
adults, the changing structure and function, both physiologic and
biomechanical, of immature bones make them susceptible to different
patterns of failure. Even the types of fracture patterns within a given
bone demonstrate temporal (chronobiologic) variations that may be
correlated with progressive anatomic changes affecting the epiphysis,
physis, metaphysis, and diaphysis at macroscopic and microscopic levels.
Fractures in children are more common and are more likely to occur
after seemingly insignificant trauma. Physeal disruptions make up about
15% of all skeletal injuries in children.126,141,142,144,150,182
Damage involving specific growth regions, such as the physis or
epiphyseal ossification center, may lead to acute or chronic growth
disturbances.140,141,194,221
The physis is constantly changing, both with active longitudinal and
latitudinal (diametric) growth and in mechanical relation to other
components. Physeal fracture patterns vary with the extent of
chondro-osseous maturation. Salter-Harris type I injuries are common in
infants, and types II, III, and IV become more common as the secondary
ossification center enlarges and physeal undulations develop. Although
joint injuries, dislocations, and ligamentous disruptions are much less
common in children, it is more likely that one of the contiguous physes
will be damaged. Changing trabecular and cortical structures affect
metaphyseal and diaphyseal fracture patterns, and the variable size of
the secondary ossification center affects susceptibility to physeal and
epiphyseal injuries.
responses in fractures and strategies for enhancing bone and physis
repair in children, the treatment options available for skeletal
injuries in children are expanding. Most notable is the introduction of
growth factors, such as the bone morphogenic proteins (BMPs),
for the induction of bone formation in nonunions and large segmental bone defects, and for the repair of cartilage defects,78 and the research and development of stem-cell-based therapy for bone and cartilage regeneration157 and physeal repair.31,81,101
Due to these new developments, it has become necessary for the
orthopaedic surgeon to understand the biological aspects of the
skeletal injury responses and new treatment options for fracture
repair. This chapter covers the basic biology and regulation of bone
growth, bone fracture repair responses, physeal injury and physeal bar
formation, roles of growth factors and cytokines in regulating
injury/repair responses, and future therapeutic strategies for
bone/articular cartilage/physis regeneration using growth factors,
tissue engineering, stem cells, and gene therapy.
four distinct, anatomic areas: the epiphysis, physis, metaphysis, and
diaphysis.95 Each region is prone to
certain patterns of injury, and the intrinsic injury susceptibility
varies with physiologic and biomechanical changes during postnatal
development. The four regions originate and become modified as a result
of the basic endochondral ossification process. Subsequently, they are
supplemented by membranous bone formation along the metaphyseal and
diaphyseal shafts. Finally, the regions are remodeled to create mature
cortical and trabecular bone.
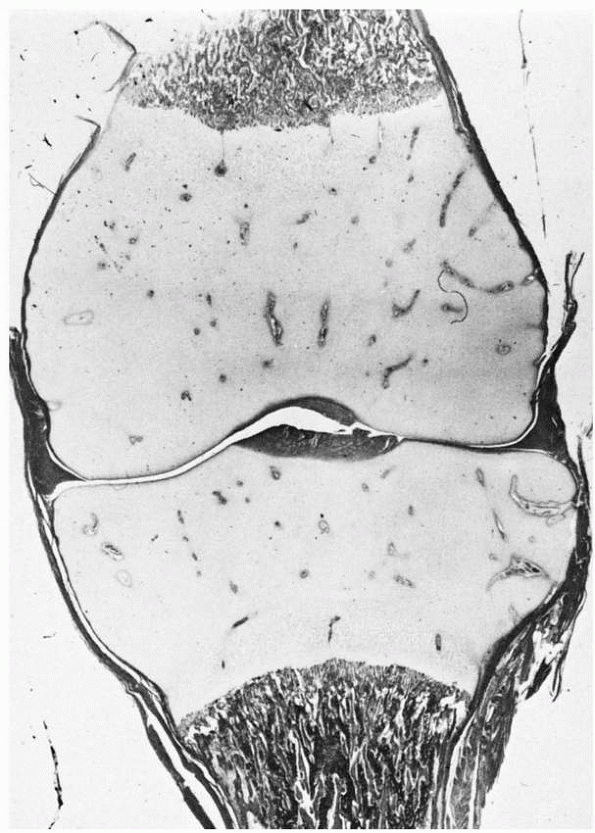
consists of a completely cartilaginous structure at the end of each
long bone (Fig. 2-1), the chondroepiphysis. At
a time characteristic for each of these chondroepiphyses, a secondary
center of ossification forms and gradually enlarges until the
cartilaginous area has been almost completely replaced by bone at
skeletal maturity. This chondro-osseous transformation is
vascular-dependent (Fig. 2-2). Only articular cartilage remains at maturity.
structural modifications. The region adjacent to the physis forms a
distinct subchondral plate parallel to the metaphysis, creating the
radiographically characteristic lucent physeal line. The appearance of
the ossification centers differ in certain chondroepiphyses, a factor
that must be considered when diagnosing fractures of these regions. The
ossification center imparts increasing rigidity to the more resilient
epiphyseal cartilage as the secondary osseous tissue expands.203
Muscle fibers, tendons, and ligaments may attach directly to the
perichondrium, which is densely contiguous with the underlying hyaline
cartilage. The perichondrium contributes to the continued centrifugal
enlargement of the epiphysis. It also blends imperceptibly into the
periosteum. This perichondrial/periosteal tissue continuity contributes
to the biomechanical strength of the epiphyseal/metaphyseal junction at
the zone of Ranvier.
forms, there are no easily demonstrable histologic differences between
the cells of the joint surface and the rest of the epiphyseal
cartilage. However, at some point, a finite cell population becomes
stabilized and physiologically different from the remaining epiphyseal
cartilage. McKibbin120 established
that these two cartilage types are different physiologically and
biochemically. If a contiguous core of articular and hyaline cartilage
is removed, turned 180°, and reinserted, the transposed hyaline
cartilage eventually will form bone at the joint surface, whereas the
transposed articular cartilage remains cartilaginous and becomes
surrounded by the enlarging secondary ossification center. Normally,
articular cartilage does not appear capable of
calcification
and ossification. As skeletal maturity is reached, a tide mark
progressively develops as a demarcation between the articular and
calcified epiphyseal hyaline cartilage.
 |
|
FIGURE 2-1
Chondroepiphyses of the distal femur and proximal tibia. These structures have an extensively developed vascular system (cartilage canals) before secondary ossification. |
 |
|
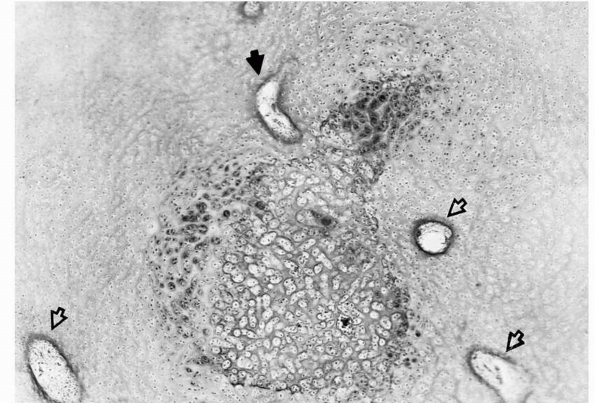
FIGURE 2-2
Early formation of the secondary ossification center within the epiphyseal cartilage. This usually occurs in a region well vascularized by cartilage canals (open arrows). One of the canals sends a branch into the hypertrophic cells (solid arrow), triggering the ossification process. |
 |
|
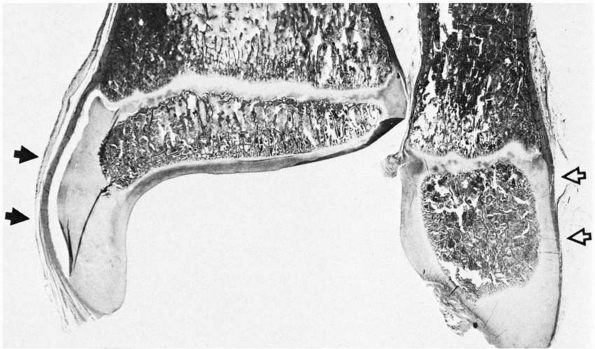
FIGURE 2-3
As the epiphysis matures, the ossification center expands and progressively follows the contours of the chondroepiphysis. The epiphyseal surface is either articular cartilage or perichondrium along the outer surfaces, as in the medial (solid arrows) and lateral (open arrows) malleoli. |
explanation of nonunion of certain fractures in which the fragment may
be rotated, causing the articular surface to lie against metaphyseal
and epiphyseal bone. Union is unlikely in such a situation because the
articular surface is incapable of a reparative osteogenic response, an
essential component of bone healing.
The primary function of the physis is rapid, integrated longitudinal
and latitudinal growth. Injuries to this component are unique to
skeletally immature patients.
except for the final stages of physiologic epiphysiodesis, its exact
location must be inferred from the metaphyseal contour, which follows
the physeal contour. The changing size of the secondary ossification
center more effectively demarcates the physeal contour on the
epiphyseal (germinal layer) side. As this center of ossification
enlarges centrifugally to approach the physis, the original spherical
shape of the ossification center flattens and gradually develops a
contour paralleling the metaphyseal contour. Similar contouring also
occurs as the ossification center approaches the lateral and
subarticular regions of the epiphysis (Fig. 2-4).
The region of the ossification center juxtaposed to the physis forms a
discrete subchondral bone plate that the essential epiphyseal blood
vessels must penetrate to reach the physeal germinal zone (Fig. 2-5). Damage to this osseous plate in a fracture may cause localized physeal ischemia.
compromised, whether temporarily or permanently, the zones of cellular
growth associated with these particular vessels cannot undergo
appropriate cell division. In contrast, unaffected regions of the
physis continue longitudinal and latitudinal growth, leaving the
affected region behind (Figs. 2-6 and 2-7).
The growth rates of the cells directly adjacent to the affected area
are more mechanically compromised than cellular areas farther away. The
differential rather than uniform growth results in an angular or
longitudinal growth deformity, or both.24,150
 |
|
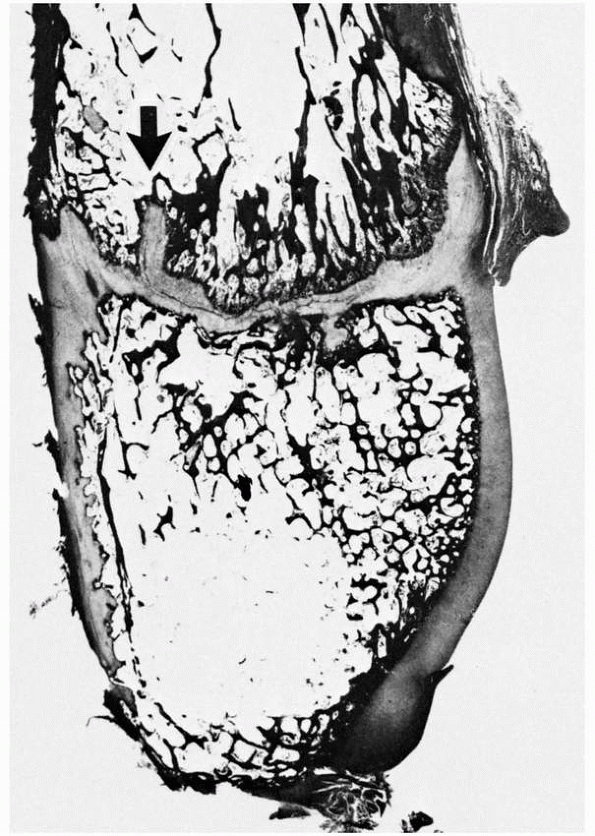
FIGURE 2-4 Distal fibula, showing the variably undulated physis, including a mammillary process (arrow).
The physeal and epiphyseal cartilage turns proximally at the medial region (lappet formation) to participate in the formation of the distal tibiofibular articulation. Note the difference in the subarticular subchondral bone, which has formed a thick plate, compared with the thin, outer subchondral bone. |
effect on chondrogenesis within the germinal zone or the sequential
cartilage maturation within the hypertrophic zone of the physis (see
Fig. 2-6). However, the subsequent
transformation of cartilage to bone (primary spongiosa) is blocked.
This causes widening of the affected area, because more cartilage is
added to the cell columns but none is replaced by invasive metaphyseal
vessels and bone. Once the disrupted metaphyseal circulation is
reestablished, this widened, calcified region of the physis is rapidly
penetrated and ossified, returning the physis to its normal width. This
is the mechanism seen in physeal and metaphyseal fractures. The
metaphyseal blood supply is temporarily blocked by separation or
impaction and requires 3 to 4 weeks for restoration. If the circulatory
compromise has been caused by a metaphyseal fracture, there also may be
a temporary halt to bone formation in the transiently ischemic portion
of the metaphysis. This leads to an apparent sclerosis when the bone is
compared with the adjacent vascularized metaphysis, which undergoes a
relative disuse osteoporosis. Compromise of the metaphyseal circulation
has minimal, if any, effect on physeal development, particularly when
compared with the major detrimental effects of epiphyseal circulatory
compromise.
 |
|
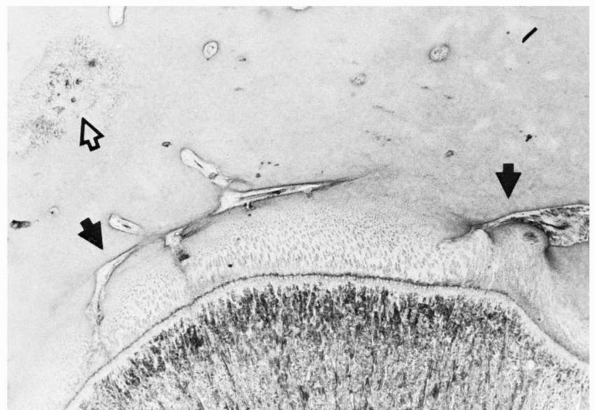
FIGURE 2-5 Epiphyseal circulation (solid arrows)
in a toddler. These supply the germinal/dividing zones of the physis. The open arrow indicates the early ossification center. As this area enlarges, it will incorporate the epiphyseal vessels. |
 |
|
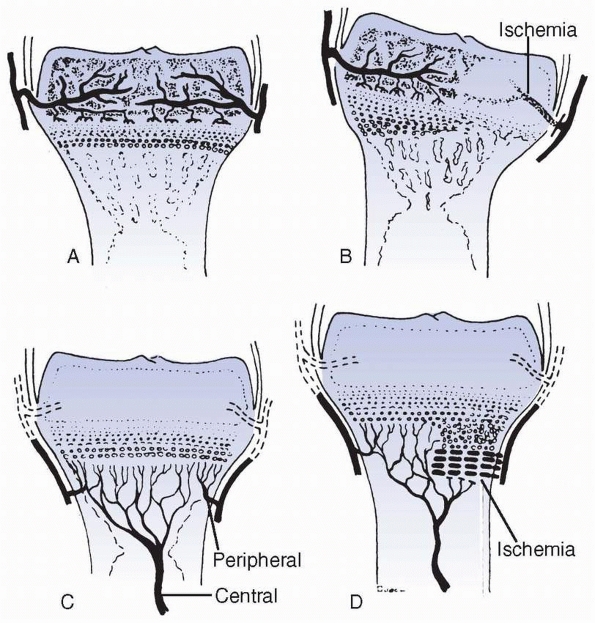
FIGURE 2-6 Patterns of response to ischemia of the epiphyseal (A,B) versus metaphyseal (C,D) circulatory systems. Metaphyseal ischemia is usually transient; epiphyseal ischemia is usually severe and permanent.
|
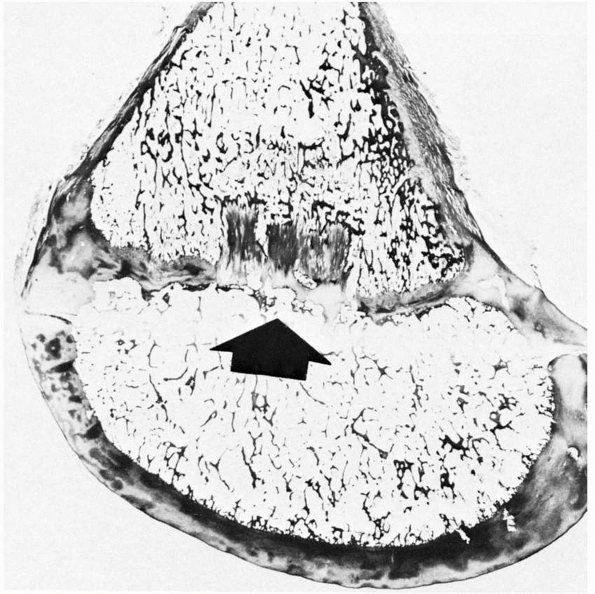 |
|
FIGURE 2-7 Histologic section showing an area of central ischemic growth arrest (arrow). The infarcted area of cartilage is left behind as the rest of the physis continues longitudinal growth.
|
Disrupting the epiphyseal circulation leads to either partial or
complete cessation of growth. The central region seems more sensitive
to ischemia than the periphery, which may have a variable capacity to
recover through continued latitudinal growth.128,138 Ischemic compromise leads to different rates of growth across the affected physis and significant changes in physeal contour.19 Some changes may be caused by venous stasis rather than arterial damage.86
of the diaphysis. Its major characteristics are decreased thickness of
the cortical bone and increased trabecular bone in the secondary
spongiosa. Extensive endochondral modeling centrally and peripherally
initially forms the primary spongiosa, which then is remodeled into the
more mature secondary spongiosa, a process that involves osteoclastic
and osteoblastic activity. Therefore, the metaphyses exhibit
considerable bone turnover compared with other regions of the bone, and
this factor is responsible for the increased uptake of radionuclides in
technetium 99m bone scans.80
with the confluent diaphysis, the metaphyseal cortex is thinner and is
more porous (trabecular fenestration; Fig. 2-8).
These cortical fenestrations contain fibrovascular soft-tissue elements
that connect the metaphyseal marrow spaces with the subperiosteal
region. The metaphyseal cortex exhibits greater fenestration near the
physis than in the diaphysis, with which it gradually blends as an
increasingly thicker, dense bone (Fig. 2-9). As
longitudinal growth continues, cortical fenestration becomes a less
dominant feature, and the overall width of the cortex increases,
creating a greater morphologic transition between the juxtaphyseal and
juxtadiaphyseal cortices. The metaphyseal region does not develop
extensive secondary and tertiary Haversian systems until the late
stages of skeletal maturation. These microscopic anatomic changes
appear to be directly correlated with changing fracture patterns and
are the reason why torus (buckle) fractures are more likely to occur
than complete metaphyseal or epiphyseal/physeal fractures.
occurs at the junction of the primary spongiosa and the hypertrophic
region of the physis. In most rapidly growing bones, the trabeculae
tend to be longitudinally oriented. However, in shorter growing bones,
such as the metacarpals and phalanges,
trabecular
formation is predominantly horizontal. As growth decelerates in
adolescence, a similar horizontal orientation may be seen in the major
long bones. These variations in trabecular orientation affect the
responsiveness of metaphyseal and physeal regions to abnormal stress
and predispose to certain fracture modes.
 |
|
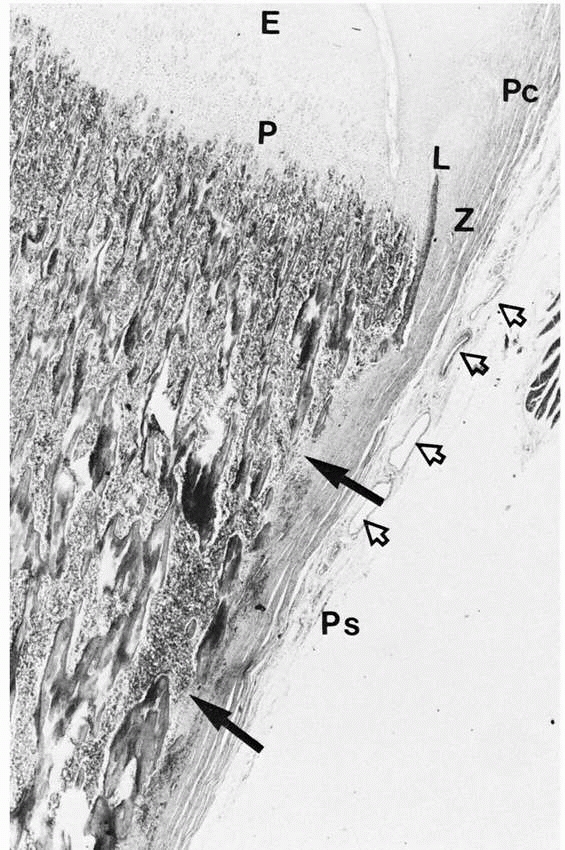
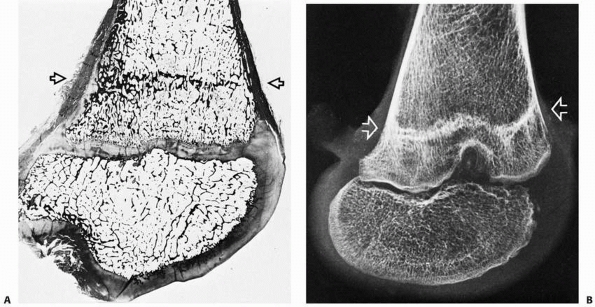
FIGURE 2-8 Cortical fenestration (solid arrows)
of a metaphysis. Note the interdigitation of periosteal (Ps) tissue with the fenestrations. The periosteum blends into the perichondrium (Pc). Extensive vascularity is often present in this region (open arrows). (E, epiphysis; P, physis; Z, zone of Ranvier; L, ring of Lacroix.) |
to the diaphysis, it is firmly fixed to the metaphysis because of the
increasingly complex continuity of fibrous tissue through the
metaphyseal fenestrations. Such intermingling of endosteal and
interosseous fibrous tissues with the periosteal tissue imparts
additional biomechanical strength to the region.198
The periosteum subsequently attaches densely into the peripheral
physis, blending into the zone of Ranvier as well as the epiphyseal
perichondrium. The fenestrated metaphyseal cortex extends to the physis
as the thin osseous ring of Lacroix.
The metaphyseal cortex is fenestrated, modified trabecular bone on
which the periosteum deposits membranous bone to thicken the cortex
progressively. Similar endosteal bone formation occurs. As this
metaphyseal region thickens, the trabecular bone is progressively
invaded by diaphyseal osteon systems, not unlike osteons traversing the
fracture site in primary bone healing. This converts peripheral
trabecular (woven or fiber) bone to lamellar (osteonal) bone, which has
different biomechanical capacities, and thus progressively transforms
metaphyseal cortex into diaphyseal cortex as longitudinal growth
continues. A torus (buckle) fracture is most likely to occur in a
metaphyseal region with a trabecular, fenestrated, compressible cortex.
 |
|
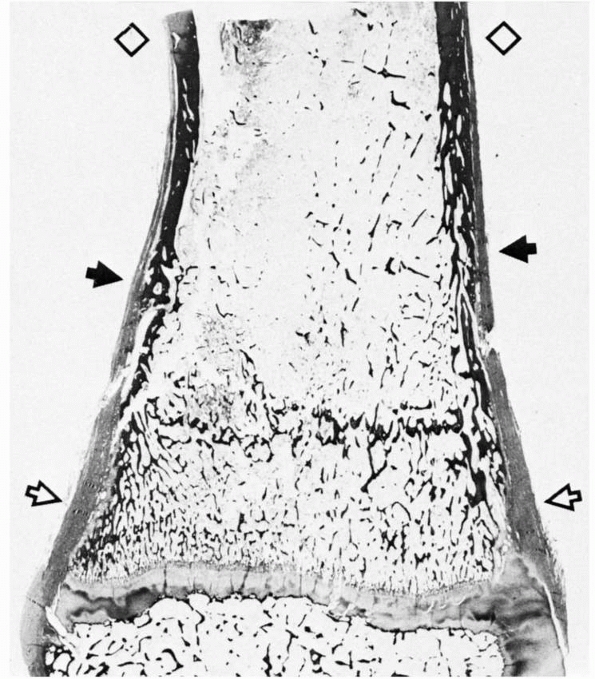
FIGURE 2-9 Section of distal tibia showing the transition (solid arrows) of cortical bone from the dense, remodeled diaphysis (diamonds) to the fenestrated metaphysis (open arrows). Note the progressive change from a relatively thin periosteum over the diaphysis to a much thicker one at the metaphysis.
|
primarily blend into the periosteum. The medial distal femoral
attachment of the adductor muscles is a significant exception. Because
of extensive remodeling and insertion of muscle and tendon in this
area, the bone often appears irregular and may be misinterpreted as
showing chronic trauma (i.e., a stress fracture), infection, or a tumor.
 |
|
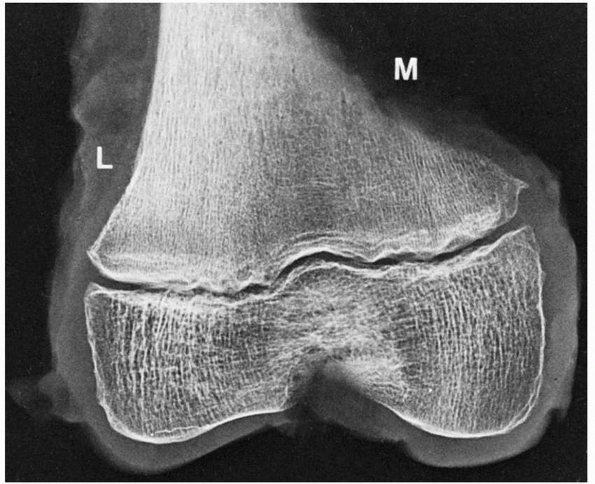
FIGURE 2-10
Extensive modeling and remodeling of the medial (M) versus the lateral (L) cortex of the distal femur may create irregularities that have been misinterpreted as fracture, stress fracture, infection, and tumor. Note the well-formed subchondral bone at the periphery of the epiphyseal ossification center. |
trabecular linear bone patterns within the metaphysis. These lines
usually duplicate the contiguous physeal contour. They may appear after
trauma, particularly when the child has been immobilized in bed (e.g.,
traction for femoral fracture), and they also may appear after
generalized illnesses or even localized processes within the bone
(e.g., osteomyelitis).1,67,160,161
The lines result from a temporary slowdown of normal longitudinal
growth after injury or illness, and they often are called Harris or
Park growth slowdown or arrest lines (Fig. 2-11).
Because of the slowdown, the trabeculae of the primary spongiosa become
more transversely than longitudinally oriented, creating a temporary
thickening in the primary spongiosa adjacent to the physis. Once the
normal longitudinal growth rate resumes, longitudinal trabecular
orientation is restored. The thickened, transversely oriented osseous
plate is left behind, and will be gradually remodeled as primary
spongiosa becomes secondary spongiosa.
symmetrically throughout the skeleton and occupy identical sites in the
corresponding bones on the two sides of the body. They are thickest in
metaphyses that grow most rapidly, such as the distal femur and
proximal tibia, as more primary spongiosa bone is formed in a
transverse orientation in these growing regions.140
In the metaphyses with slowest growth, they may not form at all, or
they are exceedingly thin and lie at the very end of the shaft,
directly under the provisional zone of calcification. These transverse
lines parallel the contours of the physeal provisional zone of
calcification. When several transverse lines are present, they tend to
be parallel. The lines nearest the end of the shaft ordinarily are the
thickest and widest, whereas lines away from the physes tend to be
thinner and less distinct and are usually broken and irregular. As
remodeling occurs, with migration of the epiphysis away from this
region, and with conversion of primary spongiosa to secondary
spongiosa, there is a gradual breakup of this transverse trabecular
orientation. As they eventually become part of the elongating
diaphysis, they disappear completely with endosteal remodeling.
 |
|
FIGURE 2-11 Histologic section (A) and x-ray study (B) of a distal femur showing a typical Harris line (arrows).
This formed during an acute illness and chemotherapy for leukemia. The child then resumed a more normal pattern of growth until her death from leukemia about 14 months later. |
with longitudinally oriented trabeculae in the juxtaphyseal region,
slower growing bones, particularly the proximal radius, metacarpals,
metatarsals, and phalanges, normally have a greater amount of
transversely oriented primary spongiosa,145
making transverse septa a normal finding. These particular bones do not
have a sufficient difference in the orientation of trabeculae to
manifest transverse lines on radiographs.
treatment in children with osteogenesis imperfecta, there are some
distinct metaphyseal bands in the growing skeleton, which may vary in
spacing according to the regimens of treatment, age of the patient,
rate of growth, and the location of the metaphysis. The bands may
reflect decreased osteoclastic activity occurring in response to drug
administration, and the spacing between the bands indicates resumption
of osteoclastic activity and linear growth of the bone between
treatments. As with growth arrest lines, the migration of these
treatment bands varies with the rate of the bone growth of the patient
and the particular physis.62
marker lines are important in analyzing the effects of a fracture on
growth. They can be measured and the sides compared to corroborate
femoral overgrowth after diaphyseal fracture and eccentric overgrowth
medially after proximal tibial metaphyseal fracture.
A
line that converges toward a physis suggests localized growth damage
that may result in an osseous bridge and the risk of angular deformity.
bone. It is principally a product of periosteal, membranous osseous
tissue apposition on the original endochondral model. This leads to the
gradual replacement of the endochondrally derived primary ossification
center and primary spongiosa; the latter is replaced by secondary
spongiosa in the metaphyseal region. At birth, the diaphysis is
composed of laminar (fetal, woven) bone that characteristically lacks
Haversian systems. The neonatal femoral diaphysis appears to be the
only area exhibiting any significant change from this fetal osseous
state to a more mature bone with osteon systems (lamellar bone) before
birth (Fig. 2-12).
formation with concomitant endosteal remodeling leads to enlargement of
the overall diameter of the shaft, variably increased width of the
diaphyseal cortices, and formation of the marrow cavity. Mature,
lamellar bone with intrinsic but constantly remodeling osteonal
patterns progressively becomes the dominant feature (Fig. 2-13).
child is extremely vascular. When analyzed in cross section, it appears
much less dense than the maturing bone of older children, adolescents,
and adults. Subsequent growth leads to increased complexity of the
Haversian (osteonal) systems and the formation of increasing amounts of
extracellular matrix, causing a relative decrease in cross-sectional
porosity and an increase in hardness, factors that constantly change
the child’s susceptibility to different fracture patterns. Certain
bones, especially the tibia, exhibit a significant decrease in
vascularity as the bone matures; this factor affects the rate of
healing and risk of nonunion.
 |
|
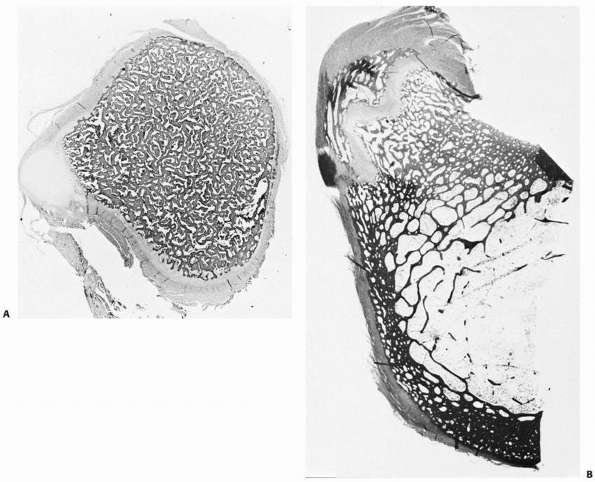
FIGURE 2-12 Sections of the femur at the level of the lesser trochanter at birth (A) and age 7 years (B).
At birth, some cortical thickening and osteon remodeling is evident laterally; the rest of the cortex is irregular. By age 7 years, extensive thickening and remodeling of the cortex has taken place. |
changes. In experimental studies, significant chronobiologic changes in
flow patterns were found in the developing canine tibia and femur.102,103,104,121,187,188,222 In particular, there was a dramatic decrease in tibial circulation with increasing skeletal maturation.188
This also occurs in humans, which helps to explain the increasing delay
in fracture healing and the increased incidence of nonunion of the
tibia in adolescents and adults. A poor vascular response could impair
the early, crucial stages of callus formation.
elevated from the diaphyseal and metaphyseal bone, and exhibits greater
osteogenic
potential than that of an adult.139
The periosteum is loosely attached to much of the shaft of the bone,
but it attaches densely into the physeal periphery (the zone of
Ranvier; Fig. 2-14) through intricate collagen meshwork, thereby playing a role in fracture mechanics and treatment of growth mechanism injuries.198
The thicker, stronger, more biologically active periosteum affects
fracture displacement, reduction, and the rate of subperiosteal callus
formation. It also may serve as an effective internal restraint in
closed reductions.
 |
|
FIGURE 2-13 Transverse sections of the tibial diaphysis in a neonate (A) and at age 2 years (B). A thick periosteum is evident in (A) (open arrows),
in association with a rapidly forming anterior cortex. At age 2 years, new subperiosteal (membranous) bone is being added to the cortex (solid arrow). |
periosteum is usually injured to some extent in all fractures in
children. However, because the periosteum more easily separates from
the bone in children, there is much less likelihood of complete
circumferential rupture. A significant portion of the periosteum
usually remains intact on the concave (compression) side of an injury.
This intact periosteal hinge or sleeve may lessen the extent of
displacement of the fracture fragments, and it also can be used to
assist in the reduction, because the intact portion contributes to the
intrinsic stability. Because the periosteum allows some tissue
continuity across the fracture, the subperiosteal new bone that forms
quickly bridges the fracture gap and leads to more rapid long-term
stability. The periosteum may be specifically damaged, with or without
concomitant injury to the contiguous bone. Such avulsion injuries may
lead to the formation of ectopic bone.147
In contrast, severe disruption of the periosteum, as in an open injury,
may impair the fracture healing response. Complete loss of a bone
segment, with the periosteal sleeve reasonably intact, may be followed
by complete reformation of the missing bone.15
While the outer fibroblast layer provides fibrous attachment to
subcutaneous connective tissue, muscles, tendons, and ligaments, the
inner cambium layer contains a pool of undifferentiated mesenchymal
cells that support bone formation and repair.193
During embryonic and postnatal bone growth, mesenchymal osteoprogenitor
cells at the inner layer differentiate directly into bone-forming cells
(osteoblasts) and form
periosteal bone collar by the intramembranous method.164
Formation of new periosteal bone keeps pace with formation of new
endochondral bone. During fracture healing, the mesenchymal cells at
the cambium layer undergo both intramembranous ossification and
chondrogenic differentiation with subsequent endochondral ossification.170
Due to the osteochondrogenic potential of these cells from the inner
periosteal layer, there has been a lot of interest surrounding the use
periosteum as graft tissues or as sources of osteochondroprogenitor
cells for repairing cartilage/bone defects or for tissue engineering.130,134,136,199,226
 |
|
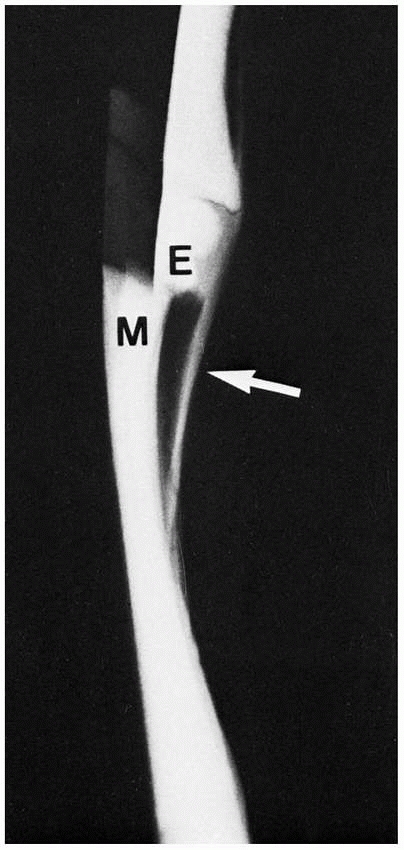
FIGURE 2-14 Simulated type 1 epiphyseal (E) displacement from the metaphysis (M). Note the thick periosteum (arrow)
and its contiguity with the cartilage of the epiphysis (radiopaque here because of the cartilage and air contrast). In the body, however, the similar soft-tissue radiodensities of cartilage, ligament, muscle, and so forth blend together, making them radiolucent. |
the origin for most muscle fibers along the metaphysis and diaphysis.
This mechanism allows coordinated growth of bone and muscle units; this
would be impossible if all the muscle tissue attached directly to the
developing bone or cartilage. Exceptions include the attachment of
muscle fibers near the linea aspera and into the medial distal femoral
metaphysis. The latter pattern of direct metaphyseal osseous attachment
may be associated with significant irregularity of cortical and
trabecular bone. Radiographs of this area often are misinterpreted as
showing a neoplastic, osteomyelitic, or traumatic response, even though
they exhibit only a variation of skeletal development.
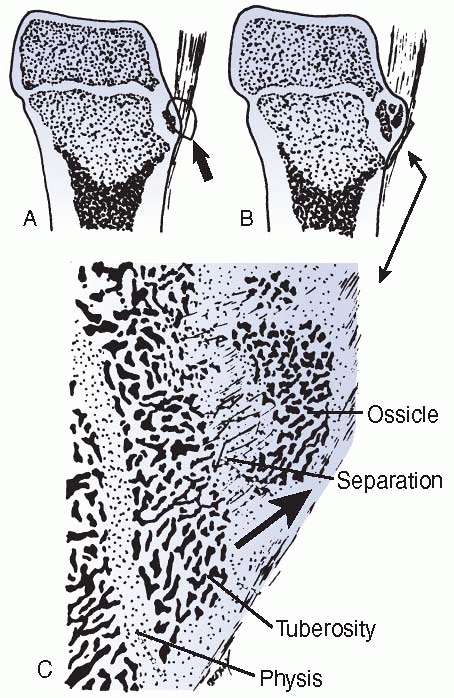
failure patterns differ from those in other physes. This area develops
primarily as a tensile-responsive structure (i.e., an apophysis).
However, the introduction of an osseous secondary ossification center,
initially in the distal tuberosity, interposes osseous tissue, which
tends to fail in tension and thus may lead to avulsion of part of this
ossification center (Fig. 2-16). Healing of the
displaced fragment to the underlying undisplaced secondary center
creates the symptomatic reactive overgrowth known as an
Osgood-Schlatter lesion.146,151 Similarly, in adolescents, excessive tensile stress may avulse the entire tuberosity during the late stages of closure.152
 |
|
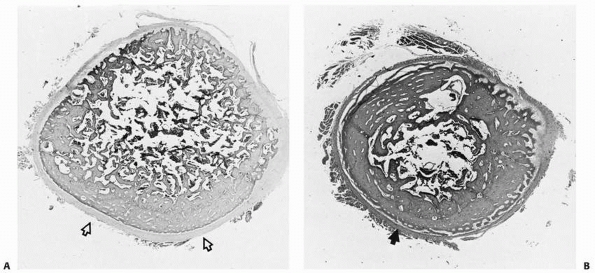
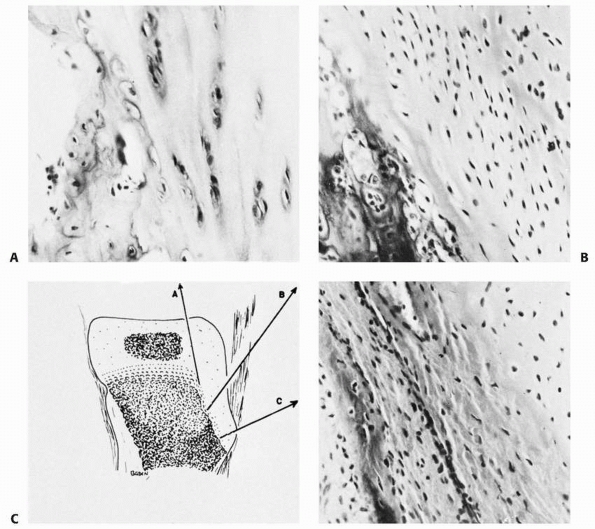
FIGURE 2-15 Histology of a typical apophysis, the tibial tuberosity (tubercle). A. Attenuated columnar cartilage adjacent to the main proximal tibial physis. B. Fibrocartilage and minimal hypertrophic matrix in the midtuberosity region. C. Fibrocartilage and membranous ossification in the distal end of the tuberosity.
|
main constituents of the cartilaginous matrix are collagens (mainly
type II) and proteoglycans. Although collagen type II provides
structural strength, the proteoglycans have structural and regulatory
effects. The structural effects of proteoglycans arise through binding
to the collagen components and the waterbinding properties that provide
resilience to compression. Regulatory effects include growth factor
interactions, cell-matrix interactions, and regulation of collagen
fibril size. Specific molecules expressed and their functions are
listed in Table 2-1.
matrix is almost entirely synthesized by osteoblasts. The composition
of the bone matrix was outlined by Buckwalter and associates.20
Briefly, bone matrix is a composite material composed of an inorganic
(mineral) portion and an organic portion. The composite structure
provides physical strength and resilience to fracture. Bone with
deficient inorganic mineral content is pliable, and bone with deficient
organic content is brittle.
 |
|
FIGURE 2-16
Avulsion (tension) failure of the developing ossification center of an apophysis. The degree of displacement determines the likelihood of healing and the symptoms and size of the final lump, typical of an Osgood-Schlatter injury. |
|
TABLE 2-1 Matrix Molecules of Cartilage
|
||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||||||||||||||||||||||||||||||||||||||||
The inorganic portion is mainly hydroxyapatite, with some carbonate and
acid phosphate groups. It has also been suggested that bone crystals do
not contain hydroxyl groups and should be termed apatite rather than
hydroxyapatite.20 The organic
portion is composed of collagen type I (90%) and noncollagenous
proteins. The noncollagenous protein portion includes a number of
proteins and proteoglycans that perform structural and regulatory
functions. Actual molecules and functions are outlined in Table 2-2 and in the following section.
provides an example of the major proteins found within bone and
cartilage matrices.
at least 19 distinct genes. Members are expressed in most tissues.
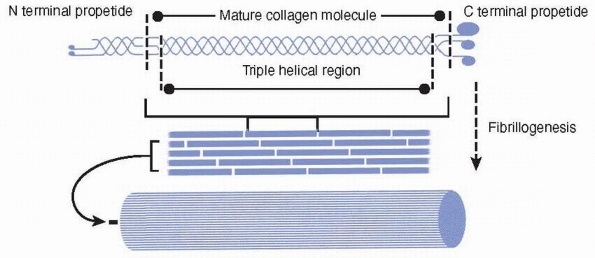
Collagens have a triple helical region that arises from the repeated
winding of three collagen molecules around a common axis. Collagens are
synthesized as propeptides that are often glycosylated. Collagen is
secreted from cells and is processed in the extracellular space. The
processed collagen forms into subunits that then undergo
fibrillogenesis (Fig. 2-17). The fact that the
final fiber is composed of many individual molecules accounts for the
dominant negative mutations that can be observed within the collagen
family.79 The incorporation of
individual molecules containing mutations that affect the packing of
the peptides into the triple helix can disturb the structure of the
whole fiber. The molecular structures that arise are in the form of
fibrils or netlike structures. In reality, the multimeric fibers
observed in vivo are often composed of a number of different collagens.8
The collagen type I fibers act as sites for initial mineralization and
provide tensile strength to the bone. Mutations in the propeptides can
cause a variety of phenotypes affecting mineralization and bone
fragility, the most severe being osteogenesis imperfecta. In contrast,
collagen type II is a triple helical molecule that is composed of three
α1(II)
polypeptides and is expressed in cartilage, particularly within the
proliferative zone of the physis. It is the main fibril-forming
collagen in cartilage. Mutations cause Langer-Saldino achondrogenesis
and spondyloepiphyseal dysplasia congenita.28,49
|
TABLE 2-2 Composition of Bone
|
||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||||||||||||||||||||||||||||||||||
Collagen type X is associated with the matrix of hypertrophic
chondrocytes and is involved with the mineralization process of
cartilage matrix.88,89,162,167 Mutation of collagen X causes spondylometaphyseal dysplasia,80 but the deletion of the encoding gene results in mild changes.80,181
 |
|
FIGURE 2-17
Collagens are synthesized as a propeptide that is often glycosylated (not shown). The collagen molecule has a triple helical region that arises from the repeated winding of three collagen molecules around a common axis. The processed collagen forms into subunits that then undergo fibrillogenesis. |
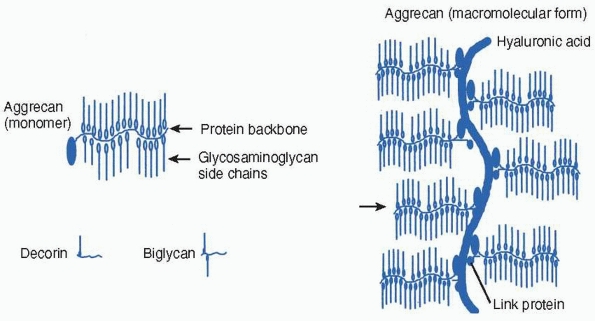
amounts within all connective tissues. Proteoglycans are proteins that
have either one or a number of polysaccharide chains linked to a
protein core. The polysaccharide’s glycosaminoglycan side chains are
heparin, heparin sulfate, chondroitin sulfate, dermatan sulfate, or
keratan sulfate. The glycosaminoglycans differ in the composition of
their constituent disaccharide structures. They can combine with other
molecules within the matrix to form macromolecular structures (Fig. 2-18).52
The proteoglycans present in the physis include large proteoglycans
like aggrecan as well as smaller proteoglycans, such as decorin,
biglycan, and possibly, fibromodulin. Decorin and biglycan have side
chains of dermatan sulfate, and betaglycan has chondroitin and heparin
sulfate chains. Fibromodulin has side chains of keratan sulfate. The
territorial capsules of the chondrocytes in the upper proliferative
region of the physis stain for biglycan, and the interterritorial
matrix stains for decorin.10 These proteoglycans have a structural role but are also known to interact with growth factors.10,72,168,189
as bone Gla protein. It has three residues of gamma-carboxyglutamic
acid that enable it to bind to hydroxyapatite. It is thought to play a
role in mineralization of the bone matrix,171,172 but the exact mechanism and function are undetermined.40,68
Osteonectin has the ability to bind calcium and collagen type I, and
may enable the process of mineralization that is initiated on the
collagen type I fibers.12 Osteopontin is thought to be critically involved with the binding of osteoclasts,74,176 the cells that degrade the bone and physeal matrix.119
Matrix Gla protein is an inhibitor of calcification. The cartilage of
mice lacking this protein undergoes spontaneous calcification.109
between neighboring cells and between cells that are separated by up to
an almost complete body length. Communication signals take the form of
diffusible molecules, which pass between the cells, or by cell
surface-bound receptor-ligand interactions.100,225 In addition, neighboring cells can pass information between one another via their gap junctions.51 These channels enable the passage of small molecules, including calcium ions,
between neighboring cells. Calcium is a key second messenger that provokes a number of cellular events.127
 |
|
FIGURE 2-18
Proteoglycans are proteins that have either one or a number of polysaccharide (glycosaminoglycan) chains linked to a protein core. Aggrecan is present in cartilage and has the ability to form macromolecular structures with hyaluronic acid and link protein. Decorin and biglycan are present in bone and cartilage matrix. |
secreted by endocrine glands and are transported by body fluids. They
coordinate body functions in complex organisms. Hormones can be in the
form of amino acid derivatives (e.g., epinephrine), polypeptides (e.g.,
somatotropin or growth hormone), glycoproteins (e.g.,
follicle-stimulating hormone), steroids (e.g., testosterone), or fatty
acids (e.g., prostaglandins).
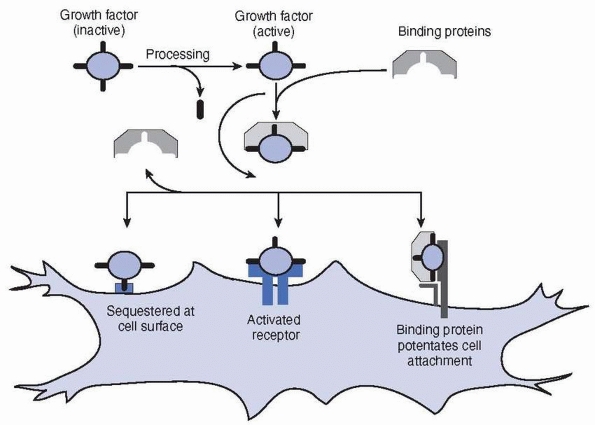
The binding of growth factors and hormones to other molecules may
facilitate their transportation to their target tissues, increase their
survival by inhibiting their proteolytic degradation, and control their
activities. Many growth factors, including the fibroblast growth
factors (FGFs), transforming growth factor-β (TGF-β),
and insulin-like growth factors (IGFs), can be bound to the matrix
molecules. Cell activation usually requires the factors to bind to
their receptors on the cell surface, although a number of hydrophobic
hormones pass directly through the outer cell membrane and bind to
intracellular receptors34,48,117,133 (Fig. 2-19).
 |
|
FIGURE 2-19
The figure shows aspects of growth factor interactions. Any particular growth factor will possess only a subset of such interactions. Growth factors may require activation (e.g., TGF-β). Binding proteins may sequester or protect the growth factor. The binding protein may also potentate the binding of the growth factor to the surface receptor (e.g., FGF and heparin). Cells may also sequester the growth factor at the cell surface. |
in the phenotype observed. A good example is the double mutant of BMP-5
and BMP-7, which is lethal during embryonic development, but a null
mutation in either one has little effect.197
fibroblast growth factors (FGFs) are widespread. FGFs are angiogenic
and can influence mitosis, differentiation, migration, and survival in
many cell types. FGFs can activate one of four high-affinity FGF
receptors (FGFRs). Point mutations of these receptors have been
implicated in a number of skeletal deformities including Pfeiffer
syndrome (FGFR1), Crouzon and Jackson-Weiss syndromes (FGFR2), and
achondroplasia (FGFR3),156 suggesting that FGFR signaling is an essential regulatory component for skeletal growth and development.132
skeleton remain largely unknown. To date, the FGF family comprises 22
members including acidic FGF (FGF-1), basic FGF (FGF-2).155 FGF-1 and FGF-2 are present in the extracellular matrix of bone.69
More recent data reveal that FGF-18 may be a physiological ligand for
FGFR-3 in regulating bone lengthening, but it may also signal through
another FGFR to regulate osteoblast growth.106
alternative forms of the specific forms of FGF-1 and FGF-2. FGF-1 is
typically 140 amino acids in length, but larger forms of 160 and 154
amino acids have been identified. FGF-2 is normally translated as a 155
amino acid molecule, but through the use of alternative start codons,
three higher molecular weight forms have been identified.
well conserved across species. Comparing the amino acid composition of
FGF-1 and FGF-2 from different species, Hearn found a 92% sequence
identity between human and bovine FGF-1. Only 2/155 and 3/155 amino
acids differ in human and bovine, and human and ovine, forms of FGF-2,
respectively.70
(IGF-1) is critical for normal bone growth as has been confirmed by the
severe growth retardation in the IGF-1 and its receptor gene knockout
mice.4,105
Genetic studies with various mutant, knock out, and congenic mice have
revealed that there is a clear relationship between circulating IGF-1
concentrations and bone volume.230
Consistently, delivery of exogenous IGF-1 stimulated growth of physeal
height in normal rats, enhanced chondrocyte maturation and thus
longitudinal growth in hypophysectomized rats,75 and stimulated longitudinal and circumferential growth and increased bone mineral density.230
on chondrocytes at all stages of differentiation in growth plate
cartilage.207 Expression of IGF-1 mRNA and protein has also been localized in all chondrocyte layers of the physis,175
suggesting that locally produced IGF-1 acts at the chondrocyte level in
a paracrine/autocrine manner to stimulate longitudinal growth. In
primary cultures, IGF-1 stimulates proliferation and matrix synthesis
in physeal chondrocytes.208
Consistently, in rodent in vivo studies, infused IGF-1 not only
stimulates physeal chondrocytes at all stages of differentiation, but
also promotes chondrogenic differentiation via its effects on resting
stem-like cells of the physis.75
deposited in bone matrix, and it has been demonstrated that the vast
majority of IGF-1 in bone is derived locally from osteoblastic
synthesis.230 Osteoblasts appear to
be the major target of IGF-1 as IGF-1 type 1 receptor is present on
osteoblasts and IGF-1 stimulates osteoblast proliferation and its
recruitment.233 Since IGF-1 is
stored within the skeletal matrix and is released during bone
resorption, IGF-1 may be a critical coupling factor that keeps bone
formation closely linked to bone remodeling230 (see Bone Remodeling section later).
and the DPP/Vg1 families have critical functions in the development of
the skeleton, its growth and maintenance, and fracture repair. The bone
morphogenic proteins (except for BMP-1) are members of the DPP/Vg1
family and are discussed in the next section.
stimulates bone formation when injected into rodent bones and induces
endochondral bone formation in adult non-human primates. Most fracture
healing studies have also demonstrated positive effects of TGF-β in stimulating bone repair and its potential usefulness in implant fixation.25
are also released in large quantities during platelet activation. The
active form is either a heterodimer or homodimer. It is thought that
the pro-region may either help in the folding of the proteins during
synthesis or control TGF-β activity. In the case of TGF-β1,
the proregion and a second glycoprotein can also bind to the active
factor to form a latent complex. Apart from the presence of the growth
factor itself, the presence or absence of the latent complex controls
the activity of TGF-β1. The active TGF-β1
complex can be released from the latent complex by extreme pH or by
catalytic methods. This is particularly important in fracture repair
and bone remodeling. The activation of latent TGF-β is likely to be critical in the induction of fracture repair and osteoblast function.
with betaglycan in binding TGF-β. Decorin has the ability to negatively regulate the activity of TGF-β.14,186
(BMPs) and their orthopaedic relevance and applications have been
reviewed previously.25,78,186
The BMPS (except BMP-1) represent a group of related growth factors
that have critical roles in the cell proliferation and differentiation
of a number of cell types including mesenchymal cells, chondrocytes,
and osteoblasts.25,30,78,90,91,182,186,216,232
They have roles in embryo and fetal development, bone growth, and
fracture repair. Several BMPs produce ectopic cartilage or bone when
implanted subcutaneously.2,219,224
factors discussed so far, the BMPs have a number of binding proteins
both in the extracellular matrix and on the cell surface. A secreted
glycoprotein termed noggin can bind and inactivate BMPs.55 Chordin is a similar protein that most likely has a similar function.166
It has been proposed that these proteins control BMP activity and may
also serve as a mechanism for establishing gradients of BMPs across the
embryo during development. Active BMPs bind to heterotetrameric
serine/threonine kinase receptors. The nonactivated receptors exist as
type 1 and 2 receptor proteins, with the type 2 receptor being able to
autophosphorylate. Once the ligand binds, the two receptors are brought
together and the receptor type 1 receptor is phosphorylated. Only after
the receptor type 1 is phosphorylated is a cellular response achieved.
Intracellular activation is via the intracellular proteins termed
SMADs, but other inhibitors can still come into play. Exposure of the
cell to a number of other growth factors (including cer-1) can inhibit
the activation of the cell by BMPs.163,186
metaphyseal vascular supply is crucial to endochondral ossification,
and fracture repair does not occur without an adequate vascular supply.
Bone fracture disrupts the marrow architecture and blood vessels within
and around the fracture site. During fracture healing, regeneration of
three normal blood supplies (the medullary, periosteal, and osseous) to
the callus and cortical bone need to be coordinated.177
neovascularization. They are essential for neovascularization during
the normal bone lengthening and fracture repair. Previous studies have
shown that the key angiogenic growth factor, vascular endothelial
growth factor (VEGF), is essential for blood vessel invasion of the
growth plate mineralized hypertrophic cartilage, cartilage remodeling,
and bone formation during normal endochondral bone lengthening.56
Endogenous VEGF also plays a key role in bone repair, as blocking VEGF
activity inhibited repair of femoral fractures and cortical defects in
rodents.200 At the bone fracture site, VEGF activity is essential for appropriate angiogenesis, callus architecture, and mineralization.25
Several studies have demonstrated that local delivery of exogenous VEGF
promotes angiogenesis and bone formation at the bone fracture site.25
and FGF-2 are sequestered in the bone matrix. Angiogenic factors act
directly (such as VEGF and FGF-2) or indirectly (such as TGF-β and tumor necrosis factor-α)
on endothelial cells, promoting proliferation and migration of the
cells into areas in which they are released at the injury site.174
Angiogenic factors acting indirectly by recruiting macrophages and
monocytes, in turn, release their own direct-acting angiogenic factors.192 During angiogenesis, while VEGF and FGF-2 induce angioblast differentiation and TGF-β1
enhances smooth muscle cell differentiation from mesenchymal cells,
PDGF-B stimulates recruitments of smooth muscle cells and pericytes
around nascent vessels.35
expansion and bone growth is achieved by adding newly synthesized bone
to existing bone by two mechanisms: endochondral ossification and intramembranous ossification.
These mechanisms are named by the intermediate structures that must be
passed to form the bone. The production of any particular bone after
initial differentiation may involve discrete, juxtaposed, or
interspersed areas of each basic pattern. Endochondral-derived bones
generally have membranous ossification by appositional bone growth from
the periosteum. Similarly, membrane-derived bones may grow and elongate
by an endochondral process.139,147
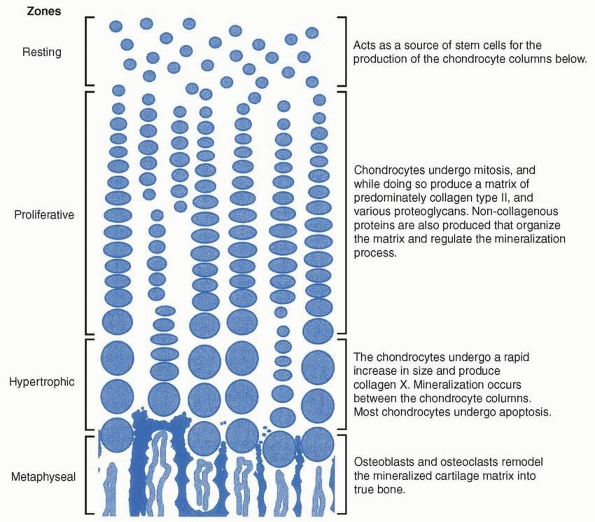
forms via a cartilaginous intermediate. The physis (or the growth
plate) best reflects this process. Physes are temporary cartilaginous
tissue situated between the primary and secondary ossification centers
of all long bones. From 9 to 10 weeks’ gestational age to skeletal
maturity at 15 to 17 years, they are responsible for the longitudinal
growth of bone. The physis can be divided into at least three zones.
The reserve or resting zone is situated on the epiphyseal side and
contains small, spherical cells in groups of two or three cells
randomly distributed throughout the zone. These cells are stem
cell-like, responsible for generating new chondrocytes of the physis.
In the adjacent proliferative zone, chondrocytes undergo mitosis and
are organized into columns running parallel to the axis of bone growth.
Cells in the proliferative zone mature and eventually increase to five
to ten times their volume in the hypertrophic region. Matrix vesicles
are also deposited within the longitudinal septa of the physis. Matrix
vesicles are membraneencapsulated structures that are thought to
concentrate calcium and phosphate. Enzymes such as alkaline phosphatase
convert organic phosphates to inorganic phosphates. The longitudinal
septum around the terminal hypertrophic chondrocytes mineralizes, and
this mineralized matrix forms the template for new bone deposition in
the metaphysis (Fig. 2-20).
and volume, the matrix in the physis also undergoes a continual
modification in content. The two major macromolecules of cartilage
matrix produced by the chondrocytes are the proteoglycans
(predominantly aggrecan and with lesser amounts of decorin, biglycan,
and fibromodulin) and the collagens (types II, IX, X, and XI). The
major change in physeal proteoglycan structure occurs as chondrocytes
organize into columns in the proliferative zone. Additional variation
occurs in the hypertrophic region, where the glycosaminoglycan
sulfation pattern demonstrates
differences
between the pericellular and extracellular spaces and the appearance of
a unique collagen (type X) is observed. The small
proteoglycans—decorin, biglycan, and fibromodulin —are also
differentially expressed across the physis, although detailed studies
of these proteoglycans have not been done (see Table 2-1).
 |
|
FIGURE 2-20
The figure shows the process of endochondral ossification within the physis. Although not as organized, endochondral ossification follows a similar pattern during fracture repair. |
tightly regulated by endocrine/paracrine/autocrine factors, such as
hormones, vitamins, transcriptional factors, and growth factors, and
involves coordinated and sequential expression of growthregulatory
factors. Although growth hormone (GH) in the circulation has a global
effect on physeal function throughout the body, many locally produced
growth factors act locally. Many hormones (such as GH, thyroid hormone,
estrogen, glucocorticoids, calcitonin), vitamins (vitamin D3,
ascorbate, retinoic acid), morphorgens (Indian hedgehog [IHH], BMPs),
growth factors (IGFs, BMPs, FGFs, parathyroid hormone-related peptide
[PTHrP], PDGF, TGF-β, VEGF) and their binding proteins (such as IGFBPs, chordin, noggin), and cytokines (tumor necrosis factor-α,
interleukin IL-1, and others) have now been shown to have important
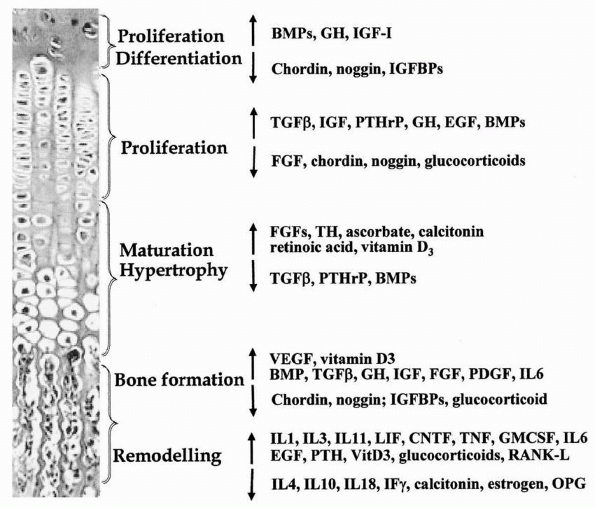
roles in regulating various processes of the endochondral ossification (Fig. 2-21).5
Cellular response is determined by parallel processing of the
intracellular signals that are induced by a number of active growth
factors binding
to
their specific receptors. Presented below is an outline of the likely
actions of a number of key growth factors on endochondral ossification.
 |
|
FIGURE 2-21
Systemic and local factors including hormones, vitamins, cytokines, and growth factors that control or influence chondrocyte differentiation, proliferation, and maturation, as well as bone formation and remodeling. |
two major factors regulating postnatal growth. In skeletal tissues,
both chondrocytes and osteoblasts synthesize IGF-1, and GH modulates
its synthesis in both cell populations. According to the original
somatomedin hypothesis, GH stimulates skeletal growth through IGF-1
that is produced in the liver under the influence of GH and secreted
into the circulation.39 Upon
reaching target tissues, IGF-1 interacts with its receptors and induces
a cellular growth signal. However, normal postnatal growth achieved in
liver-IGF-1 null mice (in which hepatic IGF-1 expression was abolished
specifically) suggests the importance of extrahepatic IGF-1 expression
and an autocrine-paracrine role for IGF-1 in normal skeletal growth.196
Therefore, both circulating and locally expressed IGF-1 play important
roles in longitudinal bone growth and the maintenance of bone mass, and
IGF-1 plays an essential role in longitudinal bone growth in response
to GH exposure.195
least partially mediated via local production and action of IGF-1 at
the stem cell zone.175 GH receptor has been localized in the physeal chondrocytes particularly in the proliferative zone,6,41,223 and it is generally accepted that GH acts at both the stem and proliferative phases of chondrocyte differentiation.75
At present, apart from the IGF-1-mediated effects on the stem cells of
the physis, GH also stimulates proliferation of the prechondrocytes in
the resting zone153 and has some
priming effect on these stem cells to promote their differentiation
independently of IGF-1, as proposed by the dual effector theory. In
addition, there is genetic evidence that supports this dual,
IGF-1-independent and IGF-1-dependent roles for GH in promoting
longitudinal bone growth.220
proliferation, differentiation, and matrix synthesis in
undifferentiated chondrocytes in the resting zone.45,90
It is believed that once the chondrocytes start differentiating, the
expression of noggin inhibits the continual outgrowth of the
undifferentiated chondrocytes.18 Once the chondrocyte has lost its resting phenotype, IGF-1 may act as a stimulator of proliferation and differentiation.135,203 Epidermal growth factor (EGF) can augment IGF stimulation by increasing the expression of the IGF-1 receptor.13 Although the chondrocytes synthesize large quantities of matrix molecules, they also synthesize FGF-1, FGF-2, TGF-β, VEGF, and a number of the BMPs.15,26,32
These molecules can act in an autocrine or paracrine manner, but many
are sequestered into the newly formed cartilage matrix. While FGF-2 in
low doses is mitogenic for the chondrocytes,110
as occurs in achondroplasia, constant activation of FGF receptor
(FGFR3) inhibits chondrocyte proliferation and accelerates terminal
differentiation of chondrocytes.93,99,111
FGF/heparin sulfate interaction is probable in the differentiation of
the physeal chondrocytes because the continuous exposure of FGF-2
inhibits chondrocyte differentiation in vitro and inhibitors of
glycosaminoglycan sulfation (including heparin sulfate) restore the
differentiation process. Additional sulfate permits glycosaminoglycan
sulfation and returns the effect of FGF-2.33
hormone-related protein (PTHrP) act to maintain the proliferative state
of and inhibit the maturation of chondrocytes. It is postulated that
two negative feedback loops involving actions of PTHrP regulate the
pace of chondrocyte differentiation in the postnatal growth plate.215
The first loop is confined to the proliferativehypertrophic transition
zone and early hypertrophic chondrocytes, which express morphogen
Indian hedgehog (IHH), its receptor Patched, and PTH/PTHrP receptor.
IHH, which stimulates chondrocyte proliferation and inhibits
hypertrophic differentiation, binds Patched in the hypertrophic zone
resulting in a stimulated production of PTHrP. PTHrP then acts on its
receptor at the proliferating chondrocytes to keep them proliferating
and, thereby to delay the production of IHH, thus closing the IHH-PTHrP
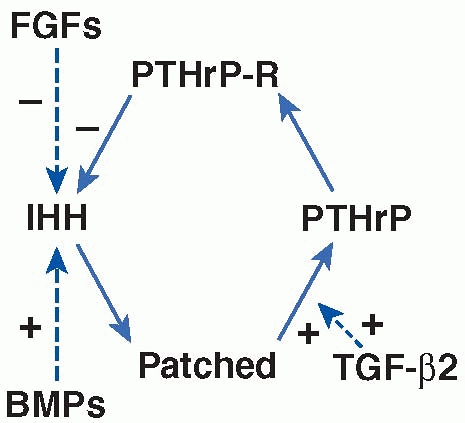
feedback loop (Fig. 2-22). In the second
feedback loop, IHH can bind Patched in the resting stem cell zone, and
this may stimulate PTHrP production, which then diffuses to its
receptor leading to IHH down-regulation. These two IHH-PTHrP signaling
cascade feedback loops limit maturation of proliferative chondrocytes
to hypertrophic form.94,215,217
regulating chondrocyte proliferation and hypertrophic differentiation
interact with the IHH-PTHrP feedback loops (Fig. 2-22).
Apart from inhibiting chondrocyte proliferation, part of the effects of
FGF signaling in positively affecting chondrocyte maturation is
mediated by the suppression of IHH expression.125 However, FGF effects on chondrocytes can occur independently of PTHrP/IHH action.185 In the physis, expression of the mRNA for BMP-2 and BMP-6 peaks in hypertrophic chondrocytes before mineralization.26
Studies have demonstrated that BMPs act on chondrocytes to induce
proliferation through the induction of IHH expression by
prehypertrophic chondrocytes, suggesting that BMP signaling modulates
the IHH/PTHrP signaling pathway that regulates the rate of chondrocyte
differentiation.93,234 In addition, in vitro studies have suggested that during embryonic development, signaling of TGF-β2 may act as a critical
signal relay between IHH and PTHrP. It mediates the effects of IHH
inhibiting hypertrophic differentiation and induces PTHrP expression,
maintaining the chondrocytes in proliferative state and slowing down
the pace of their maturation.2
 |
|
FIGURE 2-22
Schematic representation of an IHH-PTHrP feedback loop controlling the rate of chondrocyte hypertrophic maturation and its modulation by BMPs, TGF-β2, and FGFs in a postnatal growth plate. PTHrP is secreted from hypertrophic chondrocytes and acts on PTHrP receptor on proliferating chondrocytes to keep the chondrocytes proliferating. When PTHrP expression is low, IHH is produced by the maturing cells. IHH binds to its receptor Patched on the hypertrophic cells, resulting in a stimulation of PTHrP expression. Activation of PTHrP receptor by PTHrP in turn represses IHH expression, thus closing a negative feedback loop. FGFs and BMPs interact with this negative feedback loop through their activities in inhibiting and stimulating the production of IHH, respectively. Evidence has suggested that TGF-β2 mediates the function of IHH in stimulating the production of PTHrP. |
and form a cartilaginous matrix with only the epiphyseal vascular
supply, the metaphyseal vessels are critical for the mineralization
process.212 Metaphyseal vascular
invasion occurs at the hypertrophic-metaphyseal interface. The
endothelial cells invade most likely as a consequence of angiogenic
factors present in the matrix or secreted by the chondrocytes
themselves. VEGF, TGF-β, and FGFs are known
to be present in the physeal cartilage matrix and are angiogenic. It is
interesting that an oversupply of FGF-2 infused into the physis induces
vascular invasion from the metaphysis only; even if the FGF-2 is
present at the epiphyseal side of the physis, the epiphyseal vessel
will not invade.7 Apart from
providing the necessary nutrients for the mineralization process, the
metaphyseal vessels also bring in osteoblasts, osteoclasts, and other
cell types. The osteoclasts degrade the mineralized cartilage matrix
while osteoblasts lay down new bone that is also rich in growth factors
such as TGF-β, FGF-2, IGF-1, and BMPs.
involved in secondary membranous ossification. The diaphyseal cortex of
developing tubular bone is progressively formed (modeled) by the
periosteum and modified (remodeled) by the re-formation of osteons.
This peripheral periosteal process of membrane-derived ossification is
extensive and rapid in fracture healing in infants and young children.
The replacement process also may be seen when portions of the
developing metaphysis or diaphysis are removed for use as bone grafts.
cells are formed from the overlying tissue, the inner cambium layer of
the periosteum193 (see Periosteum
section earlier). The osteoprogenitor cells continue to differentiate
into osteoblasts, which produce and add new bone matrix peripherally
that later undergoes mineralization.
the metaphysis or in the fracture callus is woven bone, which is
remodeled to lamellar bone. At the metaphysis, the trabeculae of
bone-covered calcified cartilage (primary spongiosa) are resorbed by
osteoclasts and the calcified cartilage template is replaced by
lamellar bone and remodeled into more mature bone trabeculae (secondary
spongiosa). The deposited secondary bone trabeculae at the
metaphyseal-diaphyseal junction is further remodeled and incorporated
into the diaphysis, in a process in which osteoclasts remove bone from
the periphery of the metaphysis and new bone is formed at the endosteal
surfaces. Although cancellous bone can be remodeled and obtain its
nutrients from the surface, cortical bone is remodeled into a complex
structure of osteons that together form the cortical bone. Osteons are
tubular structures that interconnect. They consist of layers of ordered
lamellar bone around a central canal. The central canal contains blood
vessels, lymphatics, and in some cases, nerves.20
osteoblasts. The bone is encapsulated by bone-lining cells that have
the potential to become activated osteoblasts. The bone-lining cells
have slender cellular processes that make contact with the osteocytes
within the mineralized bone. Osteocytes are thought to arise from
osteoblasts that have become entrapped during bone formation. It has
been proposed that the bone-lining cells need to erode the osteoid that
covers the underling bone for osteoclasts to bind.122,123
Osteoclasts are bone-degrading cells that are produced from the
hematopoietic pathway. Upon activation, they bind to the surface of the
bone and secrete enzymes into the space beneath. The space is acidic
and contains many proteolytic and bone degrading enzymes.114 The acidic pH and proteases are thought to release and activate the sequestered growth factors IGF-1 and TGF-β,
resulting in the recruitment, proliferation, differentiation, and
activation of the stromal osteoprogenitor cells and bone-lining cells
to become active osteoblasts, synthesizing bone matrix and increasing
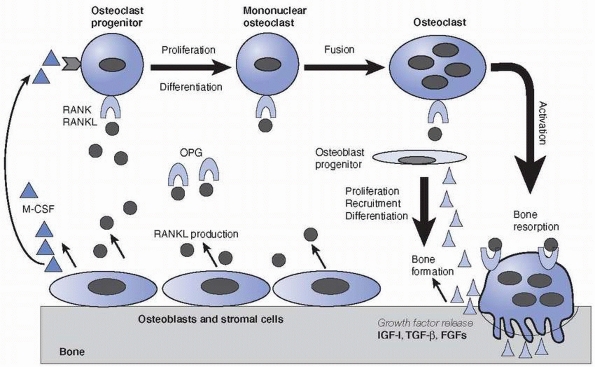
their survival (Fig. 2-23).46,47,116,118 The newly laid osteoid by osteoblasts is subsequently mineralized to become bone.
activity and osteoclast activity are linked during the bone remodeling.
On the one hand, osteoclastic activity releases growth factors IGF-1
and TGF-β stimulating osteoblastic differentiation and activity as described above (Fig. 2-23),
and on the other, cells of the osteoblast lineage provide factors
essential for the differentiation of osteoclasts. The discovery of the
interaction between the receptor activator of NF-kappaB (RANK) ligand
(RANKL) expressed by osteoblasts and its receptor RANK expressed on
osteoclast precursors confirms the well-known hypothesis that
osteoblasts play an essential role in osteoclast differentiation (Fig. 2-23).201
It is now known that two hematopoietic factors, namely RANKL and
macrophage colony-stimulating factor (M-CSF), together are necessary
and sufficient for osteoclast formation.16
Several factors have now been identified that can modulate RANK-induced
osteoclastogenesis. Although vitamin D3 metabolite, PTH, PTHrP, PGE2,
cytokines IL-1, IL-6, TNF, LIF, and IL-11, and corticosteroids have
been shown to induce the expression of RANKL in stromal/osteoblastic
cells and thus stimulate osteoclast formation, osteoprotegerin (OPG), a
decoy receptor for RANKL, blocks the RANKL-RANK interaction and
inhibits osteoclastogenesis.16,112 Recent studies have also shown that lipopolysaccharide and inflammatory cytokines such as TNF-α
and IL-1 can also directly regulate osteoclast differentiation and
function through a mechanism independent of the RANKL-RANK interaction.92 TGF-β superfamily members and interferon-gamma (INF-γ) are also shown to be important regulators in osteoclastogenesis.84 Estrogens, calcitonin, BMP2/4, TGF-β, IL-17, PDGF, and calcium have been shown to be anabolic or inhibit osteoclastogenesis16 (Fig. 2-21). In addition, EGF receptor signaling has been shown to be important for the secretion of matrix metalloproteases (MMPs)124 and maintenance of osteoclast activity,29 both of which are important for the bone remodeling.
that may be associated with bone remodeling. Osteocytes also possess
cellular processes that connect osteocytes to one another and to the
bone-lining cells above.36 It is
possible that the osteocytes are responsible for sensing bone stress;
if undue stress is detected, they favor bone deposition, whereas if a
lack of stress is detected, they favor bone resorption.
 |
|
FIGURE 2-23
The linked activities of osteoclast bone resorption and osteoblast bone formation during bone remodeling. Bone-lining cells, osteoblasts or some marrow stromal cells express RANKL, which activates receptor RANK on osteoclast progenitors of the monocyte-macrophage lineage and stimulates the osteoclast differentiation and activation. M-CSF is another essential factor for osteoclast differentiation. However, decoy receptor OPG binds RANKL and antagonizes RANK function and thus inhibits osteoclast formation. Activated osteoclasts secret acid and proteases and erode bone on the surface. During the resorption process, sequestered growth factors, such as IGF-I, TGF-β and FGFs, are released from the bone matrix and activated, which results in the recruitment, differentiation, and activation of the osteoprogenitors to become osteoblasts to initiate bone matrix synthesis and bone formation. |
fibrous, and cartilaginous tissues. Healing of these tissues differs,
depending on both the type of tissue and the temporal maturation.
fracture healing, whether in the diaphysis, metaphysis, or epiphyseal
ossification center, can be grouped conveniently into a series of
phases that occur in a reasonably chronologic sequence.120,180 Several factors that influence bone healing have been identified from clinical observation as well as experimental work160,214
and must be taken into account when treating childhood fractures on a
rational basis. However, certain areas of the developing skeleton,
particularly the physis and epiphyseal hyaline cartilage, probably do
not heal by classic callus formation. In fact, when this type of
osseous (callus) repair occurs in these cartilaginous regions,
significant growth deformities may result due to formation of an
osseous bridge between the secondary ossification center and the
metaphysis (see “Physeal Healing Patterns”).
fracture repair: primary osteonal, secondary osteonal, and nonosteonal.
Primary osteonal fracture healing occurs when cortical bone is laid
down without any intermediate, and therefore hardly any callus forms;
it is only possible if cortical bone is repositioned and fixed in close
proximity. Secondary osteonal union occurs if cortical bone is laid
down between two segments of fractured cortical bone before callus
formation. Nonosteonal union occurs through endosteal and periosteal
callus formation.183
skeleton can be divided into three closely integrated, but sequential,
phases: the inflammatory phase, the reparative phase, and the
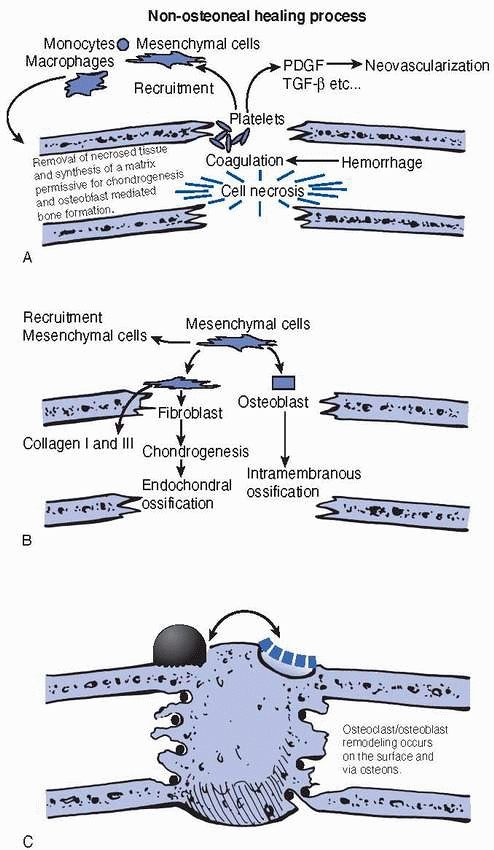
remodeling phase (Fig. 2-24). In children, the
remodeling phase is temporally much more extensive and physiologically
more active (depending on the child’s age) than the comparable phase in
adults. The remodeling phase is further modified by the effects of the
physis responding to changing joint reaction forces and biologic
stresses to alter angular growth dynamics. This occurs even when the
fracture is mid-diaphyseal.
portions of the developing skeleton (diaphysis, metaphysis, or
epiphyseal ossification center), disruption of blood vessels leads to
activation of the coagulation cascade and formation of a hematoma
enclosing the fracture area.
through the release of growth factors, cytokines, and prostaglandins.
If the fracture is localized to the maturing diaphysis, there is
bleeding from the Haversian systems, as well as from the multiple small
blood vessels of the microcirculatory systems of the endosteal and
periosteal surfaces and contiguous soft-tissue anastomoses.61
In the region of the metaphysis, this bleeding may be extensive because
of the anastomotic ramifications of the peripheral and central
metaphyseal vascular systems. A hematoma accumulates within the
medullary canal at the fracture site, beneath the elevated periosteum,
and extraperiosteally whenever the periosteum is disrupted during the
fracture. In contrast to adults, the periosteum strips away easily from
the underlying bone in children, allowing the fracture hematoma to
dissect along the diaphysis and metaphysis; and this is evident in the
subsequent amount of new bone formation along the shaft.
 |
|
FIGURE 2-24 The figure demonstrates the three phases of fracture repair (A) inflammatory phase, (B) reparative phase, and (C)
remodeling phase. The inflammatory cells remove the debris from the fracture site and, together with the fibroblastic cells, develop the site into a matrix that will support the cells that enable new bone to be formed. The mesenchymal cells are recruited by the release of growth factors in the fracture site. The mesenchymal cells may differentiate into osteoblasts that produce bone in a membranous fashion. Alternately the mesenchymal cell may become chondrogenic and produce bone by the endochondral pathway. Remodeling begins with resorption of mechanically unnecessary, inefficient portions of the callus and the subsequent orientation of trabecular bone along the lines of stress. |
the zone of Ranvier limit subperiosteal hematoma formation to the
metaphysis and diaphysis. Because the perichondrium is densely
attached, this type of hemorrhagic response is uncharacteristic of the
epiphyseal ossification center, thus limiting its contributions to
callus formation and any intrinsic stabilization effect. Further,
because of the partially or completely intracapsular nature of some
epiphyses, propagation of a fracture into the joint allows
decompression of some of the bleeding into the joint, again limiting
the potential volume for eventual callus formation.
but also produce both inflammatory mediators and angiogenic factors.
Endothelial cells respond and increase the vascular permeability, and
allow the passage of inflammatory cells (leukocytes, monocytes, and
macrophages), fibroblasts, and stem cells into the fracture site.
Neovascularization is also initiated. Angiogenic factors like
platelet-derived growth factor (PDGF), VEGF, and TGF-β also promote osteoblast recruitment and activation.
disrupted for a few millimeters on either side of the fracture,
creating juxtaposed, avascular trabecular and cortical bone60
and producing local necrosis. It is likely that the necrosis also
results in the release of sequestered growth factors (e.g., IGF-1, TGF-β,
FGF-1, and FGF-2) from the bone. These growth factors may help in
promoting differentiation of the surrounding mesenchymal cells into
bone-forming cells.
several growth factors and cytokines that have important roles in
repair. The inflammatory cells remove the debris from the fracture site
and, with the fibroblastic cells, develop the site into a matrix that
will support the cells that enable new bone to be formed. This initial
matrix often contains collagens type I, III, and V.
Fibrovascular tissue replaces the clot with a matrix rich in collagens
I, III, and V. This matrix allows chondrogenesis or intramembranous
bone formation. Such mechanisms eventually lead to mineralization and
the formation of the woven bone of the provisional (primary) callus.
Initial invasion and cell division are around the damaged bone ends but
proceed centrifugally away from the fracture site, thus placing the
most mature repair process closest to the fracture site. However, bone
formation occurs only in the presence of an intact, functional
microvascular supply. If the vascular supply is deficient, then this
modulation of cartilaginous to osseous tissue cannot readily occur.
Osteogenic cells proliferate from the periosteum to form an external
callus and, to a lesser extent, from the endosteum to form an internal
callus. However, when the periosteum is severely disrupted, healing
cells must differentiate from the ingrowth of undifferentiated
mesenchymal cells
throughout
the hematoma. By 10 to 14 days in a child, the fracture callus consists
of a thick, enveloping mass of peripheral osteogenic tissue that is
beginning to be evident radiographically. This new bone is primarily
woven (fiber) bone.177,178
During this stage, the circumferential tissues serve primarily as a
fibrous scaffold over which cells migrate and orient to induce a stable
repair. This pluripotential mesenchyme is theoretically capable of
modulation into cartilage, bone, or fibrous tissue.158
The mesenchymal cells are recruited by the released growth factors
within the fracture site. Members of the BMP family, and possibly their
inhibitors, are likely to be involved in the recruitment and
differentiation of the mesenchymal cells. The mesenchymal cells may
differentiate into osteoblasts that produce bone in a membranous
fashion or may become chondrogenic and produce bone by the endochondral
pathway. Both mechanisms usually are present in a fracture callus, and
the degree to which each is present depends on the type of bone, age,
degree of fixation, level of bone loss, and trauma. In children,
because of the osteoblastic activity, the periosteum contributes
significantly to new bone formation by accentuating the normal process
of membranous ossification to supplement the cellular organization
within the hematoma, which is going through a cartilaginous phase.64,65
The region around the fracture site thus repeats the process of
endochondral ossification, in close juxtaposition to membranous
ossification from the elevated periosteum. Similar processes occur
within the medullary cavity. An integral part of the reparative process
at this stage is microvascular invasion, which occurs very readily in
children because of the state of vascularity within the bone and
surrounding soft tissues.27 Vessels come from the periosteal region as well as from the nutrient artery and endosteal vessels.
maturation, it is still biologically plastic and, if not protected, may
gradually deform, especially in an active young child after early
release from an immobilization device. Even in a cast, this plasticity
may allow deformation from isometric muscle activity.
longer moves and is not painful to attempts at manipulation, although
it is by no means restored to its original strength at this time. With
time, the primary callus is gradually replaced. This is enhanced in the
child because as appositional growth and increasing diameter envelop
the original fracture region, the cartilage and woven bone are replaced
by mature, lamellar bone, and the fracture is consolidated and
essentially returns to most of its normal biologic standards and
response to stress.
mechanically unnecessary, inefficient portions of the callus and the
subsequent orientation of trabecular bone along the lines of stress.
The remodeling phase is the longest of the three phases and in children
may continue until (and beyond) skeletal maturation in response to
constantly changing stress patterns imposed by continued skeletal
growth and development. Initially, new bone is laid down by both the
fracture callus and subperiosteal tissue. This bone is randomly
oriented and cannot withstand all stresses imposed on it. However, as
the bone grows diametrically in the diaphyseal or metaphyseal regions,
this new bone is gradually and increasingly incorporated into the
preexisting cortical bone, aligned in accord with predominant stress
patterns, and replaced by physiologic remodeling processes. The degree
of remodeling and progressive replacement of fracture callus is greater
in younger children, who have an immense capacity for growth and change.
phases is the establishment of an intact bony bridge between the
fragments. Because this involves the joining of separated segments of
hard tissue, the whole system must become immobile. Once the bridge has
been established—provided that adequate, continued mechanical
protection is given—subsequent biologic failure is unlikely. If the two
or more fracture fragments remain connected by the periosteum or
related material, as is likely in a child, it is easy to see how
reparative activity could be conducted from one side to the other
relatively easily and rapidly.
This is much easier in children, whose skeletons are actively and
continually remodeling in response to stress, than in adults, who have
more static skeletons. The processes of replacement and repair are
continuous and concomitant in the normally developing skeleton. The
mechanisms involved in fracture healing essentially are no different
than most of the active maturational processes. These processes are
much more active in children and are more active in the metaphysis than
in the diaphysis.
cancellous bone. Both involve a process of simultaneous bone removal
and replacement by the osteoclasts and osteoblasts through the
accompanying blood vessels. In cancellous bone of the metaphysis or the
endosteal surface of the diaphysis, the cells are never very far away
from blood vessels, and the whole process of apposition and replacement
may occur on the surface of the trabeculae. However, in compact bone,
the more deeply placed cells require the presence of an adequately
functioning perfusion system that must be replaced. This is a much
longer sequence of events and is not a major method of bone repair in
children, except when the fracture involves densely cortical regions,
such as the femoral or tibial shafts. McKibbin120 presented an extensive discussion of this process, which is sometimes referred to as primary bone union because no intermediate cells are involved.
reviewed the molecular, spatial, and temporal aspects of the regulation
of bone facture healing in animal models. It is now clear that fracture
healing is a specialized postnatal repair process that recapitulates
aspects of embryological skeletal development. It is also becoming
increasingly evident that the sequential cellular responses in the
different phases of bone fracture healing are intricately regulated by
many molecules, including (a) M-CSF, IL-1, IL-6, IL-11, RANKL, OPG, INF-γ, TNF, TGF-β1,
BMP-2, and growth and differentiation factor GDF-8 after the initial
injury and during the inflammatory response; (b) M-CSF, RANKL, OPG, TGF-β2, TGF-β3,
BMP-3, BMP-4, BMP-5, BMP-6, BMP-7, BMP-8, GDF-1, GDF-10, VEGF, and
matrix metalloproteases MMP-2, MMP-8, MMP-9, MMP-13, MMP-14 during the
repair phase; and (c) all the cytokines listed above (except IL-11 and
INF-γ), GDF-10, TGF-β1, MMP-9, MMP-13, and all BMP members previously listed in the remodeling phase.
heals by increased endochondral bone and cartilage formation, and
gradual reinvasion by the disrupted metaphyseal vessels to replace the
temporarily widened physis eventually. Very little experimental work,
mostly in rats, has been directed at assessing the post-traumatic
cellular response patterns of the physis.17,97,228
may occur. First, when the fracture occurs through the cell columns,
healing occurs primarily by continued, relatively rapid increases in
the number of cells within the columns, causing moderate widening of
the physis. Because there are some small epiphyseal vessels in this
region, some damaged tissue may be resorbed early in the healing
process. These vessels also exhibit a hyperemic response, increasing
cellular proliferation rates, especially in the peripheral zone of
Ranvier. The metaphyseal response parallels this, in that an increased
rate of bone replacement of the hypertrophic cartilage also occurs.
Once the level of fracture fibrosis and debris within the physis is
encountered, the vessels rapidly invade to reach the rest of the
maturing cell columns. These cellular response patterns lead to
restoration of normal anatomy within 3 to 4 weeks.179
of hypertrophic cells to primary spongiosa (the most commonly involved
cellular level), there may be marked separation, with the gap filled by
hemorrhagic and fibroblastic tissue. This region may progressively form
disorganized cartilaginous tissue, which is similar to the initial,
disorganized cartilaginous callus around a diaphyseal fracture.
Meanwhile, cellular proliferation, cell column formation, hypertrophy,
and calcification continue on the epiphyseal side of the disorganized
callus, leading to widening of the physis. Vascular invasion of the
remnants of hypertrophic, calcified cartilage also rapidly occurs on
the metaphyseal side of the fracture. However, once metaphyseal vessel
invasion reaches the disorganized cartilaginous callus,
vascular-mediated bone replacement is temporarily slowed, because there
is no pattern of cell columns to invade in an organized fashion. As the
callus cartilage matures and calcifies, the metaphyseal vessels begin
to invade and replace the cartilage with bone irregularly.21
This callus may be variably thick, depending on the degree of
longitudinal and lateral displacement and periosteal continuity with
the physeal periphery. The callus is replaced at different rates, and
the invading metaphyseal vessels reach the normal cell columns, which
have been maturing in a normal sequence but without osseous
replacement. This widened physis is rapidly invaded by the vessels and
replaced by primary spongiosa, and normal physeal width is
progressively restored.
contributes to early stability. This region heals by vascular invasion
of the callus to form trabecular bone between the original metaphyseal
cortex and the subperiosteal membranous bone forming continuously
external to the metaphyseal cartilaginous callus. These three
microscopic bone regions progressively merge and remodel, strengthening
the bone. These initial cellular replacement processes in both
metaphyseal and physeal regions probably take 3 to 6 weeks. However,
remodeling may continue for months to years, and it enhances the
capacity for spontaneous correction of many residual deformities.
the physis, the repair processes differ slightly. Fibrous tissue
initially fills the gap between separated physeal components, whereas
typical callus formation occurs in the contiguous metaphyseal spongiosa
or epiphyseal ossification center. If large surfaces of nonossified
epiphyseal cartilage also are involved, fibrous tissue initially forms
in the intervening region. The reparative response shows irregular
healing of the epiphyseal and physeal cartilage, with loss of normal
cellular architecture. Within the central physeal regions, diametric
expansion of cell columns is minimal, so closure of a large defect by
physeal cartilage is unlikely. The gap will remain fibrous, but with
the potential to ossify. Toward the physeal periphery, diametric
expansion is more likely, but still may not lead to closure of large
cartilage gaps by progressive replacement of fibrous tissue. This
replacement process essentially requires the germinal and hypertrophic
cell regions to diametrically expand by cell division, maturation, and
matrix expansion. The intervening fibrous tissue may disappear through
growth, but only if the gap is narrow. Because blood supply is minimal
in this region, the fibrous tissue similarly is not well vascularized,
and significant cell modulation, especially to osteoblastic tissue, is
less likely in the short term. However, the larger the gap filled with
fibrous tissue and the longer the time from fracture to skeletal
maturity, the greater the likelihood of developing sufficient
vascularity to commence an osteoblastic response and to form an osseous
bridge. Further, in young children with minimal epiphyseal
ossification, the blood supply to the physeal germinal region is not as
well defined, whereas once the ossification center expands and forms a
subchondral plate over the germinal region, microvascularity probably
increases and the chances for vascularization and ossification of the
fibrous region increase. This explains the delayed appearance of the
osseous bridge.
should be present that should fill in with minimal fibrous tissue,
allowing progressive replacement of the tissue by diametric expansion
of the physis and contiguous epiphysis. However, if the fragment has
been partially or completely devascularized by either the initial
trauma or subsequent dissection to effect an open reduction, cellular
growth and diametric and longitudinal expansion may not occur. This
increases the chances of cellular disorganization, fibrosis, and
eventual osteoblastic response. Failure to correct anatomic
displacement, especially in Salter-Harris type IV growth mechanism
injuries, increases the possibility of apposition of the epiphyseal
ossification center and metaphyseal bone, and thereby enhances the risk
of forming an osseous bridge between the two regions. When the defect
was large enough and the fracture involved the whole width of the
physis extending from the metaphysis to the epiphysis, the injured
physis will have structural disorganization, formation of vertical
septa, and finally formation of a bone bridge. When the bone bridge is
large enough, particularly in Salter-Harris types III and IV injuries,
the defect will result in a growth arrest. While growth arrest at the
peripheral portions of the physis results in angular deformities,
centrally located lesions may cause longitudinal shortening.141
bridge formation at the site of physeal injury site remain largely
unknown. Using a proximal tibial drill-hole transphyseal injury model
in rats, a study from the authors’ laboratory characterized the
injury-induced responses and cellular mechanisms for the
bone bridge formation (Fig. 2-25).228
At the physeal injury site, this study demonstrated an early acute
inflammatory response (up to day 3 postinjury). Straight after this
inflammatory response, the injury site was filled by mesenchymal
infiltrate (days 3 to 14) and subsequently was repaired by bone bridge
formation (starting from day 7). Histologically, bony bridge trabeculae
appeared on day 7 (Fig. 2-25A) and became well-constructed on day 14 with marrow (Fig. 2-25B). Before and during physeal bar formation, there were no new cartilage formation, no collagen-X synthesis at the injury site (Fig. 2-25C), and no expansion of chondrocyte proliferation from adjacent physeal cartilage (Fig. 2-25D),
suggesting that the bone bridge formation did not involve endochondral
ossification in this rat model. These results are consistent with an
earlier study that reported a lack of increased expression of IHH and
collagen-2, two molecules typically involved in endochondral
ossification.98 Furthermore, Xian et al’s228 study also demonstrated infiltration of marrow-derived fibroblast-like mesenchymal cells starting from day 3 (Fig. 2-25E),
presence of osteoblast precursor cells among the mesenchymal
infiltrates and their close proximity to bone bridge trabeculae (Fig. 2-25F, Fig. 2-25G), and production of bone matrix protein osteocalcin during formation
of bone bridge trabeculae (Fig. 2-25H).
These results suggested that bone bridge formation after physeal injury
in this rat model occurs directly via intramembranous ossification
through recruitment of marrow-derived osteoprogenitor cells.228
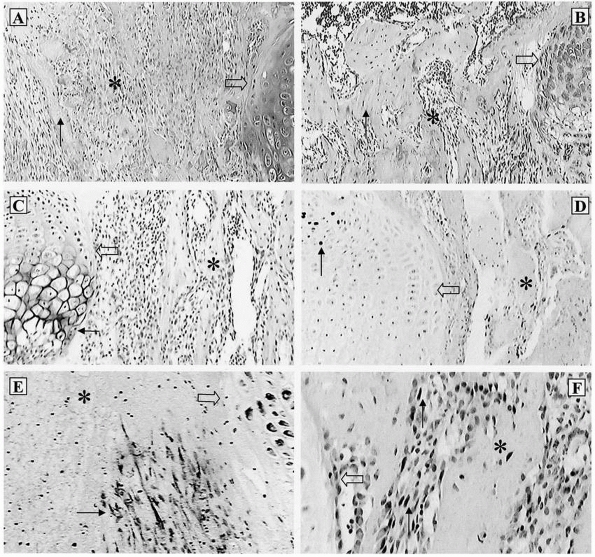 |
|
FIGURE 2-25
Intramembranous ossification mechanism for bone bridge formation at the growth plate injury site. Histologically (Barbara’s histology stain), bony bridge trabeculae start to appear on day 7 postinjury (A), and become well-constructed on day 14 with marrow (B). Prior to and during physeal bar formation, there is no new cartilage formation, no collagen-X synthesis (as examined by immunostaining) at the injury site (C), and no expansion of chondrocyte proliferation (as examined by BrdU labeling) from adjacent physeal cartilage (D). Starting from day 3 (E) until day 14, there is infiltration of marrow-derived fibroblast-like mesenchymal cells (as examined by vimentin immunostaining), some of which are osteoblast precursor cells displaying positive immunostaining for bone cell differentiation transcription factor cbf-a1 (F) and (continues) |
 |
|
FIGURE 2-25 (continued) osteoblast/osteoprogenitor maturation marker alkaline phosphatase (G).
During bone bridge formation, bone matrix protein osteocalcin is produced by osteoblasts on bone bridge trabeculae (immunostaining) (H). *, injury site; block arrow, pointing to adjacent growth plate cartilage; small arrow, pointing to bone bridge trabeculae or immunostained positive cells. This photo-composite is derived from the authors’ previous study. (From Xian CJ, Zhou FH, McCarty RC, et al. Intramembranous ossification mechanism for bone bridge formation at the growth plate cartilage injury site. J Orthop Res 2004;22:417-426; with permission from copyright 2004 Orthopaedic Research Society.) |
remodeling in the diaphysis and metaphysis (particularly the latter)
may realign initially malunited fragments, making anatomic reduction
less important than in a comparable injury in an adult. However,
although some residual angular deformities undergo spontaneous
correction, accurate anatomic reduction should be the goal whenever
possible.58,142,149
Bone and cartilage generally remodel in response to normal stresses of
body weight, muscle action, and joint reaction forces, as well as
intrinsic control mechanisms such as the periosteum. The potential for
spontaneous, complete correction is greater if the child is younger,
the fracture site is closer to the physis, and there is relative
alignment of the angulation in the normal plane of motion of the joint.
This is particularly evident in fractures involving hinge joints such
as the knee, ankle, elbow, or wrist, in which corrections are
relatively rapid if the angulation is in the normal plane of motion.
However, spontaneous correction of angular deformities is unlikely in
other directions, such as a cubitus varus deformity following a
supracondylar fracture of the humerus. Similarly, rotational
deformities usually do not correct spontaneously.
increasing the blood supply to the metaphysis, physis, and epiphysis,
and at least on an experimental basis, by disrupting the periosteum and
its physiologic restraint on the rates of longitudinal growth of the
physes.38 Such increased growth may make the bone longer than it would have been without an injury.42,213
Eccentric overgrowth may also occur; this is particularly evident in
tibia valgum following an incomplete fracture of the proximal tibial
metaphysis.
induce bone formation and have the appropriate osteoconductive matrix.
Autogenic grafts also contain osteogenic cells. Bone grafts are
effective, but there are difficulties in obtaining safe and reliable
source tissue. Although the mechanisms of fracture repair are not fully
understood, the level of understanding has enabled key molecules or
cells to be targeted as therapeutic in controlling and promoting
fracture repair. Filler compounds have been developed that stimulate
proliferation of mesenchymal cells, and/or enhance their osteoblast or
chondrocyte differentiation, leading to formation of new bone or
cartilage (osteoinductive or chondrogenic) or enabling the cells to
infiltrate and incorporate into bone (osteoconductive) or cartilage.
Further studies on repair of bone, articular cartilage, or physis are
required to understand the contribution of different cell types
(inflammatory, endothelial, chondrocytic, osteoblastic and osteoclastic
cells) to the repair process and their associated regulatory molecular
pathways. In order to develop the most optimal strategies to treat
fracture-associated complications that allow for lower effective doses
and fewer side effects, more studies should also be carried out to
identify and to test the optimal growth factors, novel factors,
inhibitors or their small-molecule mimetics, cellbased therapy
including stem cells, and/or gene therapy and their delivery systems.
ability to promote bone formation. Due to their ability to stimulate
proliferation and differentiation of mesenchymal and osteoprogenitor
cells, two bone morphogenic proteins (BMP-2 and BMP-7) have shown great
promise and acceptance for their ability to promote fracture repair.90,91,103,104,113,202,224
BMP-2 promotes bone formation and repair in critical size defects,
fractures and spinal fusions in human, and has been recently approved
for clinical use in fracture repair (Wyeth Pharmaceutical) and spinal
fusion (Medtronic Sofamor Danek, Memphis, TN). Like BMP-2, BMP-7 (also
called osteogenic protein-1, OP-1)
induces ectopic bone formation in vivo and enhances bone repair in preclinical models and clinical studies.25
Clinically, OP-1, delivered with a type-1 collagen carrier, induced
bone repair, which was found to be equivalent to autogenous bone graft
in a clinical trial of patients with tibial nonunions.54 OP-1 has now been approved for use in the treatment of established nonunions (Stryker Biotech).
It also may be of use in promoting repair in nonhealing bone defects.
PDGF also increases callus size but does not improve the fracture
mechanically.131 Growth hormone and
IGF-1 have also been tested to determine their effects on fracture
repair. Although growth hormone produces inconsistent results,3 the administration of IGF-1 increases intramembranous bone formation.205
The FGFs stimulate mitogenesis of mesenchymal cells and osteoblasts,
increase the callus size and mineral content, and improve mechanical
stability at early stages of fracture repair.77,85,218 FGF-2 also increases osteoclastic bone remodeling.131 In addition, it is possible that the effects of FGFs22
and of a number of the other growth factors are a result of the
angiogenic properties of such growth factors, as most osteogenic
factors also stimulate angiogenesis, if not directly, then indirectly,
through production of angiogenic molecules, such as VEGF. The potential
synergism between potent proangiogenic factors (such as VEGF) and
strong osteoinductive factors (such as BMPs) suggest that combination
therapies might produce optimal results, particularly for individuals
at risk of delayed repair or nonunions.25
and some successful clinical applications of BMP-2 and OP-1 in inducing
bone formation and repair, more research is required to establish the
most effective delivery devices for these growth factors.76
Apart from the requirements of biocompatibility and effectiveness, the
growth factor carriers or delivery matrix systems or devices need to
make the growth factor delivery cost-effective and practical in their
clinical applications to induce bone formation in vivo, which should
allow the application of relatively low doses of growth factors for
optimal bone or cartilage regeneration in clinical contexts.
interest in tissue engineering and mesenchymal stem cells for bone
reconstruction of nonunion defects and for articular cartilage
regeneration. Using tissue-engineering technologies, it is now possible
to enhance bone repair and/or replace bone defect; and using the
patient’s own articular chondrocytes retrieved during arthroscopy and
expanded in vitro, it is now possible to repair full-thickness
articular cartilage defects with satisfactory clinical results.137
Researchers worldwide are working to prepare the three fundamental
components for the successful orthopaedic tissue engineering: (a)
appropriate biological or artificial carriers or delivery systems or
extracellular matrix scaffolds, and (b) the right set of viable
responding cells (such as mesenchymal stem cells) in combination with
(c) appropriate soluble inductive signal molecules or growth factors
that, once transplanted, will ensure bone repair and/or cartilage
regeneration.173
vitro, the less stringent ethical and regulatory issues and the lack of
immunologic implications, mesenchymal stem cells derived from
autologous bone marrow stroma have been the research focus for their
potential applications, which have offered a new perspective for bone
and cartilage tissue engineering. There have been some excellent
advances in understanding the stem cells, their interactions with their
matrix, particularly stimuli or signal molecules controlling their
proliferation (such as LIF, FGF-2, HGF, Wnt and Dickkopt-1), osteogenic
differentiation (such as IHH, Notch-1 and PPARgamma), and chondrogenic
differentiation (such as BMP-4 and TGF-β3).157
However, preclinical and clinical evaluations of the stromal stem cells
or their derived osteoblasts or chondrocytes or the engineered bone or
cartilage tissues will yet have to prove their efficacy,
biocompatibility, safety, practicality, and reproducibility in bone and
cartilage regeneration.108
opened novel treatment avenues for the regeneration or repair of
damaged articular cartilage, as gene transfer provides the capability
to deliver bioactive proteins or gene products to sites of tissue
damage locally and in a sustained manner. Previous research has already
convincingly demonstrated the principle of gene delivery to synovium,
chondrocytes and mesenchymal progenitor cells, and efficacy studies
provide optimism that this gene transfer approach can be used to
enhance articular cartilage repair.206
They unfortunately found that these treatments under the experimental
conditions were not preventative of bony bridge formation. Similarly,
in a rat tibial physeal fracture model, Gruber et al63
found that interposed periosteum at the fracture site could not enhance
healing of the fractured physis, and in a rabbit tibial physeal injury
model, Lee et al97 observed that
BMP-2 gene therapy delivered via adenoviral vectors within muscle
implant caused increased osteogenic activity in the injured physis.
Previous work has demonstrated that implantation of cultured
chondrocytes embedded in agarose into physeal defects resulted in a
partial correction of angular deformity and a significant reduction in
growth arrest in a rabbit model,96 and muscle-based gene therapy with adenoviral vectors encoding for IGF-1 restored the injured physeal cartilage.97
More recently, direct transfer of periosteum-derived mesenchymal stem
cells embedded in agarose into the tibial physeal defect resulted in
regeneration of the physis, preventing growth arrest or angular
deformity of the tibia.31,101 The search for the optimal treatment options to achieve correction of angular deformity and to prevent
limb length discrepancy using tissue engineering will continue and is not clinically applicable at this time.
Growth Foundation (Australia), Channel-7 Children’s Research Foundation
of South Australia, National Health and Medical Research Council of
Australia, Skeletal Educational Association, and the Foundation for
Musculoskeletal Research and Education. The authors would like to
acknowledge the contribution of Drs. Edward W. Johnstone, John A.
Ogden, Timothy M. Ganey, and Dali A. Ogden, writers of the previous two
editions, of which parts have been updated and carried forward.
RM. Effects of starvation, septicaemia, and chronic illness on the
growth cartilage plate and metaphysis of the immature rat. J Anat
1959;93:123-130.
J, Sohn P, Zeng X, et al. TGFbeta2 mediates the effects of hedgehog on
hypertrophic differentiation and PTHrP expression. Development
2002;129:1913-1924.
R, Haynes KM, Werther GA, et al. The ontogeny of growth hormone
receptors in the rabbit tibia. Endocrinology 1988;122:2562-2569.
J, Klein KO, Yanovski JA, et al. Induction of growth plate cartilage
ossification by basic fibroblast growth factor. Endocrinology
1994;135:2790-2793.
JF, Lamande SR, Ramshaw JAM. Collagen superfamily. In: Comper WD, ed.
Extracellular matrix. Amsterdam: Harwood Academic Publishers,
1996:22-67.
P, Fisher LW, Young MF, et al. Expression and localization of the two
small proteoglycans biglycan and decorin in developing human skeletal
and nonskeletal tissues. J Histochem Cytochem 1990;38:1549-1563.
DE, Fitch JM, Babiarz JP, et al. Collagen fibrillogenesis in vitro:
interaction of types I and V collagen regulates fibril diameter. J Cell
Sci 1990;95:649-657.
ME, Young MF, Fisher LW, et al. Osteonectin cDNA sequence reveals
potential binding regions for calcium and hydroxyapatite and shows
homologies with both a basement membrane protein (SPARC) and a serine
proteinase inhibitor (ovomucoid). Proc Natl Acad Sci USA
1998;85:2919-2923.
LJ, Trippel SB. Interaction of epidermal growth factor and insulin-like
growth factor-I in the regulation of growth plate chondrocytes. Exp
Cell Res 1997;234:1-6.
WA, Noble NA, Yamamoto T, et al. Natural inhibitor of transforming
growth factor-beta protects against scarring in experimental kidney
disease. Nature 1992;360: 361-364.
BD, Schwartz Z, Park-Snyder S, et al. Latent transforming growth
factor-beta is produced by chondrocytes and activated by extracellular
matrix vesicles upon exposure to 1,25-(OH)2D3. J Biol Chem
1994;269:28374-28381.
HR Jr. Epiphyseal fractures—a microscopic study of the healing process
in rats. J Bone Joint Surg Am 1959;41A:1055-1064.
LJ, McMahon JA, McMahon AP, et al. Noggin, cartilage morphogenesis, and
joint formation in the mammalian skeleton. Science 1998;280:1455-1457.
RW, Ogden JA. Patterns of ischemic necrosis of the proximal femur in
nonoperatively treated congenital hip disease. In: Nelson CL, ed. The
hip: Proceedings of the Hip Society, vol 6. St. Louis: CV Mosby,
1978:43-63.
JA, Glimcher MJ, Cooper RR, et al. Bone biology. I: structure, blood
supply, cells, matrix, and mineralization. Instr Course Lect
1996;45:371-386.
M, Sherman BS, Sobel AE. Observations on the influence of chondroitin
sulphate on the rate of bone repair. J Bone Joint Surg 1962;44B:675-687.
WH, Mehlman T, Marshak DR, et al. Structural evidence that endothelial
cell growth factor is the precursor of both endothelial cell growth
factor and acidic fibroblast growth factor. Proc Natl Acad Sci USA
1986;83:7216-7220.
S, van Rooden JC, Foster BK. Structural changes in the large
proteoglycan, aggrecan, in different zones of the ovine growth plate.
Calcif Tissue Int 1997;60:71-78.
RA, Gilmer WS. Proliferation, regeneration, and repair of articular
cartilage of immature animals. J Bone Joint Surg 1962;44A:431-455.
DE, Liu X. Expression of bone morphogenetic protein-6 messenger RNA in
bovine growth plate chondrocytes of different size. J Bone Miner Res
1995;10:401-405.
D, Taylor TK, Cole WG. Characterization of an arginine 789 to cysteine
substitution in alpha-1(II) collagen chains of a patient with
spondyloepiphyseal dysplasia. J Biol Chem 1993;268:15238-15245.
SY, Wong RW. Expression of epidermal growth factor in transgenic mice
causes growth retardation. J Biol Chem 2000;275:38693-38698.
S, Li IW, McCulloch CA, et al. Influence of osteogenic protein-1
(OP-1,P-7) and transforming growth factor-beta 1 on bone formation in
vitro. Connect Tissue Res 1996;35:71-78.
F, Hui JH, Chan WK, et al. Cultured mesenchymal stem cell transfers in
the treatment of partial growth arrest. J Pediatr Orthop
2003;23:425-429.
SK, Miller RR, McDevitt CA. Basic fibroblast growth factor binds to
heparan sulfate in the extracellular matrix of rat growth plate
chondrocytes. Arch Biochem Biophys 1994;310:180-186.
SK, Miller RR, McDevitt CA. Role of heparan sulfate in the terminal
differentiation of growth plate chondrocytes. Arch Biochem Biophys
1995;316:227-234.
S, Ushiro H, Stoscheck C, et al. A native 170,000 epidermal growth
factor receptor-kinase complex from shed plasma membrane vesicles. J
Biol Chem 1982;257: 1523-1531.
SR, Baker NL, Oh J, et al. Growth hormone receptor abundance in tibial
growth plates of uremic rats: GH/IGF-I treatment. Kidney Int
2000;58:62-70.
P, Syversen SM. Overgrowth of the femur after fractures of the shaft in
childhood. J Bone Joint Surg Br 1976;58:339-346.
DM, Harris SE, Dean DD, et al. Recombinant bone morphogenetic protein
(BMP)-2 regulates costochondral growth plate chondrocytes and induces
expression of BMP-2 and BMP-4 in a cell maturation-dependent manner. J
Orthop Res 1997;15: 371-380.
A, Derynck R. Increased expression of TGF-beta 2 in osteoblasts results
in an osteoporosis-like phenotype. J Cell Biol 1996;132:195-210.
A, Filvaroff EH, Ye JQ, et al. Osteoblastic responses to TGF-beta
during bone remodeling. Mol Biol Cell 1998;9:1903-1918.
DR, Upton MP, Shapiro FD, et al. Nonexpression of cartilage type II
collagen in a case of Langer-Saldino achondrogenesis. Am J Hum Genet
1986;39:52-67.
A, Hardingham T. Matrix proteoglycans. In: Comper WD, ed. Extracellular
matrix. Amsterdam: Harwood Academic Publishers, 1996:200-229.
BK, Hansen AL, Gibson GJ, et al. Reimplantation of growth plate
chondrocytes into growth plate defects in sheep. J Orthop Res
1990;8:555-564.
GE, Perry CR, Cole JD, et al. Osteogenic protein-1 (bone morphogenetic
protein-7) in the treatment of tibial nonunions. J Bone Joint Surg Am
2001;83A(Suppl 1):S151-S158.
E, Gangji V, Canalis E. Bone morphogenetic proteins induce the
expression of noggin, which limits their activity in cultured rat
osteoblasts. J Clin Invest 1998; 102:2106-2114.
HP, Vu TH, Ryan AM, et al. VEGF couples hypertrophic cartilage
remodeling, ossification and angiogenesis during endochondral bone
formation. Nat Med 1999;5: 623-628.
LC, Cullinane DM, Barnes GL, et al. Fracture healing as a postnatal
developmental process: molecular, spatial, and temporal aspects of its
regulation. J Cell Biochem 2003;88:873-884.
RG, Ivins JC. Fractures of the distal part of the forearm in children:
correction of deformity by growth. Minn Med 1952;35:744.
G, Lin DL, Francki K, et al. Type X collagen is colocalized with a
proteoglycan epitope to form distinct morphological structures in
bovine growth cartilage. Bone 1996;19:307-315.
L. Vascular reactions in experimental fractures: microangiographic and
radioisotope studies. Acta Chir Scand Suppl 1961;284(Suppl):1-34.
HE, Phieffer LS, Wattenbarger JM. Physeal fractures, part II: fate of
interposed periosteum in a physeal fracture. J Pediatr Orthop
2002;22:710-716.
HA. The growth of long bones in childhood with special reference to
certain bony striations of the metaphysis and to the role of vitamins.
Arch Intern Med 1926; 38:785-806.
PV, Lian JB, Cole DE, et al. Osteocalcin and matrix Gla protein:
vitamin K-dependent proteins in bone. Physiol Rev 1989;69:990-1047.
PV, Mavrakos AE, Iafrati MD, et al. Growth factors in bone matrix.
Isolation of multiple types by affinity chromatography on
heparin-Sepharose. J Biol Chem 1986; 261:12665-12674.
MT. Structure and function of the heparin-binding (fibroblast) growth
factor family. Baillieres Clin Endocrinol Metab 1991;5:571-593.
E, Heinegård D. Binding of fibromodulin and decorin to separate sites
on fibrillar collagens. J Biol Chem 1993;268:27307-27312.
A, Romarís M, Rasmussen LM, et al. Interaction of the small
interstitial proteoglycans biglycan, decorin, and fibromodulin with
transforming growth factor beta. Biochem J 1994;302:527-534.
BL, Blessing M, Winnier GE, et al. Growth factors in development: the
role of TGF-beta related polypeptide signalling molecules in
embryogenesis. Dev Suppl 1994; 53-60.
K, Reinholt FP, Heinegård D, et al. Osteopontin: a ligand for the alpha
v beta 3 integrin of the osteoclast clear zone in osteopetrotic (ia/ia)
rats. Ann N Y Acad Sci 1995;760:315-318.
EB, Wagner J, Zapf J. Differential effects of insulin-like growth
factor I and growth hormone on developmental stages of rat growth plate
chondrocytes in vivo. J Clin Invest 1994;93:1078-1086.
OE, Feldmann CP. Stimulation of fracture healing by local application
of humoral factors integrated in biodegradable implants. Eur J Pediatr
Surg 1998;8:251-255.
K, Maeda M, Sano A, et al. Local application of basic fibroblast growth
factor minipellet induces the healing of segmental bony defects in
rabbits. Calcif Tissue Int 1998;63:490-495.
PS, DiCesare PE. Recent advances toward the clinical application of
bone morphogenetic proteins in bone and cartilage repair. Am J Orthop
2003;32:429-436.
O, Ito S, Olsen BR. Skeletal and hematopoietic defects in mice
transgenic for collagen X. Ann N Y Acad Sci 1996;785:278-280.
O, LuValle PA, Olsen BR. Spondylometaphyseal dysplasia in mice carrying
a dominant negative mutation in a matrix protein specific for
cartilage-to-bone transition. Nature 1993;365:56-61.
EW, McArthur M, Solly PB, et al. The effect of osteogenic protein 1 in
an in vivo physeal injury model. Clin Orthop Relat Res 2002;395:234-240.
FS, Hayes WC, Keaveny TM, et al. Form and function of bone. In: Simon
SR, ed. Orthopaedic basic science: American Academy of Orthopaedic
Surgeons. Rosemont, IL: Port City Press, 1994:127-184.
T, Kawaguchi H, Hanada K, et al. Single local injection of recombinant
fibroblast growth factor-2 stimulates healing of segmental bone defects
in rabbits. J Orthop Res 1998;16:654-659.
SW, Kelly PJ. The effect of venous stasis on intraosseous pressure and
longitudinal bone growth in the dog. J Bone Joint Surg Am
1965;47A:539-544.
DR, Oxford JT, Morris NP. Ultrastructural localization of collagen
types II, IX, and XI in the growth plate of human rib and fetal bovine
epiphyseal cartilage: type XI collagen is restricted to thin fibrils. J
Histochem Cytochem 1995;43:967-979.
T, von der Mark K. Isolation of bovine type X collagen and
immunolocalization in growth-plate cartilage. Biochem J
1990;265:453-459.
J, Louwerse RT, Heyligers IC, et al. Osteogenic protein (OP-1, BMP-7)
stimulates cartilage differentiation of human and goat perichondrium
tissue in vitro. J Biomed Mater Res 1998;40:614-620.
J, Semeins CM, Mulder JW, et al. Stimulation of cartilage
differentiation by osteogenic protein-1 in cultures of human
perichondrium. Tissue Eng 1998;4: 305-313.
K, Takahashi N, Jimi E, et al. Tumor necrosis factor alpha stimulates
osteoclast differentiation by a mechanism independent of the
ODF/RANKL-RANK interaction. J Exp Med 2000;191:275-286.
HM, Lanske B, Kovacs CS, et al. Functional analysis of the PTH/PTHrP
network of ligands and receptors. Recent Prog Horm Res 1998;53:283-301;
discussion 301-303.
EH, Chen F, Chan J, et al. Treatment of growth arrest by transfer of
cultured chondrocytes into physeal defects. J Pediatr Orthop
1998;18:155-160.
CW, Martinek V, Usas A, et al. Muscle-based gene therapy and tissue
engineering for treatment of growth plate injuries. J Pediatr Orthop
2002;22:565-572.
MA, Nissen TP, Otsuka NY. Utilization of a murine model to investigate
the molecular process of transphyseal bone formation. J Pediatr Orthop
2000;20:802-806.
L, Benoist-Lasselin C, Delezoide AL, et al. Fibroblast growth factor
receptor 3 mutations promote apoptosis but do not alter chondrocyte
proliferation in thanatophoric dysplasia. J Biol Chem
1998;273:13007-13014.
R, Hamburger V. Selective growth stimulating effects of mouse sarcoma
on the sensory and sympathetic nervous system of the chick embryo. J
Exp Zool 1951; 116:321-362.
L, Hui JH, Goh JC, et al. Chitin as a scaffold for mesenchymal stem
cells transfers in the treatment of partial growth arrest. J Pediatr
Orthop 2004;24:205-210.
TR, McKinstry MP, Schnitzer J, et al. Bone blood flow: regional
variation with skeletal maturation. In: Arlet J, Ficat RP, Hungerford
DS, eds. Bone circulation. Baltimore: Williams & Wilkins, 1984.
M. Growth factor stimulation of bone healing. Effects on osteoblasts,
osteomies, and implants fixation. Acta Orthop Scand Suppl 1998;283:2-37.
JP, Baker J, Perkins AS, et al. Mice carrying null mutations of the
genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF
receptor (Igf1r). Cell 1993;75:59-72.
Z, Xu J, Colvin JS, et al. Coordination of chondrogenesis and
osteogenesis by fibroblast growth factor 18. Genes Dev 2002;16:859-869.
E, Donati D, Cenacchi A, et al. Bone reconstruction of large defects
using bone marrow derived autologous stem cells. Transfus Apheresis Sci
2004;30:169-174.
G, Ducy P, McKee MD, et al. Spontaneous calcification of arteries and
cartilage in mice lacking matrix GLA protein. Nature 1997;385:78-81.
AM, Wroblewski J, Pawlowski A. Effects of IGF-I, rGH, FGF, EGF and NCS
on DNA-synthesis, cell proliferation and morphology of chondrocytes
isolated from rat rib growth cartilage. Cell Biol Int Rep
1989;13:259-270.
EE, De Luca F, Uyeda JA, et al. Effects of fibroblast growth factor-2
on longitudinal bone growth. Endocrinology 1998;139:2900-2904.
SC. Birth and death of bone cells: basic regulatory mechanisms and
implications for the pathogenesis and treatment of osteoporosis. Endocr
Rev 2000;21: 115-137.
MD, Cogan AG, Taylor M, et al. Maxillary sinus augmentation in the
non-human primate: a comparative radiographic and histologic study
between recombinant human osteogenic protein-1 and natural bone
mineral. J Periodontol 1998;69:911-919.
J, Weis-Garcia F. Serine/threonine kinase receptors: mediators of
transforming growth factor beta family signals. Cancer Surv
1996;27:41-64.
MD, Nanci A. Osteopontin at mineralized tissue interfaces in bone,
teeth, and osseointegrated implants: ultrastructural distribution and
implications for mineralized tissue formation, turnover, and repair.
Microsc Res Tech 1996;33:141-164.
P, Schnitzer JE, Light TR, et al. Relationship of 99mTC-MDP uptake to
regional osseous circulation in skeletally immature and mature dogs.
Skeletal Radiol 1982;8:115-121.
MC, Bord S, Hembry RM, et al. Human osteoblasts in culture synthesize
collagenase and other matrix metalloproteinases in response to
osteotropic hormones and cytokines. J Cell Sci 1992;103:1093-1099.
MC, McGarrity AM, Thomson BM, et al. Bone-derived growth factors
modulate collagenase and TIMP (tissue inhibitor of metalloproteinases)
activity and type I collagen degradation by mouse calvarial
osteoblasts. Bone Miner 1991;12:41-55.
PJ, Chin JR, Shum L, et al. Epidermal growth factor receptor function
is necessary for normal craniofacial development and palate closure.
Nat Genet 1999;22: 69-73.
E, Kreschel C, Naski MC, et al. Interaction of FGF, Ihh/Pthlh, and BMP
signaling integrates chondrocyte proliferation and hypertrophic
differentiation. Dev Cell 2002; 3:439-449.
T, Benson WM, Foster BK, et al. Statistical analysis of the incidence
of physeal injuries. J Pediatr Orthop 1987;7:518-523.
H. The chondrocyte, architect of cartilage. Biomechanics, structure,
function, and molecular biology of cartilage matrix macromolecules.
Bioessays 1995;17:1039-1048.
H, Goldberg VM, Caplan AI. Culture-expanded human periosteal-derived
cells exhibit osteochondral potential in vivo. J Orthop Res
1991;9:465-476.
TJ, Howlett CR, Martin C, et al. Effect of platelet-derived growth
factor on tibial osteotomies in rabbits. Bone 1994;15:203-208.
G, Gospodarowicz D. The identification and partial characterization of
the fibroblast growth factor receptor of baby hamster kidney cells. J
Biol Chem 1985;260: 13860-13868.
A, Ohlsson C, Isaksson OG, et al. Hormonal regulation of longitudinal
bone growth. Eur J Clin Nutr 1994;48(Supp l)1:S150-S158; discussion
S158-S160.
JA. An anatomical and histological study of the factors affecting
development and evolution of avascular necrosis in congenital
dislocation of the hip. In: Harris WH, ed. The hip: proceedings of the
Hip Society, vol 2. St. Louis: CV Mosby, 1974:125-153.
JA. Chondro-osseous development and growth. In: Urist MR, ed.
Fundamental and clinical bone physiology. Philadelphia: JB Lippincott,
1980.
JA. Injury to the immature skeleton. In: Touloukian R, ed. Pediatric
Trauma, 2nd ed. New York: John Wiley & Sons, 1990.
JA. The development and growth of the musculoskeletal system. In:
Albright JA, Brand RA, eds. The Scientific Basis of Orthopaedics. New
York: Appleton-Century-Crofts, 1979.
JA, Grogan DP, Light TR. Postnatal skeletal development and growth of
musculoskeletal system. In: Albright JA, Brand RD, eds. The scientific
Basis of Orthopaedics. New York: Appleton & Lange, 1987.
JA, Rosenberg LC. Defining the growth plate. In: Uhthoff HK, Wiley JJ,
eds. Behavior of the Growth Plate. New York: Raven Press, 1988.
C, Nilsson A, Isaksson O, et al. Growth hormone induces multiplication
of the slowly cycling germinal cells of the rat tibial growth plate.
Proc Natl Acad Sci USA 1992;89:9826-9830.
DM, Marie PJ. FGF signaling pathways in endochondral and
intramembranous bone development and human genetic disease. Genes Dev
2002;16:1446-1465.
WR, Rao J. Tomorrow’s skeleton staff: mesenchymal stem cells and the
repair of bone and cartilage. Cell Prolif 2004;37:97-110.
EP, Jacenko O, Olsen B, et al. The role of type X collagen in
endochondral ossification as deduced by Fourier transform infrared
microscopy analysis. Connect Tissue Res 1996;35:371-377.
DG, Kujawa MJ, Caplan AI. Morphology of bone development and bone
remodeling in embryonic chick limbs. Bone 1986;7:459-472.
B, Ronzière MC, Hartmann DJ, et al. Ultrastructural organization of
type XI collagen in fetal bovine epiphyseal cartilage. Histochemistry
1993;100:231-239.
S, Sasai Y, Lu B, et al. Dorsoventral patterning in Xenopus: inhibition
of ventral signals by direct binding of chordin to BMP-4. Cell
1996;86:589-598.
AR, Matsui Y, Hinek A, et al. Cartilage macromolecules and the
calcification of cartilage matrix. Anat Rec 1989;224:167-179.
AR, Webber C, Pidoux I, et al. Localization of a dermatan sulfate
proteoglycan (DS-PGII) in cartilage and the presence of an
immunologically related species in other tissues. J Histochem Cytochem
1986;34:619-625.
M, Ritsilä V. The osteogenic capacity of free periosteal and
osteoperiosteal grafts. A comparative study in growing rabbits. Acta
Orthop Scand 1979;50:491-499.
PA, Williamson MK. Primary structure of bovine matrix Gla protein, a
new vitamin K-dependent bone protein. J Biol Chem 1985;260:14971-14975.
LN, Matsaba TN, Teare J, et al. Tissue engineering: TGF-beta
superfamily members and delivery systems in bone regeneration. Expert
Rev Mol Med 2002;4: 1-11.
DA, Mark D, Banda MJ, et al. Wound macrophages express TGF-alpha and
other growth factors in vivo: analysis by mRNA typing. Science
1988;241:708-712.
M, Schmid AC, Heyberger-Meyer B, et al. Effect of growth hormone and
insulin-like growth factor I (IGF-I) on the expression of IGF-I
messenger ribonucleic acid and peptide in rat tibial growth plate and
articular chondrocytes in vivo. Endocrinology 2000;141:2847-2853.
FP, Hultenby K, Oldberg A, et al. Osteopontin—a possible anchor of
osteoclasts to bone. Proc Natl Acad Sci USA 1990;87:4473-4475.
FW, Phillips RS, Steel WM, et al. Microangiography and bone healing.
II. Displaced closed fractures. J Bone Joint Surg Am 1968;50(4):643-662
passim.
P, Slaetis P. The repair of experimental fractures during long-term
anticoagulant treatment. An experimental study on rats. Acta Orthop
Scand 1964;35:21-38.
R, Horan GS, Pinero GJ, et al. Normal long bone growth and development
in type X collagen-null mice. Nat Genet 1994;8:129-135.
LJ, Sugai JV, Trippel SB. Expression of collagens I, II, X, and XI and
aggrecan mRNAs by bovine growth plate chondrocytes in situ. J Orthop
Res 1994;12:1-14.
E, Provot S. PTHrP, PTH, and the PTH/PTHrP receptor in endochondral
bone development. Birth Defects Res Part C Embryo Today 2003;69:352-362.
JM, Hwang K, Winn SR, et al. Bone morphogenetic proteins: an update on
basic biology and clinical relevance. J Orthop Res 1999;17:269-278.
JE, McKinstry P, Light TR, et al. Quantitation of regional
chondro-osseous circulation in canine tibia and femur. Am J Physiol
1982;242:H365-H375.
JE, McKinstry P, Light TR, et al. Quantitation of regional osseous
circulation in the maturing canine tibia and femur. Surg Forum
1980;31:509-511.
E, Broszat M, Brandan E, et al. Decorin core protein fragment
Leu155-Val260 interacts with TGF-beta but does not compete for decorin
binding to type I collagen. Arch Biochem Biophys 1998;355:241-248.
E, Hausser H, Beavan L, et al. Decorin-type I collagen interaction.
Presence of separate core protein-binding domains. J Biol Chem
1995;270:8877-8883.
P, Hall BK. Differentiative ability of the tibial periosteum for the
embryonic chick. Acta Anat (Basel) 1980;106:129-140.
NA, Clement-Lacroix P, Da Ponte F, et al. Bone homeostasis in growth
hormone receptor-null mice is restored by IGF-I but independent of
Stat5. J Clin Invest 2000; 106:1095-1103.
K, Liu JL, Blad K, et al. Liver-derived insulin-like growth factor I
(IGF-I) is the principal source of IGF-I in blood but is not required
for postnatal body growth in mice. Proc Natl Acad Sci USA
1999;96:7088-7092.
MJ, Robertson EJ. Early embryonic lethality in Bmp5;Bmp7 double mutant
mice suggests functional redundancy within the 60A subgroup.
Development 1999; 126:1753-1768.
DP. Collagenous architecture of the growth plate and perichondral
ossification groove. J Bone Joint Surg Am 1982;64A:399-407.
MM, Qanadilo HF, Langer R, et al. A rapid-curing alginate gel system:
utility in periosteum-derived cartilage tissue engineering.
Biomaterials 2004;25:887-894.
J, Bao M, deGuzman L, et al. Vascular endothelial growth factor
stimulates bone repair by promoting angiogenesis and bone turnover.
Proc Natl Acad Sci USA 2002; 99:9656-9661.
T, Takahashi N, Udagawa N, et al. Modulation of osteoclast
differentiation and function by the new members of the tumor necrosis
factor receptor and ligand families. Endocr Rev 1999;20:345-357.
T, Kobayashi M, Suzuki R, et al. Recombinant human bone morphogenetic
protein-2 stimulates osteoblast differentiation and suppresses matrix
metalloproteinase-1 production in human bone cells isolated from
mandibulae. J Periodontal Res 1998; 33:476-485.
SB, Van Wyk JJ, Foster MB, et al. Characterization of a specific
somatomedinc receptor on isolated bovine growth plate chondrocytes.
Endocrinology 1983;112: 2128-2136.
SB, Wroblewski J, Makower AM, et al. Regulation of growth-plate
chondrocytes by insulin-like growth-factor I and basic fibroblast
growth factor. J Bone Joint Surg Am 1993;75:177-189.
J, Amato MP. The vascular contribution to osteogenis. III. Changes in
the growth cartilage caused by experimentally induced ischaemia. J Bone
Joint Surg Br 1960;42B: 571-587.
J, Morgan JD. The vascular contribution to osteogenesis. I. Studies by
the injection method. J Bone Joint Surg Br 1960;42B:97-109.
H, Suren EG. Fehlstellungen, wachstumsstorungen, and pseudoarthrosen
nach kindlichen frakturen. Langenbecks Arch Chir 1976;342:299-304.
der Eerden BC, Karperien M, Gevers EF, et al. Expression of Indian
hedgehog, parathyroid hormone-related protein, and their receptors in
the postnatal growth plate of the rat: evidence for a locally acting
growth restraining feedback loop after birth. J Bone Miner Res
2000;15:1045-1055.
SW, Luvalle P, Leask T, et al. A BMP responsive transcriptional region
in the chicken type X collagen gene. J Bone Miner Res 1998;13:1521-1529.
A, Lee K, Lanske B, et al. Regulation of rate of cartilage
differentiation by Indian hedgehog and PTH-related protein. Science
1996;273:613-622.
JS. Basic fibroblast growth factor for stimulation of bone formation in
osteoinductive or conductive implants. Acta Orthop Scand Suppl
1996;269:1-33.
EA, Rosen V, D’Alessandro JS, et al. Recombinant human bone
morphogenetic protein induces bone formation. Proc Natl Acad Sci USA
1990;87:2220-2224.
J, Zhou J, Cheng CM, et al. Evidence supporting dual, IGF-I-independent
and IGF-I-dependent, roles for GH in promoting longitudinal bone
growth. J Endocrinol 2004;180:247-255.
DT, Kelly PJ, Owen CA Jr. Blood flow in bone distal to a femoral
arteriovenous fistula in dogs. J Bone Joint Surg Am 1964;46A:1676-1682.
GA, Haynes KM, Barnard R, et al. Visual demonstration of growth hormone
receptors on human growth plate chondrocytes. J Clin Endocrinol Metab
1990;70: 1725-1731.
K, Tsai DC, Nam EK, et al. Ectopic bone formation via rhBMP-2 delivery
from porous bioabsorbable polymer scaffolds. J Biomed Mater Res
1998;42:491-499.
RJ, Janik-Schmitt B, Renauer D, et al. Contact-dependent inhibition of
growth of normal diploid human fibroblasts by plasma membrane
glycoproteins. Biochimie 1988;70:1661-1671.
T, Byers S, Byard RW, et al. The implantation of cartilaginous and
periosteal tissue into growth plate defects. Int Orthop 1994;18:220-228.
JB. Acute changes in femoral arterial blood flow after closed tibial
fracture in dogs. J Bone Joint Surg Am 1964;46A:1262-1268.
CJ, Zhou FH, McCarty RC, et al. Intramembranous ossification mechanism
for bone bridge formation at the growth plate cartilage injury site. J
Orthop Res 2004;22: 417-426.
T, Bianco P, Fisher LW, et al. Targeted disruption of the biglycan gene
leads to an osteoporosis-like phenotype in mice. Nat Genet
1998;20:78-82.
S, Rosen CJ. From mouse to man: redefining the role of insulin-like
growth factor-I in the acquisition of bone mass. Exp Biol Med (Maywood)
2003;228:245-252.
Y, Mann DM, Ruoslahti E. Negative regulation of transforming growth
factor-beta by the proteoglycan decorin. Nature 1990;346:281-284.
N, Akiyama S, Katagiri T, et al. Smad1 and smad5 act downstream of
intracellular signalings of BMP-2 that inhibits myogenic
differentiation and induces osteoblast differentiation in C2C12
myoblasts. Biochem Biophys Res Commun 1997; 238:574-580.
J, Froesch ER. Insulin-like growth factor I actions on somatic growth.
In: Kostyo JL, ed. Handbook of Physiology. New York and Oxford: Oxford
University Press, 1999.
D, Schwarz EM, Rosier RN, et al. ALK2 functions as a BMP type I
receptor and induces Indian hedgehog in chondrocytes during skeletal
development. J Bone Miner Res 2003;18:1593-1604.