
Editors: Frassica, Frank J.; Sponseller, Paul D.; Wilckens, John H.
Title: 5-Minute Orthopaedic Consult, 2nd Edition
Copyright ©2007 Lippincott Williams & Wilkins
> Table of Contents > Marfan Syndrome
Marfan Syndrome
Paul D. Sponseller MD
Description
-
Marfan syndrome is a familial disorder of
elastic connective tissue that is characterized by aortic root
dilatation and dissection, valvular insufficiency, lens dislocation,
and arachnodactyly, among other findings. -
It affects the cardiovascular, ocular,
skeletal, and neurologic systems and has caused sudden death in many
patients, including several prominent basketball and volleyball players. -
Although the disorder is inherited at
birth, some of the manifestations, such as aortic dilatation,
scoliosis, and sternal deformity, take time to develop. -
This delayed manifestation may cause the diagnosis to be delayed until later in childhood.
-
Classification (1):
-
Classic Marfan syndrome (2):
-
MASS (Mitral prolapse, Aortic dilatation, Skin and Skeletal findings) phenotype, which is a forme fruste of the syndrome
-
Contractural arachnodactyly (Beals syndrome), disorder of fibrillin-2 (2)
-
Epidemiology
Incidence
Males and females are affected equally.
Prevalence
~1 in 5,000 persons is affected (2).
Risk Factors
-
Positive family history of Marfan syndrome
-
Family history of aortic dissection or unexplained sudden death
-
Tall, slender habitus
Genetics
-
The syndrome is autosomal dominant with variable expressivity.
-
Some patients are affected by a de novo mutation, and they are more likely to have severe cases or be diagnosed in the neonatal period.
-
Even among families with high penetrance, manifestations may vary from member to member.
Etiology
-
Marfan syndrome results from a defect in the fibrillin-1 gene, which is found on chromosome 15.
-
Multiple different deletions have been found that result in Marfan syndrome and probably explain the condition’s heterogeneity.
-
Fibrillin is found in the zonules that suspend the lens of the eye, as well as in the arterial walls.
-
The explanation for other findings is still being sought, but it may involve molecular signaling and structural differences.
Signs and Symptoms
-
The diagnosis is made mainly by clinical (Ghent) criteria (1).
-
The patient must have at least 2 systems
involved, at least 2 major criteria (ascending aortic enlargement or
dissection, ectopia lentis, dural ectasia, positive family history,
proven mutation, or 4 skeletal findings), and 1 minor criterion. -
Symptoms:
-
Relatively few; rarely the means for diagnosis
-
Delay in walking or coordination, fatigability, poor vision, chest pain (at aortic dissection)
-
-
Signs:
-
Tendency to tall stature
-
Long limbs in relation to the trunk (3)
-
Scoliosis
-
Kyphosis
-
Multiple foot deformities
-
Pectus excavatum or carinatum
-
Slender cranium
-
Joint hypermobility, which usually is moderate and is considerably less than in Ehlers-Danlos syndrome
-
Often, a positive thumb sign, in which the clenched thumb protrudes beyond the ulnar border of the closed fist (2)
-
Physical Exam
-
Measure the patient’s height: The upper:lower segment ratio (head to symphysis over symphysis to floor) is <0.85.
-
Check for kyphosis, scoliosis, and pectus deformity.
 Fig.
Fig.
1. The thumb sign is positive when the entire distal phalanx of the
thumb protrudes beyond the ulnar border of the clenched fist. The wrist
sign is positive when the thumb can completely overlap the 5th
fingernail when wrapped around the wrist. -
Assess thumb and wrist signs (4) (Fig. 1).
-
Asses leg-length inequality.
-
Slit-lamp examination by an experienced
ophthalmologist also is helpful in making the diagnosis and in
following the patient’s course.
Tests
Lab
-
Results of routine laboratory tests are normal, but genetic testing for mutations in fibrillin is available.
-
False-negative results are still possible with this test.
-
Therefore, genetic testing is not used routinely in clinical practice (2,5).
Imaging
-
Echocardiography is key to assessing main structures at risk in this syndrome, such as the heart valves and the ascending aorta.
-
This test should be performed as a
baseline at the time of diagnosis and periodically afterward, according
to the judgment of the cardiologist.
-
-
MRI is useful for imaging entire aorta; it also can be used to evaluate the spine for dural ectasia (6,7,8) (Fig. 2).
-
Radiography:
-
Plain radiographs of the spine are used for the diagnosis of scoliosis, if it is suspected.
-
Patients also should have an AP radiograph of the pelvis to evaluate for protrusio acetabulae (Fig. 3), which is an excessive deepening of the hip sockets (9).
-
Pathological Findings
-
The aortic root shows dissection of the medial layer in some patients.
-
The dura of the lower lumbar spine
sometimes shows dilatation and saclike protrusions from the sides and
front of the spinal canal.![]() Fig. 2. Dural ectasia is enlargement of the dural sac, usually in the lowest portion of the spine.
Fig. 2. Dural ectasia is enlargement of the dural sac, usually in the lowest portion of the spine.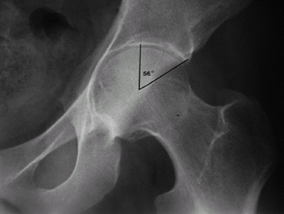 Fig. 3. Protrusio acetabulae is considered as a center-edge angle >50°.
Fig. 3. Protrusio acetabulae is considered as a center-edge angle >50°.

P.249
Differential Diagnosis
-
Congenital contractural arachnodactyly
(caused by a fibrillin-2 defect coded on chromosome 5), which is
distinguished by multiple joint contractures in the presence of
arachnodactyly -
Stickler syndrome (hereditary arthro-ophthalmopathy)
-
Homocystinuria (characterized by mental delay and inferior dislocation of the lens)
-
Ehlers-Danlos syndrome (generalized ligamentous laxity more extreme than in Marfan syndrome, with more cutaneous laxity)
-
MASS phenotype
-
Familial aortic dissection
General Measures
-
Medication for aortic dilatation
-
Genetic counseling
-
Periodic cardiology follow-up with ultrasound, as indicated (2)
-
If aortic enlargement is suspected, a β-2
blocking drug may be started to minimize pressure in the aorta; this
medication has been shown to be effective in slowing dilatation in
clinical trials. -
Atenolol is the β-2 blocker most commonly used for this syndrome.
-
-
Patients should avoid sports that cause high impact or cardiac stress.
-
Bracing or surgery for spinal deformities occasionally may be appropriate (10).
-
Procedures with which to address a partially dislocated lens or a retinal detachment are available.
-
No current drug or therapy can correct the basic defect in fibrillin.
Activity
-
Persons with Marfan syndrome should keep active, but they should avoid high-impact or high-stress sports.
-
A geneticist or cardiologist should be consulted if specific questions arise.
Special Therapy
Physical Therapy
-
May help an infant who is severely delayed in walking or achieving other motor milestones
-
Also useful as part of a nonoperative program for back pain
Surgery
-
Spinal fusion (correction with
instrumentation and bone graft for fusion) occasionally is indicated
for severe scoliosis or kyphosis (7). -
Spinal fusion sometimes is indicated for severe or symptomatic spondylolisthesis of L5 on S1.
-
Hip replacement occasionally is indicated for arthritis related to protrusio acetabuli (9).
-
Aortic root and valve repair or
replacement with a composite graft are indicated when the dilatation
reaches a certain size; this procedure is highly successful and is
preferable to waiting for aortic dissection to occur!
Disposition
Issues for Referral
If Marfan syndrome is suspected, the patient should be referred to a cardiologist or geneticist.
Prognosis
-
Without modern cardiovascular management, the life expectancy for patients with this condition would be <50 years.
-
Lifespan can be prolonged substantially, and many patients live into or past the 7th decade.
Complications
-
Aortic dissection
-
Valvular insufficiency
-
Retinal detachment
-
Spontaneous pneumothorax
-
Chronic musculoskeletal pain
Patient Monitoring
-
Coordinate care by a geneticist and a cardiologist.
-
See other specialists as needed.
-
Routine checkups may prevent catastrophes.
References
1. De Paepe A, Devereux RB, Dietz HC, et al. Revised diagnostic criteria for the Marfan syndrome. Am J Med Genet 1996;62:417–426.
2. Judge DP, Dietz HC. Marfan’s syndrome. Lancet 2005;366:1965–1976.
3. Jones KB, Sponseller PD, Hobbs W, et al. Leg-length discrepancy and scoliosis in Marfan syndrome. J Pediatr Orthop 2002;22:807–812.
4. Pradhan BB, Bhasin M, Otsuka NY. A metatarsal equivalent to the metacarpal index in Marfan syndrome. Foot Ankle Int 2005;26:881–885.
5. Dietz HC, Loeys B, Carta L, et al. Recent progress towards a molecular understanding of Marfan syndrome. Am J Med Genet 2005;139C:4–9.
6. Foran JRH, Pyeritz RE, Dietz HC, et al. Characterization of the symptoms associated with dural ectasia in the Marfan patient. Am J Med Genet 2005;134A:58–65.
7. Jones KB, Erkula G, Sponseller PD, et al. Spine deformity correction in Marfan syndrome. Spine 2002;27:2003–2012.
8. Sponseller PD, Ahn NU, Ahn UM, et al. Osseous anatomy of the lumbosacral spine in Marfan syndrome. Spine 2000;25:2797–2802.
9. Sponseller
PD, Jones KB, Ahn NU, et al. Protrusio acetabulae in Marfan syndrome:
age-related prevalence and associated hip function. J Bone Joint Surg 2006;88A:486–495.
PD, Jones KB, Ahn NU, et al. Protrusio acetabulae in Marfan syndrome:
age-related prevalence and associated hip function. J Bone Joint Surg 2006;88A:486–495.
10. Sponseller PD, Bhimani M, Solacoff D, et al. Results of brace treatment of scoliosis in Marfan syndrome. Spine 2000;25:2350–2354.
Codes
ICD9-CM
759.82 Marfan syndrome
Patient Teaching
-
Describe the warning signs of aortic dissection and retinal detachment.
-
Stress the importance of taking β-blockers when prescribed.
-
Offer genetic counseling.
-
Evidence suggests that the risk of aortic dissection or critical dilatation may be decreased by cardioselective β-blockade.
-
Support group in the United States: National Marfan Foundation, Port Washington NY (tel. 800-4-MARFAN)
FAQ
Q: Are braces effective for the scoliosis seen in Marfan syndrome?
A: Braces rarely influence the scoliosis seen in Marfan syndrome unless started early.
Q: Are braces indicated for flat feet in Marfan syndrome?
A: They do not change the shape of the feet. In most cases, they are not indicated unless pain is severe.