
Editors: Tornetta, Paul; Einhorn, Thomas A.; Damron, Timothy A.
Title: Oncology and Basic Science, 7th Edition
Copyright ©2008 Lippincott Williams & Wilkins
> Table of Contents > Section
II – Specific Bone Neoplasms and Simulators > 7 – Congenital and
Inherited Bone Conditions > 7.1 – Familial Adenomatous Polyposis
II – Specific Bone Neoplasms and Simulators > 7 – Congenital and
Inherited Bone Conditions > 7.1 – Familial Adenomatous Polyposis
7.1
Familial Adenomatous Polyposis
Kathryn Palomino
Familial adenomatous polyposis (FAP) is characterized by
early onset of multiple colorectal polyps. These polyps are
premaligmant lesions that are highly likely to progress into
carcinomas. Its orthopaedic importance lies in the association of
osteomas with FAP and in the fact that Gardner syndrome, previously
considered a distinctly different entity, is now considered to be a
subset of FAP in which patients, in addition to having polyps, may
develop osteomas, epidermoid cysts, desmoid tumors, and dental
abnormalities.
early onset of multiple colorectal polyps. These polyps are
premaligmant lesions that are highly likely to progress into
carcinomas. Its orthopaedic importance lies in the association of
osteomas with FAP and in the fact that Gardner syndrome, previously
considered a distinctly different entity, is now considered to be a
subset of FAP in which patients, in addition to having polyps, may
develop osteomas, epidermoid cysts, desmoid tumors, and dental
abnormalities.
Pathogenesis
Etiology
-
Genetic defect for both FAP and Gardner syndrome: mutation of the adenomatous polyposis coli (APC) gene (5q21-22 region)
-
Creates premature stop codon resulting in production of a truncated nonfunctioning protein
-
-
Inheritance: autosomal dominant with high but variable penetrance
-
De novo mutations: 10% to 20% of cases
-
Phenotype varies by mutation site.
P.213
Epidemiology
-
Incidence: 1 in 8,000 to 1 in 10,000 people
-
Accounts for 1% of all cases of colorectal cancer
Pathophysiology
-
Gene for FAP has been cloned, but function is unknown, as is the biological basis of the syndrome.
-
Abnormalities in colonic epithelium and skin fibroblasts are present.
Classification
-
Classic FAP
-
Polyps (Fig. 7.1-1)
-
Colonic and duodenal adenomas
-
Gastric fundic hyperplasia
-
Jejunal and ileal adenomas
-
Ileal lymphoid polyps
-
-
Extraintestinal lesions
-
Mandibular osteomas
-
Dental abnormalities
-
-
-
Gardner variant
-
Polyps
-
Same as FAP
-
-
Extraintestinal lesions
-
Osteomas of long bones, mandible, and skull
-
Epidermoid and sebaceous cysts
-
Lipomas
-
Fibromas
-
Desmoid tumors
-
-
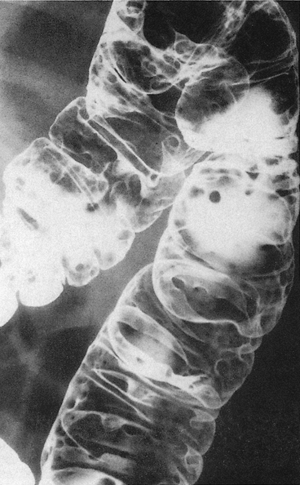 |
|
Figure 7.1-1
Familial polyposis. Small uniform polyps virtually carpet the colon. This patient had familial adenomatous polyposis. (From Swischuk LE. Imaging of the Newborn, Infant and Young Child, 4th ed. Philadelphia: Lippincott Williams & Wilkins, 1997:486.) |
Clinical Features
-
Endoscopically detectable lesions develop in the second decade.
-
Natural history in untreated patients: symptoms at average age of 33 years; cancer at 39 years; death at average age of 42 years
-
Extraintestinal features (Table 7.1-1)
-
Osteomas
-
Do not evolve into malignant lesions
-
-
Impacted or supernumerary teeth
-
Epidermoid lesions (Gardner)
-
Lipomas (Gardner)
-
Fibromas (Gardner)
-
Desmoid tumors (Gardner)
-
Occur in 8% to 13% of cases
-
Most lethal complication of syndrome after carcinoma
-
-
-
Extraintestinal manifestations are frequently detectible before adenomas and may enable early diagnosis of FAP.
|
Table 7.1-1 Bone and Soft Tissue Lesions Most Commonly Seen in Gardner Syndrome
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
P.214
Diagnosis
-
FAP diagnosis: must meet one of the following criteria
-
At least 100 colorectal adenomas
-
Gene mutation of APC gene
-
Family history of FAP with one of the following:
-
Epidermoid cyst
-
Osteoma
-
Desmoid tumor
-
-
Treatment
-
Polyposis
-
Annual endoscopic screening beginning at age 10
-
Antioxidants and nonsteroidal anti-inflammatory drugs may reverse small lesions and are in trial.
-
Only reasonable option for colon in FAP
-
Prophylactic colectomy is performed.
-
Timing and extent decided on an individual basis
-
-
-
-
Orthopaedic manifestations
-
Desmoid tumors
-
Surgical wide excision is first-line treatment for resectable desmoid tumors.
-
Nonoperative options
-
Nonsteroidal anti-inflammatory agents: sulindac may reduce size of desmoid tumors and is under investigation
-
Hormones (tamoxifen)
-
Steroids (prednisone)
-
-
-
Osteomas
-
Nonoperative care indicated except in rare cases
-
-
Suggested Reading
Boland CR, Kim YS. Gastrointestinal polyposis syndromes. In: Sleisenger JS, Fordtran JS, eds. Gastrointestinal Disease: Pathophysiology, Diagnosis, Management. Philadelphia: WB Saunders, 1993:1430–1438.
DeVita Jr V, Hellman S, Rosenberg S, eds. Cancer: Principles and Practice of Oncology, 6th ed. Philadelphia: Lippincott Williams & Wilkins, 2001:1221.
Nilbert M, Coffin CM. Familial adenomatous polyposis. In: Fletcher CDM, Unni KK, Mertens F, eds. Tumours of Soft Tissue and Bone: Pathology and Genetics. World Health Organization Classification of Tumours. Lyon: International Agency for Research on Cancer Press, 2002:352–354.