to isolate these
variables and do provide some insight into appropriate treatment.
selective review of 19 patients (mean age at review, 21 years). Six
patients with low lumbar lesions were treated for bilaterally
dislocated hips. At follow-up they found that only one hip was painful
and all patients with low lumbar functioning were ambulatory, although
44% had dislocated hips at follow-up (119).
These researchers and others have concluded that a level pelvis and
good range of motion are more important parameters of ambulatory
ability than an anatomically located hip (111). In addition, in a review of 55 patients with L-3 and L-4 motor function followed over a 17-year period, Fraser et al. (112), found that hip location or the lack thereof made no difference in the ability to walk (112).
challenge as regards abnormal gait (limb length discrepancy), pelvic
obliquity, and sitting imbalance. The literature is less consistent
about recommendations for reduction and stabilization in patients with
unilaterally dislocated hips (109,117,126,257,262,266,277). Fraser et al. (279)
have demonstrated, in a long-term follow-up, that a unilaterally
dislocated hip has little effect in patients with thoracic or high
lumbar paralysis who were predominantly wheelchair dependent (277). They reported that ambulatory patients with limb length discrepancy had abnormal gait that could
have been addressed by reduction. On the other hand, Sherk et al. found
that residual discrepancy could be expected after reduction of a
unilaterally dislocated hip because of concurrent femoral shortening
and congenital differences in the lengths of the extremities (126). Crandall et al. found no increase in the rates of decubitus ulcers in patients with unilaterally dislocated hips (109).
than 25 to 30 degrees) and external rotation contractures of the hip
may require treatment in order to improve their walking and standing
potential with the use of RGOs or HKAFOs (110,117,119,143,266,278). Feiwell et al. (111) were of the opinion that range of motion of the hip without pelvic obliquity is very important for ambulation (274).
In such patients, hip or knee contracture greater than 25 degrees
greatly increases the energy needed for stance because of the inability
to lock joints in a stable upright position. Hip flexion contractures
may lead to extreme lumbar lordosis that becomes fixed and may increase
back discomfort as well as affect the ability to transfer. Some have
suggested that release of hip flexion contractures prior to spine
surgery may decrease failure rates (278). On
the other hand, active hip flexion may be desirable if an RGO is to be
used. Proponents of this orthosis believe that the psoas should not be
released and that contractures of the hip up to 35 to 40 degrees may be
tolerated (141,142,143,279).
analysis in 17 patients with unilateral hip subluxation and three
patients with hip dislocation. They concluded that asymmetric
contractures were more likely to alter gait, independent of the
presence of unilateral hip displacement. They suggest that restoration
of symmetric motion is more crucial than surgical reduction of the
unilaterally displaced hip. Unfortunately, the low numbers of
dislocated hips in their study limits the application of their
conclusions regarding the appropriateness of surgical relocation of
unilaterally dislocated hips (279).
contractures, because there are differences between individual patients
in regard to levels of functioning and orthosis requirement. If a hip
contracture precludes the attainment of desired functional goals,
surgical release should be considered. Surgical lengthening or release
of the sartorius, iliopsoas, rectus femoris, tensor fascia lata, and
hip capsule may reduce hip-flexion contracture and allow more
successful standing programs (108,261,278). Prior to such procedures, it is important to determine whether concurrent knee flexion contractures require treatment (117).
Recurrence of hip flexion deformity is likely if severe fixed lordosis
of the lumbar spine is present prior to hip-flexion contracture
release. After the procedure, patients should be started on early
range-of-motion exercises and standing in order to prevent
osteoporosis. The benefits of hip contracture release or iliopsoas
recession include improved bracing, ease of standing, improved social
life (108), and potential decrease in hip subluxation (123).
cause of hip displacement and hip dislocation is disproportional
weakness of the hip extensors and abductors. The psoas muscle remains
strong in the upper-lumbar-level patients, and is considered to be a
major deforming force. Historically, surgeons have developed methods to
address muscle imbalance by transferring functioning muscles, which
benefit by removing a deforming force or by augmenting weakness in hip extension and abduction.
Currently, isolated psoas transfer is not performed because the
procedure does not generate enough power for ambulation. In addition,
the procedure is associated with failure to maintain hip reduction and
with the incidence of complications such as hip flexor weakness, risk
of extension contracture, fracture, and avascular necrosis of the
femoral head (125,132,263,264,284). Some have proposed or advocated simple release or recession of the psoas muscle in order to prevent dislocation (97,123).
Lindseth et al., at Indiana University, developed a constellation of
muscle transfers for patients who develop progressive hip instability (268,272). Termed the triple transfer,
this procedure consists of posterior transfer of the adductor longus
and brevis to the hamstring origin (to increase hip extension power);
transfer of the external oblique muscle to the greater trochanter; and
posterior transfer of the tensor fascia lata (the latter two designed
to increase hip abductor power). At this institution good results have
been obtained at maintaining location of the hip, as verified
radiographically (272). In a later follow-up
study, the authors concluded that these transfers were successful at
improving gait by decreasing the use of bracing (268).
not uniformly acknowledged, and it is a matter of controversy whether
the transferred muscles are actively firing or simply producing a
tenodesis caused by scarring (269). Others have
suggested that psoas transfer may have an untoward effect on gait such
as diminishing hip flexor power and decreasing the range of motion (100).
Either way, clear, consistent proof (randomized trials) of either
improved or worsened gait (through gait analysis and oxygen consumption
studies), comparing similarly affected individuals with and without
muscle transfers, has not been provided in the literature (97,98).
to maintain hip stability, some surgeons have determined that these
procedures should be done only in combination with bony reconstruction
in younger patients (194). Such procedures include combinations of psoas transfer with open reduction and femoral osteotomy (267,280,290) as described in the McKay procedure (269,291).
When surgical management of hip dislocations is indicated, treatment
may also include open reduction and capsular repair, femoral osteotomy (114,292,293), and/or pelvic osteotomy (125,194,294,295,296). Lee and Carroll (125)
have demonstrated that such procedures, when combined with psoas
transfer, are successful in stabilizing subluxated or dislocated hips
in patients with L-4 level of function (125). Results from these combined procedures are less positive in patients with higher levels of paralysis and complications.
myelomeningocele at all levels of function is important in order to
promote the ability to stand and transfer, along with the physiologic
and psychosocial benefits previously outlined (106).
Soft tissue releases of the hips and knees can be accomplished with a
minimum of morbidity in younger patients. With time, however, the
benefits of standing decrease and the personal costs of maintaining
upright posture in adolescence tend to increase. The latter is due to
the faster locomotion available with wheelchairs, increased weight of
the patient, and the extensive surgery and therapy often needed for
correcting contractures.
certainly patients with dislocated hips who ambulate, and those with
located hips who cannot ambulate. It is therefore tempting to state
that no patient with a dislocated hip, or a hip with progressive
subluxation, requires stabilization surgery. Unfortunately, what is not
known is whether a patient with hip displacement would walk or function
better if the hips were reduced and whether
the morbidity caused by the surgery can be justified by the benefit
obtained. It is probably reasonable to consider these operations in
patients with less motor deficit and greater potential to benefit.
Families should be fully informed of these issues, and their input is
needed in the planning of the treatment.
bilateral hip dislocations should not undergo surgery for reduction
unless they have strong L-5 or sacral level of function at the very
least. Treatment of unilateral hip dislocation may be indicated in
patients with L-4 to sacral-level motor function in both lower
extremities (119,125,277).
Surgical methods require open reduction and muscle transfers and
variable use of femoral osteotomy (to facilitate reduction) and pelvic
osteotomy (to improve acetabular dysplasia) (Fig. 16.13).
in tandem with femoral and pelvic osteotomies, muscle-balancing
surgeries should not be prophylactically performed in radiographically
normal hips in young patients with myelodysplasia purely on the basis
of a suspected motor level of function (97). It
is very challenging to adequately assess motor function in these
patients. Muscle transfers may be indicated in heretofore normal hips
that develop progressive subluxation in ambulatory patients with low
lumbar or sacral level of function (125,269,297). These hips may be stabilized against future displacement by using procedures such as those described by Dias and Hill (292), McKay (269), and Lindseth et al. (268,272).
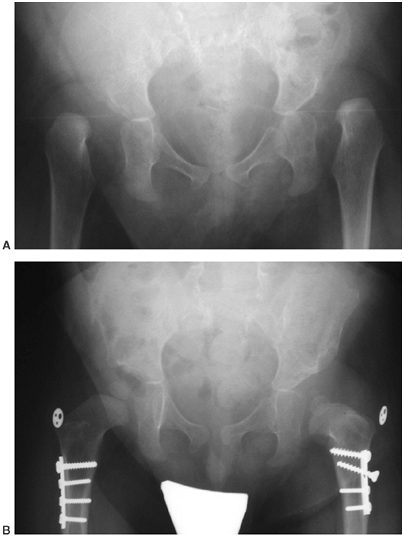 |
|
Figure 16.13
A child with L-5 motor level of function with bilateral hip dislocations. Although the indication for surgery is controversial, he underwent bilateral open reductions, femoral shortening, and triple transfer. The latter procedure is needed in order to prevent redislocation due to muscle imbalance. At 2 years his hips remain located. |
includes congenital dislocation or hyperextension deformities,
knee-flexion contractures, knee valgus and, later, instability and pain
with ambulation (157,298,299,300).
rarely, in patients with myelomeningocele because of breech positioning
and also because of unopposed quadriceps function from paralytic
hamstring muscles (299,301).
This pattern is seen in patients with L-3 and L-4 levels of function,
and affected individuals may also have ipsilateral hip dislocation and
clubfoot deformity (94). Anatomically the knee
may be simply hyperextended, with associated anterior displacement of
the hamstring muscles and shortening of the quadriceps muscle and
tendon. Dislocation of the knee is extremely rare but, when present,
the joint capsule is usually atrophic and stiff, with a small
suprapatellar pouch that may be adherent to the distal femur.
difficult because children have to lie prone while the repair of the
spinal defect is healing. The eventual methods of correction and
results of these treatments depend to a great degree on the severity of
pathology and presence of clubfoot deformity or hip dislocation. In the
latter case, manipulations of the knee are easier as the dislocated hip
and the foreshortened femur lead to relative quadriceps lengthening.
Congenitally hyperextended knees may respond to serial manipulations
and casting in a progressively more flexed position. Concurrent
clubfoot treatment with manipulation and casting may be performed if
the clubfoot is amenable to manipulation. At least 90 degrees of knee
flexion is desired; when obtained, the correction may be maintained
with a KAFO or a Pavlick harness until the clubfoot reconstruction is
done.
anterior dislocation or with anterior subluxation of the hamstring
muscles. In these cases anterior quadricepsplasty, capsular release,
and posterior displacement of the hamstrings are performed at 1 year of
age (302).
Ipsilateral clubfoot release is performed 6 weeks prior to knee
reconstruction, and the foot reconstruction is cast in as much flexion
as possible. At 6 weeks the cast is removed, knee release is performed,
and then the limb is cast in 90 degrees of knee flexion for 6 weeks,
totaling 12 weeks of casting for the clubfoot release.
capsular release, and transposition of the hamstrings, as needed; the
quadriceps is repaired at 90 to 100 degrees in order to prevent
weakness and an extensor lag (Fig. 16.14).
After surgery the child is placed in a KAFO flexed at 90 degrees during
the night and when not engaged in physiotherapy and standing programs.
The problems associated with this surgery include recurrence,
stiffness, and extensor lag that may require a locked KAFO when
standing (302). In rare instances, fixed
knee-extension contractures can persist into late childhood and prevent
adequate ability to sit; in these cases simple tenotomy of the patellar
tendon will increase knee flexion (303).
 |
|
Figure 16.14 The V-Y quadricepsplasty for hyperextension contracture of the knee. A:
In addition to the detachment of the rectus femoris tendon from the muscle of the rectus femoris, the vastus medialis, and the vastus lateralis muscles, the vastus medialis and lateralis muscles are separated from the iliotibial band, the lateral hamstrings, the medial hamstrings, and the sartorius muscles. B: When the knee is flexed, the hamstring muscles and tensor fascia lata slip posterior to the knee axis, restoring their normal function. The quadriceps muscles are then repaired in the lengthened position. (From Lindseth RE. Extension contracture of the knee. In: McLaurin RL, Oppenheimer S, Dias L et al., eds. Spina bifida: a multidisciplinary approach. New York: Praeger, 1986:40, with permission.) |
levels of paralysis and tend to progress in many patients who are
predominantly nonambulatory and wheelchair bound (301,304). Contractures with larger magnitude are usually present in thoracic-level patients as opposed to lumbar-level patients (301).
This may be due to weakness of the quadriceps muscle, and because the
legs persistently rest in a “frog” position. Further flexion deformity
may occur in these and other patients because of fracture malunion (304,305).
Thoracic-level patients have the highest incidence of flexion
contractures, and it is debatable whether hamstring spasticity
predisposes further flexion of the knee (301,304).
Finally, knee-flexion contractures can occur in ambulatory patients
because of lever arm dysfunction and an inability to obtain full
extension in stance (305).
These deformities usually occur as patients grow older; they and their
families must decide how aggressively these should be treated in order
to maintain current or better levels of function and standing. Greater
degrees of knee-flexion contracture are tolerated in patients who are
predominantly wheelchair dependent or have hip-flexion contractures
that are severe enough (greater than 45 degrees) to be the more
limiting deformity for standing and ambulation (117).
Surgery is generally recommended for knee-flexion contractures if they
are progressive and preclude appropriate bracing. These patients will
have decreased walking endurance and progressive crouch (306).
It is prudent to coordinate these surgeries, as other procedures may be
needed at the hip or foot level. Posterior release is of benefit in
younger patients with mild deformities but may additionally cause
weakness of the hamstrings and decreased hip extension power. In
thoracic-level patients, the posterior capsule, hamstring and
gastrocnemius muscles, and posterior cruciate ligament are released (304,306);
in sacral-level patients, the hamstring tendons are z-lengthened to
maintain knee flexor power and hip extensor power. Care is needed in
assessing and protecting the neurovascular structures, which are often
the last remaining structures. Casts are often used to maintain
reduction; however, a high incidence of sores and wound sloughing
persists despite the use of great care in application (304).
for correcting flexion contracture in order to prevent recurrence from
remodeling (304). This can be done with rigid
internal fixation, which allows early standing and prevents
osteoporosis and secondary fractures. Osteotomy can be performed with
soft-tissue release (310) in such a manner as
to correct residual coronal plane malalignment (valgus) or internal
rotation of the femur. Anterior hemiepiphyseal stapling has been
described and may be efficacious in growing individuals with minimal
deformities (309).
In ambulatory patients with thoracic- and low- lumbar-level paralysis,
additional valgus knee instability is common. This is a result of the
adaptive gait from knee flexion and hip and knee weakness. Valgus
appears to be more significant in patients with concordant knee flexion
deformity in stance because of weakness of the abductors, quadriceps,
and gastrocsoleus (157). The knee is driven
into valgus from above because of several forces. Some patients may
have persistent and excessive femoral anteversion that internally
rotates the femur in stance phase (156). This is accentuated as the ipsilateral hemi pelvis is internally rotated to accommodate for the weak hip abductors (312).
Valgus is further promoted from above as the hemi pelvis displaces
laterally to accommodate hip abductor weakness, and this motion moves
the joint’s reactive force lateral to the center of the knee and
increases the adductor moment arm of the knee (156). Knee valgus can also be promoted from below because of persistent external tibial rotation (155,312).
If the tibia or foot is externally rotated, lever arm dysfunction and a
decrease in the plantar flexion–knee extension couple will occur,
promoting knee flexion and increasing the valgus stress in the knee (155).
the joint and degenerative changes in the lateral joint surface. Knee
pain and anatomic laxity of the joints in ambulatory adults with lumbar
or sacral levels of function can approach 30% (155,157,298,313).
This may be a consequence of degenerative changes as well as of the
increased patellar forces. These patellar forces arise from lateral
subluxation and persistent knee flexion that increase the joint’s
reactive force on the patellar cartilage surface (157).
Surgical treatment of external tibial torsion has been shown to
decrease the valgus stress at the knee and improve knee pain (166).
to decrease the Trendelenburg gait and, subsequently, the lateral
moment arm of the knee. In addition, the patients may benefit from
KAFOs with medial and lateral uprights that help stabilize the knee (115,156).
Surgical treatment may be indicated in patients with severe valgus
caused by anatomic deformity in addition to the dynamic deformity from
muscle weakness and accommodative gait maneuvers. Distal femoral
osteotomy producing varus, extension, and external rotation may be
helpful in reorientating the knee if anatomic femoral valgus deformity
is present (94,298,299).
Tibial derotational osteotomy may be needed in cases of external tibial
torsion that prevents anterior placement of the foot and efficient use
of a floor-reaction AFO (155). Internal fixation is used for these procedures, and immediate standing is encouraged to prevent osteoporosis.
individuals with myelomeningocele, and each patient requires
individualized treatment depending on the level of paralysis and
expected ambulatory potential (Fig. 16.15). For
instance, the nonambulatory child will require adequate positioning of
the foot to accept shoes, prevent pressure sores, and be a candidate
for standing programs. In general, ambulatory patients require
alignment of the foot for the most favorable transfer of the floor
reaction force through the center of the ankle, and to produce
stability of the knee and hip during the stance phase of gait. The
motion of the joints in the foot should be preserved as much as
possible. This will allow preservation of the shock-absorptive capacity
of the foot and lessen the possibilities of degeneration of the joints
and skin ulceration.
the context of other orthopaedic conditions. For instance, severe
congenital foot deformity in a patient with thoracic-level paralysis
(nonambulatory patient) takes precedence over the possible presence of
a dislocated hip. On the other
hand,
severe kyphosis, occasionally seen in thoracic-level patients, should
be corrected prior to foot deformities. Correction of the spinal
deformity provides more significant overall improvement. In general,
foot deformities in patients with lower lumbar involvement (with good
ambulatory potential) take high priority in order to facilitate
ambulation. The goal of treatment in the ambulatory patient with
myelomeningocele is maintaining a braceable, ulcer-free, plantigrade
foot that can be cleared during swing phase and yet is an effective
lever arm during late stance phase. Shoe inserts and modifications are
rarely successful in providing correction of alignment and position
deformities of the foot.
 |
|
Figure 16.15
Clinical photograph of an 11-month-old child with myelomeningocele with L-5 motor level of function on the right and subsequent development of a calcaneal valgus foot because of strong anterior tibialis power. He has weak L-4 motor level of function on the left, with a stiff clubfoot. Surgical treatment will include simple release of the anterior tibialis and long toe extensors on the right and posterior medial release on the left at 14 months of age. |
be needed to balance and realign the foot. Surgical correction of foot
pathology requires treatment of structural deformity and balance of the
extrinsic and intrinsic foot muscles without producing undue stiffness.
For correcting muscle imbalance the options include: (a) removal of the
muscle force by excision or lengthening of the tendon; or (b) transfer
of the functioning force to another location. The power and the phasic
activity of the muscle during gait need to be considered in determining
whether the transfer will be sufficient to control the floor reaction
forces during the stance phase of gait. If the child will need an
orthosis to walk despite muscle transfer, then muscle control of the
foot is of little importance and simple release may be just as
efficacious. Additionally, transfer of a tendon to correct a deformity
may result in muscle weakness and production of a secondary problem.
For example, transfer of the anterior tibialis to a more lateral
position in the foot may correct dynamic foot supination and inversion,
yet result in a drop foot caused by weakness after transfer.
somewhat related to the level of paralysis and characteristic patterns
of muscle imbalance (314). However, within each
level of paralysis, there is a variable incidence of different foot
deformities. Therefore, it is difficult to attribute the occurrence of
these deformities to simple imbalance of voluntary muscles (315,316,317,318).
Common foot deformities include clubfoot (midlumbar levels of
paralysis), congenital vertical talus, calcaneal valgus deformities
(low lumbar and sacral lesions), ankle valgus, and cavus deformities (316).
Neuropathic foot problems such as destruction of a joint or skin
ulceration may result from inadequate treatment of these presenting
conditions.
deformities and is the most common congenital foot abnormality
requiring surgical treatment in myelomeningocele (314,317,319,320).
It can present at all levels of paraplegia but is more common in
patients without motor function below the fourth lumbar level. The
clubfoot deformity is significantly stiffer and more resistant to
manipulation in comparison to idiopathic clubfeet. Although difficult,
an attempt to manipulate and cast the deformity during the newborn
period may be worthwhile in the extremely rare instance when the foot
is supple and can be manipulated into a satisfactory position prior to
the casting (321,322). The cast can only hold the correction obtained by manipulation and cannot be used to obtain correction, because pressure sores may develop (314,317).
In uncommon cases of satisfactory correction, the infant must then be
placed in orthotics to prevent recurrence of the deformity. The braces
must be worn continuously until standing age.
patients with severe deformity, or if the foot correction is not making
satisfactory progress by the time the infant is 3 months of age.
Although some have recommended primary talectomy (323)
in myelomeningocele, most surgeons perform extensive posteromedial and
lateral release as a primary method of correction. Performing the
surgery at an age when the child is able to stand utilizes weight
bearing as well as an orthosis to maintain correction. The surgical
procedure for clubfoot in myelomeningocele is different from the one
performed in idiopathic clubfoot. In a patient with myelomeningocele,
tissues are stiffer, joints are tighter, and soft tissue coverage of
the posteromedial aspect of the foot presents a major problem during
surgical correction. After skin incision, all of the paralyzed and
spastic or unbalanced tendons should be resected for 1 to 2 cm rather
than lengthened (319,324).
The surgical correction of the clubfoot in myelomeningocele is also
different from idiopathic clubfoot in that a complete circumferential
subtalar and midfoot release is required to correct the residual
deformity. Following surgery, the patient is placed into long leg
splints and casts until healing is complete, and eventually fitted with
AFOs that are worn during the day.
The treatment of the recurrent deformity is dependent on several
factors including the age and the functional level of the patient as
well as the previous treatment of the clubfoot. In toddlers, recurrent
clubfoot may be treated with a repeat posteromedial release (324).
Occasionally, residual forefoot adduction requires further surgery,
such as metatarsal or midfoot osteotomy, when the child is 3 to 5 years
of age (319).
require talectomy. In early reports, good short-term results have been
obtained (320,323,326,327,328). Talectomy certainly has a place in the treatment armamentarium for severe recurrent deformities in the older child (328);
in these cases other options are limited. Although it is considered to
be effective in reducing a majority of deformities, talectomy rarely
allows full correction of residual forefoot deformity (317,320,324,325).
Uneven distribution of pressure on the plantar surface may persist
(even when the foot is plantargrade) and predispose these patients to
ulcerations (315,323). Additionally, the short distance from the medial malleolus to the plantar aspect of the foot makes
orthotic fitting difficult. In the long term, the beneficial results of
talectomy tend to decrease with time because of recurrence of hindfoot
deformity (320,324,327,329,330) and adjacent osteoarthrosis (326). Long-term use of AFOs is needed in these patients in order to decrease the chance of recurrence (324).
deformity include hindfoot and midfoot osteotomies, and triple
arthrodesis (331,332).
The use of a triple arthrodesis should be limited to extreme cases
because of the risk of pseudoarthrosis, the danger of joint
degeneration, and the risk of skin ulceration related to the stiffness
of the foot (333,334).
In this deformity, there is weakness and contracture of the posterior
muscles; the intrinsic muscles fail to develop; and the anterior crural
muscles (which may function) cause dorsiflexion of the midfoot with bow
stringing of the anterior tibialis and peroneal tendons (336,337).
The congenital vertical talus is rigid and cannot be treated by
nonoperative means. With time, the untreated valgus deformity becomes
difficult to brace; foot pressure sores may develop under the prominent
head of the talus. An aggressive surgical approach is needed for
releasing the deforming tendons and capsules. The anterior tibial
tendon can either be resected or transferred back to the neck of the
talus. Occasionally an extraarticular subtalar arthrodesis is needed
later to stabilize the subtalar joint (338).
Care is needed in selecting these patients because there may be a high
rate of occurrence of later skin problems after arthrodesis (339).
and are caused by the unopposed voluntary or spastic pull of the
anterior tibial muscle, the toe extensor muscles, or the peroneal
muscles. The calcaneal deformity may be neutral, valgus, or varus
depending upon the predominant muscle activity. With time the deformity
progresses, and the calcaneus eventually becomes vertical underneath
the talus, promoting excessive pressure under the calcaneus and
preventing the forefoot from contacting the floor. This results in
decreased knee extension moment and poor knee stabilization in stance
(causing the child to walk in a flexed-knee or crouched gait);
interference with balance because of reduced surface area of the foot;
and increased risk of sores because of lack of sensation. The deformity
leads to heel ulcerations in middle and late adolescence (340).
In cases of severe deformity, and in children older than 4 years, the
anterior tibialis tendon may be transferred (provided its muscle
strength is at least 4 out 5) through the interosseus membrane and
attached to the os calcis (340,343,344,345,346,347,348).
This transfer will likely arrest further calcaneal deformity,
especially if the remaining contracted anterior structures are released
(343,347,349). Anterior tibialis transfer may be supplemented with peroneal transfer (314) or Achilles tenodesis (344,350,351).
This transfer is designed to make the foot braceable and ulcer free;
unfortunately the transferred tendon does not uniformly provide enough
power for braceless ambulation, and secondary equinus deformity or foot
drop may develop (343,344,347). Better results are likely in sacral-level patients with 5 out of 5 muscle power (340,345). Combined anterior tibialis transfer and Achilles tenodesis may be more effective in decreased knee flexion in stance phase (344).
Therefore, simple resection of the offending extensor tendons at an
early age may be preferred over transfer, if brace wear is to be
expected ultimately. Release may be performed in the child younger than
5 years and allows the foot to be brought into satisfactory position
for bracing (343). This procedure is more
predictable than transfer in patients with weaker anterior tibialis
function, or when a high degree of uncontrolled spasticity is present (343).
may occur in the subtalar joint, in the ankle, or in both sites.
Radiographs taken with the patient in weight-bearing position
demonstrate valgus of the ankle; there is a lateral tilt to the ankle
mortise with shortening of the fibula and wedging of the tibial
epiphysis (162,352).
When radiographic evidence of ankle valgus is lacking, the deformity is
present in the subtalar joint. Radiographs of the foot taken with the
patient standing will demonstrate a widened talocalcaneal angle, and
axial views of the weight-bearing heel will also confirm deformity to
be present in the subtalar joint. Supramalleolar osteotomy may be
indicated in patients with severe ankle valgus that is predominantly at
the distal tibial level. Alternatively, a medial tibial epiphysiodesis
with a screw (353) or a staple (162) may be worth considering in isolated cases of ankle valgus deformity (161).
If the valgus deformity is mild and associated with a calcaneus
deformity, a calcaneal fibular Achilles tenodesis can prevent
progression of the deformity in approximately 70% of the patients, and
theoretically may prevent excessive ankle dorsiflexion during toe off (161,350).
Complications of this procedure include stretching and loss of the
tenodesis effect and the possible development of equinus contracture,
especially if the procedure is combined with posterior transfer of the
anterior tibialis (347).
subtalar and triple arthrodesis have been suggested for severe subtalar deformity (163,338),
arthrodesis is generally avoided because it increases the risk of skin
sores and Charcot-like changes in the ankle joint as the child matures.
The remaining options include medial displacement calcaneal osteotomy
or a calcaneal lengthening procedure. In the latter procedure, a wedge
of bone is placed in an osteotomy through the calcaneus at a level
between the anterior and medial facets. The effect is that the lateral
column of the foot is lengthened relative to the medial column, and the
talonavicular joint reduces. This procedure is beneficial as it also
corrects an accompanying midfoot abduction deformity, which can
accentuate external foot progression angle. Often tendo-Achilles
lengthening is required after calcaneal lengthening, and good results
have been noticed in most patients (354).
The surgeon must remember that the healing rate of the supramalleolar
osteotomy is prolonged in comparison to calcaneal osteotomies, and
pseudoarthrosis may develop with premature cast removal. Weight bearing
must be delayed for 8 to 10 weeks, and then gradually begin with the
protection of a well-fitted cast or rigid ankle-foot orthosis.
The latter are caused by paralysis of the intrinsic muscles of the foot
and imbalance of the extrinsic muscles. Progressive forefoot deformity
will result in ulcerations on the toes and under the metatarsal heads.
Varus deformity may also be seen because of muscle imbalance and
resultant ankle instability and lateral column overload.
with severe calluses or ulcerations and ankle instability due to
hindfoot varus. Orthotic treatment can redistribute the foot pressures
and prevent ankle instability. Surgical treatment may be indicated if
orthotic wear is ineffectual or if the deformities are progressive. In
the young child with mild deformation, a plantar fascia release
followed by an ankle-foot orthosis may provide satisfactory correction
of the cavus deformity. Rigid cavus due to midfoot deformity can be
corrected by a metatarsal or midfoot osteotomy combined with plantar
release. A calcaneal osteotomy is occasionally necessary for excessive
calcaneal pitch or fixed varus. Muscle transfers may be used to
rebalance the deforming forces, provided the transferring muscles are
strong enough and no fixed deformity exists. Such procedures may
include lateral transfer of the anterior tibialis tendon (to correct
persistent supination) or recession of the extensor tendons of the toes
to help elevate the metatarsal heads and prevent recurrent cavus.
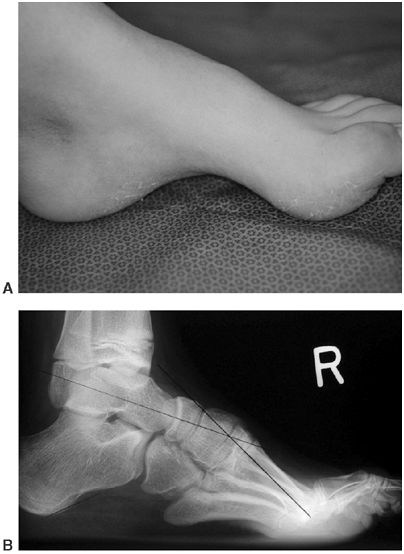 |
|
Figure 16.16
A 12-year-old boy with a sacral-level myelomeningocele, with a cavus foot. Radiographs demonstrated a plantarflexed first metatarsal and clawing of the toes. |
neuropathic problems because of the aforementioned hip and knee
dysfunction that alter normal gait and may lead to asymmetric
distribution of foot pressures. Examples include concentrated forces on
the plantar lateral foot surface from residual varus deformity or
residual calcaneus deformity (Fig. 16.17). On
the other hand, valgus deformity in the knee may lead to excessive
pressure over the head of the talus. Stiffness of the foot decreases
the ability of the joints to absorb shock, and that increases the
possibility of Charcot joint degeneration and skin ulceration (339). Case reports of osteonecrosis and collapse have also been noted in the knee, and these cases likely followed axial trauma (356,357).
sensory level that is above the motor level, and therefore lack
protective sensation in the joints of the lower extremity
The
L-4—L-5 patient appears to be the most vulnerable; however, patients
with the sacral level of paraplegia may also have Charcot changes (Fig. 16.18). Failure to recognize these problems often results in pressure sores and inability to use braces or shoes.
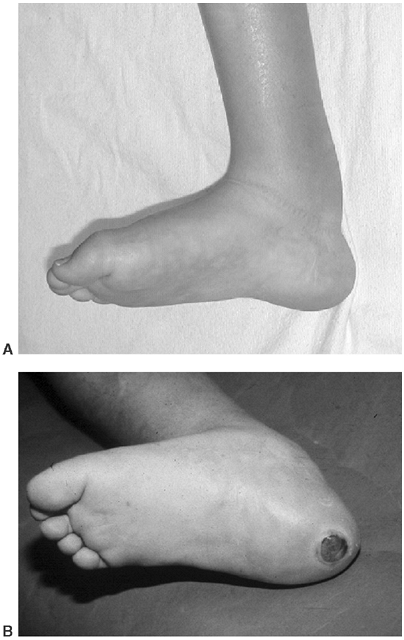 |
|
Figure 16.17
A teenager with L-5 function and a calcaneus deformity with a neuropathic ulcer under the point of the calcaneus. (Courtesy of Charles T. Price, MD) |
be quite mild or occurs after surgery and immobilization, at which time
the bones are osteoporotic. There is usually no severe pain but there
may be some minor discomfort that the patient pays little attention to.
Following initial trauma there is swelling and redness, which gives the
appearance of cellulitis or deeper infection. Because the initial x-ray
films are often unremarkable, the patient may be given antibiotics for
the mistaken diagnosis of infection. Progressive arthropathy occurs
after the patient resumes walking, leading to further microfractures
and joint destruction. For Charcot arthropathy to be successfully
treated, the condition must be suspected and the diagnosis made early,
rather than waiting for radiographic confirmation. Treatment consists
of complete joint protection by applying a splint or cast in order to
ensure that there is no weight bearing. If the early treatment is
successful, radiographic changes may never be identified. Protection
must be resumed for a longer period of time if redness and swelling
recur after the onset of weight bearing. A polypropylene orthosis with
accommodative lining is usually an effective combination for
maintaining alignment of the joint and equal pressure distribution.
patients grow older because the increase in weight is relatively more
rapid than concurrent increases in pedal surface area (357). With time, the foot may also change with regard to deformity and stiffness following treatment such as arthrodesis (339). Treatment of these conditions is difficult and expensive (358); minor foot ulcerations require
initial healing with wound care and total contact casting or non–weight
bearing, followed by primary correction of the foot deformity.
Concurrent treatment of spinal cord tethering, where present, may be of
benefit in those patients with persistent skin breakdown (359).
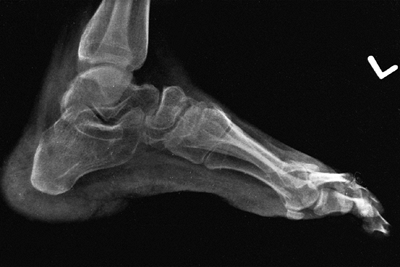 |
|
Figure 16.18
Lateral radiograph of an L-5 paraplegic patient shows the vertical position of the os calcis. Note the hypertrophy of the heel pad. |
fat pad must be restored in order to prevent recurrence of the
ulceration. Treatment consists of secondary excision of the prominent
part of the os calcis and the ulcer, with primary closure by means of a
local flap. Simply allowing the ulcer to heal by secondary intention
will not provide reconstitution of the normal weight-bearing fat pad,
and repeated ulceration can be expected. Posthealing orthotic wear is
important, and compliance is essential. If unprotected walking is
allowed and recurrent ulceration occurs, osteomyelitis of the calcaneus
may develop. This is a difficult problem that may require additional
resection of the calcaneus and flap coverage, or in some cases require
a below-knee amputation to restore ambulatory status.
with myelodysplasia are a common occurrence after minimal or
unrecognized trauma. Approximately 10% to 30% of patients with
myelomeningocele will suffer such an injury, and these usually occur
between the ages of 5 and 13 years (360,361,362,363,364,365).
This age range may be because of the early age at which many
myelodysplastic individuals undergo surgical intervention for
orthopaedic conditions, and consequently develop disuse osteopenia (363,366,367,368).
Higher rates of fracture are noted in patients with higher levels of
paralysis who likely have disuse osteoporosis from limited ambulation (365), and in whom decreased bone mineral density has been documented (369). Although this is rarely mentioned in the literature (367),
one should always consider child abuse as a causative agent, especially
if there are multiple fractures at various states of healing; if
lower-extremity fractures occur simultaneously with upper-extremity
fractures; or if posterior rib or spine fractures are present. A subset
of fractures relates to neonates with severe contractures, who may
undergo treatment in the perinatal period with casting or physiotherapy
(364). Necropsy studies of infants demonstrate bony changes consistent with cortical atrophy and osteoporosis (370).
are easily detected on radiographs with deformity and exuberant
formations of fracture callus. Patients with these fractures are more
likely to have a history of trauma than individuals with physeal
fractures do, but they will have little to no pain (87,365,371,372,373,374). Symptoms such as erythema, swelling, and low-grade fever may lead to mistaken diagnosis of infection (367,375). Because elevations in sedimentation rate and white blood count may occur (375,376,377),
evaluation of the inflamed area with plain radiographs is essential;
when fracture lines are seen, the diagnosis of fracture is confirmed.
fairly straightforward. However, it is important to realize that rigid
cast immobilization is likely to further increase the osteoporosis that
may have been an inciting factor in the first place. Furthermore, these
fractures usually occur without pain. In the healthy child, pain leads
to a natural restriction of activity, and therefore of motion, at the
fracture site. Despite these factors, immobilization in rigid casts,
splints, or bulky soft dressings is needed for 3 to 6 weeks (365).
If rigid cast material is initially used, it should be replaced with
more flexible material in 2 to 3 weeks, at which time weight bearing is
also increased. Great care is needed when applying casts on patients
with no sensation, because the rates of occurrence of skin sores and
cast burns is higher in such patients (365). In general, these fractures heal without difficulty or long-term sequelae (363,364,372,373,375,378).
Operative stabilization is rarely needed in metaphyseal or diaphyseal
fractures; indications for potential surgery might include recurrent
fractures or severe malalignment in the lower extremity in adolescent
individuals with good ambulatory abilities (379).
fractures, tend to occur in low-lumbar-level individuals with
relatively higher ambulatory ability, and may be due to repetitive
microtrauma (87,361,372,380). Rodgers et al. (205,381)
hypothesize that this injury is a result of “chronic stress or trauma
to the poorly sensate limb (which) produces micromotion at the zone of
hypertrophyleading to fracture” (381). Acute
physeal fractures are rarely associated with a history of trauma, and
are particularly hard to detect because they may not produce the
systemic signs that are seen in metaphyseal or diaphyseal fractures (361,372).
Common locations of physeal injury include the distal tibia and femur;
the less common sites are the proximal tibia and femur (362). These fractures typically do not show much displacement, and the widening of the physis, although present (373), is often hard to detect. Stress radiographs (375) or MRI may be helpful in the diagnosis of acute physeal fractures in select cases (381).
fractures demonstrate considerable bone formation and may increase the
differential diagnosis by adding sarcoma (381,382), fibrous dysplasia, and metaphyseal dysplasia (Fig. 16.19). Historical diagnostic considerations also include scurvy, syphilis, and rickets (87,376).
Exuberant bone formation may extend up or down the bone and is probably
caused by delayed diagnosis and fracture instability (374).
Significant elevation of the periosteum could be present because of
poor muscle mass or tone. Such significant periosteal stripping is
absent in normal children with physeal injuries. Irregardless of the
pathophysiology, the bone formation tends to persist for many years and
does not remodel at the same rate as in normal, sensate children (383). When it does remodel, the bone may have the classic Erlenmeyer-flask appearance of Gaucher disease (87).
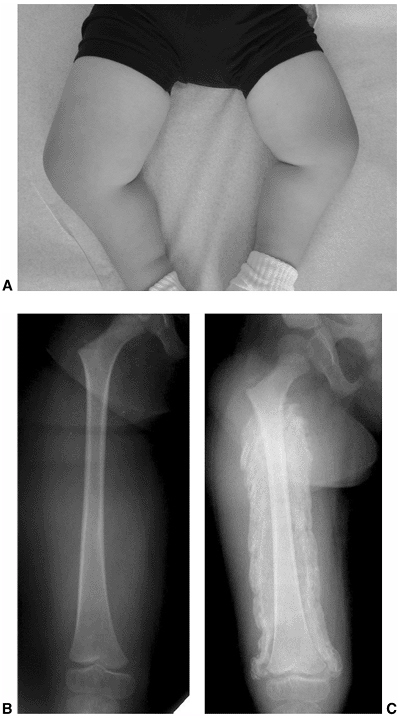 |
|
Figure 16.19 A: Clinical picture of a 7-year-old boy with upper lumbar level paralysis, who presented with a warm and swollen right leg. B: Radiographs of this child demonstrate a physeal fracture with extensive periosteal reaction. (Courtesy of David Skaggs, MD)
|
being acute and may be treated with more standard immobilization and
activity modification. Subacute or chronic physeal fractures are harder
to care for and may require more prolonged treatment in order to avoid
displacement, growth arrest, and deformity (361,371,372,373,377,380,381,384).
The duration of immobilization required for healing is unknown and
should be individually determined for each patient. It is prudent to
immobilize these fractures until swelling and warmth have completely
resolved, and the width of the growth plate is equal to that on the
contralateral side (372). Some discordance may
be expected between the clinical and radiographic methods of estimating
healing, and this may be a factor in the reported differences in
treatment (373). Extended durations of immobilization for up to 18 months are sometimes required (381). The average duration of immobilization ranges from 1 to 5 months (361,372,380,381).
fractures may be needed for delayed healing and is effective in
promoting healing. However, the potential for premature growth arrest
is likely increased (362,381).
The intermediate and long-term sequelae of chronic physeal fractures
include stiffness, growth arrest, and other complications (361,380,381).
The significance of growth arrest should be assessed separately for
each patient; for instance, it is likely of less consequence in
nonambulatory patients.
characterized by lack of portions of the distal portion of the spine.
Affected individuals have variable deficiencies in motor function below
the lowest level of the remaining normal spine. Sensation tends to be
preserved at much more caudal levels. In more severe cases of sacral
agenesis, a part or whole of the lumbar spine may be absent, with the
potential for pelvic-spinal instability (385,386,387,388,389,390).
Other abnormalities of the musculoskeletal system include
lower-extremity deformities such as pterygium. As in patients with
congenital scoliosis, affected individuals may have cardiac anomalies
and renal or visceral abnormalities (391,392).
patients according to the amount of sacrum remaining and the
characteristics of the articulation between the spine and the pelvis (Fig. 16.20).
Type I has either total or partial unilateral agenesis of the sacrum.
Type II has partial sacral agenesis with a bilaterally symmetrical
defect, a normal or hypoplastic first sacral vertebra, and a stable
articulation between the ilia and the first sacral vertebra. Type III
has variable lumbar and total sacral agenesis, with the ilia
articulating with the sides of the lowest vertebra present. Type IV has
variable lumbar and total sacral agenesis, with the caudal end plate of
the lowest vertebra resting above either the fused ilium or an iliac
amphiarthrosis. The level of involvement in sacral agenesis is best
described by the most caudal normal vertebrae, and this usually
corresponds with the motor level of disability in affected children.
and appears to occur with equal frequency according to gender. Most
cases involve absence of the sacrum and sometimes some lumbar vertebrae
as well. The exact cause of sacral agenesis is unknown; however, an
increased association with maternal diabetes has been
noted by some (395,396).
Some have documented the rate of sacral agenesis in infants whose
mothers have diabetes (0.25% to 1%) to be higher than that in the
general population (394,397).
However, because of possible selection bias in the studies, it is
possible that the true incidence of maternal diabetes and the extent of
its increased risk cannot be firmly established (398).
Other researchers have reported the incidence of sacral agenesis in
infants who have no background of maternal diabetes. Also, cases of
familial sacral agenesis have been reported. For instance, Type 1
sacral agenesis is often inherited in either an autosomal or a
sex-linked dominant pattern (399,400).
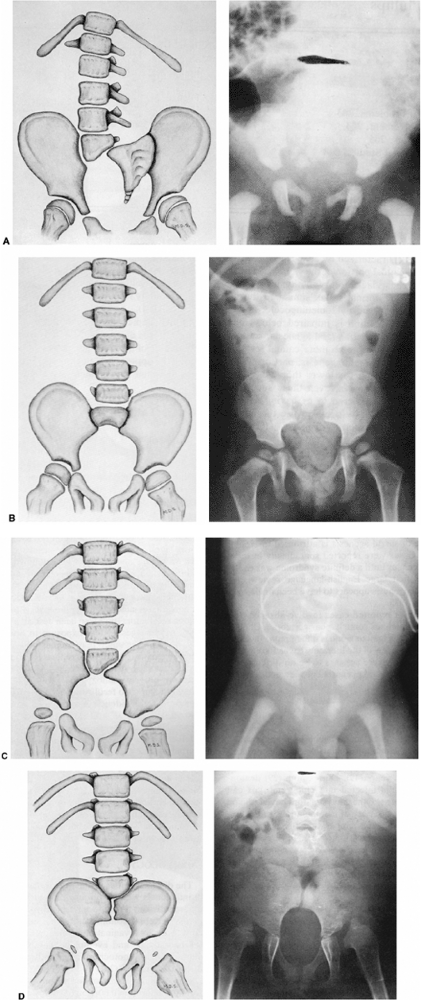 |
|
Figure 16.20 Renshaw classification of sacral agenesis. A: Type I sacral agenesis—unilateral agenesis of the sacrum. B:
Type II sacral agenesis—partial sacral agenesis with a bilaterally symmetrical defect, a normal or hypoplastic first sacral vertebra, and a stable articulation between the ilia and the first sacral vertebra. C: Type III sacral agenesis—variable lumbar and total sacral agenesis with the ilia articulating with the sides of the lowest vertebra present. D: Type IV sacral agenesis—variable lumbar and total sacral agenesis with the caudal end plate of the lowest vertebra resting above either the fused ilia or an iliac amphiarthrosis. (From Renshaw TS. Sacral agenesis: a classification and review of twenty-three cases. J Bone Joint Surg Am 1978;60:373,374,375,376,377,378,379,380,381,382,383, with permission.) |
associated myelomeningocele is usually within one level of the last
normal-appearing vertebral body, and these patients will often have
more normal sensation distal to the level of motor involvement. Almost
all individuals with sacral agenesis have affected sacral roots, and
will subsequently develop a neurogenic bladder (401,402).
As opposed to individuals with myelomeningocele, the motor level in
patients with sacral agenesis does not usually change with growth.
Patients who experience increasing motor deficit should be evaluated
for cord tethering (403,404) or lipomas (386).
Appropriate neurosurgical referral is needed, because surgical
correction of spinal dysraphia may result in improvement in previously
stable deficits (405). When sacral agenesis is
associated with myelomeningocele, the sensory and motor deficits
correspond to the level of the myelomeningocele.
appears to parallel the severity of spinal involvement. Children with
sacral agenesis above the first lumbar vertebra tend to have the most
severe contractures. Associated problems include foot deformities such
as clubfoot; hip dysplasia and teratologic hip dislocation (390); knee-flexion contractures and pterygium; limb-length discrepancy; and
congenital anomalies of the spine, such as congenital scoliosis and Klippel-Feil syndrome (390,393,406,407).
In severe sacral agenesis (Types III and IV), the spine and pelvis are
poorly associated, thereby resulting in lumbar-sacral kyphosis and
increased motion between spine and pelvis. These patients sit with
knees flexed (because of knee contractures), and the hips are
externally rotated. With increased spinal-pelvic instability they prop
sit with support from the upper extremities, which fortunately are
spared significant involvement. In severe cases, ambulation is
difficult because of combinations of paralysis, spinal-pelvic
instability, and severe joint contractures.
predict from the literature, and depends on the severity of the
condition. However, patients with severe sacral agenesis eventually
become wheelchair-dependent because of lower limb contractures, which
are associated with greater neurologic deficit. Because of the comorbid
anomalies of the lower extremity, the problems associated with
spinal-pelvic instability rarely lend themselves to surgical
stabilization. Like patients with thoracic-level myelomeningocele, most
of the patients with severe sacral agenesis are dependent on others for
assistance with the activities of daily living.
each patient’s requirements, and depends on distal motor function,
stability of the spinal-pelvic articulation, and the presence and
severity of lower limb contractures. Treatment at each level must be
considered in the context of the degree of associated pathology at
adjacent joints. For instance, moderate clubfeet may not necessarily
need to be corrected if the affected child has severe knee pterygium
and spinal-pelvic instability that effectively preclude ambulation.
instability, including dependence on the arms as props while sitting,
compression of the intraabdominal contents from collapsing
lumbar-sacral kyphosis, and skin breakdown over the kyphosis. In an
effort to overcome these problems, some have attempted to stabilize the
spinal-pelvic junction (197,408,409,410,411,412).
been reported, resulting in improved sitting and gastrointestinal
function. Although it seems intuitive that achieving such stability is
desirable, the benefits must be weighed against the potential
complications from these surgeries, which are very extensive (390,413).
In addition, some surgeons have had to reoperate to increase length of
the spine and to supplement atrophic fusion mass with additional bone
graft. Complications from this treatment may include infection, back
pain, increased hip problems, and a failure to really improve the
quality of life of the affected patients, many of whom might have
developed improved stability with time if left unattended (390,413).
In summary, although some studies have demonstrated that fusion is
attainable, it is probably not routinely indicated in patients without
problems.
surgical release of severe contractures is doomed to failure in
patients with spinal-pelvic instability. This instability will fail to
render an efficient lever arm during postoperative therapy. Because of
this, recurrence of contracture is likely. On the other hand,
spinal-pelvic instability can accommodate hip contractures and make
ambulation possible. The indications for hip surgery in patients with
dislocated hips are similar to those in patients with myelomeningocele.
Bilateral hip dislocations in children with high-level sacral agenesis
and with severe associated deformities in the lower limb do not require
reduction, because these patients are not likely to be good ambulators.
Rare unilateral dislocations in milder forms of sacral agenesis may
benefit from surgical reduction. As many of these patients have
anatomic limb-length discrepancy, proximal dislocation of the hip does
accentuate this problem and necessitate extensive orthotic wear. Muscle
transfers may be needed in order to prevent recurrence of dislocation
in patients with weak hip abductors and hip extensors.
reveal muscles with striking similarity to muscles from children
afflicted with arthrogryposis (394,406).
The degree of knee contracture appears to correlate with the level of
quadriceps functioning. In severely involved individuals with
pterygium, the neurovascular bundle tends to be subcutaneous at the
apex of the web. The knee contractures can be greater than 120 degrees
and are difficult to correct because of subcutaneous location of the
neurovascular bundle (which tends to be the limiting factor), muscle
stiffness, and joint fibrosis.
straightening knee contractures and thereby improving orthotic fitting
in patients who could be ambulatory. Soft-tissue releases, femoral
osteotomy, and the use of the Ilizarov device have all been attempted
to correct these contractures. After surgical correction, prolonged
splinting is also needed in order to avoid recurrence as the patient
grows. Because many of the methods mentioned in the preceding text are
fraught with complications, high rates of recurrence, and the need for
further procedures, some surgeons have recommended amputation through
the knee and prosthetic fitting in children with severe hip and knee
flexion contractures.
as congenital vertical talus and clubfeet tend to be stiff and similar
to those seen in arthrogryposis (414,415,416,417).
As mentioned earlier, the rationale for correcting foot deformities
depends on the ambulatory potential of the child. Standard surgical
release of the kind that is used in other neurologic foot deformities
is usually sufficient. Fortunately, these individuals have good distal
sensation, and so the incidence of Charcot arthropathy and neurogenic
sores is lower than that seen in myelomeningocele.
agenesis is challenging, and the treatment should focus on improving
the natural history of these complicated cases. The improvement depends
on realistic expectations of function and is balanced by the morbidity
associated with these treatments.
understanding and improvements in the care of affected individuals with
myelodysplasia and spinal dysraphism. The medical community has
transformed a uniformly fatal disorder into one where patients can
expect multiple treatment modalities to improve the residual morbidity
associated with myelomeningocele. Orthopaedic surgeons have also kept
pace with advances in orthotics, surgical techniques, and
instrumentation. More importantly, they have increased their
understanding of the natural history of these conditions and are better
able to selectively indicate appropriate surgical and nonsurgical
treatments. However, it is important to realize that myelodysplasia as
a disease entity will be with us for the foreseeable future, despite
the use of folic acid. Furthermore, it is currently not known whether in utero
surgery and repair of the defect will alter the incidence and severity
of the residual morbidity. Orthopaedists must continue to study and
question what they can do to help these individuals obtain the greatest
possible functional improvement and independence.
guidance from Drs. Martin Kaefer and Bermans Iskander, whose respective
expertise in pediatric urology and pediatric neurosurgery were
invaluable. The author also recognizes Dr. Richard Lindseth, whose
previous accomplishments, expertise, and writings in this field guided
the current version of this chapter.
EH, Churchill BM, Jayanthi VR, et al. The sensitivity of pressure
specific bladder volume versus total bladder capacity as a measure of
bladder storage dysfunction. J Urol 1978; 119:551.
JM, McLone DG, Storrs BB, et al. Analysis of 153 patients with
myelomeningocele or spinal lipoma reoperated upon for a tethered cord. Pediatr Neurosurg 1993;19:243–249.
R, Strehl E, Wenzel D, et al. Does growth hormone (GH) enhance growth
in GH-deficient children with myelo-meningocele? J Clin Endocrinol Metab 2000;85(8):2740–2743.
DH, Tchernoukha K, Bazmi B, et al. Change in spinal curvature following
release of tethered spinal cord associated with spina bifida. Pediatr Neurosurg 1994;20:30–42.
PR, Ragheb J, Sevald J, et al. Cerebrospinal fluid shunt complications
after urological procedures in children with myelodysplasia. Neurosurgery 2002;50(2):313–320.
CA, Albright AL, Sutton LN, et al. Dermoid inclusion cysts and early
spinal cord tethering after fetal surgery for myelomeningocele. N Engl J Med 2002;347(4):256–259.
EM, Shaw GM. Impact of prenatal diagnosis and elective termination on
prevalence and risk estimates of neural tube defects in California. Am J Epidemiol 1996;144:473–479.
for Disease Control and Prevention. Recommendations for the use of
folic acid to reduce the number of cases of spina bifida and other
neural tube defects. MMWR Morb Mortal Wkly Rep 1992;RR-14:41.
JR, Ferguson-Smith MA, Shenkin A, et al. Is disordered folate
metabolism the basis for the genetic predisposition to neural tube
defects? Clin Genet 1987;31(5):279–287.
VL, Seeds JW, Albright SG, et al. Role of ultrasound and informed
consent in the evaluation of elevated maternal serum alpha-fetoprotein.
Am J Perinatol 1991;8(2):73–76.
LD, Feuchtbaum L, Filly R, et al. The California maternal serum
alpha-fetoprotein screening program: the role of ultrasonography in the
detection of spina bifida. Am J Obstet Gynecol 1992;166(5):1328–1329.
DA, Wardinsky T, Shurtleff DB, et al. Cesarean section before the onset
of labor and subsequent motor function in infants with myelomeningocele
diagnosed antenatally. N Engl J Med 1991;324:662–666.
JP, Tulipan N, Paschall RL, et al. Fetal surgery for myelomeningocele
and the incidence of shunt-dependent hydrocephalus. JAMA 1999;282:1819–1825.
N, Sutton LN, Burner JP. The effect of intrauterine myelomeningocele
repair on the incidence of shunt-dependent hydrocephalus. Pediatr Neurosurg 2003;38(1):27–33.
M, Meuli-Simmen C, Hutchins GM, et al. The spinal cord lesion in human
fetuses with myelomeningocele: implications for fetal surgery. J Pediatr Surg 1997;32:448–452.
LN, Adzick NS, Bilaniuk LT, et al. Improvement in hindbrain herniation
demonstrated by serial fetal magnetic resonance imaging following fetal
surgery for myelomeningocele. JAMA 1999;282:1826–1831.
M, Pabby A, Kelly M, et al. Improved bladder function after
prophylactic treatment of high risk neurogenic bladder in newborns with
myelomeningocoele. J Urol 1999;162:1068–1069.
A, Essakalli N, Faik M, et al. Continent urostomy with hydraulic ileal
valve in 136 patients: 13 years of experience. J Urol 1989;142:46.
FB, Bradley WE, Timm GW, et al. Treatment of incontinence secondary to
myelodysplasia by an implantable prosthetic urinary sphincter. South Med J 1973;66:987.
A, Cima LG, Kim W, et al. Injectable alginate seeded with chondrocytes
as a potential treatment for vesicoureteral reflux. J Urol 1993;150:745.
L, Smith E, Parrott T, et al. Submucosal bladder neck injection of
bovine dermal collagen for stress urinary incontinence in the pediatric
population. J Urol 1996;156:633.
R, Padron OF, Singla AK, et al. The gastric augment single pedicle tube
catheterizable stoma: a useful adjunct to reconstruction of the urinary
tract. J Urol 1994;152:2005.
GN, Westhoven VC, Shapera Phillips W, et al. A multimethod,
multi-informant, and multidimensional perspective on psychosocial
adjustment in preadolescents with spina bifida. J Consult Clin Psychol 2003;71(4):782–796.
PK, Dsida RM, Grayhack JJ, et al. Do latex precautions in children with
myelodysplasia reduce intraoperative allergic reactions? J Pediatr Orthop 1996;16:799–802.
PI, Merguerian PA, Klein RB, et al. Evaluation and risk factors of
latex allergy in spina bifida patients: is it preventable? J Urol 1993;150:691–693.
R, Stehl E, Wenzel D, et al. Arm span, serum IGF-I and IGFBP3 levels as
screening parameters for the diagnosis of growth hormone deficiency in
patients with myelomeningocele —preliminary data. Eur J Pediatr 1998;157:451–455.
D, Reigel DH. Effect of growth hormone treatment on muscle strength,
obesity and growth symmetry for children with myelomeningocele
[Abstract]. Endocrinol Metab 1997;4(Suppl):56.
CM, Jaffe KM, Shurtleff DB. Assessment of muscle strength in children
with myelomeningocele; accuracy and stability of measurements over
time. Arch Phys Med Rehabil 1986; 67:855–861.
M, Albertini G, Romei M, et al. Gait analysis in children affected by
myelomeningocele: comparison of the various levels of lesion. Funct Neurol 2002;17(4):203–210.
S, Moore C, Statler K, et al. The influence of forearm crutches on
pelvic and hip kinematics in children with myelomeningocele: don’t
throw away the crutches. Dev Med Child Neurol 1997;39:614–619.
RC, Birkebak RC, Winter RB. The role of hip location and dislocation in
the functional status of the myelodysplastic patient: a review of 100
patients. Orthopedics 1989;12(5): 675–684.
CW, Ramsey PI. Myelodysplasia: the influence of the quadriceps and hip
abductor muscles on ambulatory function and stability of the hip. J Bone Joint Surg Am 1978;60-A:432–443.
CM, Jaffe KM, Mosca VS, et al. Ambulatory outcome of children with
myelomeningocele: effect of lower extremity muscle strength. Dev Med Child Neurol 1991;33:482–490.
A, Vankoski SJ, Dias L, et al. Independent ambulators with high sacral
myelomeningocele: the relation between walking kinematics and energy
consumption. Dev Med Child Neurol 2001;43:16–21.
Müller E, Nordwall A, von Wendt L. Influence of surgical treatment of
scoliosis in children with spina bifida on ambulation and motoric
skills. Acta Paediatr 1992;81:173–176.
HH, Uppal GS, Lane G, et al. Treatment versus non-treatment of hip
dislocations in ambulatory patients with myelo-meningocele. Dev Med Child Neurol 1991;33:491–494.
GS, Shurtleff DB, Bloss JW, et al. Mobility aids for children with
high-level myelomeningocele: parapodium versus wheelchair. Dev Med Child Neurol 1992;34:787–796.
LK, Nielsen DH. Ambulation of children with myelo-meningocele:
parapodium versus parapodium with Orlau swivel modification. Dev Med Child Neurol 1986;28:489–497.
D, Field RE, Broughton NS, et al. Reciprocating orthoses for children
with myelomeningocele: a comparison of two types. J Bone Joint Surg Br 1995;77-B:110–113.
DE, Haideri N, Song K, et al. Comparative study of conventional
hip-knee-ankle-foot orthoses versus reciprocating gait orthoses for
children with high-level paraparesis. J Pediatr Orthop 1997;17:377–386.
F, Burke S, Roberts JM, et al. Functional ambulation in myelodysplasia:
the effect of orthotic selection on physical and physiologic
performance. J Pediatr Orthop 1986;6:661–665.
TJ, Freeling RP, Sienko Thomas S, et al. Energy consumption in children
with myelomeningocele: a comparison between reciprocating gait orthosis
and hip-knee-ankle-foot orthosis ambulators. Dev Med Child Neurol 1997;39:239–242.
JD, Ounpuu S, Davis RB, et al. The effects of ankle-foot orthoses on
the ankle and knee in persons with myelomeningocele: an evaluation
using three-dimensional gait analysis. J Pediatr Orthop 1999;19:27–33.
RC, Vankoski SJ, Dias LS. Internal derotation osteotomy of the tibia:
pre- and postoperative gait analysis in persons with high sacral
myelomeningocele. J Pediatr Orthop 2000;20:623–628.
P, Hopf C, Voth D. Development of scoliosis in myelomeningocele.
Differences in the natural history caused by idiopathic pattern. Neurosurg Rev 1993;16:301–306.
TS, Cail WS, Maggio WM, et al. Progressive spasticity and scoliosis in
children with myelomeningocele. Radiological investigation and surgical
treatment. J Neurosurg 1985;62:367–375.
ER, Thomson JD, Smith BG, et al. Results and morbidity in a consecutive
series of patients undergoing spinal fusion for neuromuscular
scoliosis. Spine 1998;23(21):2308–2317.
JV, Park SM. Improvement in pulmonary function in patients having
combined anterior and posterior spine fusion for myelomeningocele
scoliosis. Spine 1983;8(7):765–770.
A, Holger H, Muthu K, et al. Is sacral instrumentation mandatory to
address pelvic obliquity in neuromuscular thoracolumbar scoliosis due
to myelomeningocele? Spine 2001; 26(14):325–329.
YL, Lonstein JE, Winter RB, et al. Luque-Galveston procedure for
correction and stabilization of neuromuscular scoliosis and pelvic
obliquity: a review of 68 patients. J Spinal Disord 1991;4:399–410.
J, Menelaus MB, Dickens DR, et al. Efficacy of surgical management for
scoliosis in myelomeningocele: correction of deformity and alteration
of functional status. J Pediatr Orthop 1986;6:568–575.
WT, Wenger DR, Roach JW. Surgical correction of myelomeningocele
scoliosis: a critical appraisal of various spinal instrumentation
systems. J Pediatr Orthop 1989;9:262–268.
WB, Williams MS, Schwend RM, et al. Spinal deformity in
myelodysplasia—correction with posterior pedicle screw instrumentation.
Spine 1997;22:2435–2443.
A, Saraste H. Anterior fusion insufficient for scoliosis in
myelomeningocele: 8 children 2–6 years after the Zielke operation. Acta Orthop Scand 1993;64:22–24.
GR, Raducan V, Bednar DA, et al. Anterior and posterior spinal fusion:
comparison of one-stage and two-stage procedures. CJS 1993;36(5):468–473.
TML, Soorani-Lunsing IJ, De Jong TPVM, et al. Urological problems after
surgical treatment of scoliosis in children with myelomeningocele. J Urol 1996;155:1066–1069.
PD, LaPorte DM, Hungerford MW, et al. Deep wound infections after
neuromuscular scoliosis surgery: a multi-center study of risk factors
and treatment outcomes. Spine 2000;25(19):2461–2466.
S, Eysel P, Hopf C, et al. Sagittal static imbalance in
myelomeningocele patients: improvements in sitting ability by partial
and total gibbus resection. Eur Spine J 1999;8:451–457.
T, Walker JL, van den Brink K, et al. The progression of untreated
lumbar kyphosis and the compensatory thoracic lordosis in
myelomeningocele. Dev Med Child Neurol 1997;39:326–330.
MT, Sarwark JF, Vora A, et al. A kyphectomy technique with reduced
perioperative morbidity for myelomeningocele kyphosis. Spine 2002;27(16):1807–1813.
JS, Gillespie R. Management of myelomeningocele kyphosis in the older
child by kyphectomy and segmental spinal instrumentation. Spine 1987;12:37–41.
J, Kimar SJ, Guille JT, et al. Congenital kyphosis in myelomeningocele:
results following operative and nonoperative treatment. J Pediatr Orthop 1994;14:323–328.
K, Hall J, Johnson D, et al. Acute elevation of intracranial pressure
following transaction of non-functional spinal cord. Clin Orthop 1977;128:41–44.
APT, Vankoski SJ, Dias LS, et al. Gait analysis in low lumbar
myelomeningocele patients with unilateral hip dislocation or
subluxation. J Pediatr Orthop 2003;23:330–334.
LS, Thomas SS, Robinson C, et al. Hip dislocation in spina bifida—the
external oblique transfer: a gait analysis evaluation. Orthop Trans 1992–93;16:624–625.
JJ, Rosenthal RK, Dabrowski S, et al. The adductor transfer in the
high-risk hip in myelodysplasia: a preliminary report. Clin Orthop 1978;132:108–114.
VL, Perry J, Garrett A, et al. Paralytic dislocation of the hip. In:
Proceedings of The American Academy of Orthopaedic Surgeons. J Bone Joint Surg Am 1966;48-A:1021.
LS, Hill JA. Evaluation of treatment of hip subluxation in
myelomeningocele by intertrochanteric varus derotation femoral
osteotomy. Orthop Clin North Am 1980;11(1):31–37.
H, Fairclough JA, Jones DG. Stabilization of the hip in
myelomeningocele: comparison of posterior iliopsoas transfer and
varus-rotation osteotomy. J Bone Joint Surg Br 1988;70-B:29–33.
PS, Broughton NS, Menelaus MB. Tenotomy of the ligamentum patellae in
spina bifida: management of limited flexion range of the knee. J Bone Joint Surg Br 1995;77-B(5): 832–833.
MH, Smith CF, Oppenheim WL. Supracondylar femoral extension osteotomies
in the treatment of fixed flexion deformity of the knee. Clin Orthop 1982;171:87–93.
CA, Dias LS, Vankoski SJ, et al. Valgus stress as the knee joint in
lumbo-sacral myelomeningocele: a gait analysis evaluation. Dev Med Child Neurol 1996;38:2–3.
LS, Stern LS. Talectomy in the treatment of resistant talipes
equinovarus deformity in myelomeningocele and arthrogryposis. J Pediatr Orthop 1987;7:39–41.
LS, Mann DC, Feiwell E, et al. Equinovarus deformity in arthrogryposis
and myelomeningocele: evaluation of primary talectomy. Foot Ankle 1989;10(1):12–16.
HH, Marchinski LJ, Clancy M, et al. Ground reaction forces on the
plantar surface of the foot after talectomy in the myelomeningocele. J Pediatr Orthop 1989;9:269–275.
R, Fernandez SA, Colon C, et al. Transfer of the tibialis anterior for
calcaneus deformity in myelodysplasia. J Bone Joint Surg Am 1992;74:1038–1041.
SJ, Cowell HR, Townsend P. Physeal, metaphyseal and diaphyseal injuries
of the lower extremities in children with myelomeningocele. J Pediatr Orthop 1984;4:25–27.
AT, Loder RT, Hensinger RN. Telescoping intramedullary rodding with
Bailey-Dubow nails for recurrent pathologic fractures in children
without osteogenesis imperfecta. J Pediatr Orthop 1998;18:4–8.
WB, Schwend RM, Jaramillo D, et al. Chronic physeal fractures in
myelodysplasia: magnetic resonance analysis, histologic description,
treatment and outcome. J Pediatr Orthop 1997;17:615–621.
CTE, Seaver L, Carrion C, et al. Anatomical evaluation of the caudal
regression syndrome (lumbosacral agenesis) with magnetic resonance
imaging. Neurosurgery 1989;25:462–465.
H, Beaver BL, Colombani PM, et al. Neonatal diagnosis of a presacral
mass in the presence of congenital anal stenosis and partial sacral
agenesis. J Pediatr Surg 1989;24:1076–1078.
RB. Congenital absence of the lumbar spine and sacrum: one-stage
reconstruction with subsequent two-stage spine lengthening. J Pediatr Orthop 1991;11:666–670.
C, Damsin J, Forin V, et al. Lumbosacral agenesis: three cases of
reconstruction using Cotrel-Dubousset or L-rod instrumentation. Spine 1993;18:1229–1235.
J, Kalamchi A, MacEwen GD. Sacral agenesis: a clinical evaluation of
its management, heredity, and associated anomalies. Clin Orthop Relat Res 1979;139:52–57.