
Editors: Tornetta, Paul; Einhorn, Thomas A.; Damron, Timothy A.
Title: Oncology and Basic Science, 7th Edition
Copyright ©2008 Lippincott Williams & Wilkins
> Table of Contents > Section
II – Specific Bone Neoplasms and Simulators > 7 – Congenital and
Inherited Bone Conditions > 7.5 – Rothmund-Thomson Syndrome
II – Specific Bone Neoplasms and Simulators > 7 – Congenital and
Inherited Bone Conditions > 7.5 – Rothmund-Thomson Syndrome
7.5
Rothmund-Thomson Syndrome
Kathryn Palomino
Rothmund-Thomson syndrome is an autosomal recessive congenital syndrome (Box 7.5-1)
associated with potential development of osteosarcoma. Since the
patient usually arrives in the orthopaedic surgeon’s office with the
diagnosis, the key for the orthopaedic surgeon is simply to recall the
risk of development of osteosarcoma and to evaluate all musculoskeletal
complaints in that context.
associated with potential development of osteosarcoma. Since the
patient usually arrives in the orthopaedic surgeon’s office with the
diagnosis, the key for the orthopaedic surgeon is simply to recall the
risk of development of osteosarcoma and to evaluate all musculoskeletal
complaints in that context.
Pathogenesis
Etiology
-
Genetic defect: subset associated with defect in RECQL4 helicase gene at locus 8q24
-
Inheritance: autosomal recessive (AR)
Epidemiology
-
Rare
-
Incidence unknown
-
-
Male:female 2:1
Pathophysiology
-
Known genetic defect (RECQL4) present in 40% of cases
-
Normal expression highest in thymus and testes
-
Homology with genes that cause syndromes with some clinical overlap
-
Bloom syndrome: BLM gene defect on 15q26 with AR inheritance and increased incidence of osteosarcomas
-
Werner syndrome: WRN gene defect on 8p11-12 with AR inheritance and increased risk of various bone and soft tissue tumors
-
-
Diagnosis
Although no specific criteria have been established,
generally the diagnosis is first suspected in the neonate based upon
the characteristic rash that occurs within the first 6 months of life.
Genetic testing may identify the characteristic defect in up to 40% of
cases. Similar disorders, such as Werner syndrome, should be excluded
(e.g., urinary testing for the characteristically elevated hyaluronic
acid in Werner syndrome).
generally the diagnosis is first suspected in the neonate based upon
the characteristic rash that occurs within the first 6 months of life.
Genetic testing may identify the characteristic defect in up to 40% of
cases. Similar disorders, such as Werner syndrome, should be excluded
(e.g., urinary testing for the characteristically elevated hyaluronic
acid in Werner syndrome).
Box 7.5-1 Components of Rothmund-Thomson Syndrome
-
Various skin abnormalities
-
Skeletal defects
-
Juvenile cataracts
-
Premature aging
-
Predisposition to tumors
-
Osteosarcoma
-
Skin cancer
-
Others
-
History and Physical Examination
Physical examination reveals the key finding in
Rothmund-Thomson syndrome, the sun-sensitive erythematous facial rash
that spreads to buttocks and extremities and eventually leaves a
characteristic mixed hyper- and hypopigmented rash (poikiloderma) for
which the syndrome received its other name, poikiloderma congenitale.
Other nonorthopedic and orthopaedic manifestations may be present.
Rothmund-Thomson syndrome, the sun-sensitive erythematous facial rash
that spreads to buttocks and extremities and eventually leaves a
characteristic mixed hyper- and hypopigmented rash (poikiloderma) for
which the syndrome received its other name, poikiloderma congenitale.
Other nonorthopedic and orthopaedic manifestations may be present.
-
Nonorthopaedic clinical features
-
Poikilodermatous skin changes
-
Develop in first 6 months of life
-
Usually appear in sun-exposed areas and around buttocks (Fig. 7.5-1)
-
Photosensitivity and blister formation
-
-
Thin hair and/or alopecia
-
Short stature
-
Juvenile cataracts
-
Malignancies, particularly of the skin
-
-
Orthopaedic clinical features
-
Osteoporosis
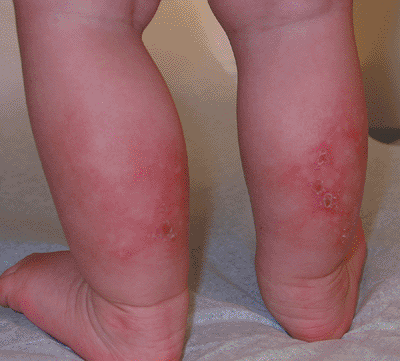 Figure 7.5-1 Rothmund-Thomson syndrome: posterior leg poikiloderma. (Image provided by Stedman’s.)
Figure 7.5-1 Rothmund-Thomson syndrome: posterior leg poikiloderma. (Image provided by Stedman’s.) -
Clavicular hypoplasia
-
Syndactyly
-
Patella aplasia
-
Genu valgum
-
Benign osseous defects
-
Osteosclerosis
-
Bone cysts
-
Metaphyseal chondrodysplasia
-
-
Sarcomas
-
Second most frequently occurring malignancy
-
P.227 -
Differential Diagnosis
-
Werner syndrome
-
Bloom syndrome
Treatment
-
No specific treatment for Rothmund-Thomson syndrome exists.
-
Symptomatic management of conditions as they arise
-
Surgical treatment not advised for height, alignment, or nonmalignant osseous abnormalities due to poor bone and wound healing
-
Hypervigilance in investigating bone pain
and or soft tissue swelling is mandatory due to increased risk of
malignancy, including multiple primary malignancies.
Suggested Reading
Anbari KK, Ierardi-Curto LA, Silber JS, et al. Two primary osteosarcomas in a patient with Rothmund-Thomson syndrome. Clin Orthop Rel Res 2000;(378):213–223.
Behrman RE, Kliegman RM, Arvin AM, et al, eds. Nelson Textbook of Pediatrics, 15th ed. Philadelphia: WB Saunders, 1996:1862.
Lindor NM. Rothmund-Thomson syndrome. In: Fletcher CDM, Unni KK, Mertens F, eds. Tumours of Soft Tissue and Bone: Pathology and Genetics. World Health Organization Classification of Tumours. Lyon: International Agency for Research on Cancer Press, 2002:365.