
characterized by weakness of muscle. The pattern and severity are
variable and depend on the type of dystrophy.
genes, the protein products of which play important roles in muscle
function (Table 20.1-1). The first dystrophy in
which a mutation was identified is Duchenne muscular dystrophy (DMD),
the most common type. This disorder is inherited in an X-linked
recessive manner and is caused by a mutation in a gene located at Xp21,
encoding the dystrophin protein. Becker
muscular dystrophy is due to a defect in the same gene, but has a
milder phenotype (the clinical course tends to be less severe).
neuromuscular disorders, and the most common form of muscular dystrophy
is DMD, with an incidence among boys of 1 in 3,500 and a prevalence of
60 per million. The others are less common:
-
Congenital muscular dystrophy: 25 per million
-
Becker muscular dystrophy: 16 per million
-
Limb girdle muscular dystrophy: 8 per million
-
Fascioscapulohumeral muscular dystrophy: 8 per million.
effects, but has a predominant role maintaining muscle membrane
stability. Skeletal and cardiac muscle are lacking the protein
dystrophin in DMD. Lack of dystrophin leads to membrane damage during
contraction, activating the inflammatory cascade, resulting in muscle
cell death, subsequent fibrosis, and loss of function. The damaged
muscle is replaced by fat and fibrous tissue. The role in other cell
types, such as in the central nervous system, seems to be related to
its cell signaling function, and this may be responsible for findings
such as a relatively low IQ in some affected boys.
-
Sex-linked:
-
□ DMD
-
□ Becker
-
□ Emery-Dreyfus (mostly)
-
-
Autosomal dominant:
-
□ Fascioscapulohumeral
-
□ Distal
-
□ Ocular
-
□ Oculopharyngeal
-
□ Myotonic dystrophy
-
-
Autosomal recessive:
-
□ Limb girdle
-
□ Infantile fascioscapulohumeral
-
□ Congenital muscular dystrophy
-
|
TABLE 20.1-1 TYPES OF MUSCULAR DYSTROPHY WITH PROTEIN AFFECTED AND GENE DEFECT
|
|||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
years of age or older. It is not uncommon for an orthopaedic surgeon to
be the first medical professional to entertain this potential diagnosis.
-
Pregnancy, including lack of in utero movement
-
Birth history (e.g., floppy baby)
-
Developmental delays, especially milestones (e.g., slow to sit, walk, talk)
-
Specific current symptoms:
-
□ Toe-walking: often benign, but always check creatine phosphokinase level to exclude DMD
-
□ Clumsiness
-
□ Inability to keep up with other children
-
□ Easily fatigued
-
□ Waddling
-
□ Abnormal gait
-
-
Other systems: cardiac problems, ocular, seizures, skin
-
Family history
-
Look:
-
□ General appearance and morphology
-
□ Posture: sitting and standing
-
□ Winging of scapula (fascioscapulohumeral dystrophy)
-
□ Excessive lumbar lordosis (to compensate for pelvic girdle muscle weakness)
-
□ Muscle wasting
-
□ Pseudohypertrophy of calves
-
□ Scoliosis (often the first sign of neuromuscular disease)
-
□ Gait—waddling (differential diagnosis: developmental dysplasia of the hip)
-
□ Skin—facial expression, skin markings
-
-
Feel:
-
□ Muscle quality, tone
-
□ Contractures:
-
□ Hip abduction, fixed flexion (later)
-
□ Ankle equinus
-
-
-
Move:
-
□ Gross and fine motor skills
-
□ Joint range of movement
-
□ Pattern of motor weakness (proximal/distal/generalized)
-
-
Specific signs:
-
□ Gower: weak pelvic girdle muscles—child climbs up legs from sitting on the floor.
-
□ Meryon: weak shoulder girdle—child slips through when lifted by encircling arms
-
-
Transmission of DMD is sex-linked,
therefore only boys are affected (though women with the mutated gene
are carriers and may show subtle clinical findings). -
Usually normal during early infancy with normal milestones.
-
Start to develop waddling gait at age 4.
-
Proximal lower limb muscles first to be affected; therefore, difficulty ascending stairs.
-
Toe-walking may be presenting complaint: need to exclude DMD in all boys with toe-walking due to tightness of Achilles tendon.
-
“Clumsy”: frequent falls.
-
Pseudohypertrophy of muscles, especially calf, due to replacement of muscle by fat and fibrous tissue.
-
Muscle feels rubbery and is larger than would be expected.
-
Weakness of hip extensors leads to compensatory lumbar lordosis to maintain upright posture.
-
Weakness of upper limbs develops later.
-
Distal muscles are the last to be affected.
-
Ability to walk is lost between 12 and 14 years of age.
-
Fixed contractures develop quickly after loss of ambulation.
-
Progressive scoliosis.
-
Sphincter control and ability to eat maintained.
-
Pulmonary function deteriorates after age 13.
-
Intercostal muscles weaken and cardiomyopathy develops.
-
Death occurs from respiratory failure or cardiac failure in late teens or early 20s.
appropriate supportive therapy (including part-time use of a
respirator) boys may survive up to their fourth decade. Recent work has
suggested that use of the steroid deflazacort may significantly slow
progression of the disease, particularly in terms of the pulmonary
function and scoliosis, thus allowing patients to remain active and to
live longer.
-
Blood:
-
□ Creatine phosphokinase is significantly
raised to 20 to 300 times normal (i.e., a minimally raised level is
unlikely to represent muscular dystrophy) -
□ Aspartate aminotransferase (AST) is mildly elevated.
-
dystrophy has been made, and particularly if the creatine phosphokinase
level is high, referral to a pediatric neurologist for further
investigation is the appropriate next step. He or she will most likely
involve a clinical geneticist to help with further diagnosis and
genetic counseling.
-
DNA testing:
-
□ The diagnosis can be confirmed using DNA testing from white blood cells in many cases.
-
-
Radiologic:
-
□ Generally not helpful.
-
□ May show osteopenia, enlarged heart in cardiomegaly.
-
-
Neurophysiologic:
-
□ Electromyelography should show a myopathic rather than a neuropathic picture.
-
□ Invasive and painful; use in children only if necessary.
-
-
-
Cardiac:
-
□ Electrocardiography may show conduction defects or arrhythmias.
-
□ Echocardiography may show cardiomyopathy.
-
-
Respiratory:
-
□ Pulmonary function tests show reduced
functional vital capacity (FVC). This is particularly relevant in
planning timing of scoliosis surgery (discussed later in the chapter).
-
-
Muscle biopsy:
-
□ Histopathology
-
□ Monoclonal antibody staining
-
□ Electrophoresis (Western blot)
-
cases. If performed, the biopsy should be taken from a muscle that is
not yet significantly weakened (since involved muscle will be replaced
with fibrous tissue, making the histologic interpretation difficult).
undertaken in a multidisciplinary framework, with the input from
pediatricians, neurologists, pulmonologists, physiotherapists,
occupational therapists, and orthotists.
-
Pediatrician:
-
□ To coordinate the care and treat medical complications (cardiac and respiratory).
-
-
Physiotherapist:
-
□ To prolong muscle strength and prevent contractures.
-
□ Also to help with functional testing to assess progression of the disease.
-
-
Occupational therapist:
-
□ To help maintain independence within the home as long as possible.
-
-
Orthotist:
-
□ To provide ankle-foot orthoses (AFOs)
or knee—ankle—foot orthoses (KAFOs) to assist with ambulation and
possibly slow the onset of contracture formation. -
□ Orthoses can also be helpful in allowing the patient to transfer to a wheelchair after loss of ambulation occurs.
-
vital in the provision of an appropriately molded custom wheelchair to
help with comfortable upright sitting and therefore mobility after
ambulation is lost.
function or to decrease pain. In some cases, surgery can help maintain
ambulation, and when the patient becomes non-ambulatory, surgery may
help to allow comfortable sitting and wheelchair use.
ambulation is lost due to disuse of osteopenia. Management is generally
with reduction and cast fixation although internal fixation is
occasionally warranted.
patients to extend ambulation. These take the form of Achilles tendon
lengthening,
gastrocnemius
recession (Vulpius), or tibialis posterior transfer for equinovarus
foot deformity; the Yount procedure (iliotibial band and tensor fascia
lata release) for knee contracture; and anterior hip release/tensor
fascia lata release for hip flexion and abduction contracture. No
definitive evidence is available to support this; patients tend to
become wheelchair dependent soon after, and there is a risk of
decreasing the time the patient has walking prior to loss of ambulation
by placing him in a cast.
not recommend routine release of lower limb contractures in ambulatory
boys with DMD.
comfortable wheelchair placement, but there is no evidence to support
the routine release of contractures in asymptomatic patients.
tend to interfere with function and surgical release is very rarely
required.
progressive scoliosis that is unresponsive to brace therapy. Surgery is
indicated early when the Cobb angle reaches 30 degrees or possibly even
less, so that the patients have sufficient residual pulmonary function
to allow safe spinal surgery. FVC below 30% is associated with a very
poor outcome in terms of postoperative recovery, and the FVC should
ideally be above 45%. Early operation also allows for stabilization
before significant deformity has occurred, making the surgery less
complex.
instrumentation and grafting with allograft, stabilizing from high
thoracic to the sacrum. Select cases may be managed with fusion to L5,
rather than the sacrum, although there is a risk of progressive pelvic
obliquity, especially in cases where the apex of the curve is in the
lumbar spine. For this reason, we recommend instrumentation and fusion
to the sacrum.
-
Similar to DMD but less severe, with onset after age 7
-
Progresses more slowly; orthotics may be of benefit
-
Treatment otherwise as for DMD
-
Life expectancy greater than with DMD; some patients survive into later adulthood
-
Characterized by early contractures
(ankle equinus, elbow flexion, neck extension, tightness of lumbar
paravertebrals), slow, progressive weakness in a humeroperoneal
distribution (upper limb proximal, lower limb distal), and subsequent
cardiomyopathy with conduction defects -
Conduction defects often asymptomatic; high incidence of sudden death due to arrhythmias
-
□ Early pacemaker fitting recommended
-
-
Scoliosis may be a feature but tends not to progress
-
Creatine phosphokinase elevation mild to moderate
-
Treatment by physiotherapy, with surgical heel cord release occasionally indicated
-
Characterized by weakness of facial and shoulder girdle muscles
-
Usually presents in later childhood or early adulthood
-
Progression often slow or stop-start
-
Heart and central nervous system are normal
-
Life expectancy is normal
-
Orthopaedic problems:
-
□ Loss of forward flexion and abduction
-
□ Winging of the scapula
-
-
Surgical intervention:
-
□ Posterior scapulocostal fusion (Jakab
and Gledhill) to stabilize the scapula and restore mechanical advantage
to the deltoid and rotator cuff muscles
-
-
Rare
-
Late onset (after 45 years of age)
-
Starts in the intrinsic muscles of the hands
-
May involve the calf muscles
-
Rare
-
Starts during the adolescent years, with diplopia
-
May eventually involve the proximal upper extremities and pelvis
-
Myopathy is of a mitochondrial origin.
-
Largely a disease of French Canadians
-
Onset in the third decade, with dysarthria, dysphagia, and ptosis
-
Group of disorders characterized by the inability of skeletal muscle to relax after a strong contraction
-
Three forms:
-
□ Myotonic dystrophy (Steinert disease):
-
□ Comprises myotonia with progressive muscle weakness, heart defects, frontal baldness, gonadal atrophy, and dementia.
-
□ Onset is usually in late adolescence.
-
□ The protein abnormality has been
identified as a mutation in the dystrophia myotinician protein kinase
gene on chromosome 19. The defect results in an amplified trinucleotide
repeat in the 3′ untranslated region of the gene. The phenotypic
severity of the disorder is directly related to the number of repeats
an individual has. Normal is 5 to 30 copies; mildly affected
individuals have 50 to 80 copies; and in severe cases, there may be
more than 2,000 copies. Amplification tends to increase with generation
and transmission. Extreme amplification is not transmitted along the
male line, however, which is why the most severe congenital form of the
disorder (see below) is almost always passed from mother to child.
P.215 -
-
□ Congenital myotonic dystrophy:
-
□ Relatively common, with variable expression that tends to increase with the generations.
-
□ Although autosomal dominant, it tends to be passed on by the mother. The mother often has a forme fruste of the disorder, and shaking her hand may give you an early indication of what to expect in the examination of the child.
-
□ Sufferers have a long, narrow face, are hypotonic, and often have difficulty feeding.
-
□ 40% have severe involvement or die in infancy; 60% may be affected later.
-
□ Patients may suffer from severe
clubfoot. They are often resistant to nonoperative treatment, and later
develop teratologic hip dysplasia and scoliosis.
-
-
□ Congenital myotonia:
-
□ Presents later (after age 10) although present at birth
-
□ Early myotonia decreases with repetitive movement
-
□ Variable clinical expression
-
□ May present as low back pain or decreased physical ability
-
□ No associated systemic abnormalities, no significant orthopaedic features; life span is normal
-
-
-
Relatively common
-
Clinically and genetically heterogeneous (15 forms at last count)
-
May be relatively benign, with a similar presentation to Becker, but with a normal dystrophin, or with a scapulohumeral form
-
Onset tends to be later in childhood, with a more benign prognosis (survival to around age 40).
-
Treatment tends to be as for DMD, but scoliosis less of a feature due to later onset
-
More severe than the autosomal dominant form
-
Earlier in onset with a more rapid clinical course, and loss of ambulation by the second decade
-
Facial diplegia is noted in infancy with loss of hearing by age 5
-
Weak glutei, resulting in a severe lumbar lordosis (pathognomic for this disorder)
-
Weakness of shoulder girdle muscles so severe that stabilization is of little value
-
A variety of disorders are considered in this category.
-
They present at birth, often with a floppy baby.
-
Several forms are recognized, with a spectrum of severity.
-
Joint stiffness and contractures may be a feature.
-
Some types (e.g., Fukuyama) are rapidly
progressive and may be fatal in the first decade, but most cases are
not and survival into adulthood is common. -
Orthopaedic problems:
-
□ Hip dysplasia and dislocation—treat as for idiopathic disease, but beware of recurrence.
-
□ Clubfoot—treat vigorously.
-
□ Scoliosis—try bracing, but surgical stabilization is likely to be required.
-
BA, Kim HK. Pelvic obliquity after fusion of the spine in Duchenne
muscular dystrophy. J Bone Joint Surg (Br) 1999;81: 821-824.
JR, McKeon J. Orthopedic surgery and rehabilitation for the
prolongation of brace-free ambulation of patients with Duchenne’s
muscular dystrophy. Am J Phys Med Rehabil 1991;70:323-330.
F, Specht L. Current concepts review: the diagnosis and orthopaedic
treatment of inherited muscular diseases of childhood. J Bone Joint
Surg (Am) 1993;75:439-454.
PJ, Wagner PT, Kartlinchak B, et al. Evaluation of a program for
long-term treatment of Duchenne muscular dystrophy. J Bone Joint Surg
(Am) 1996;78:1844-1852.
a symptom complex that consists of contractures of the upper and lower
limbs and spine. These contractures may be in flexion or extension at
any of the involved joints. The pattern of the deformity is related to
the innervation levels of the involved musculature. There are many
forms of arthrogryposis that cause contractures and contractions of the
same anatomic areas of the extremities and spine. These are similar in
clinical appearance to AMCN but in histologic, etiologic, or neurologic
involvement are essentially very different entities.
recently the gene loci for several forms of AMCN have been mapped to
the q region of chromosome 5. The heredity pattern appears to be
recessive and incomplete penetrance. Distal arthrogryposis is autosomal
dominant or, rarely, X-linked, and maps to the p regions of chromosome
9, 11, or the X chromosome. AMCN has not been shown to be infectious or
to develop after birth. It is possible however, that some forms are
acquired in utero through the fetal
circulation carrying viral or bacterial organisms that specifically
destroy the anterior horn cells of the spinal cord of the fetus. (This
is the same pathophysiology that causes poliomyelitis.) It is also
possible that there is an embryologic defect responsible for the
malformation of the anterior horn cells. The final common pathway leads
to the destruction of the motor neural innervation of muscles.
|
TABLE 20.2-1 SUSCEPTIBILITY TO PARALYSIS OF MUSCLES OF THE LOWER EXTREMITIES COMPARED WITH LENGTH OF ANTERIOR HORN CELL COLUMN
|
|||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||||||||||||||||||||||||||||||
directly correlated with the segmental innervation of the involved
muscles. The lengths of the anterior horn cell columns show distinct
correlation patterns when compared to the ratio of partial paralysis to
paralysis in children with AMCN (Table 20.2-1). These patterns correlate well with the specific levels of neurosegmental innervations (Figs. 20.2-1 and 20.2-2).
The shorter the length of the anterior horn cell column that innervates
a motor nerve, the more likely deformities involving those muscles is
to occur in the child with AMCN. Also, the length of the anterior horn
cell column inversely correlates with the severity of the contractures.
This is because a destructive lesion or agenesis of the anterior horn
cells is more likely
to
be able to affect a greater percentage of the cells when the column is
shorter and has fewer cells. Therefore, a typical example would be the
common fixed extension contracture of the knees. This is attributed to
the short anterior horn cell column of the hamstrings at the level of
L5-S1, which is more likely to be absent or largely destroyed than the
longer anterior horn cell column of the quadriceps at the level of
L2-L4.
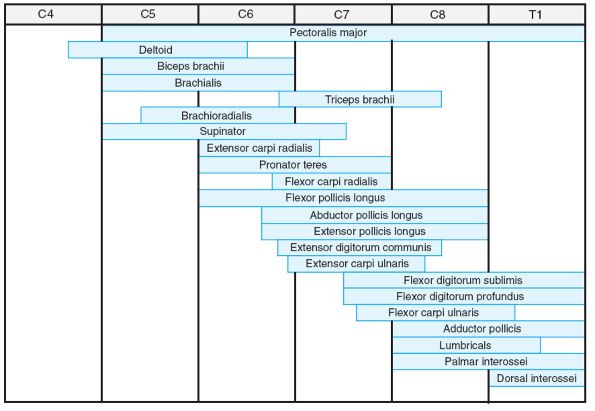 |
|
Figure 20.2-1
Segmental innervation of upper limb muscles. (From Brown LM, Robson MJ, Sharrard WJW. The pathophysiology of arthrogryposis multiplex congenita neurologica. J Bone Joint Surg (Br) 1980;62:291-296.) |
deformities. The most common deformities are type I in the upper
extremities and type III in the lower extremities. See Table 20.2-2 for the classification of AMCN and Figures 20.2-3 and 20.2-4 for case examples.
and evaluation of the deformed individual. Using physical diagnosis and
keeping the types of deformity in mind, one can make an accurate
diagnosis and assessment usually without the need for many other tests.
-
Genetic consultation will usually exclude most known syndromes.
-
Electromyelogram and nerve conduction velocities may be of some help in evaluating the patient’s levels of innervation.
-
If the correct levels and areas of muscle
biopsy are performed, the muscles on the active motor side of the
deformed joint should show normal fibers while the muscles not
functioning on the opposite side of the deformed joint will show the
pathologic features of denervation.-
□ An example would be normal-appearing
quadriceps muscles in a knee in fixed extension while the hamstrings
have fatty replacement and fibrosis as well as other signs of
denervation. -
□ The muscle belly of the quadriceps may only deform extremely proximally in the anterior leg if the paralysis is severe.
-
□ Biopsy of the quadriceps more distally would show fibrosis as well.
-
arthrogryposis or arthrogrypotic-like syndrome a patient has. Certain
syndromes (Box 20.2-1) may mimic some of the
features of true AMCN but have other known causes and known sequelae
that require different treatment. The term amyoplasia in the literature is probably synonymous with
AMCN, if the levels of involvement in these published patients are consistent with the known types of deformities (Table 20.2-2).
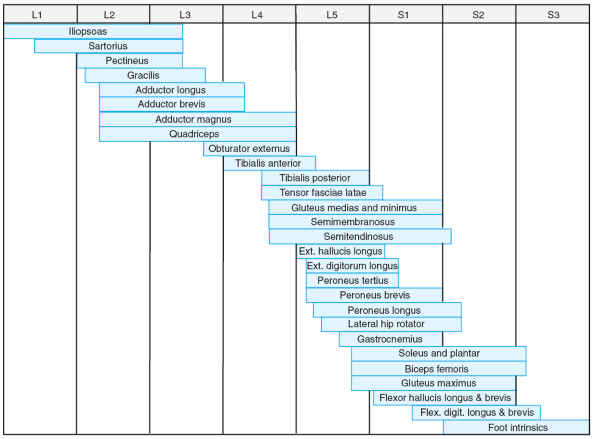 |
|
Figure 20.2-2
Segmental innervation of lower limb muscles. (From Brown LM, Robson MJ, Sharrard WJW. The pathophysiology of arthrogryposis multiplex congenita neurologica. J Bone Joint Surg (Br) 1980;62:291-296.) |
|
TABLE 20.2-2 DEFORMITIES FOUND IN ARTHROGRYPOSIS MULTIPLEX CONGENITA NEUROLOGICA
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||
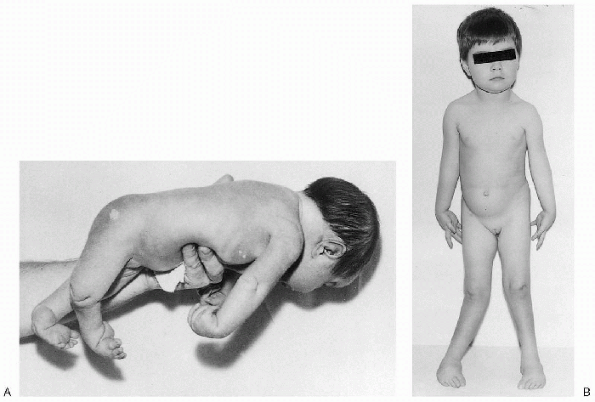 |
|
Figure 20.2-3 Neurogenic arthrogryposis. (A)
Patient at birth exhibiting type I (upper limb) and type III (lower limb) deformities. There is adduction of the shoulders, extension of the elbows, pronation of the forearms, flexion of the wrists, flexion and adduction deformity of the hips, extension of the knees, equinovarus deformities of the feet, and dimples over elbows and knees. (B) Same patient at 4 years of age. (From Brown LM, Robson MJ, Sharrard WJW. The pathophysiology of arthrogryposis multiplex congenita neurologica. J Bone Joint Surg (Br) 1980;62:291-296.) |
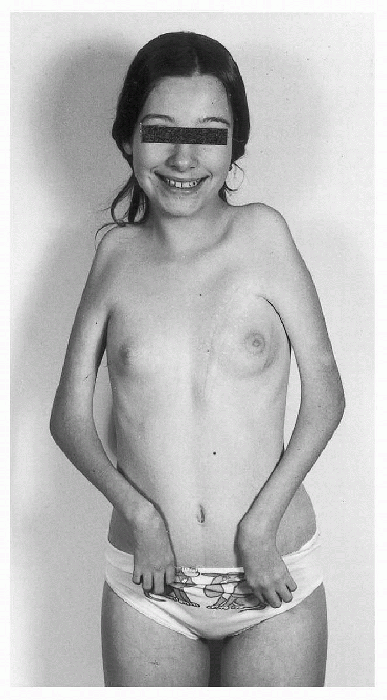 |
|
Figure 20.2-4
Type II deformity. There is adduction and medial rotation of the shoulders, flexion deformity of the elbows, flexion and ulnar deviation of the wrists, and weak intrinsic muscles of the hands in a teenage girl with only upper body involvement. Deformity of the chest is secondary to a right-sided pectoralis muscle transfer. (From Brown LM, Robson MJ, Sharrard WJW. The pathophysiology of arthrogryposis multiplex congenita neurologica. J Bone Joint Surg (Br) 1980;62:291-296.) |
pathophysiology of arthrogryposis multiplex congenita neurologica. J
Bone Joint Surg (Br) 1980;62:291-296.
mobility, self-care, and lower extremity alignment which is optimal for
standing and walking. If the patient is first seen at birth, when the
paralysis is present but not complete, there is considerable joint
range of motion that can be gained and maintained with frequent passive
range of motion therapy by the parent and physical therapist. If the
degree of paralysis is severe, and the joint has not moved with fetal
motion since the limb developed, less motion can be gained.
-
The initial goal is anatomic reduction of any dislocated or malpositioned joints.
-
The entire situation of the individual child first needs to be taken into account.
-
□ For example, if both hips cannot be
reduced, it is detrimental to reduce only one hip. All joints involved
need to be treated simultaneously.
-
-
If it is possible to loosen a contraction
by passive stretch, then later osteotomies will require a less severe
angular correction and, therefore, result in less juxtaarticular
deformity. -
The longer a functional position can be
maintained by therapy and bracing, the longer an osteotomy can be
delayed at the major joints. -
Bracing and splinting will most likely be necessary through the child’s lifetime.
-
Foot deformities of AMCN are either a clubfoot (equinovarus and adductus deformity) or rarely vertical talus (valgus deformity).
-
Goal of treatment is plantigrade feet for standing.
-
Almost all severe vertical tali and
clubfeet not attributed to other known conditions may in fact be
arthrogryposis limited to these peripheral locations. -
Serial casting should be started at birth and manipulated more often than an idiopathic clubfoot deformity.
-
After the maximum correction is obtained by stretch casting, it is maintained by physical therapy and removable splints.
-
Open reconstruction is performed between 6 and 12 months of age.
-
A high recurrence of some deformity
through the growing years of life is expected, and other osteotomies
and surgery may need to be performed to maintain plantigrade feet.-
□ Soft tissue releases, medial open-wedge
first cuneiform with lateral cuboid closing-wedge autograft
osteotomies, midtarsal dorsal wedge osteotomies, metatarsal
osteotomies, and potentially, triple arthrodesis for salvage.
-
-
If the child has an untreated or extreme
deformity that is resistant to the standard treatment procedures for
the clubfeet or vertical tali feet, partial or complete talectomy is
the treatment of choice.
-
Either extension contracture or flexion contracture may be present.
-
Flexion contractures initially can be somewhat corrected by serial casting or physical therapy of the knees to extension.
-
□ Care should be taken to not displace the physes during the manipulations or to cause laxity to the collateral ligament.
-
-
After maximum correction is obtained by
passive stretching, surgically lengthening the distal hamstrings and
soft tissue may be of help. -
If the knees are not at less than 30
degrees from full extension at this point, supracondylar extension
osteotomies are needed to get the legs straight enough to brace for
standing. -
Extension contractures are also treatable by serial flexion casting.
-
If this does not correct the problem by allowing knee flexion, a quadricepsplasty can be performed.
-
Care must be taken to not lose the
initial posture ability as well (i.e., to not overlengthen the quads to
get flexion of the knee and therefore lose the opposing extension
posture).
-
Teratologic dysplasia of the hips is a frequent finding in AMCN.
-
About 65% of patients manifest some degree of hip dysplasia.
-
If initially there is a unilateral dislocated or subluxed hip, it should be reduced and splinted.
-
If the hip is rigidly dislocated, then aggressive traction and therapy may allow it to reduce eventually.
-
Surgical muscle releases of hip contractures may aid in the reductions and will help in later ambulation.
-
Doing these as a first stage may shorten the time of the initial closed reduction attempts.
-
If the hip fails to reduce, then an
early, rather than late, open reduction of the hip with femoral
shortening or acetabular reconstruction (as one would do for any
developmental hip dysplasia) is performed. -
Less postoperative immobilization than
usual is advisable (to reduce incidence of further contracture) and
surgical intervention may allow this. -
If the child presents later in life, it is imperative to try to correct a unilateral dislocation.
-
Bilateral dislocated hips in the older
child should be left dislocated, and femoral osteotomies and soft
tissue releases performed to correct the alignment if necessary.
-
Correction of upper extremity deformities are usually performed after 3 years of age.
-
Only functional reconstruction is done to the upper extremities.
-
Fixed severe deformity only will require an osteotomy.
-
Most deformities are acceptable and functional.
-
If there is a fixed extension deformity, serial casting at birth may improve the position.
-
If function is not acceptable, osteotomies and muscle releases are then performed.
-
Flexion can be obtained by triceps
lengthening, and if not sufficient, supracondylar osteotomy is
performed to obtain enough flexion so that the child can care for the
head and neck area as well as performing self-feeding. -
Extension can be obtained by biceps
tendon release. If not sufficient, then a supracondylar osteotomy to
extension may be performed unilaterally to allow for lower body care. -
Children with upper extremity arthrogryposis develop many trick maneuvers to allow limb positioning for functional use.
-
If needed to allow active flexion of one elbow, the pectoralis major may be transferred (Fig. 20.2-4).
-
□ This should not be done bilaterally
because of the need for one arm to extend to allow for lower body care
and to allow for the use of a cane for walking. -
□ There must be 90 degrees of passive flexion before a pectoralis major transfer is undertaken.
-
-
If the child has severe scoliosis or a
spinal deformity, fusions should be performed as would be for any
patient with neuromuscular disease. -
In an adolescent with a curve under 40 degrees, a plastic thoracolumbosacral brace works well.
-
Congenital scoliosis secondary to malformed vertebrae most likely is not AMCN.
LM, Robson MJ, Sharrard WJW. The pathophysiology of arthrogryposis
multiplex congenita neurologica. J Bone Joint Surg (Br) 1980;62:291-296.
JG, Reed SD, Dricoll EP. Part I: Amyoplasia, a common, sporadic
condition with congenital contractures. Am J Med Genet 1983;15: 571-590.
LT, Chew DE, Elliott JS, et al. Management of hip dislocations in
children with arthrogryposis. J Pediatr Orthop 1987;7:681-685.
an abnormal proliferation of cells from the neural crest. Clinical
manifestations involve the skin, nervous tissue, bones, and soft
tissues. There are two types of neurofibromatosis, NF-1 (peripheral)
and NF-2 (central). NF-1, also referred to as von Recklinghausen
disease, is more common and has orthopaedic manifestations, whereas
NF-2 does not. NF-1 is characterized by café-au-lait macules,
intertriginous freckles, neurofibromas, Lisch nodules, optic gliomas,
bony dysplasias, and learning disabilities.
50% of cases are due to spontaneous mutations. Penetrance is close to
100%. The gene for NF-1 has been localized to chromosome 17 at band
q11.2. The protein product of this gene loci is neurofibromin.
Neurofibromin acts as a tumor suppressor by down-regulating a protein
(Ras) that enhances cell growth and proliferation. A point mutation or
deletion of this gene results in diminished function of neurofibromin.
disease affects approximately 1 in 4,000 persons, and at least 1
million people throughout the world. NF-1 is seen in all racial and
ethnic groups and affects the sexes equally.
diminished function of neurofibromin as a tumor suppressor may be
related to the occurrence of certain tumors in neurofibromatosis. It is
unclear how the gene mutation results in other disease manifestations.
Spinal deformities may be due to osteomalacia, localized
neurofibromarelated erosion and infiltration into bone, endocrine
disturbances, or mesodermal dysplasia.
neurologic, skeletal, and neoplastic manifestations. These
manifestations include café-au-lait macules, intertriginous freckles,
neurofibromas, Lisch nodules, optic gliomas, bony dysplasias, and
learning disabilities.
affects roughly 1 in 40,000 persons. The mutation occurs on chromosome
22. NF-2 is characterized by schwannomas of cranial nerve VIII,
meningiomas, and ependymomas.
Development Conference on Neurofibromatosis-1 concluded that the
diagnosis can be established when two of seven criteria are met (Box 20.3-1).
The criteria may be problematic for diagnosis in infants since 46% of
patients with spontaneous mutations and 30% of all patients have met
only one criterion by the age of 1 year. Ninety-seven percent of
patients meet two or more criteria by age 8. Diagnostic testing may
include x-rays, computed tomography (CT)/magnetic resonance imaging
(MRI), electroencephalogram, visual evoked responses, skin lesion
biopsies, slit lamp exams, and developmental/neuropsychiatric testing.
Prenatal testing is available but requires chorionic villus sampling, a
more complicated procedure than amniocentesis.
neuromas are present. The diagnosis can also be made when a
first-degree relative has NF-2 and the patient has either a unilateral
cranial nerve VIII palsy or has two of following present: neurofibroma,
meningioma, glioma, schwannoma, or juvenile posterior subcapsular
lenticular opacity.
-
Six or more café-au-lait macules, size >5 mm in prepubertal patients or >15 mm in postpubertal patients
-
Two or more neurofibromas or one plexiform neurofibroma
-
Freckling in axillary or inguinal region
-
Presence of optic glioma
-
Two or more Lisch nodules (iris hamartomas)
-
Distinctive osseous lesion (i.e., sphenoid dysplasia, thinning of long bone cortex, pseudarthrosis)
-
First-degree relative with neurofibromatosis-1 by above criteria
the manifestations of NF-1. Patients and parents should be asked about
skin lesions, pain, cognitive or psychomotor problems, progressive
neurologic deficits, constipation, growth problems (precocious puberty
or hypogonadism), orthopaedic problems, and hypertension. The family
history should include first-degree relatives, grandparents, great
aunts, and great uncles. The physical examination should include a
blood pressure measurement, a thorough skin exam, ophthalmic exam,
musculoskeletal exam, and neurologic exam; pubertal status should be
assessed. Particular portions of the exam may be specific to specialty
or clinic.
to predict which signs will manifest in which patients with the
disease. See Table 20.3-1 for the clinical manifestations of NF-1.
-
Hyperpigmented tan macules with smooth well-defined borders.
-
Lesions are present at birth or develop during infancy and are seen in 99% of cases by age 1.
-
Lesions are found in skin areas not exposed to sun.
-
Four types are recognized: cutaneous, subcutaneous, nodular plexiform, and diffuse plexiform.
-
Cutaneous neurofibromas are mixed cell
tumors rich in Schwann cells, but also contain fibroblasts, endothelial
cells, and glandular elements. They have no malignant potential. -
Subcutaneous neurofibromas can be painful and tender to palpation.
-
Nodular and plexiform neurofibromas can
also be painful and have the potential to dedifferentiate into
malignant neurofibrosarcomas. -
Nodular plexiform neurofibromas may involve dorsal nerve roots.
-
Diffuse plexiform neurofibromas may invade muscle, bone, or viscera.
-
Lesions are seen in 48% of patients by age 10 and in 84% of patients by age 20.
-
The lesions increase during pregnancy and puberty.
|
TABLE 20.3-1 CLINICAL FEATURES OF NEUROFIBROMATOSIS
|
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
-
Large soft tissue masses with rough, raised villous skin.
-
Underlying bone is typically dysplastic.
-
Skin overgrowth with thickening.
-
Skin integrity can be compromised by superficial infection or by weeping in skin folds causing maceration.
-
Found in 90% of patients by age 7.
-
Asymptomatic iris hamartomas.
-
The nodules, specific for NF-1, are seen in 90% of NF-1 patients more than 6 years old.
-
Most common tumor in children with NF-1
-
Only occur in 1% of patients by age 1 year and in 4% of patients by age 3 years
-
Small percentage can cause exophthalmus and visual impairment
-
10% to 60% of patients with NF-1 have scoliosis.
-
Deformity of the spine may be associated with intraspinal abnormalities.
-
Of all children and adolescents with scoliosis, 2% to 3% of cases are due to NF-1.
-
Dystrophic and nondystrophic changes occur in vertebral bodies.
-
Nondystrophic changes include wedging, angulation, and rotation.
-
See Box 20.3-2 for dystrophic changes in the spine.
-
Pseudoarthrosis is more common after spinal fusion for dystrophic and nondystrophic curves.
-
□ Risk of pseudarthrosis further increases with kyphotic curves.
-
□ Neurofibromas are often found at pseudarthrosis sites.
-
-
Scalloping of vertebral bodies
-
Severe rotation of the apical vertebrae
-
Vertebral wedging
-
Widening of the spinal canal
-
Enlargement of neural foramina
-
Widened interpediculate distance
-
Defective pedicles
-
Presence of a paravertebral soft tissue mass
-
Spindling of transverse process or ribs
-
Penciling of ribs
-
Curve characteristics are similar to
idiopathic scoliosis, but a higher percentage have progression of
deformity and postoperative pseudarthrosis. -
Deformations have ability to modulate
(transform by acquiring dystrophic features). The curves are more
likely to modulate if they manifest before 7 years of age. -
Likelihood of progression increases when
a curve acquires either three penciled ribs or a combination of three
dystrophic features.
-
Curves are characterized by short curves
(four to six segments), dystrophic changes, and the tendency to
progress to severe curves. -
Risk factors of early progression include:
-
□ Early age of onset
-
□ High Cobb angle at presentation
-
□ Kyphosis more than 50 degrees, apex in middle to lower thoracic area
-
□ Penciling of one or more ribs on concave or both sides
-
□ Penciling of four or more ribs, rotation more than 11 degrees at apex
-
□ Severely notched anterior vertebral body
-
-
Natural history demonstrates high rate of progression.
-
Defined as kyphosis more than 50 degrees with associated scoliosis
-
Associated with neurologic injuries
-
Further increased risk of pseudarthrosis after surgery
-
Curves with sagittal plane curvature less than normal curvature
-
Associated with mitral valve prolapse and decreased pulmonary function
-
Also associated with dystrophic deformities and dural ectasia
-
Expands spinal canal at the expense of bony and ligamentous elements.
-
Results of expansion can lead to
destabilization of vertebrae, spontaneous dislocation, and penetration
of the spinal canal by the ribs.
-
Protrusion of meninges through intervertebral foramen or a bony defect in vertebrae
-
Typically asymptomatic
-
May surround spine and impede operative approach in patients with NF-1
-
Abnormalities present with head and neck mass, torticollis, or dysphagia.
-
Most common abnormality is asymptomatic kyphosis.
-
More frequently associated with dysplastic changes
-
44% of NF-1 patients with scoliosis and 9% without scoliosis have cervical spine abnormalities.
-
Anteroposterior and lateral cervical
spine x-rays should be obtained prior to performing traction or general
anesthesia in patients with NF-1.
-
Cases due to NF-1 have occurred, but incidence lower than that in general population
-
Typically due to dural ectasia with meningoceles or neurofibroma with involvement of lumbosacral nerve root
-
Etiology must be acutely defined.
-
Typically due to neoplasm in the older
patient. In younger patients, the paraplegia is more often due to
spinal deformity, instability, dural ectasia, or rib penetration into
the spinal canal. -
Kyphosis contributes to neurologic changes more than scoliosis.
-
Intraspinal lesion, extradural dumbbell
neurofibroma, and intradural extramedullary neurofibroma should be
included in the differential diagnosis.
-
Dumbbell appearance results when a neurofibroma is constricted as it exits the foramina.
-
Can be intraspinal or extraspinal
-
Rare in NF-1.
-
Can occur with little clinical or radiographic warning
-
Diagnosis should be considered in NF-1 patients with unexplained neck and back pain.
-
Preferred term over congenital pseudoarthrosis
-
Anterolateral bowing of the tibia can occur with or without pseudoarthrosis.
-
Seen in 1% to 2% of NF-1 cases, compared with 1 per 140,000 in the general population.
-
Typically involves the middle or lower third of the tibia or fibula
-
Often diagnosed in infancy, and fracture typically occurs before age 2.5 years.
-
Occurs spontaneously or after fracture
-
See Table 20.3-2 for radiographic types.
-
Extremely difficult to resolve, as fracture and re-fracture are common
-
Pseudoarthrosis also occurs in ulna, radius, humerus, femur, clavicle.
-
Segmental hypertrophy is most common in the extremities but may also occur in the skull, mandible, and pelvis.
-
The cystic lesions that are often seen consist of nonspecific fibrous tissue.
-
Periosteal dysplasia results in a loose attachment to bone and predisposes to subperiosteal hematoma.
-
Limb length discrepancy
-
Short stature
-
Relative macrocephaly
-
Sphenoid dysplasia, bony defects in skull
-
Postaxial and preaxial polydactyly
-
Protrusio acetabuli
-
Can involve any organ system.
-
Most commonly occurs in the renovascular system with resultant hypertension
-
Neurofibrosarcoma: presents as a symptom complex of unexplained pain, neurofibroma enlargement, focal neurologic deficit
-
Leukemia
-
Rhabdomyosarcoma of the urogenital tract
-
Wilms tumor
-
Parathyroid adenoma
-
Sipple syndrome: triad of neurofibromas, bilateral pheochromocytomas, medullary thyroid carcinoma
|
TABLE 20.3-2 FOUR RADIOGRAPHIC TYPES OF PSEUDARTHROSIS OF THE TIBIA
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||||
-
50% of patients have learning disorders.
-
Unidentified bright objects (seen as high
signal-intensity lesions on a T2-weighted MRI) may correlate with the
cognitive deficits.
thoracolumbar, and sacral spine x-rays should be taken because of the
potential for occult deformities in each location. Each film should be
examined for abnormal lordosis, kyphosis, and dystrophic changes. A
CT/MRI should be obtained preoperatively to rule out the presence of an
intraspinal lesion. Other radiographs are acquired as dictated by signs
and symptoms.
-
McCune-Albright syndrome
-
Watson syndrome
-
Bannayan-Riley-Ruvalcaba syndrome
-
Epidermal nevus syndrome
-
Proteus syndrome
-
Multiple lentigines syndrome
-
Multiple café-au-lait lesions
primary care physician or neurofibromatosis specialty clinic. Treatment
involves both management of known symptoms of disease and prompt
detection of new manifestations.
-
Arrange for serial eye exams.
-
Perform neurologic exams for signs of neural tumors.
-
Inquire about milestone delays, learning disabilities, speech impairment, school performance problems, retardation.
-
Check for scoliosis/kyphosis, pseudarthrosis, signs of tumors
-
Educate about risk of progression during puberty, pregnancy.
-
Evaluate first-degree relatives.
-
Ketotifen can be prescribed for pruritus
-
Surgical excision or carbon dioxide laser excision as needed
-
Observe curves less than 20 degrees. Follow radiographs for signs of modulation and progression.
-
Brace curves 20 to 35 degrees.
-
Curves more than 35 degrees require posterior fusion with segmental instrumentation.
-
Curves more than 60 degrees require anterior release with bone graft as well as posterior fusion and instrumentation.
-
Curves less than 20 degrees should be reevaluated every 6 months.
-
Bracing has not been effective for dystrophic scoliosis and aggressive early surgery is recommended.
-
For curves 20 to 40 degrees, posterior
spinal fusion and segmental instrumentation should be considered.
Posterior spinal fusion is only appropriate in selected patients with
curves involving at least five segments and kyphosis less than 50
degrees. A CT should be obtained 6 months postoperatively for signs of
pseudarthrosis. -
Curves more than 40 degrees require anterior and posterior fusion.
-
Consider postoperative bracing.
-
The treatment of kyphosis less than 50 degrees depends upon the degree of the scoliosis.
-
Anterior and posterior spinal fusion
should be performed for a kyphosis of more than 50 degrees. A strong
anterior strut graft such as the fibula is recommended. -
Curves more than 70 degrees may require bracing even after surgical intervention.
-
A laminectomy alone for cord compression in kyphoscoliosis is contraindicated.
-
These curves require fusion and
instrumentation well above the lordosis because of the possible
development of a junctional kyphosis at the cervicothoracic junction.
-
Requires posterior spinal fusion and a postoperative hyperextension cast
-
Posterior spinal fusion should be performed for cervical spine deformities with instability.
-
Multiple treatment options are available:
-
□ Ankle-foot orthoses (AFOs) should be started prior to ambulation. Orthotics should be maintained until maturity.
-
□ Autogenous bone graft with intramedullary rod fixation from proximal tibia to calcaneus
-
□ Vascularized autogenous graft, such as contralateral fibula, iliac crest, rib
-
□ Resection of nonunion followed by distraction osteogenesis using the Ilizarov method
-
-
A Boyd or Syme amputation should be
considered after three unsuccessful surgeries or when limb length
discrepancy or foot and ankle deformity compromises function. These
amputations allow the weightbearing surface of the foot to be
maintained. -
Treatment of pseudarthrosis of fibula, ulna, os pubis, clavicle similar to tibia.
-
Empirical and individualized treatment is
necessary, usually a combination of epiphysiodesis, debulking, and
neurofibroma resection.
T, Mirowski G, Caldemeyer K. Neurofibromatosis type 1. Part II.
Non-head and neck findings. J Am Acad Dermatol 2001; 44:1027-1029.
AH. Neurofibromatosis. In: Weinstein SL, ed. The pediatric spine:
principles and practice. Philadelphia: Lippincott Williams &
Wilkins, 2001:471-490.
K, Szudek J, Friedman JH. Use of the National Institutes of Health
criteria for diagnosis of neurofibromatosis 1 in children. Pediatrics
2000;105:608-614.
developmental disturbances that result in excessive focal overgrowth of
mature tissues. This excessive overgrowth can be responsible for a
variety of dysmorphic features affecting the musculoskeletal system,
such as hemihypertrophy and macrodactyly, as well as other orthopaedic
manifestations. In general, hamartomatous disorders occur as rare
sporadic events. The true incidence and prevalence of these disorders
are unknown. There does not appear to be specific predilection for race
or sex or inheritance pattern. Germ line cells are not affected.
musculoskeletal system. The differential diagnosis of these disorders
is presented in Box 20.4-1. Neurofibromatosis (Chapter 23) and Maffucci (Chapter 23)
syndrome are presented in other chapters. This chapter focuses on
congenital hemihypertrophy, macrodactyly, Proteus syndrome, and
Klippel-Trenaunay syndrome.
The former is the most common type. The etiology of nonsyndromic
hemihypertrophy is unknown. Although numerous hypotheses have been
proposed, including abnormalities in vascular or lymphatic flow,
chromosomal mutations, localized endocrine or paracrine malregulation,
and defects in embryologic development, there is insufficient evidence
to lend credence to any proposed hypotheses of hemihypertrophy. In
addition, epidemiologic studies have not clearly differentiated
nonsyndromic from syndromic hemihypertrophy; thus, true incidence of
nonsyndromic hemihypertrophy is unknown. Prevalence is estimated to be
1 per 50,000 individuals.
-
Idiopathic hemihypertrophy/hemihyperplasia
-
Idiopathic macrodactyly
-
Proteus syndrome
-
Klippel-Trenaunay syndrome
-
Parkes-Weber syndrome
-
Maffucci syndrome
-
Neurofibromatosis, type 1
-
Epidermal nevus syndrome
-
Bannayan-Riley-Ruvalcaba syndrome
-
Hemihyperplasia/lipomatosis syndrome
-
Familial lipomatosis
-
Symmetric lipomatosis
-
Encephalocraniocutaneous lipomatosis
right and left sides of the body with 5% or greater difference in limb
length or circumference (Fig. 20.4-1).
Hemihypertrophy is easily diagnosed when severe discrepancies are
clinically apparent; however, when discrepancies are mild, the
distinction between hemihypertrophy and hemihypotrophy becomes
difficult. Diagnosis of hemitrophy is made by comparison of measured
limb lengths with expected lengths in relation to normal body
proportions. Trunk height can be used to determine the patient’s trunk
height percentile, which can subsequently be used to determine the
patient’s expected limb lengths.
-
Nonsyndromic
-
Total organ involvement
-
Limited
-
Classic (ipsilateral upper and lower limbs)
-
Segmental (single limb)
-
Facial
-
Crossed (contralateral upper and lower limbs)
-
-
-
Syndromic
 |
|
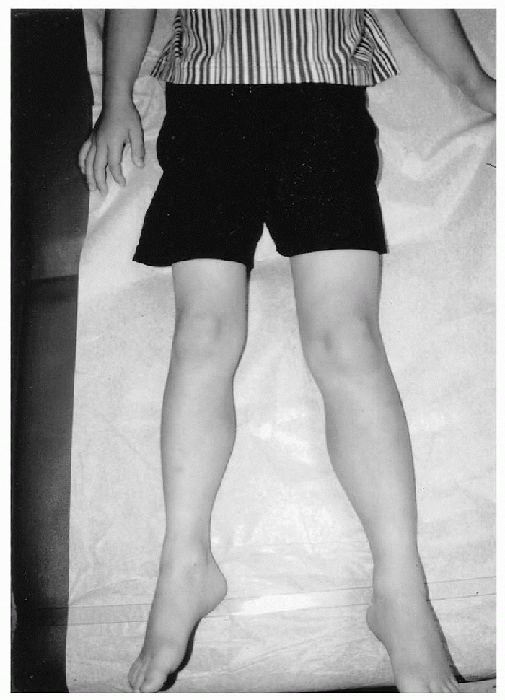
Figure 20.4-1
Eight-year-old boy with congenital hemihypertrophy involving the right side of the body. Observe the discrepancy in length and circumference of the left lower extremity. |
-
Hemihypertrophy is rarely apparent at birth but clinically manifests during subsequent growth.
-
Anatomic structures, including the eye, ear, tongue, thorax, abdomen, head on one side of the body, enlarge asymmetrically.
-
The skin may be thicker and there may be more hair on the affected side.
-
Ipsilateral internal organs are also increased in size.
-
In contrast to syndromic hemihypertrophy,
where the growth is irregular and unpredictable, the growth pattern in
nonsyndromic hemihypertrophy is regular and proportionate with growth
of the patient.-
□ This allows for improved prediction of eventual lower extremity length discrepancy and appropriate timing of interventions.
-
□ Lower extremity length discrepancy rarely exceeds 5 cm by skeletal maturity.
-
-
Nonsyndromic hemihypertrophy is not associated with cutaneous or vascular lesions.
-
The presence of vascular or cutaneous anomalies, in the setting of hemihypertrophy, indicates a generalized overgrowth syndrome.
-
Patients with nonsyndromic hemihypertrophy typically have normal mental capabilities.
-
Hemihypertrophy can be clinically
differentiated from hemihypotrophy/hemiatrophy, which may be
characterized by the presence of mental retardation, muscle
hypotrophy/atrophy, focal neurologic abnormalities, or joint
abnormalities. -
Genitourinary abnormalities are commonly
associated with nonsyndromic hemihypertrophy, including inguinal
hernia, renal cysts, cryptorchidism, sponge kidney, and horseshoe
kidney. -
Other associated orthopaedic findings
include scoliosis, macrodactyly, syndactyly, lobster-claw hand,
developmental dysplasia of the hip, and clubfoot.
increased risk of malignancy, such as Wilms tumor, adrenal carcinoma,
pheochromocytoma, hepatoblastoma, and leiomyosarcoma. Periodic
abdominal ultrasound screening remains controversial because the
benefits of early detection are unproved, and extraabdominal tumors
remain undetectable by abdominal ultrasound. Despite these issues,
current recommendations include abdominal ultrasound every 3 months
until 7 years of age followed by physical examination every 6 months
until skeletal maturity.
Hypotheses include genetic mutations, neuroinduction, and localized
endocrine or paracrine malregulation. None of these hypotheses have
been adequately proven, although the frequent enlargement of digits in
the sensory distribution of a major peripheral nerve provides abundant
circumstantial evidence to the hypothesis of neuroinduction. In all
cases of true idiopathic macrodactyly, hypertrophy of the digital
nerves and median or ulnar nerve coincide with hypertrophy of the
digits within the distribution of innervation.
macrodactyly, although, there appears to be a slightly greater male
predominance with a 3:2 male-to-female ratio. Some 90% to 95% of cases
are unilateral with multiple adjacent digit involvement. Macrodactyly
is equally likely to occur in the hands as in the feet. The second ray
is the most commonly involved followed by third, first, fourth, and
fifth in decreasing frequency of occurrence.
fibrous bands and adipose tissue consistently infiltrate muscles and
nerves. There is tumorlike proliferation of adipose tissue resembling
that of adult adipose tissue. In addition, digital nerves, median, or
ulnar nerves in the distal third of the forearm are prominent resulting
from fibrolipomatous proliferation of endoneurium, perineural, and
epineural tissues. There is diffuse periosteal fibromatosis resulting
from proliferation of fibroblastic tissue underlying the periosteum
with increased osteoblastic and osteoclastic activity that may account
for phalangeal and metacarpal overgrowth.
single digit or several adjacent digits of the hand or foot with
variable enlargement of the involved hand or foot proximal to the
digits. True macrodactyly must be differentiated from
pseudomacrodactyly where there is isolated enlargement of soft tissue
without enlargement of bone. Pseudomacrodactyly can occur secondary to
hemangiomas, congenital arteriovenous fistulas, or congenital
constriction band syndrome.
-
True macrodactyly presents without any evidence of cutaneous manifestations.
-
There is maximal enlargement of the digit distally, which tapers proximally.
-
Because the areas of enlargement lie in a
particular regional peripheral nerve distribution, it is also known as
nerve territory-oriented macrodactyly. -
The median nerve distribution is most commonly involved.
-
Enlargement of the nerve extends well
proximal to the macrodactyly, often beginning at the distal third of
the forearm with gradual enlargement toward the tip of the involved
digits.-
□ Enlarged median and ulnar nerves are susceptible to compressive neuropathy.
-
□ Carpal tunnel syndrome is a common finding in adults with macrodactyly.
-
-
The palmar or plantar surface is
disproportionately hypertrophied in relation to the dorsal surface,
resulting in hyperextension deformities of the
metatarsophalangeal/metacarpophalangeal and interphalangeal joints.-
□ The increased palmar or plantar soft tissue bulk results in limitation of flexion with ensuing joint stiffness.
-
□ Clinodactyly of the involved digit is common when half the digit is involved.
-
-
In mild cases, enlargement is isolated to
the digits; however, in severe cases, metatarsal/metacarpal bones are
affected with hypertrophy of the interosseous muscles and widening of
the affected hand or foot. -
The static form of macrodactyly is often present at birth.
-
□ The involved digits are 50% larger in
length and width of normal digits and continue to enlarge
proportionately to the rate of growth. -
□ Patients can retain reasonable function
if the condition is localized to one or two digits, and thus tend to
present in adolescence for treatment.
-
-
The progressive form is typically normal
or nearly normal at birth, with unremitting and disproportionate
digital overgrowth by 2 to 3 years of age. -
In both forms, longitudinal overgrowth
continues until physeal closure or arrest, but transverse growth and
soft tissue overgrowth may continue even after skeletal maturity. -
Documentation of hand and digit size with
anteroposterior hand radiographs is essential to determine the growth
pattern and differentiate the static and progressive forms of
macrodactyly. -
Macrodactyly is associated with syndactyly in 10% of cases and can also be associated with polydactyly and cryptorchidism.
-
Idiopathic macrodactyly
-
Static
-
Progressive
-
-
Syndromic macrodactyly
-
Static
-
Progressive
location, type, and severity. Primary consideration in management of
macrodactyly in the hand is function. Significant increases in girth
and length of digits in the
hand
are tolerable if there is only minor functional impairment. On the
other hand, the goals of treatment in the foot are for proper shoe
fitting and painless weightbearing. This is more difficult to achieve
since a small increase in width of the foot will prevent adequate shoe
accommodation.
-
Treatment options include nerve excision
and grafting, soft tissue debulking, physeal arrest and
hemiepiphysiodesis, osteotomies, digital shortening, amputation, ray
resection, or a combination of procedures. -
Mild to moderate macrodactyly can usually
be managed by epiphysiodesis of the involved bones and staged soft
tissue debulking in children less than 6 to 8 years of age or digital
shortening and staged debulking in older children and adolescents. -
This strategy is ineffective in the foot
because it does not adequately address girth that may be more properly
addressed with ray resection. -
Ray resection is not an option when the first ray is involved because of its unique role in balance and weightbearing.
-
More extensive involvement of the foot may require a midfoot or Syme amputation.
adequately addressing the challenges of macrodactyly. Patients and
parents must be informed of the complexities involved and the potential
number of surgeries required as good cosmetic result will be difficult
to achieve and functional result decreases with each successive surgery.
capillary, lymphatic, and venous malformations in the extremities
without involvement of the arterial system. The increased capillary and
lymphatic flow is thought to cause secondary hypertrophic effects on
the extremities. Primary mesodermal abnormality may also be present
because macrodactyly has been found to occur in the “uninvolved” limb.
Somatic mutation for a factor critical to vasculogenesis during
embryonic development is thought to be the causative factor; however,
the underlying molecular basis of Klippel-Trenaunay syndrome has not
been clearly elucidated.
95% of patients and the upper limbs in 5% of patients with 15% of
patients having combined upper and lower limb involvement. Only 10% of
patients will have discrepancies of more than 3 cm by skeletal maturity.
mesodermal abnormality resulting in the malformation of combined
capillary, venous, and lymphatic channels. The vascular channels in
these combined capillary, venous, and lymphatic malformations are lined
by a single layer of endothelial cells in distinction from hemangiomas
and other vascular tumors that are characterized by endothelial
hyperplasia. Venous fibromuscular dysplasia with a hypertrophied,
irregular, or absent medial layer results in dilation of the
superficial venous system. Deep venous system is absent in 14% of
patients.
-
Combined vascular malformations of the capillary, venous, and lymphatic types
-
Varicosities in unusual distributions in infancy or childhood
-
Limb (bony and soft tissue) hypertrophy.
malformations characteristic of Klippel-Trenaunay syndrome and the
fast-flow arteriovenous malformations of Parkes-Weber syndrome, in
which lymphatic malformations are absent.
-
Clinical diagnosis of Klippel-Trenaunay
syndrome needs to be confirmed by magnetic resonance imaging (MRI) with
gadolinium to distinguish between lymphatic, venous, and arterial
malformations. -
The characteristic lateral venous anomaly and any deep venous abnormalities should be documented by MRI or venography.
-
The combination of capillary malformation and hemihypertrophy is insufficient to make the diagnosis.
-
Klippel-Trenaunay syndrome is commonly
characterized by the classic lateral varicosities (vein of Servelle) in
the lower extremity beginning as a venous plexus from the dorsolateral
aspect of the foot, extending in a superolateral fashion with variable
termination proximally. -
Full leg distribution of varicosities with drainage into gluteal veins is found in 33% of patients.
-
Characteristic lymphatic vesicles and
venous flares originating from the lateral varicosities appear on the
surface as cutaneous capillary malformations. -
Abnormalities of the deep venous systems
such as agenesis, atresia, hypoplasia, valvular incompetence, or
aneurysmal dilatation are common. -
Other clinical manifestations include
complications secondary to distal venous hypertension and congestion
depending on the regional anatomy affected:-
□ Lymphedema, chronic venous ulceration,
gangrene, thromboembolism, thrombophlebitis, hematochezia, hematuria,
vaginal bleeding, esophageal varices, chronic consumptive coagulopathy
secondary to localized intravascular coagulation, and rapidly
progressive limb/trunk overgrowth
-
-
In contrast to Proteus syndrome, the
overgrowth observed in Klippel-Trenaunay syndrome is present and severe
at birth, although ultimate limb length discrepancy is not as
pronounced as that found in Proteus syndrome and typically does not
exceed 5 cm by skeletal maturity.
present with less severe hemihypertrophy and limb length discrepancy,
the orthopaedic intervention tends to be more successful in patients
with Klippel-Trenaunay syndrome. Treatment is generally supportive
until the symptoms become intolerable or functional impairment becomes
severe. There is recent evidence to suggest that early surgical
treatment of the vascular malformation in Klippel-Trenaunay syndrome
may prevent long sequelae of distal venous hypertension including
hemihypertrophy; surgical treatment of vascular malformations however
remains controversial.
 |
|
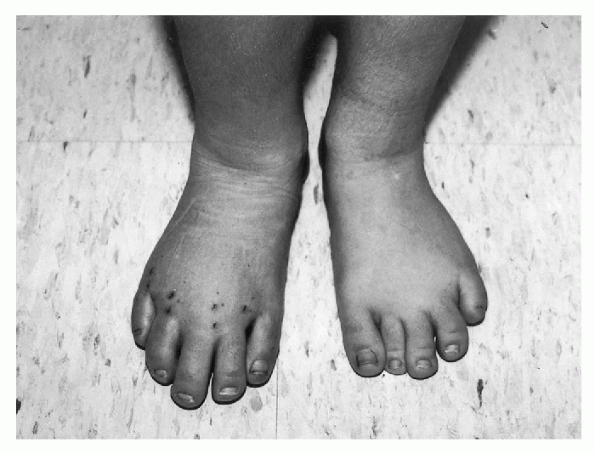
Figure 20.4-2
Postoperative photograph of a 7-year-old girl with Klippel-Trenaunay syndrome. She has disproportionate macrodactyly primarily of her right second, third, and forth toes and, to a lesser extent, the left third and fourth toes. The involved toes on the right foot have been treated with a percutaneous epiphysiodesis of the metatarsals and proximal phalanges to allow for progression shortening with subsequent growth. |
postzygotic somatic mutations in multiple cell lineages that occur only
in the mosaic state. This mosaicism accounts for the variability of
presenting clinical features. The hypothesized somatic mutations in
Proteus syndrome are presumed to be lethal in the nonmosaic state such
that there is no risk of recurrence or inheritance. There is recent
evidence to suggest a somatic mutation in the PTEN tumor suppressor
gene as the principal cause of many overgrowth syndromes including
Proteus syndrome and Proteus-like syndromes.
three germ layers. There is irregular overgrowth of multiple tissues
and cell lines resulting in highly variable phenotypic manifestations.
Proteus syndrome is mainly characterized by connective tissue nevi,
epidermal nevi, and hyperostoses. Commonly, there is excessive growth
of mature tissues including epidermis, connective tissue, endothelium,
adipose tissue, and bone. Overgrowth of tissues is progressive but
plateaus after the adolescent growth spurt.
clinical manifestations; however, general syndromic criteria must be
met regardless of the presence of multiple specific criteria. The
diagnosis of Proteus syndrome must include the following:
-
Mosaic pattern of distribution of tissue overgrowth
-
Evidence of the progressive nature of the disease
-
Lack of any mode of inheritance or transmission.
-
Epidermal nevus
-
Disproportionate overgrowth
-
Gigantism of limbs
-
Hemihypertrophy (arms/legs)
-
Macrodactyly (hands/feet/digits)
-
-
Skull hyperostosis
-
External auditory meatus hyperostosis
-
Megalospondylodysplasia
-
Viceromegaly (spleen/thymus)
-
-
Specific tumors before end of second decade
-
Bilateral ovarian cystadenomas
-
Parotid monomorphic adenoma
-
-
Dysregulation of adipose tissue
-
Lipomas
-
Regional absence of fat
-
-
Vascular malformations
-
Capillary malformations
-
Venous malformations
-
Lymphatic malformations
-
-
Facial phenotype
-
Dolichocephaly
-
Long facies
-
Minor downslanting of palpebral fissures and/or minor ptosis
-
Low nasal bridge
-
Wide or anteverted nares
-
Open mouth at rest
-
-
Connective tissue nevi result from dense
overgrowth of the underlying collagen resulting in the appearance of
“cerebriform or gyriform” lesions. -
These connective tissue nevi most commonly occur in the plantar and palmar surfaces.
-
Epidermal nevi, vascular malformations, and lipomas are also common findings in patients with Proteus syndrome.
-
Macrodactyly may occur in any combination
of digits in the hand, feet, or both and is not always ipsilateral with
hemihypertrophy (Fig. 20.4-3). -
It is usually minor or absent at birth but rapidly progresses in the first few years of life.
-
The degree of macrodactyly usually results in severe cosmetic and functional impairment.
-
Growth of the affected digits typically
decreases to a more proportionate rate in late childhood and plateaus
at skeletal maturity. -
Hemihypertrophy can be partial, complete, or crossed (Fig. 20.4-4).
-
Hemihypertrophy of the upper extremity
usually does not result in severe functional impairment; however,
involvement of the lower extremity can result in severe limb length
discrepancy. -
In combination with macrodactyly of the
foot and hemihypertrophy of the pelvis, leg length discrepancy in
excess of 10 cm can be expected.
 |
|
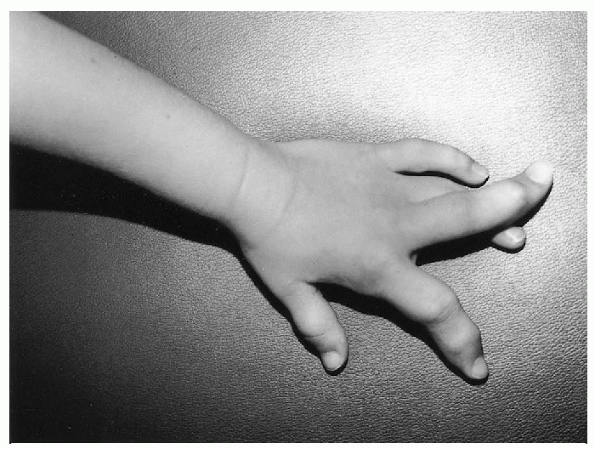
Figure 20.4-3
Severe macrodactyly of the index and long fingers of the left hand in a 5-year-old girl with Proteus syndrome. The fingers are essentially nonfunctional and were amputated. She also has ovarian abnormalities, macrodactyly of several toes, and other fibrous lesions of bone. |
 |
|
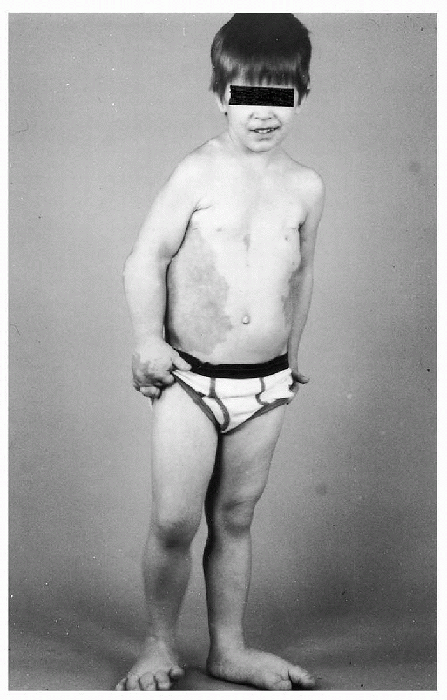
Figure 20.4-4
Four-year-old boy with Proteus syndrome. An extensive vascular malformation involves the right chest and abdomen, his right hand and both feet are enlarged, and there is hemihypertrophy of the right lower extremity. |
-
Spinal deformity (scoliosis, kyphoscoliosis, megalospondylodysplasia, gazelle neck)
-
Angular limb deformities (coxa valga/vara, knee and ankle valgus/varus)
-
Developmental dysplasia of the hip
-
Exostosis of hands, feet, or skull
-
Syndactyly, polydactyly, clinodactyly
-
Joint contractures/ligamentous laxity
-
Talipes equinovarus
-
Calcaneovalgus
affecting variable organ systems, patients with Proteus syndrome are
often cared for by multiple subspecialists. In general, management for
Proteus syndrome is individualized and directed toward each specific
manifestation. Hemihypertrophy and macrodactyly are the most common
reasons for orthopaedic consultation, however, there is dissociated or
delayed skeletal age making growth prediction unreliable and treatment
difficult.
successful for limb or digit length equalization, soft tissue bulk and
digital and pedal girth continue to be problems. Surgical options
include soft tissue debulking, epiphysiodesis, ray resection, and
amputation. Many patients with Proteus syndrome with severe
disproportionate overgrowth and multiple recurrences will go on to
multiple amputations at increasingly more proximal levels.
V, Coletti M, Cipolat L, et al. Early surgical management of
Klippel-Trenaunay syndrome in childhood can prevent long-term
haemodynamic effects of distal venous hypertension. J Pediatr Surg
2002;37:232-235.
LG, Happle R, Mulliken JB, et al. Proteus syndrome: diagnostic
criteria, differential diagnosis, and patient evaluation. Am J Med
Genet 1999;84:389-395.
D, Hager J, Nikolaides N, et al. Proteus syndrome: musculoskeletal
manifestations and management: a report of two cases. J Pediatr Orthop
1992; 12:106-113.
WG, Gabos PG. Localized disorders of bone and soft tissue. In:
Morrissey RT, Weinstein ST, eds. Lovell and Winter’s pediatric
orthopaedics, 5th ed. Philadelphia: Lippincott Williams & Wilkins,
2001:343-355.
AA, Gloviczki P, Cherry KJ, et al. Surgical treatment of venous
malformations in Klippel-Trenaunay syndrome. J Vasc Surg 2000;
32:840-847.
T. Macrodactyly. In: Buck-Gramcko D, ed. Congenital malformations of
the hand and forearm. London: Churchill Livingstone, 1998:183-197.
M, Spitz L. Klippel-Trenaunay syndrome: clinical features,
complications and management in children. Br J Surg 1995;82: 757-761.
S. Musculoskeletal manifestations of Proteus syndrome: report of two
cases with literature review. J Pediatr Orthop 1992; 12:667-674.