
Editors: Frassica, Frank J.; Sponseller, Paul D.; Wilckens, John H.
Title: 5-Minute Orthopaedic Consult, 2nd Edition
Copyright ©2007 Lippincott Williams & Wilkins
> Table of Contents > Muscular Dystrophies
Muscular Dystrophies
Paul D. Sponseller MD
Description
-
Muscular dystrophies are a group of
inherited disorders characterized by progressive degeneration and
weakness of skeletal muscle without apparent cause in the nervous
system. -
Skeletal and cardiac muscles are affected, and secondary effects occur in the lungs, skeleton, and many other systems.
-
These conditions have been categorized by
clinical distribution, severity of muscle weakness, and pattern of
genetic inheritance. -
Because of limited space, only Duchenne muscular dystrophy is described in detail (Fig. 1).
-
Classification:
-
Sex-linked muscular dystrophy: Duchenne muscular dystrophy, Becker, Emery-Dreifuss
-
Autosomal-recessive muscular dystrophy: Limb-girdle, infantile fascioscapulohumeral
-
Autosomal-dominant muscular dystrophy: Fascioscapulohumeral, distal, ocular, oculopharyngeal
-
Epidemiology
Incidence
-
Duchenne muscular dystrophy occurs in young boys.
-
Duchenne muscular dystrophy occurs in 1 in 3,500 live male births (1).
-
Becker dystrophy occurs in ~1 in 30,000 live male births (1).
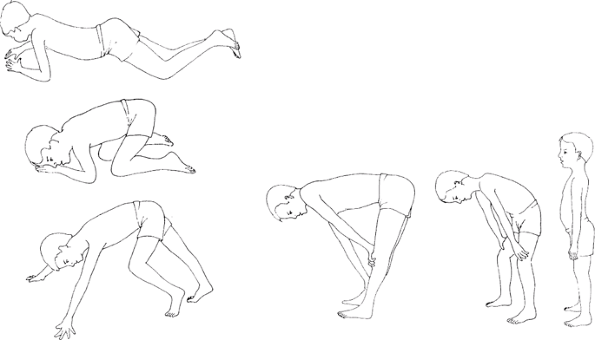 Fig. 1. This series of 6 drawings illustrates the Gower maneuver of a 7-year-old child with Duchenne muscular dystrophy.
Fig. 1. This series of 6 drawings illustrates the Gower maneuver of a 7-year-old child with Duchenne muscular dystrophy.
Risk Factors
Male gender
Genetics
-
Duchenne muscular dystrophy is sex-linked, as is Becker-type tardive dystrophy.
-
Other dystrophies are autosomal recessive and autosomal dominant.
Etiology
-
A single gene defect in the short arm of
the X chromosome has been identified as being responsible for Duchenne
muscular dystrophy and Becker muscular dystrophy.-
The gene encodes the protein dystrophin, which is a component of the cell membrane cytoskeleton.
-
Signs and Symptoms
-
Duchenne muscular dystrophy:
-
The disease occurs only in males, and it usually becomes evident at 3–6 years of age.
-
Common presentations include:
-
Delayed walking
-
“Waddling,” Trendelenburg gait, or lordotic gait
-
Frequent tripping and falling
-
Inability to hop and jump
-
-
Progressive weakness occurs in the
proximal muscle groups, including the gluteus, quadriceps, abdominal
muscles, and shoulder girdle muscles -
Pseudohypertrophy and contracture of calf muscles is common.
-
Most patients have cardiac involvement, most commonly tachycardia and right ventricular hypertrophy.
-
Many also have static encephalopathy with mental retardation.
-
Death from pulmonary and cardiac failure occurs during the 2nd or 3rd decade of life.
-
Because of hip muscle weakness, patients
compensate by carrying the head and shoulders behind the pelvis during
gait, thus producing an anterior pelvic tilt and increased lumbar
lordosis. -
Weakness in the shoulder girdle occurs 3–5 years after presentation.
-
It is difficult to lift the patient under the arms because of the weakness.
-
This weakness has been termed the “Meryon” sign.
-
-
No sensory deficits are detected.
-
Children usually are unable to ambulate effectively beyond 10 years of age.
-
-
Becker muscular dystrophy:
-
Similar to Duchenne muscular dystrophy in clinical appearance and distribution of weakness, but less severe
-
The onset usually occurs after the age of 7 years.
-
The rate of progression is slower than in Duchenne muscular dystrophy
-
-
Many more types of muscular dystrophy exist (not described here).
P.267
Physical Exam
-
History, physical examination,
measurement of creatine phosphokinase and dystrophin, and
electromyography help in making the diagnosis. -
Electromyography shows a myopathic pattern, with reduced amplitude, short duration, and polyphasic muscle action potentials.
-
Muscle biopsy also may be performed.
-
Evaluate muscle bulk to assess for pseudohypertrophy of the calves.
-
Observe the patient’s gait and look for Trendelenburg gait.
-
Starting proximally, look for muscle weakness.
-
Evaluate the patient’s ability to stabilize the shoulder; test for Meryon sign.
-
Note contracture, developing later, followed by scoliosis.
Tests
Lab
-
Serum creatine phosphokinase markedly is elevated in the early stages of Duchenne muscular dystrophy.
-
It may be 200 times normal, but it later declines as muscle degeneration becomes complete.
-
-
Dystrophin levels are completely absent in Duchenne muscular dystrophy; they are less than normal in Becker dystrophy.
Pathological Findings
-
Muscle degeneration, with subsequent loss of fibers
-
Variation in fiber size
-
Proliferation of connective tissue
Differential Diagnosis
-
Peripheral neuropathy
-
Anterior horn cell disease
-
Poliomyelitis
General Measures
Most patients with Duchenne muscular dystrophy die in
their 2nd or 3rd decade of life; therefore, orthopaedic treatment
should be designed to improve or maintain the functional capacity of
the involved adolescent.
their 2nd or 3rd decade of life; therefore, orthopaedic treatment
should be designed to improve or maintain the functional capacity of
the involved adolescent.
Activity
-
No restrictions on activity.
-
Activity is to be encouraged as much as possible.
Special Therapy
Physical Therapy
-
Test muscle strength to assess the rate of deterioration.
-
Use ankle-foot orthoses for correctable deformities.
-
The best treatment for fractures is closed reduction and immobilization.
-
Fractures of the lower extremities occur
frequently in children with Duchenne muscular dystrophy, especially in
children who are wheelchair bound. -
Contractures of both lower and upper extremities may occur.
-
Surgical release of contractures sometimes is indicated to improve function.
-
-
~95% of patients with Duchenne muscular dystrophy develop progressive scoliosis (2).
-
Surgical correction of scoliosis improves sitting balance and minimizes pelvic obliquity.
-
Posterior spinal fusion is recommended for curves of >20–30°.
-
-
Programs of vigorous respiratory therapy
and the use of home negative-pressure and positive-pressure ventilators
may promote life extension. -
Proper diagnosis and early genetic
counseling may help parents to be aware of the risk of additional male
infants with Duchenne muscular dystrophy.
Medication
-
No drugs have been proved effective.
-
Steroids have some benefit (delaying
scoliosis and prolonging function), but they also are associated with
long-term problems, including weight gain and osteoporosis.
Surgery
-
Contracture release (Achilles, fascia lata) may be indicated.
-
Correction of scoliosis involves fusion of nearly the entire thoracic and lumbar spine (T2–L5 or sacrum).
-
Rods are used to straighten and hold the spine.
-
This intervention should be performed for curves of ≥30°.
-
Prognosis
-
Duchenne muscular dystrophy is fatal in the 2nd or 3rd decade of life.
-
Becker dystrophy is more slowly progressive, and life expectancy is greater.
Complications
-
Respiratory failure
-
Cardiac failure
-
Fracture
-
Scoliosis
Patient Monitoring
Patients must be followed frequently (every 4–6 months) by a neurologist to assess their progression.
References
1. Alman BA, Raza SN, Biggar WD. Steroid treatment and the development of scoliosis in males with Duchenne muscular dystrophy. J Bone Joint Surg 2004;86A:519–524.
2. Biggar WD, Gingras M, Fehlings DL, et al. Deflazacort treatment of Duchenne muscular dystrophy. J Pediatr 2001;138:45–50.
Codes
ICD9-CM
359.1 Duchenne muscular dystrophy
Patient Teaching
Genetic counseling is important, to warn of the risk of additional affected infants.
FAQ
Q: What is the benefit of scoliosis surgery in patients with Duchenne muscular dystrophy?
A: It improves sitting balance and prevents discomfort that develops as the spine collapses.