
means to run together. When several relatively uncommon anomalies occur
in the same individual, it may be nothing more than coincidence.
However, if all the anomalies result from the same cause, or occur in
the same pattern in other children, that particular combination of
birth defects is called a syndrome. A
syndrome should be suspected if a characteristic orthopaedic
malformation (e.g., radial clubhand) is encountered, if all four
extremities are affected, if limb deformities are symmetric, if there
are several associated nonorthopaedic anomalies, or if the patient has
a familiarly dysmorphic face (1,2,3,4). Children who have syndromes look more like one another than like their parents.
physician to recognize that a child has characteristics of a syndrome.
In such cases, appropriate referrals should be made to a geneticist to
assist in syndrome identification, order appropriate confirmatory
tests, and arrange for management of the nonorthopaedic manifestations
of the syndrome. The evaluation of a child for a syndrome includes a
family history, a systems review, and a search for minor dysmorphic
features, such as abnormal palm creases or abnormal shape of digits or
toes. These evaluation processes may not be of immediate orthopaedic
importance, but they are the clues to look further.
activated in a coordinated manner to allow cells to divide,
differentiate, move, and die off, ultimately resulting in a normally
formed individual. These cell signaling pathways play roles in the
development of multiple organs. It is not surprising that dysregulation
of such developmentally important pathways can cause the malformation
of a number of organs, resulting in several otherwise uncommon
abnormalities occurring together, producing a syndrome. Such pathways
can be dysregulated by a mutation in a key pathway member, by fetal
environmental factors (e.g., a teratogen, such as in fetal alcohol
syndrome), or both.
and the cause of a syndrome is not always as simple as one would wish.
Even within a family in which all the members carry the identical
causative gene mutation, some individuals are minimally affected,
whereas others have all of the findings of the syndrome. This may be
due to the presence of modifying genes, which may not be inherited in
the same way as the gene mutation that causes the syndrome, or due to
fetal environmental factors that modify the manner in which the
pathways are activated. In addition, different mutations in the same
gene can cause different syndromes. Such is the case with the
dystrophin gene, which causes both Duchenne and Becker muscular
dystrophies. This occurs because the products of different mutations
have different cellular functions.
important, because it has implications for the parents as to the risk
of recurrence in subsequent pregnancies, and may hold the key to the
development of novel treatments. The rapid pace of basic research in
developmental biology and genetics makes it difficult for a traditional
textbook to provide the most up-to-date information about syndrome
etiology. The Internet is becoming an excellent source for such
information. One useful site is the On-line Mendelian Inheritance in
Man (OMIM), administered by the National Institutes of Health. This
site can be accessed at http://www.ncbi.nlm.nih.gov/Omim/, and can be searched by syndrome name, causative gene, or clinical findings (5).
Discussions about the risk of subsequent pregnancies are in the realm
of the genetic counselor. Parents often assume that if the condition
has a name, it is treatable or curable. This, sadly, is not the case.
The importance of understanding syndromes is in recognizing that
associated medical abnormalities may adversely influence orthopaedic
outcomes, and may influence surgical timing and management. The
orthopaedic surgeon also needs information from the geneticist.
Associated conditions may influence the outcome of orthopaedic problems
and can affect anesthesia (e.g., cardiac or renal anomalies). Even if
parents are not planning subsequent pregnancies, and if there are no
plans for their child to undergo surgery in the near future, genetic
evaluation is still important for proper syndrome diagnosis. Correct
diagnoses are essential for research into syndrome etiology and
treatment. Patients should be given the opportunity to participate in
such research, especially in cases of relatively rare syndromes.
because a single syndrome may have several names. Eponyms are not
descriptive of the syndrome, nor do they give information about
etiology. Many syndromes are caused by a mutation in a gene, and the
causative gene has been identified in most such syndromes. Classifying
syndromes by the causative gene alone can be problematic because some
genes cause more than one syndrome; some syndromes are caused by more
than one gene; and some syndromes are not caused by a gene mutation.
Furthermore, gene names are frequently unrelated to clinical findings
associated with a given syndrome. A numbering system is used by
computer databases; the most widely used is that of the OMIM (5),
but this is helpful only for database searches. An ideal nomenclature,
which would give information about clinical findings and etiology, has
yet to be developed.
supplant the need for the clinician to know the phenotypic features of
individual syndromes (7). For many syndromes,
molecular genetic tests are not available, or are available only at a
very high cost. As such, it is impractical to test a given patient for
every known genetic condition (8). A thorough
study of the patient’s history and a physical examination give clues as
to which supportive tests to order, such as radiographs. This
information is used for narrowing down the diagnosis to only a handful
of syndromes. In many cases, the ultimate diagnosis can be made on the
clinical and radiographic findings alone [e.g., neurofibromatosis (NF)
type I]. For syndromes in which molecular genetic tests are available,
these are usually performed to confirm a diagnosis rather than to make
a diagnosis, and should only rarely be ordered by an orthopaedist
before consultation with a clinical geneticist or genetic counselor.
gene mutations into groups broadly categorized by the function of the
causative gene (9,10).
Such syndromes can be broadly classified into those caused by mutation
in genes encoding one of the following types of proteins: structural
proteins; proteins that regulate developmentally important signaling
pathways; proteins implicated in neoplasia; proteins such as enzymes
that play a role processing molecules; and proteins that play a role in
nerve or muscle function (7). Syndromes within
each broad group share similarities in the mode of inheritance and
clinical behavior. For instance, syndromes caused by mutations in genes
encoding structural proteins tend to be inherited in an autosomal
dominant manner and result in skeletal structures that wear out with
time, for which corrective surgery has a high recurrence or failure
rate. Spondyloepiphyseal dysplasia is an example of such a disorder,
which is caused by a mutation in type II collagen. It is inherited in
an autosomal dominant manner, and surgery to correct hip deformity is
associated with a rather high recurrence rate. Using such clinical
findings, one can also predict, in the case of an inherited syndrome
whose etiology has not yet been identified, which category of causative
gene it likely belongs to. Most of the disorders in this chapter will
be grouped using this functional genetics scheme. The one exception is
the contracture syndromes, which are considered as a separate group.
Although the genetic etiology of many of the contracture syndromes has
been identified, it is easiest, from a practical standpoint, to
consider them as a few subgroups based on clinical and treatment
similarities.
connective tissues, including the bones, articular cartilage,
ligaments, and skin. Mutations in such genes disrupt the structural
integrity of the connective tissues in which they are expressed. In
most cases, the phenotype is absent or there are only minor
manifestations present at birth; the phenotype evolves with time,
because the abnormal structural components slowly fail or wear out with
time as the individual grows. Deformity often recurs after surgery,
because the structural components are abnormal and will wear out again.
In cases where the structural abnormality involves cartilage, there may
be growth abnormality caused by physeal mechanical failure, or early
degenerative disease of the joints caused by articular cartilage
failure. When a protein that is important for ligament or tendon
strength is affected, joints often subluxate. There can be substantial
heterogeneity in the severity of the phenotype, depending upon the
exact way in which the mutation alters the protein function. In
patients with mild disease, life expectancy is normal; however, in
patients with more severe disease, life expectancy may be shortened
because of secondary effects of the structural defects on vital organs.
These disorders tend to be inherited in an autosomal dominant manner (9, 10).
Many of the disorders caused by mutations in genes that encode
structural proteins, including osteogenesis imperfecta and
spondyloepiphyseal dysplasia, are covered in other sections of this
textbook.
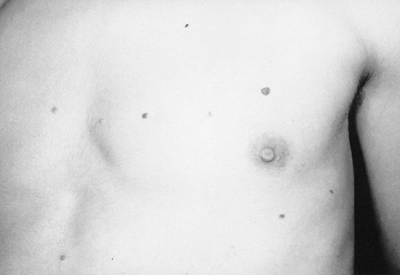
this syndrome in 1896, as a condition associated with long limbs and
involvement of the cardiovascular, ocular, and skeletal systems (11).
Although some authorities believe that Abraham Lincoln had Marfan
syndrome, there remains considerable controversy surrounding this, and
a decision was made against using some of the DNA from his remains to
test for this diagnosis (12). This is one of
the few syndromes of orthopaedic importance associated with tall
stature. Patients can be recognized by the characteristic tall stature,
arachnodactyly (abnormally long and slender digits), dolichostenomelia
(long, narrow limbs), pectus deformities, and scoliosis. Stria can be
seen in the skin (Fig. 9.1). In the
cardiovascular system, aortic regurgitation, aortic dilatation,
aneurysms, and mitral valve prolapse can occur. Ocular findings are
myopia and superior displacement of the lens. It is important for an
orthopaedist to recognize this condition, since undiagnosed patients
with Marfan syndrome not infrequently present to an orthopaedist with a
diagnosis of scoliosis. Recognizing this syndrome allows for referral
for management of the cardiovascular abnormalities, early treatment of
which can prevent premature mortality.
clinical findings, including abnormalities in the ocular, cardiac, and
musculoskeletal systems (13). Several meetings
have been held recently in an attempt to come to a consensus about
diagnostic criteria. Unfortunately, controversy
remains
as to the best set of diagnostic criteria to employ: the more stringent
criteria exclude many individuals who are currently managed as Marfan
syndrome patients. The less stringent Berlin criteria and more
stringent Ghent criteria are outlined in Table 9.1 (14,15,16).
To further confound issues of diagnosis, dural ectasia, which is a
major diagnostic criterion, is defined as a dural volume greater than 7
cm3 below the inferior L5 endplate; this measurement
requires a magnetic resonance image (MRI) study to be performed, using
a technique that allows for this volume to be measured (17,18).
In addition, the normal dural volume in younger, growing children is
not known, and the reliability and reproducibility of volume measures
when used outside of clinical investigative groups is not known.
 |
|
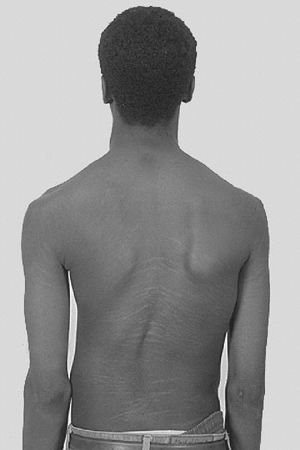
Figure 9.1 Stria in a boy with Marfan syndrome, who initially presented for evaluation of scoliosis.
|
are suggestive of Marfan syndrome and, although not considered part of
the diagnostic criteria, should alert one to consider this diagnosis.
It was thought that the physical finding of an arm span longer than
height would be diagnostic of Marfan syndrome; however, population
studies have shown that this is not the case. The ratio of upper
segment (head to pubic symphysis) to lower segment (pubic symphysis to
plantar surface), which is normally 0.93 in a mature individual, is
often decreased in Marfan syndrome to 0.85 or less (19).
Two clinical findings associated with arachnodactyly are a thumb that
protrudes past the ulnar border of the hand when it is held in a
clenched fist (Steinberg sign) and overlap in the thumb and index
finger when they are wrapped around the opposite wrist (20).
that are frequently present in patients with Marfan syndrome, these are
not pathopneumonic. Spinal morphology suggestive of dural ectasia and
pedicle dysplasia are suggestive of this disorder. The use of
measurements from spine radiographs in making this diagnosis (an
interpedicular distance at L5 greater than or equal to 36.0 mm; a
sagittal diameter at L5 greater than or equal to 13.5 mm; a transverse
process-to-vertebral width ratio at L3 greater than or equal to 2.25
mm) yields a high sensitivity, but a relatively poor specificity (16).
Arachnodactyly is defined, for purposes of radiographic readings, as an
increase in the ratio of length to width of the second to fifth
metacarpals (Fig. 9.2). The average ratio of
the lengths of the second to fifth metacarpals, divided by the widths
of the respective diaphyses, is greater than 8.8 in male patients and
greater than 9.4 in female patients with Marfan syndrome (21).
There are no studies, however, that determine the sensitivity and
specificity of the use of these measures to make a diagnosis of Marfan
syndrome.
Thirty percent of the cases, however, are sporadic. The current
evidence points to a dominant-negative effect, in which expression of
the mutant gene product inactivates the function of the normal gene
product. As such, this condition could potentially be treated by the
use of therapies that decrease the expression of the mutant gene (23).
The fibrillin protein plays a role in maintaining the normal mechanical
properties of the soft tissues, especially in resistance to cyclic
stress (24). The clinical findings of laxity
and subluxation of the joints, and weakening of arterial walls with
resultant aortic dilatation, are easy to understand on the basis of the
function of fibrillin. The tall stature and arachnodactyly associated
with the syndrome are seemingly difficult to attribute to the fibrillin
mutation. However, the extracellular matrix also contains growth
factors, which are bound to extracellular matrix proteins. Recent
investigations have found that fibrillin mutations cause some of these
extracellular growth factors, such as transforming growth factor β to
become more readily accessible to cell receptors (25).
The increased growth factor availability likely causes increased
cellular growth and rapid longitudinal bone growth, resulting in long,
thin fingers and toes and tall stature. Although molecular diagnosis
for fibrillin gene mutations is possible, such use of molecular data is
usually not required in making the diagnosis, as physical findings are
generally sufficient for this purpose.
problems in Marfan syndrome, including subluxation of joints, a
predisposition to sprains, and even scoliosis.
|
TABLE 9.1 DIAGNOSTIC CRITERIA FOR MARFAN SYNDROME: A COMPARISON OF THE BERLIN AND GHENT DIAGNOSTIC CRITERIA
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
referred to the orthopaedist. Smaller curves can be managed in a manner
similar to that for idiopathic scoliosis, with bracing considered for
select curves in skeletally immature individuals. Although bracing is
often prescribed, it seems to be less effective than in idiopathic
scoliosis (26). This has led some orthopaedists
to suggest that bracing only delays the need for surgical treatment.
There are no well-controlled studies comparing brace treatment with
observation or any other type of management in these patients.
Therefore, brace treatment remains controversial. Despite this, we
offer brace treatment using the same principles as for idiopathic
scoliosis for children with Marfan syndrome. Curves will often be
relatively short and associated with deformity of vertebrae termed
dysplastic (Fig. 9.3). The spinal deformity is
often associated with kyphosis, especially in the lumbar spine region.
Surgery is considered for rapidly progressive curves in skeletally
immature individuals, or for large progressive curves in skeletally
mature individuals. Surgery in these patients is challenging, with
higher complication rates reported than for idiopathic scoliosis.
Infection, instrumentation fixation failure, pseudarthrosis, or coronal
and sagittal curve decompensation occur in 10% to 20% of patients.
Infection is often associated with a dural tear and decompensation is
associated with extreme correction. Although surgery can be safely
undertaken in most patients, there is a report of a postoperative death
from valvular insufficiency. To avoid such complications, the
cardiopulmonary condition of patients with Marfan syndrome should be
evaluated
preoperatively (27,28,29,30,31,32,33,34).
Computerized tomography (CT) scan to assess bony anatomy, especially of
the pedicles, is quite useful in preoperative planning of hook and
screw placement. Avoidance of extreme correction, extension of the
fusion to vertebrae that are neutral and stable in both planes, and
fusing both primary and secondary curves may prevent curve
decompensation, hardware failure, and pseudarthrosis. We recommend
these techniques in the operative management of this condition. Other
unusual spinal deformities can occur, such as subluxation of vertebrae (35).
 |
|
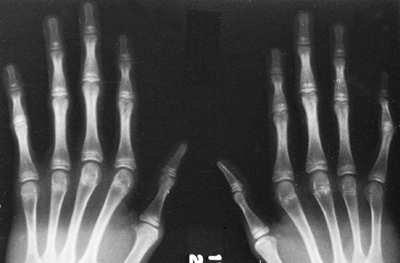
Figure 9.2 Hands showing arachnodactyly. Notice the long, thin metacarpals and phalanges.
|
 |
|
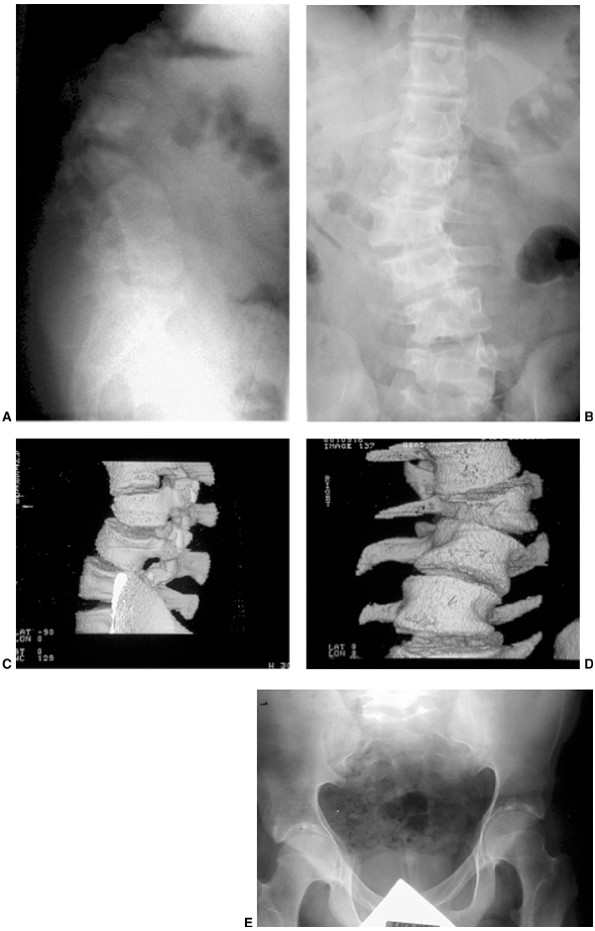
Figure 9.3 Scoliosis (A, B) and protrosia of the hips (E) in a patient with Marfan syndrome. C, D:
Deformity of the apical vertebrae is shown in a three-dimensional reconstruction of a computerized tomographic scan image. (Courtesy of Chris Reily, MD, Vancouver, British Columbia, Canada.) |
syndrome, and seems to increase in severity with age. Its severity is
not related to the severity of other clinical findings; for instance,
there is no association between aortic dilatation and dural ectasia (36).
Although there is a slightly higher incidence of back pain in patients
with dural ectasia than in those without, a 40% incidence of back pain
in patients with Marfan syndrome without dural ectasia suggests that
dural ectasia itself is not the cause of the pain. One should thus
evaluate patients with Marfan syndrome for other causes of back pain
even in the presence of dural ectasia.
may be caused in part by the fibrillin abnormality disrupting the
normal extracellular matrix structure of bone, and in some cases it may
be related to relative physical inactivity. Susceptibility to fracture
does not seem to be a problem, and it is therefore not clear whether
intervention for the decreased bone density is warranted (37,38).
Protrusio acetabula is present in about one-third of patients with
Marfan syndrome. It is not related to bone mineral density and is
usually asymptomatic (39), and thus prophylactic fusion of the triradiate cartilage is probably not warranted.
can lead to premature death. Indeed, many cases of sudden death during
athletic activities in the young are in individuals with Marfan
syndrome. Despite this, there are no universally accepted criteria for
restricting physical activity in individuals with Marfan syndrome.
Early intervention using β-blockers can reduce the development of
aortic dilatation. Individuals with aortic dilation may also benefit
from earlier cardiac surgical intervention. Lens dislocation requires
ophthalmologic intervention. In Marfan syndrome the lens is dislocated
in a superior direction, whereas in homocystinuria there is an inferior
dislocation. Homocystinuria shares many clinical features with Marfan
syndrome, but is also associated with a coagulation disorder. As such,
it is crucial that an individual suspected of having Marfan syndrome be
evaluated for cardiovascular problems, and that the possibility of
homocystinuria be excluded before the patient undergoes surgery.
distinguish patients with homocystinuria from those with Marfan
syndrome, as patients with homocystinuria often present to the
orthopaedists with a clinical picture suggesting Marfan syndrome.
Unlike Marfan syndrome, homocystinuria is associated with a
coagulopathy, which can be fatal if unrecognized, especially during
surgery. Although homocystinuria is not caused by a mutation in a gene
encoding a structural protein, it shares phenotypic similarities with
Marfan syndrome, and it is therefore being discussed here. It is caused
by a defect in one of the enzymes that is important in the production
of cysteine from methionine, thereby resulting in the accumulation of
intermediate metabolites in the blood (homocysteine and homocystine)
and in the urine (homocystine) (40,41). There are several subtypes, and patients with type I have a phenotype similar to that of Marfan syndrome (42).
Affected individuals are tall with long limbs and may have
arachnodactyly and scoliosis. Dislocation of the lens of the eye is
common but, in contrast to Marfan syndrome the displacement is
inferior. Osteoporosis is often more severe in type I homocystinuria
than in Marfan syndrome. Vertebral osteoporosis can produce biconcavity
and flattening of vertebral bodies, whereas in Marfan syndrome the
vertebral bodies are either normal or excessively long. Widening of the
epiphyses and metaphyses of long bones is more typically seen in
homocystinuria. Mental retardation is not a feature of Marfan syndrome,
but occurs in approximately half of all patients with homocystinuria (43).
Patients with type I homocystinuria have an abnormality in clotting,
which leads to venous and arterial thromboembolic episodes (44).
Such episodes can complicate surgery; therefore, such patients should
be managed by a hematologist when surgical intervention is considered.
cystathionine synthetase, which normally catalyzes the chemical union
of homocysteine and serine to form cystathionine. The enzyme uses
pyridoxine (vitamin B6) as a cofactor. Blood levels of
methionine are increased, and thus screening of patients with Marfan
syndrome for homocystine in the urine (with the cyanide nitroprusside
test) can differentiate type I homocystinuria from Marfan syndrome.
Types II and III homocystinuria are biochemically distinct. Because the
errors cause blocks at other points, blood levels of methionine are
normal, and other clinical findings such as skeletal changes and
thromboses are absent.
type I, the typical course is methionine restriction and pyridoxine
supplementation (44). For types II and III,
methionine restriction is harmful. Treatment with cofactors also varies
for these other types. Vitamin B12 is suggested in the management of type II, and folic acid for type III.
subtypes, each of a different genetic etiology. As such, it is really a
collection of different disorders that are associated with the common
phenotypic findings of hyperextensibility of the skin and hypermobility
of the joints. Easy bruisability of soft tissue, fragility of bone,
calcification of soft tissues, and various degrees of osteopenia are
associated with the various subtypes. The hyperlaxity allows affected
individuals to have impressively large ranges of motion of the joints.
Contortionists are often individuals with this syndrome. Although the
first detailed clinical description of the syndrome was reported by
Tschernogobow in 1892, the condition derives its name from reports by
Edward Ehlers, a Danish dermatologist, in 1901 and by Henri-Alexandre
Danlos, a French physician, in 1908. These two individuals combined the
pertinent features of the condition to provide a detailed description
of the phenotype (45).
and fragile, bruisable skin that heals with peculiar “cigarette-paper”
scars and may show changes resulting from multiple bruises (Fig. 9.4).
Children with this condition may be born prematurely because of
premature rupture of fetal membranes, because these membranes are
derived from the fetus itself. The fragile soft tissues can also cause
problems such as “spontaneous” carotid-cavernous fistula, ruptures of
large vessels, hiatus hernia, spontaneous rupture of the bowel,
diverticula of the bowel, rupture of the colon, aortic dilatation, and
retinal detachments (46,47,48,49,50).
There are at least six additional subtypes of EDS; these are very rare,
often being reported as a single family. Although an understanding of
the genetic cause of the rare types provides important information
about how various proteins contribute to the maintenance of the
mechanical integrity of the soft tissues, the infrequency of their
occurrence makes their incorporation into a general classification
scheme less useful to the clinician.
 |
|
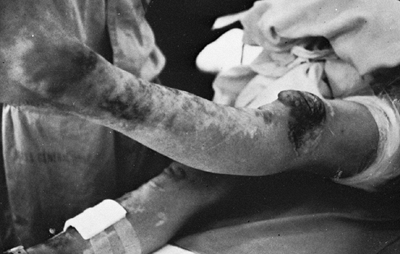
Figure 9.4
Patient with Ehlers-Danlos syndrome, type I. The knees and pretibial regions have been subjected to recurrent injury and have accumulated heme pigmentation. (Courtesy of Michael G. Ehrlich, MD, Providence, Rhode Island.) |
in a gene that produces a protein that processes collagen. The types of
EDS that are caused by a mutation in collagen are inherited in an
autosomal dominant manner, whereas those caused by a protein processing
defect (kyphoscoliotic and dematosparaxis types) are inherited in an
autosomal recessive pattern. Since collagen is the main structural
component of a variety of connective tissues, it is easy to understand
how these mutations cause the associated changes in soft tissue
mechanics (45,53,54).
The hypermobility type, which is characterized by multiple dislocations
of joints, is also associated with a delay in achieving developmental
milestones, perhaps because of the dislocations. Individuals with this
type have the greatest functional disability. The vascular type is
associated with ruptures of vessels or viscera. Such events are rare in
childhood, but by the age of 20, one fourth of those with the condition
will have had some vascular or visceral complication. Teenage boys may
be at a high risk for this during their prepubertal growth spurt (57).
Early death occurs, most commonly because of vascular rupture, with the
median age of survival being less than 50 years. Individuals with the
kyphoscoliosis type often present as “floppy” infants, and this
diagnosis should therefore be considered in such children. Children
with clinical features suggestive of EDS should be referred to a
geneticist. Although molecular diagnosis is possible for some of the
subtypes, these are usually not needed for making the diagnosis. There
are no universally accepted criteria for restricting participation in
physical activity in patients with EDS, so recommendations to limit
activity should be made on an individual basis.
|
TABLE 9.2 A MODIFIED CLASSIFICATION SCHEME FOR EHLERS-DANLOS SYNDROME
|
||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
common occurrences in the various subtypes. The chronic pain that such
individuals complain of is often attributed to these subluxations. The
management of the subluxations is problematic, and a multidisciplinary
effort, including pharmacologic and physical therapeutic approaches, is
often required. Since the matrix components that provide the mechanical
properties to the soft tissues are defective, surgical approaches
focusing on ligaments and tendons (e.g., soft tissue procedures around
the shoulder) have a low success rate. Procedures that involve surgery
to the bones to better contain the joints have a higher success rate. A
variety of such operations are reported, such as osteotomies, which
change the direction and location of insertion of tendons or capsules
or that provide a larger joint area (tibial tubercle transfer
operations for patellar dislocations, and femoral and pelvic
osteotomies for hip subluxation). In particularly problematic cases, it
may be necessary to place a bone graft to limit motion and prevent
dislocation (e.g., a posteriorly placed graft at the elbow).
Arthrodesis may be required as a last resort in those cases that remain
symptomatic despite other managements (58,59,60).
the same principles as those for idiopathic scoliosis, despite a lack
of studies showing this management approach to be efficacious. Surgical
management can be
problematic in the vascular type, in which vessel ruptures can occur during surgery (61).
It is important not to place undue stretch on vessels during surgery,
and it is probably safest to have a vascular surgeon available in case
a major disruption is encountered. Cardiac valve problems can occur in
EDS, so patients should have a cardiac evaluation before undergoing
surgery. Low bone density is identified in EDS; however, when one
corrects for the activity level of these patients, the bone density may
not be so abnormal (62). Pharmacologic treatment for low bone density should be considered only in rare instances.
pathways that are important in regulating cell reproduction or
proliferation. A mutation that results in dysregulation of such
pathways can cause increased cell proliferation, resulting in
overgrowth of a cell type or organ. Such pathways are frequently
dysregulated in neoplasia. In some inherited conditions, when a single
copy (one allele) of a gene that is important in regulating cell
proliferation is mutated in the germ-line, the result is an overgrowth
phenotype, but when the second copy becomes mutated in a somatic
manner, the result is the development of neoplasia. Since these
disorders are usually caused by one copy of the defective gene, they
tend to be inherited in an autosomal dominant manner. The type of
tissue or organ involved depends on the cell type in which the gene is
expressed. In many syndromes, such as NF, the tissues of the
musculoskeletal system are affected, resulting in obvious bone or soft
tissue abnormality. There is a risk of malignant progression, which
develops over time as the cells are subjected to genetic damage (second
hits), causing the loss of the normal copy of the causative gene.
Recurrence of a deformity after surgery is not unusual, because the
underlying genetic defect that causes abnormal cell growth cannot be
corrected by any surgical procedure. Many children present with
limb-length discrepancy, but most of these conditions will not be
related to a syndrome and can be managed as described in Chapter 29
on limb length inequality. It is important to understand the various
associated syndromes so that appropriate referrals can be made for
nonorthopaedic problems.
|
TABLE 9.3 NEUROFIBROMATOSIS TYPE I: DIAGNOSTIC CRITERIA
|
|
|---|---|
|
are type I and type II (NF1 and NF2). Orthopaedic manifestations are
common in NF1, which is also called von Recklinghausen disease, whereas they are rare in NF2, which is also called central neurofibromatosis or familial acoustic neuroma.
The clinical findings in NF1 are quite variable, and many of these
findings develop over time. Children may exhibit none of the typical
findings at birth, but the diagnosis can be made as they grow older and
develop the characteristics necessary to confirm a diagnosis of NF1 (63,64).
Although a causative gene for NF1 has been identified, this diagnosis
is made by identifying at least two of the clinical findings in Table 9.3.
In patients with NF these spots often appear after 1 year of age, and
then they steadily increase in number and size. The spots have a smooth
edge, often described as similar to the coast of California, as opposed
to the ragged edge of spots associated with fibrous dysplasia, which
are described as similar to the coast of Maine. The spots vary greatly
in number, shape, and size, and six lesions greater than 1 cm in size
are required for the diagnostic criteria. Axillary and inguinal
freckling are common and serve as good diagnostic markers, because such
freckling is exceptionally rare except in people with NF.
Hyperpigmented nevi are dark brown areas that are sensitive to the
touch; they typically overlie a deeper plexiform neurofibroma.
common is the cutaneous neurofibroma, composed of benign Schwann cells and fibrous connective tissue (Fig. 9.6).
This type of neurofibroma may occur anywhere, but is usually just below
the skin. These neurofibromas may not be detectable until 10 years of
age, and with puberty there is a rapid increase in their number. When
many are grouped together on the skin, it is known as a fibroma molluscum.
Plexiform neurofibromas are usually present at birth and are highly
infiltrative in the surrounding tissues. The overlying skin is often
darkly pigmented. They are highly vascular and lead to limb giantism,
facial disfigurement, and invasion of the neuroaxis (Figs. 9.7 and 9.8).
 |
|
Figure 9.5
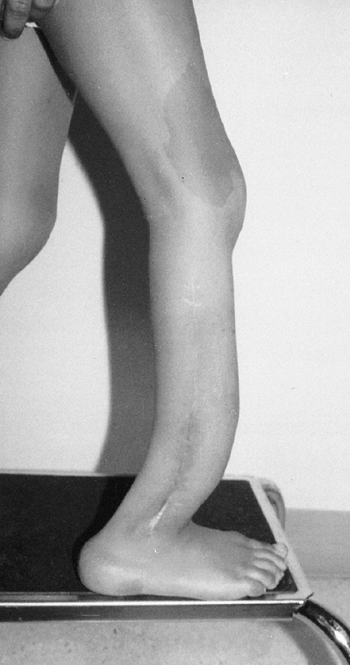
Neurofibromatosis in a 6-year-old child. Notice the large café-au-lait spot on the thigh and the anterior bowed tibia typical of pseudarthrosis. (From Goldberg MJ. The dysmorphic child: an orthopedic perspective. New York: Raven Press, 1987, with permission.) |
of an unusual scoliosis, overgrowth of a part, or a congenital
pseudarthrosis lesion seen on radiographs should alert the physician to
consider a diagnosis of NF (65). There are a
variety of anomalies of bone observed in radiographic images, ranging
from a scalloping of the cortex, to cystic lesions in long bones that
look much like nonossifying fibromas, to permeative bone destruction (Fig. 9.9). These radiographic findings may mimic benign or malignant bone lesions (66,67,68).
Radiographs of the pelvis usually show various degrees of coxa valga,
and in nearly 20% of patients there is radiographic evidence of
protrusio acetabuli (69,70).
are present in 50% of all 5-year-olds with NF1, and in all adults with
NF1. It is unusual for Lisch nodules to be present in individuals who
do not have NF1, and so the detection of these nodules can aid in
making this diagnosis. However, it may be difficult to detect these
lesions, and patients should be sent to an experienced ophthalmologist
for this diagnosis. The lesions do not cause any visual disturbances.
Once the diagnosis is established, further ophthalmologic evaluation is
not necessary (71,72).
NF1 is an autosomal dominant disorder with 100% penetrance, but
one-half of cases are sporadic mutations and are associated with an
older-than-average paternal age. The most well-known patient who was
presumed to have had NF, Joseph Merrick, also called the Elephant Man,
probably did not have this condition; his clinical profile better fits
Proteus syndrome (76). The NF1 gene is located on chromosome 17 (77). Its protein product, neurofibromin, acts as a tumor suppressor (78). There are also other potential genes located in introns within the NF1 gene, whose functional significance is unclear.
signal transduction proteins. These proteins convey messages from cell
surface receptors to cytoplasmic effectors. Neurofibromin plays such a
role in the Ras signaling system, which is involved in the control of
cell growth (79). Mutations in the NF1
gene cause a disruption in its normal regulatory function of Ras
signaling. This gives the affected cells an abnormal growth pattern.
Neurofibromin is expressed at higher levels in the neural crest during
development. Cells from the neural crest migrate to become pigmented
cells of the skin, parts of the brain, spinal cord, peripheral nerves,
and adrenals, thus explaining the common sites of abnormalities in the
disorder. The gene defect gives a clue to potential novel therapies,
because pharmacologic agents that block Ras signaling could be used to
treat the disorder. Disruption of the normal Ras signaling cascade is
probably responsible for the malignant potential of this disorder. Only
one of the two copies of the NF1 gene is
mutated in affected patients; however, tumors from such individuals
have been found to have only the mutated gene because of loss of the
normal copy (80,81,82,83).
Farnestyl transferase inhibitors block the downstream effects of Ras
signaling activation and thus have the potential to be used in the
treatment of some of the more severe
manifestations of NF. There are several ongoing early-phase trials of such drugs (84,85).
 |
|
Figure 9.6
Neurofibromatosis in a 14-year-old patient. Cutaneous neurofibromas make their appearance with the onset of puberty. (From Goldberg MJ. The dysmorphic child: an orthopedic perspective. New York: Raven Press, 1987, with permission.) |
to an orthopaedist, one should be aware of these types because
musculoskeletal malformations are occasionally present. Patients with
NF2 present with acoustic neuromas, central nervous system tumors, and
rare peripheral manifestations. There are usually fewer than six
café-au-lait spots, and no peripheral neurofibromata. These patients
are very unlikely to present with an orthopaedic deformity. There are
two much less common types of NF, type three and type four (NF3 and
NF4), in which patients are more likely to develop a problem requiring
orthopaedic intervention. Individuals with NF3 present with some of the
characteristics of NF1, but also have acoustic neuromas, which are
characteristic of NF2. These individuals often have spinal deformity,
especially in the cervical region. NF4 presents with the same clinical
findings as NF1, except that one of the cardinal features of NF1,
namely, Lisch nodules of the iris, is absent (63,64).
It is important to distinguish these types from NF1, because they are
probably caused by mutations in a gene different from the one that
causes NF1 (this has already been demonstrated in NF2), and will
therefore not be diagnosed by DNA testing for NF1.
 |
|
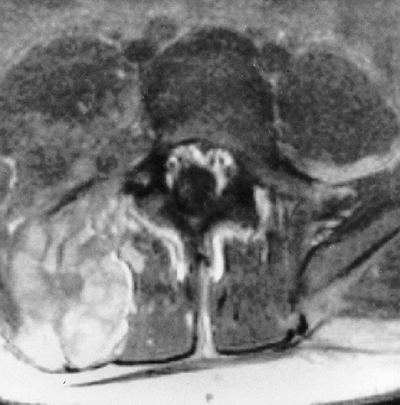
Figure 9.7
Neurofibromatosis in a 16-year-old patient. The magnetic resonance image at the level of L4–L5 demonstrates a large plexiform neurofibroma that invades the neural axis. It extends from the level of L3 to the sacrum. |
scoliosis, overgrowth of the limbs, pseudarthrosis, and radiographic
appearances of lesions, enable the initial diagnosis to be made if the
syndrome is kept in mind. Patients with NF often exhibit overgrowth,
ranging from a single digit to an entire limb and from mild anisomelia
to massive giantism. As such, the possibility of NF should be
considered in a child with focal giantism, such as macrodactyly. When
NF is compared with the more symmetric idiopathic hemihypertrophy,
there is disproportional overgrowth involving the skin and subcutaneous
tissue more than the bone (Fig. 9.8).
categories: a dystrophic curve and an idiopathic curve. Most curves in
NF resemble idiopathic scoliosis curves.
Their
relation to NF is not understood, and their precise incidence is still
being debated. These curves can be managed like any other idiopathic
curve.
 |
|
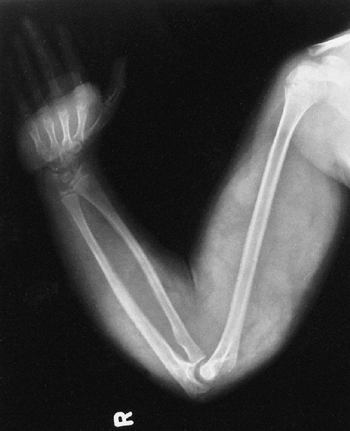
Figure 9.8
Neurofibromatosis in a 10-year-old patient. Hypertrophy affects the arm from the shoulder to the fingertips; the major component is soft tissue. Nodular densities throughout the upper arm are consistent with a plexiform neurofibroma. Notice the lack of skeletal overgrowth and some attenuation of the radius and ulna, caused by external compression by the neurofibroma. (From Goldberg MJ. The dysmorphic child: an orthopedic perspective. New York: Raven Press, 1987, with permission.) |
It is associated with deformity of the ribs and vertebrae. The onset is
early in childhood, and it is relentlessly progressive. Curves that
initially appear to be idiopathic in children under age 7 have almost a
70% chance of becoming dystrophic over time, although there may be
subtle clues, for example, mild rib penciling (thinning of the ribs in
a shape similar to a pencil point near the vertebrae), suggesting that
the curve is actually dystrophic. The most important risk factors for
progression are an early age of onset, a high Cobb angle, and an apical
vertebra that is severely rotated, scalloped (concave loss of bone),
and located in the middle-to-lower thoracic area (90).
The combination of curve progression and vertebral malformation mimics
congenital scoliosis in appearance and behavior. Dystrophic curves are
refractive to brace treatment. Sagittal plane deformities may occur,
including an angular kyphosis (i.e., gibbus) and a scoliosis that has
so much rotation that curve progression is more obvious on the lateral
than on the anteroposterior radiograph (90). In
those with angular kyphosis, there is a risk of paraplegia. Dystrophic
curves are difficult to stabilize, and it is best to intervene with
early surgery involving both anterior and posterior fusion (90,94,95,96).
Kyphotic deformities are often the most difficult to manage surgically,
and strut grafts across the kyphosis anteriorly may be necessary. In
rare severe cases, the spine can even seem to be “dislocated” because
of the kyphosis and scoliosis. In cases with extremely severe
deformity, halo-femoral or halo-gravity traction may be necessary to
safely straighten the spine to a more acceptable deformity without
producing neurologic sequelae. Other reported techniques include
inserting a bone graft without instrumentation and then gradually
straightening the curve using a cast postoperatively (97).
In rare severe cases in which there is a vertebral “dislocation” one
can use instrumentation to achieve an overall alignment of the back,
while leaving the vertebrae “dislocated” (98). Unusual complications have been reported in the management of such dystrophic curves, such as a rib head
migrating into the neural canal resulting in spinal cord compromise (99).
 |
|
Figure 9.9
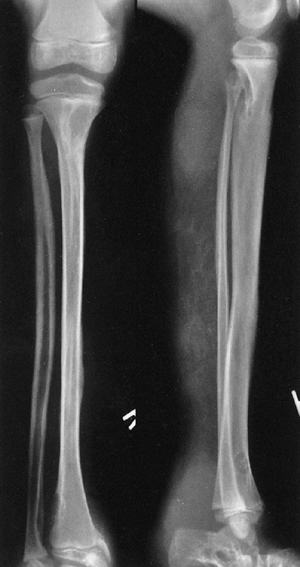
Neurofibromatosis in a 10-year-old patient. The radiograph shows an array of cystic and scalloped skeletal lesions in the tibia and os calcis of the right leg. Some of the lesions are characteristic of neurofibromatosis. Other lesions, occurring in isolation, can mimic benign fibrous tumors. Scalloped cortical erosion at the upper end of the femur, permeative bone destruction in the region of the os calcis, and metaphyseal cystic lesions are other features. (From Goldberg MJ. The dysmorphic child: an orthopedic perspective. New York: Raven Press, 1987, with permission.) |
 |
|
Figure 9.10
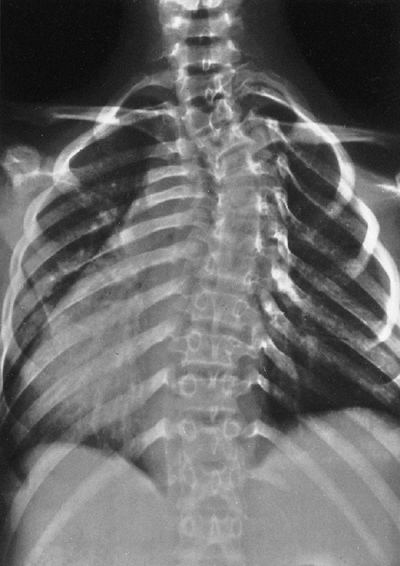
Neurofibromatosis in a 14-year-old patient. The dystrophic curve is produced by a short-segment scoliosis. Ribboned ribs show cystic irregularities. (From Goldberg MJ. The dysmorphic child: an orthopedic perspective. New York: Raven Press, 1987, with permission.) |
radiographs. These include scalloping of the posterior body,
enlargement of the neural foramina, and defective pedicles,
occasionally with a completely dislocated vertebral body (100,101,102,103,104).
Such findings may mean that there is a dumbbell-shaped neurofibroma in
the spinal canal, extending out through a neural foramina. The dura in
NF patients behaves like the dura in patients with a connective tissue
disorder, and dural ectasia is common, with pseudomeningoceles
protruding through the neural foramina. Unlike neurofibroma, dural
ectasia is an outpouching of the dura, without an underlying tumor or
overgrowth of spinal elements (Fig. 9.11) (105,106,107,108). The incidence of anterolateral mening-oceles was underestimated until asymptomatic patients were screened with MRI (65,109).
The erosion of the pedicles may lead to spinal instability, especially
in the cervical spine. In rare cases, this can even lead to dislocation
of the spine (110,111).
MRI and CT scans are helpful preoperatively in delineating the presence
of defective vertebrae or dural abnormalities, and may assist in
choosing the levels on which to place instrumentation.
 |
|
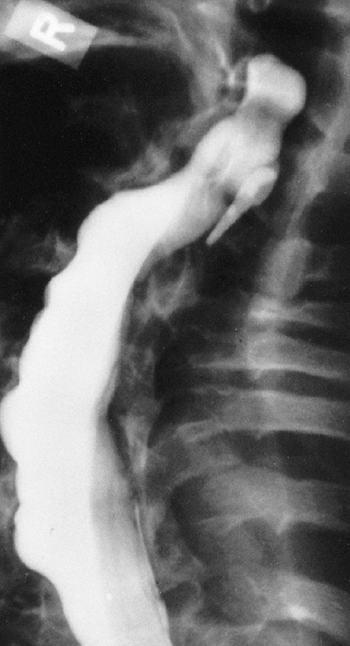
Figure 9.11 Myelogram of a young adult with neurofibromatosis and scoliosis with pseudomeningoceles and dural ectasia.
|
Fracture usually follows, with spontaneous union being rare and
surgical union presenting a challenge. An anterolateral bowed tibia is
routinely managed with a total-contact orthosis to prevent fracture,
although there are no well-designed studies showing that this is indeed
effective. Intramedullary rod fixation seems to offer the best results
for the initial management of a pseudarthrosis. Recent studies have
shown the importance of achieving neutral tibial alignment in the
healing of a tibial pseudarthrosis. The presence of an intact fibula is
associated with a lower healing rate, perhaps because of associated
tibial malalignment (114). The cause of the
pseudarthrosis is not known; however, neurofibromas have not been
identified at the pseudarthrosis site. The pseudarthrosis process may
affect the ulna, radius, femur, or clavicle (115,116,117,118,119,120,121).
In each of these locations, there is a course similar to that in the
tibia, with bone loss and difficulty in achieving union (Fig. 9.13). Not all pseudarthroses of the forearm require treatment (122), but if they are symptomatic, the available options include proximal and distal synostosis to produce a single-bone
forearm, the use of a vascularized fibula graft, or resection of the
pseudarthrosis with shortening of the forearm and internal fixation (123).
 |
|
Figure 9.12
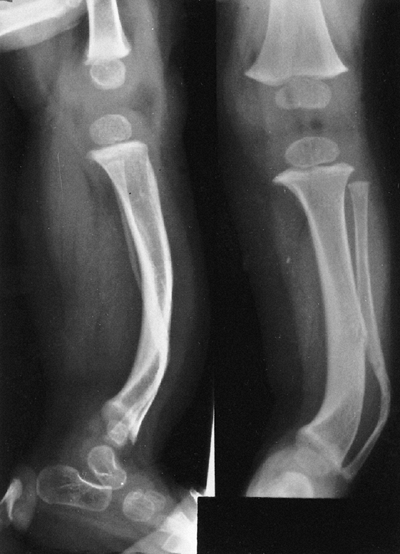
Neurofibromatosis in a 1-year-old patient. The anterolateral bow of the tibia and fibula warrant concern about impending fracture and pseudarthrosis. (From Goldberg MJ. The dysmorphic child: an orthopedic perspective. New York: Raven Press, 1987, with permission.) |
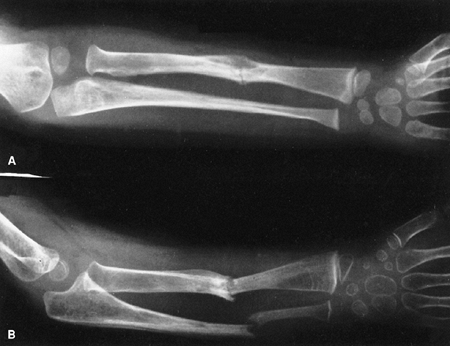 |
|
Figure 9.13
Neurofibromatosis in a 3-year-old patient. The radiograph shows progressive pseudarthrosis of the radius and ulna after a pathologic fracture. A: Fracture through the cystic lesion of the radius and thinning of the mid-ulna. B: After 10 months of cast immobilization, pseudarthrosis affects the radius and ulna. (From Goldberg MJ. The dysmorphic child: an orthopedic perspective. New York: Raven Press, 1987, with permission.) |
processes that affect individuals with NF1. Most neurofibromas do not
require treatment, but symptomatic lesions may require excision.
Plexiform neurofibromas that become symptomatic are very difficult to
manage. Their vascularity and infiltrative nature make complete
extirpation almost impossible, with a substantial risk of
uncontrollable hemorrhage and neurologic deficit. Although speculative,
the use of angiogenesis inhibitors, such as interferon, or some
experimental agents that modulate the effect of the causative gene
mutation, such as farnesyl transferase inhibitors, may be beneficial (124,125).
The most common tumor location is in the central nervous system, with
lesions such as optic nerve glioma, acoustic neuroma, and astrocytoma (131).
There is a risk of malignant degeneration of a neurofibroma to a
neurofibrosarcoma. This process can occur in a central or peripheral
neurofibroma (132,133,134,135).
It can be quite difficult to distinguish a malignant lesion from a
benign one. CT scans show areas of low-enhancing density in
neurofibrosarcomas (136), but there are no
studies confirming the sensitivity and specificity of this finding.
Similar patterns can also be visualized using MRI. Routine surveillance
for sarcomatous change is impossible because of the large number of
neurofibromas. Lesions that increase in size or develop new
characteristics should be investigated. There is a propensity for
children with neurofibroma to develop other malignancies, such as Wilm
tumors or rhabdomyosarcomas.
pheochromocytoma is reported regularly, as is a curious type of
metabolic bone disease similar to hypophosphatemic osteomalacia (137,138). Hypertension is a major
risk factor for early death (130). Precocious puberty may occur because of an intracranial lesion (139).
Affected children are short, but tend to have large heads.
Approximately 50% have an intellectual handicap. Although the mean IQ
is low, the range of IQ is quite wide (140).
More than the low IQ, it is the difficulty in concentrating (which is
common in this condition) that may interfere with the learning process (141). The concentration problems can sometimes be managed pharmacologically.
The incidence is 1 in 14,000, and it is probably an autosomal dominant
trait of variable expression. Patients are large, although this feature
is not always noticed at birth (143). The child
is in the 97th percentile for size by 1 year of age. The tongue is
gigantic at birth, and although it tends to regress, hemiglossectomy is
sometimes needed. Omphalocele is common, and 15% of the babies born
with omphaloceles have Beckwith-Wiedemann syndrome. The abdominal
viscera are enlarged, and a single-cell hypertrophy accounts for the
large organs: in the adrenals, giant cortical cells; in the gonads, an
increased number of interstitial cells; and in the pancreas, islet cell
hyperplasia. This underlies the 10% risk of developing benign or
malignant tumors. Wilm tumor is the most common.
11p15, which is near the Wilm tumor gene (11p13) and the insulin-like
growth factor gene (11p15.5) (144). There may be some paternal genomic imprinting (145,146).
The closeness of the Beckwith-Wiedemann gene locus and these embryonal
tumor gene loci accounts for the dysregulation of the tumor-related
genes and the associated overgrowth and higher incidence of tumors seen
in this syndrome.
It is crucial that the neonatologist diagnose this syndrome early so as
to prevent the consequences of hypoglycemia. If it is not managed
properly, seizures occur at day 2 or 3. Central nervous system damage
from the hypoglycemia leads to a cerebral palsy-like picture. The
cerebral palsy-like findings confuse the diagnosis of this syndrome,
and make the management of these patients more complex. The diagnosis
can occasionally be made prenatally by ultrasound (147,148).
the presence of this disorder is the unusual combination of two
otherwise common problems: spastic cerebral palsy and hemihypertrophy (Fig. 9.14).
The spasticity is thought to be a result of the neonatal hypoglycemic
episodes, especially if accompanied by neonatal seizures, but spastic
hemiplegia is most commonly seen. In general, children with cerebral
palsy tend to be small; Beckwith-Wiedemann syndrome should be suspected
if a large child has spastic cerebral palsy. Asymmetric growth affects
about 20% of the patients. It is usually true hemihypertrophy, but it
can be significant if the spastic hemiplegia affects the smaller side.
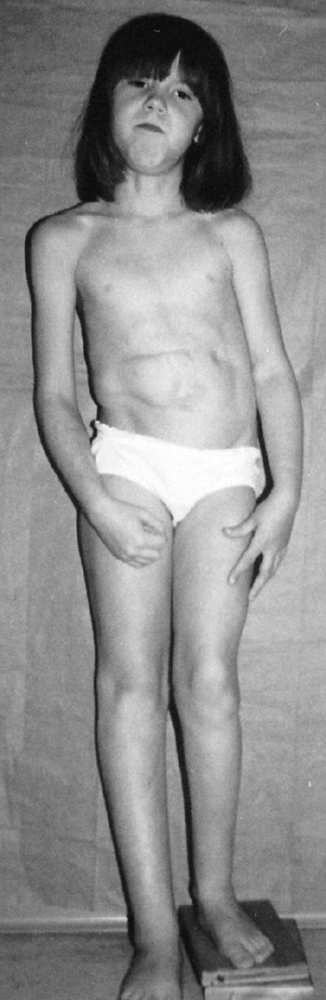 |
|
Figure 9.14
Beckwith-Wiedemann syndrome in an 8-year-old patient. Hemihypertrophy on the right, a part of this syndrome, is combined with hemiatrophy on the left, caused by acquired encephalopathy secondary to hypoglycemic seizures as a newborn, leading to a significant leg-length discrepancy of 4.6 cm. Abdominal scars are a consequence of omphalocele repair. (From Goldberg MJ. The dysmorphic child: an orthopedic perspective. New York: Raven Press, 1987, with permission.) |
predisposed to a variety of neoplasms, most notably Wilm tumor.
Abdominal ultrasounds at regular intervals until the age of 6, to
screen for Wilm tumor, are advocated. A series comparing a screened
population (ultrasounds every 4 months) with a population that was not
screened showed that none of the children in the screened group
presented with late-stage Wilm tumor, whereas one-half of the children
who developed Wilm tumor in the nonscreened group presented with
late-stage disease. This study suggests that screening every 4 months
will identify early disease. However, a larger study is needed to
determine whether screening improves patient survival (148,149).
may be insignificant morphogenic variations, such as 13 ribs. It is
managed in the same way as any idiopathic curve. Other orthopaedic
findings include cavus feet, dislocated radial heads, and occasional
cases of polydactyly (150,151).
clinically as a short child with body asymmetry and a characteristic
facial shape (152,153,154) (Fig. 9.15).
The diagnostic characteristics include (i) a birth weight less than or
equal to two standard deviations below the mean, (ii) poor postnatal
growth, less than or equal to two standard deviations from the mean at
diagnosis, (iii) preservation of occipitofrontal head circumference,
(iv) classic facial features, and (v) asymmetric growth (155).
Poor feeding is also a common occurrence. The cause of the disorder is
unclear; although some cases are associated with uniparental disomy,
there is a suggestion of autosomal dominant inheritance, and there is
some evidence implicating an abnormal intrauterine environment (153,154).
The associated genitourinary malformations and the variation in the
pattern of sexual maturation chemically (increased gonadotropin
secretion) or clinically (precocious sexual development) suggest that
hypothalamic or other endocrine disturbances may contribute to the
pathogenesis. Affected children are small at birth and remain below the
third percentile throughout growth, with a marked delay in skeletal
maturation. Body asymmetry with hemihypertrophy affects 80% of them.
The asymmetry averages approximately 2 cm at maturity, but can be as
much as 6 cm. Regardless of the magnitude of the discrepancy, it is
clinically more apparent because the child is small. The face is
characteristically triangular and seemingly too small for the cranial
vault. There have been several reports of variations in sexual
maturation pattern, and malformations of the genitourinary system.
orthopaedic findings, but it is not clear which form part of the
syndrome and which are coincidental (156,157,158,159,160).
Scoliosis is usually idiopathic. Hand and foot abnormalities include
clinodactyly, polydactyly, and hallux varus. Developmental hip
dysplasia, avascular necrosis of the femoral head, and slipped capital
femoral epiphysis may be present. Many radiographic changes, such as
the minor hand abnormalities, suggest a disturbed morphogenesis.
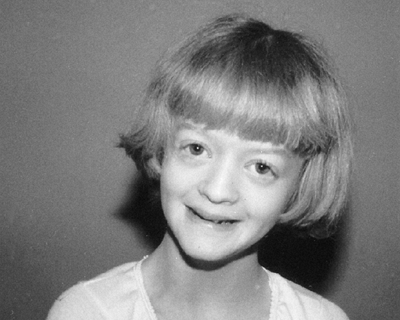 |
|
Figure 9.15 Russell-Silver syndrome. The triangular face is seemingly small for the size of the skull.
|
can be difficult because individual growth curves may vary, the
skeletal age is very retarded, and puberty may be very abnormal. It is
easy to miss the appropriate timing for epiphysiodesis. Growth hormone
has been administered in an attempt to improve stature. Although the
use of growth hormone will increase growth velocity, it is not yet
known whether the ultimate height is increased (161).
and 17, but most patients have anomalies involving chromosome 7.
However, no single causative gene has yet been identified. It is not
known whether screening for Wilm tumor, as is performed in other forms
of hemihypertrophy, is necessary. Despite early evidence that the
insulinlike growth factor receptor, which plays a causative role in
Wilm tumor, is involved in this syndrome, more comprehensive molecular
genetic investigations have not found any abnormalities in this gene.
However, there is a case report of Wilm tumor developing in an affected
patient (162), leading some to recommend screening for Wilm tumor in these patients as one would in any other hemihypertrophy.
there is a bizarre array of abnormalities that include hemihypertrophy,
macrodactyly, and partial giantism of the hands or feet, or both. The
key to this diagnosis is worsening of existing symptoms and the
appearance of new ones over time. Unlike in other overgrowth syndromes,
an increased incidence of malignancy has not been reported in Proteus
syndrome (163,164,165,166,167).
are case reports of familial occurrence, the vast majority of cases are
sporadic (168,169,170).
It is most likely due to a gene that is mutated in a mosaic manner
(mutated in the affected tissues but not in the normal tissues),
similar to McCune-Albright syndrome (polyostotic fibrous dysplasia).
Such a mutation can occur very early in development in a single cell,
which will divide to ultimately form various structures throughout the
body.
demigod who could change appearance and assume different shapes. The
progressive nature of the deformities seen in this syndrome can lead to
grotesque overgrowth, facial disfigurement, angular malformation, and
severe scoliosis (171). Joseph Merrick, called the Elephant Man, is now believed to have had this syndrome rather than NF (172).
hamartomatous overgrowth conditions, such as idiopathic
hemihypertrophy, Klippel-Trenaunay syndrome, Maffucci syndrome, and NF.
However, unlike these other syndromes, the features here are more
grotesque and involve multiple tissue types
and sites. Proteus can be differentiated from NF1 by the lack of café-au-lait spots and Lisch nodules (173).
A rating scale, which assigns points on the basis of clinical findings
(macrodactyly, hemihypertrophy, thickening of the skin, lipomas,
subcutaneous tumors, verrucae, epidermal nevus, and macrocephaly) may
be used to assist in diagnosis (174). However, the finding of worsening overgrowth features over time is usually sufficient to make this diagnosis.
it as part of Proteus syndrome. In these sporadic cases, an isolated
digit is involved or, when multiple digits are involved, these are
located adjacent to each other. Macrodactyly affecting nonadjacent toes
or fingers or opposite extremities is almost always due to Proteus
syndrome. There is a characteristic thickening and deep furrowing of
the skin on the palms of the hands and soles of the feet. The array of
cutaneous manifestations includes hemangiomas and pigmented nevi of
various intensities, and subcutaneous lipomas (Fig. 9.16).
Varicosities are present, although true arteriovenous malformations are
rare. There are cranial hyperostoses, and occasionally exostosis of the
hands and feet.
terminal branches of a peripheral sensory nerve. Digital involvement in
the hand favors the sensory distribution of the median nerve (1).
The index is the most frequently affected finger, followed by the long
finger and the thumb. It is the second toe that is most commonly
macrodactylous. The regional sensory nerve is greatly increased in
size, taking a tortuous route through the fatty tissue.
Angular malformations of the lower extremities, especially genu valgum,
are common. Because the genu valgum is often associated with restricted
range of motion, flexion contractures, and pain in the joints, it is
postulated that an intraarticular growth disturbance contributes to the
angular malformation. Hip abnormalities that show up in
roentgenographic tests, acetabular dysplasia for example, are
frequently discovered in asymptomatic patients. Deformities in the
hindfoot are frequent and are usually heel valgus, but congenital
equinovarus and “Z-foot” deformities have also been described (173,176).
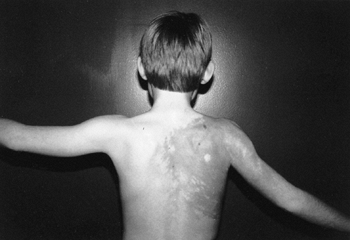 |
|
Figure 9.16
Proteus syndrome. Notice the cutaneous markings, large hemangioma of the shoulder, and lightly pigmented area on the back. There is some atrophy of the shoulder and arm muscles and a fixed contracture of the elbow. |
case reports or case series, and there are no data comparing results of
different types of treatments. In addition, recurrences after surgical
intervention are very common. This is probably due to an underlying
growth advantage in affected tissues that cannot be corrected
operatively. Thus, musculoskeletal deformities caused by Proteus
syndrome are very difficult to manage.
because of macrodactyly, it is best managed by ablation rather than
debulking (178). Anisomelia is best managed
with epiphysiodesis. Osteotomies can correct angular malformations, but
the decision to undertake surgical correction must take into account
the possibility of a rapid recurrence of the deformity after corrective
surgery (175,176). In
some cases, a sudden overgrowth of the operative limb has been
reported. There are anecdotal reports of soft tissue procedures to
“debulk” overgrown lesions; however, there are no series in the
literature reporting results of these procedures, and our experience
with them is that the results are only temporary. In rare cases, nerve
or spinal cord impingement can occur. Nerve compression can be managed
using decompression, but spinal cord compression
is difficult, if not impossible, to successfully treat operatively (179,180). Scoliosis can occur and seems to be caused by overgrowth of one side of the spine (181).
Mixed results are obtained from surgical treatment in this disorder,
and operative treatment should be reserved for individuals who have
exhausted nonsurgical management. Sometimes the operative procedures
can be used as a temporizing measure, and patients may need to have
repeat procedures performed throughout life.
The life expectancy is unknown, but many adult patients have been
reported. Intubations can be difficult because of overgrowth of
structures surrounding the trachea.
activated in a coordinated manner to cause cells to proliferate, move,
and undergo programmed cell death, so as to allow the organism to
pattern normally and develop into an adult. Normal patterning is
altered by mutations in the genes that encode proteins that play roles
in these pathways. Environmental events such as exposure to a teratogen
can also dysregulate these same pathways, resulting in a phenotype
similar to that of a gene mutation. Such events occurring in a pathway
that is important for skeletal development can result in a
musculoskeletal malformation. These disorders can be identified at
birth, because the problem is present at the start of development.
Despite this, sometimes the abnormalities do not become obvious to
parents or physicians until the child is older. Because these are
generally patterning problems, surgery to correct malalignment is
usually quite successful. There are frequently manifestations in other
organ systems, because the same developmental signaling pathways play
important roles in the development of multiple organs. These disorders
are not associated with an increased rate of neoplasia. Symptoms from
the malformations often increase with age because the abnormally shaped
structures cannot sustain the stresses of normal activity. This results
in the early development of degenerative problems. These disorders are
usually inherited in an autosomal dominant manner, although the
inheritance pattern is more variable than in disorders caused by genes
encoding for structural proteins or for proteins implicated in
neoplasia.
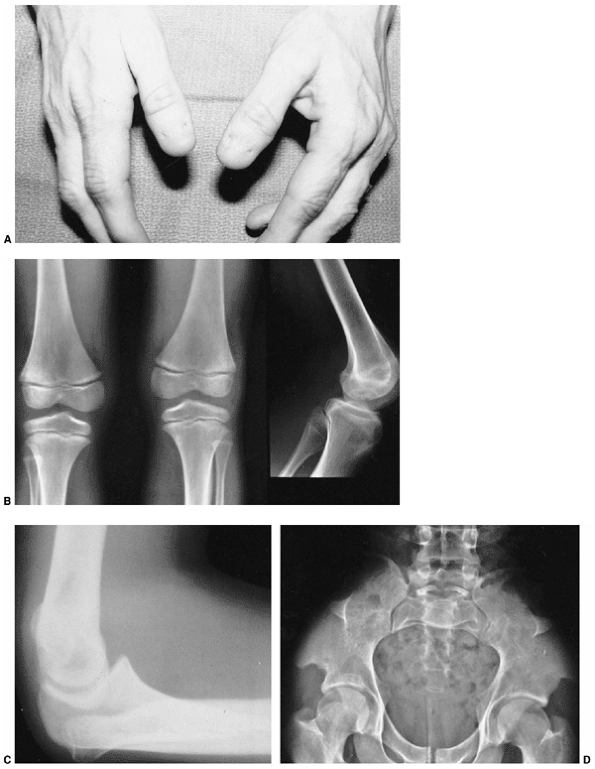
findings that include nail dysplasia, patellar hypoplasia, elbow
dysplasia, and iliac horns (183). The most prominent feature is dystrophic nails (Fig. 9.17A). The nail may be completely absent, hypoplastic, or have grooves and distortions in its surface (184).
The thumb is more involved than the small finger, and the ulnar border
more involved than the radial. The hands are often very symmetric, and
fingernails are more involved than toenails.
Where present, they are unstable, and may be found in a position of
fixed dislocation. The patellar abnormality highlights the total knee
dysplasia, with an abnormal femoral condyle and a peculiar septum
running from the patella to the intercondylar groove (septum
interarticularis), dividing the knee into two compartments.
Abnormalities in varus and valgus alignment occur, with valgus more
common because of the small, flat lateral femoral condyle (185).
The elbow joint is dysplastic, with abnormalities in the lateral
humeral condyle, in many ways mimicking the dysplasia of the knee. The
trochlea is large and the capitellum is hypoplastic, creating an
asymmetric shape that may predispose the radial head to dislocation.
gene. This gene is a homeodomain protein, which plays a role regulating
transcription in limb patterning during fetal development. Mutation in
the gene will disrupt normal limb patterning and alter kidney
formation, resulting in deformities in the extremities and an
associated nephropathy (188).
height being between the third and tenth percentiles. There may be a
shoulder girdle dysplasia, and a variety of abnormalities of the
glenoid and humeral head are possible. These, however, merely represent
curious radiographic features and not any significant functional
disability (189). There is a foot deformity that is sometimes the chief presenting complaint of children with nail-patella syndrome (185,190). The foot deformities include variations of stiff calcaneal valgus, metatarsus adductus, and clubfeet.
affect several large joints; these include knee-flexion deformities and
external rotation contracture of the hip. When these contractures are
severe and accompanied by stiff clubfeet, the condition may be
misdiagnosed as arthrogryposis multiplex congenita. Madelung deformity,
spondylolysis, and in some adults, inflammatory arthropathy may be
present (183,191,192).
of quadriceps dysfunction and the dislocated patella. At long-term
follow-up, knee pain is the main musculoskeletal complaint in patients
with nail-patella syndrome (193). Small femoral
condyles make it difficult to achieve patellar stability. As a rule,
limited soft tissue or capsular releases are
ineffective, but combined proximal and distal patella realignments have an overall favorable outcome (185,194).
A contracted and fibrotic quadriceps may result in a knee extension
contracture, and in such cases quadricepsplasty is indicated along with
the patella realignment. More commonly, an associated knee-flexion
deformity may require hamstring release and posterior capsulotomy,
although results have been inconsistent (185).
Residual deformity, which is usually related to flexion or rotation, is
managed by femoral osteotomy towards the end of the first decade of
life. Osteochondritis dissecans of the femoral condyle is relatively
common (Fig. 9.17B). An intraarticular septum makes arthroscopic management difficult, but the septum can be removed arthroscopically.
 |
|
Figure 9.17 Nail-patella syndrome. The classic quartet of features consists of dystrophic nails (A), absent patellae (notice the region of osteochondritis dissecans on the lateral film) (B), posterior dislocation of the radial head (C), and iliac horns (D).
|
children, but may become symptomatic with time. In symptomatic
individuals, excision of the radial head will improve symptoms arising
from the prominent lateral bump, but the range of motion is rarely
improved. Although traditional teaching advocates performing radial
head excision after skeletal maturity, earlier excision in symptomatic
children does not seem to be associated with significant problems (185). Dislocated hips (195) and clubfeet can occur, and can be managed using techniques similar to those in idiopathic cases.
failure. The nephropathy of nail-patella syndrome causes significant
morbidity, affecting the patient’s longevity. There is great
variability in the age at onset and severity of the nephropathy (196).
All patients should be referred for a nephrology evaluation when this
diagnosis is made. Patients may go on to chronic renal failure,
requiring long-term nephrology management.
It is not a rare syndrome, with an estimated incidence of 1 in 5,600
births. The cause is unknown, but marked geographic variation and
segregation analysis suggests a genetic disorder (200).
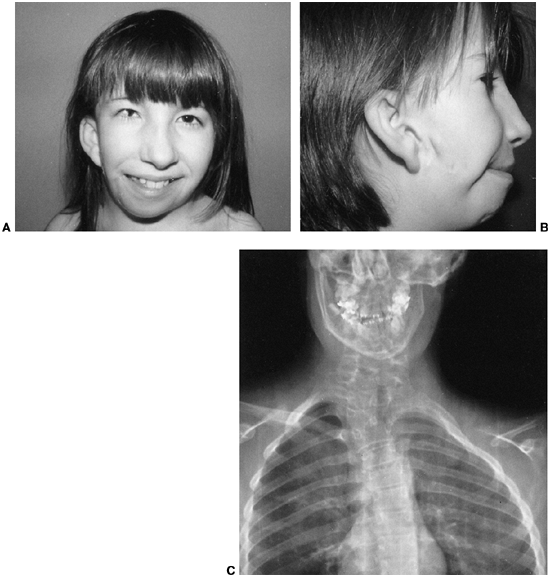
In some patients, the ear may be hypoplastic or absent. The eye and ear
anomalies are unilateral in 85% of these children, and facial asymmetry
is the result of a hypoplastic mandibular ramus, invariably on the same
side as the ear anomalies (Fig. 9.18C).
in Goldenhar syndrome, although the lower cervical and upper thoracic
locations predominate (Fig. 9.18C).
Hemivertebrae are the most common defect, with an occasional block
fusion. Neural tube defect occurs more often than in the general
population, and it may involve lumbar spine, cervical spine, or the
skull (i.e., encephalocele). Approximately one-half of the patients
have clinically detectable scoliosis (201). An
idiopathic, compensatory curve below the congenital curve is often more
troublesome than the congenital curve itself. Sprengel deformity and
rib anomalies may be present in association with the congenital curves
in the cervical–thoracic region. Orthotic management of scoliosis is
difficult, and has no effect on the congenital portion of the curve.
The location of the scoliosis is often too high for brace management.
Early fusion should be performed when there is progression of the
congenital curve. Preoperative CT scan and MRI are recommended to
delineate the anatomy of the congenital curve and determine whether
there is any intraspinal pathology. There may be occult posterior
element defects that will also be identified on CT scan.
Mental retardation, affecting 10% to 25% of patients, is usually
limited to cases involving microphthalmia or an encephalocele (204).
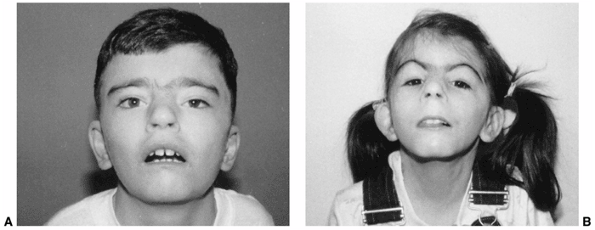
growth retardation makes the clinical diagnosis of Cornelia de Lange
syndrome reasonably reliable (205). The face
has immediately recognizable down-turned corners of the mouth, eyebrows
meeting in the midline (synophrys), elongated philtrum, and long
eyelashes (206,207) (Fig. 9.19).
Notch plays a major role in central nervous system development, hence
the associated mental retardation. Duplication or deletion of the
chromosome band 3q25-29 produces a phenotype similar to Cornelia de
Lange syndrome (210,211).
In these instances, the mother is always the transmitting parent,
suggesting genomic imprinting. The syndrome is relatively common,
occurring in 1 in 10,000 live births, and it is possible to make a
prenatal diagnosis by ultrasound (158,159,212,213).
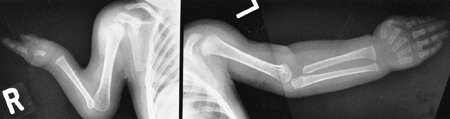
They form a curious constellation of a small hand, a proximally placed
thumb, clinodactyly of the small finger, and decreased elbow motion,
usually caused by a dislocated radial head. This combination rarely
causes any disability. Some patients, however, have severe deformities
of the upper extremity in the form of an absent ulna and a monodigital
hand, a condition that can be unilateral or bilateral (Fig. 9.20).
 |
|
Figure 9.18 Goldenhar syndrome. A: Facial asymmetry and epibulbar dermoid of the right eye. B: Malformed ears with preauricular tags and sinuses. C:
x-ray film demonstrates the congenital anomalies of the lower cervical and upper thoracic spine. Hypoplasia of the ascending ramus of the mandible accounts for the facial asymmetry. The clavicle is absent on the same side as the deformation of the face. (From Goldberg MJ. The dysmorphic child: an orthopedic perspective. New York: Raven Press, 1987, with permission.) |
 |
|
Figure 9.19
Cornelia de Lange syndrome. Notice the classic facial features of heavy eyebrows meeting in the midline, upturned nose, downturned corners of the mouth, and long eyelashes in a 13-year-old boy (A) and a 7-year-old girl (B). (From Goldberg MJ. The dysmorphic child: an orthopedic perspective. New York: Raven Press, 1987, with permission.) |
 |
|
Figure 9.20
Cornelia de Lange syndrome: a child with a severely affected upper extremity on her right side (i.e., absent ulna and fingers) and a mildly affected arm on her left (i.e., short thumb and dysplasia of proximal radius). (From Goldberg MJ. The dysmorphic child: an orthopedic perspective. New York: Raven Press, 1987, with permission.) |
cords and other cerebral palsy-like contractures are seen occasionally.
These can be managed similarly to cerebral palsy, but there seems to be
a higher rate of recurrence (220). Syndactyly
of the toes is fairly constant. Aplasia of the tibia has been reported
rarely. There is possibly a higher incidence of Legg-Perthes disease,
approaching about 10%. Scoliosis can occur, and should be managed
similarly to scoliosis in cerebral palsy. Most of the skeletal
deformities in Cornelia de Lange syndrome are asymptomatic, and
probably do not benefit from surgical intervention (220).
retardation. Children remain small, with a delayed skeletal age. The
mortality rate in the first year of life is high because of defective
swallowing mechanisms (221), gastroesophageal reflux (222),
aspiration, and respiratory infections. If the children survive their
first year, they usually do well, but the long-term outcome is unclear.
Almost all of them walk, but their milestones are delayed. There is
retarded mentation, but the added features of no speech and no
interactions cause major disability (223). Self-mutilating behavior can be an obstacle to orthopaedic care (224,225).
develop speech, and paucity of social interactions raises questions
about the suitability of these patients for some types of orthopaedic
treatment. Braces, physical therapy, and surgery for tight heel cords
are justifiable. Upper-extremity surgery is not indicated unless
improved performance capacity is ensured. Patients with Cornelia de
Lange syndrome do not use upper-extremity prostheses. Lower-extremity
prostheses, however, should be prescribed for the rare case with tibial
deficiency. Because the gastroesophageal reflux and swallowing
disorders may persist well past the first year, there is a higher risk
of anesthesia complications (226).
can share similarities with conditions caused by genes that encode
proteins that are important in normal development. Many teratogenic
agents modulate the same pathways that are dysregulated by the
mutations that cause such syndromes. A good example of this is
holoprosencephaly, a midbrain patterning disorder. This can be caused
by mutations in a gene called sonic hedgehog,
and can also be caused by teratogenic agents that block the hedgehog
signaling pathway, such as derivatives found in the plant Veraculum californium (227,228).
delineated in children of alcoholic mothers. The full-blown syndrome is
usually seen only in children of chronic alcoholics who drink
throughout pregnancy. Lesser manifestations of the syndrome, known as
fetal alcohol effects, may be related to more moderate alcohol
ingestion (229). Although the risk to alcoholic
mothers is known, there is substantial difference of opinion about the
effects of moderate alcohol use during pregnancy (230,231,232). Alcohol is the most likely teratogen for a mother to encounter (233). The overall incidence of full-blown fetal alcohol syndrome is 0.33 per 1000 live births (234,235). For an alcoholic mother, there is a 30% risk for fetal alcohol syndrome in her child.
children have intrauterine growth retardation, small weight, and small
length at birth, and these limitations remain despite good nutrition
during childhood (236,237) (Fig. 9.21).
Their smallness and a loss of fat suggest a search for endocrine
dysfunction; the patients often look similar to those who are deficient
in growth hormone. The second cardinal feature is disturbed central
nervous system development. Many children with fetal alcohol syndrome
are found in cerebral palsy clinics. The typical child has a small
head, a small brain, and delayed motor milestones. Accomplishing fine
motor skills is also delayed. Hypotonia is present early but many
develop
spasticity
later. The typical face has three characteristic features: short
palpebral fissures (i.e., the eyes appear small), a flat philtrum
(i.e., no groove below the nose), and a thin upper lip (238,239) (Fig. 9.21).
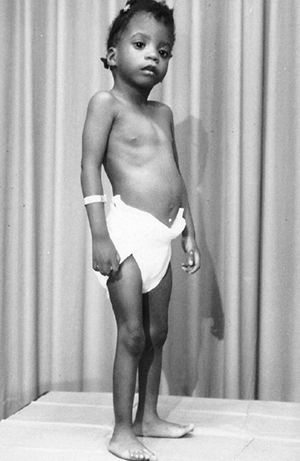 |
|
Figure 9.21 The 3-year-old patient is small and has the characteristic face of fetal alcohol syndrome. (From Goldberg MJ. The dysmorphic child: an orthopedic perspective. New York: Raven Press, 1987, with permission.)
|
At birth, the range of motion is restricted, especially of the hands
and feet, and occasionally these contractures are fixed. The
contractures typically respond well to physical therapy, although
residual stiffness in the proximal interphalangeal joints may remain.
Clubfoot is common, and approximately 10% of these patients have
developmental dysplasia of the hip. The clubfoot is usually not rigid (243). Cervical spine fusions, usually involving C2 and C3, may be seen on radiographs (242,244,245,246,247,248).
These may resemble the picture seen in Klippel-Feil syndrome, but there
are usually none of the other findings associated with that syndrome.
Synostoses are also common in the upper extremity, with fusions
involving the radial-ulnar articulation and the carpal bones, all
without any resultant disability (244,247,249). Stippled epiphyses may be seen in the lower extremities, but rarely in the upper extremities (250).
syndrome can be managed in the same way as in children without this
syndrome. The future for children with fetal alcohol syndrome is dim,
despite placement away from the alcoholic home. Intellect remains
retarded, with little catch up. Social services departments should be
involved in these children’s care.
The letters of VACTERLS in this syndrome’s name constitute an acronym
for the systems and defects involved: vertebral, anal, cardiac,
tracheal, esophageal, renal, limb, and single umbilical artery. The
physician does not need to find examples of all seven categories of
anomalies in order to diagnose the syndrome. The syndrome can be
diagnosed prenatally by visualizing several of the malformations on
ultrasound. The most obvious physical finding at birth is the radial
ray defect. Between 5% and 10% of radial club hands are associated with
VACTERLS.
The current thought is that these structures are either all formed at
the same time, or are all patterned by the same developmental signaling
pathway. An event occurring during fetal development that disrupts
either the common signaling pathway, or any of a variety of susceptible
pathways operating at the same time, is probably responsible for the
associated malformations.
Thoracic anomalies are worse in those with tracheoesophageal fistula,
and lumbar anomalies are more common in those who have an imperforate
anus. Occult intraspinal pathology is common (254,255),
and a screening MR study of the spine is recommended, especially in
patients who require operative management of their scoliosis. The
curves can be managed like other types of congenital scoliosis.
these patients. A ventricular septal defect is the most common problem.
Duodenal atresia may be found in this syndrome. The VACTERLS patient
often has a single kidney. Other collecting-system anomalies occur
frequently among this group.
radial club hand. The defect may be unilateral or bilateral; bilateral
defects are always asymmetric (253). The legs are spared 80% of the time. When the lower extremities are involved, a duplicated hallux is the most common finding.
The absence of an artery, detectable only at the time of delivery or in
the immediate newborn period, reflects the broad range of morphologic
defects dating back to placental formation.
be the consequence of skeletal anomalies of the arms, scoliosis, and
surgery for gastrointestinal or genitourinary malformations.
Nevertheless, several central nervous system malformations (e.g.,
encephalocele hydrocephalus) may be associated with VACTERLS, and must
be excluded (255,256).
If the patient survives the gastrointestinal anomalies and correction
of the cardiac defects, the prognosis for a normal life is excellent.
Each orthopaedic abnormality can be treated as an isolated problem. The
sections in this chapter that deal with congenital scoliosis and radial
clubhand contain detailed information. The key point is to recognize
this association and to identify other abnormalities that might
interfere with treatment.
can be caused by genes that encode for proteins that are important for
nerve or muscle function. The course of these disorders is variable;
however, several show progressive weakening effects over time. Various
inheritance patterns are possible, but frequently these disorders are
linked to the X chromosome. Duchenne muscular dystrophy and Rett
syndrome are two such disorders that are inherited in an X-linked
recessive manner and X-linked dominant manner, respectively.
is an autosomal recessive disorder occurring primarily in Jews who
trace their ancestry to Eastern Europe. Among such individuals, the
incidence is estimated to be about 1 in 3700. The clinical
manifestations are caused by defective functioning of the autonomic
nervous system and sensory system. The autonomic dysfunction causes
labile blood pressure, dysphagia, abnormal temperature control, and
abnormal gastrointestinal motility. Infants have difficulty swallowing,
with misdirected fluids going to the lungs, resulting in pneumonia.
There is a poor suck response and a curious absence of tears. During
childhood, the autonomic dysfunction becomes more apparent, with wide
swings in blood pressure and body temperature. There are cyclic
vomiting episodes; these crises often last hours or days. Swallowing
remains poor. The skin is blotchy. There is relative insensitivity to
pain, and poor hot-cold distinction. Intelligence is normal, but the
children exhibit emotional liability, and may have unusual personality
development, especially in the teenage years. The diagnosis is made on
clinical findings and on the basis of the presence of five signs: (a)
lack of axon flare after intradermal injection of histamine, (b)
absence of fungiform papillae on the tongue, (c) miosis of the pupil
after conjunctival installation of methacholine chloride, (d) absence
of deep tendon reflexes, and (e) diminished tear flow (257,258,259).
of kappa light polypeptide gene enhancer in B cells. The protein
product of this gene plays a role in the phosphorylation of other
signaling proteins, but the mutant form is expressed only in select
tissue types, primarily affecting cells in the autonomic nervous system
(260,261). Since the
mutation is expressed only in certain tissue types, one approach to
treatment would be to change the tissue-specific expression of the
mutant form by using drugs that regulate the expression of only the
mutant variant. Such a potential treatment has been proposed using
tocotrienols, which are members of the vitamin E family (262,263).
Pathologic anatomy reveals a paucity of neurons in cervical sympathetic
ganglia, dorsal sensory roots, and abdominal parasympathetic nerves (264).
A number of small axons are depleted from the sensory nerves and the
dorsal columns. Because of a primary failure to develop axons, the
symptoms are present at birth, and there is a loss of nerve cells and
progression of symptoms as the patient grows older.
fracture susceptibility, avascular necrosis, and a Charcot joint-like
process. Scoliosis affects a majority of patients, and approximately
one-fourth will need operative intervention (265,266,267,268,269,270,271).
It has an early onset, and progression is often rapid. Kyphosis,
accentuated by tight anterior pectoralis muscles, appears in
approximately one-half of the patients. Bracing does not work well,
because of the underlying gastrointestinal and emotional problems.
Anesthesia can be challenging in individuals with such autonomic
liability, but with proper techniques, operative intervention is
successful. Surgery seems to give better results if performed early in
the course of the disease (268,269,270).
The physician should be suspicious of occult fractures in patients who
have had trauma and swelling but experience minimal tenderness.
Fractures usually heal quite well, but early diagnosis and avoiding
displacement is the goal.
There are Legg-Perthes changes in the hips. Osteochondritis dissecans
of the knees is often extensive, involving both femoral condyles (Fig. 9.22).
It may be difficult to determine whether the ossification changes in
the knee are because of osteochondritis dissecans or the early stage of
Charcot joint (274,275). Hip dysplasia may be seen in patients with this syndrome.
characterized by a relatively high mortality rate in infancy,
attributed to aspiration pneumonia (259). Sudden death in
childhood and adolescence occurs because the child is unable to respond
appropriately to stress or hypoxia. Early recognition of this syndrome
and appropriate care lead to a life expectancy of many decades.
Management of the gastrointestinal problems and the use of gastrostomy
and fundoplications have been extremely successful in such patients.
There have been successful pregnancies brought to term in mothers with
the syndrome (276,277).
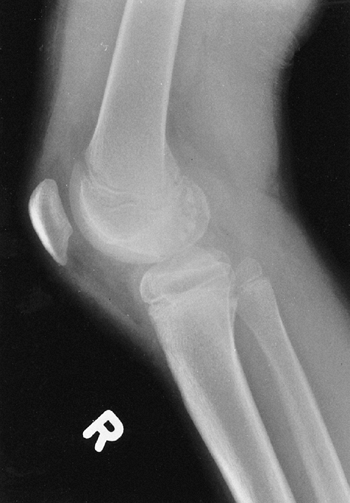 |
|
Figure 9.22 Familial dysautonomia. Irregular ossifications of the distal femoral epiphysis mimic osteochondritis dissecans.
|
present almost exclusively in girls, and is characterized by normal
development for the first 6 to 18 months, followed by rapid
deterioration of higher brain functions. This is accompanied by
dementia, autism, loss of purposeful use of the hands, and ataxia.
After the initial rapid decline, the deterioration slows dramatically,
so that affected individuals may have a relatively stable picture for
several decades (278). There is variability in
the severity of the decline, so that some girls are still walking as
teenagers, whereas others stop ambulating in early childhood (279).
A hand radiograph may help with the diagnosis, because 60% will have
either negative ulnar variance or a short fourth metacarpal (280,281).
have cerebral palsy with a movement disorder. Andreas Rett, a
pediatrician practicing in Austria, noted that these girls all had
normal development in the first month of life, and was thus able to
separate them from those with cerebral palsy. It occurs with an
incidence of 1 in 40,000. In some patients, it is caused by a mutation
in the MECP2 gene, which encodes X-linked
methyl-CpG-binding protein 2. This protein plays an important role in
regulating gene expression during development, especially in the
central nervous system (282). X-linked dominant
diseases are more severe in boys, and Rett is probably fatal in the
vast majority of male patients, though few such cases have been
reported (283). Genetic testing and prenatal diagnosis (284)
are possible, but as in other syndromes, a careful physical examination
and history can be used to make the diagnosis in most cases (285).
with a clinical picture similar to that of a cerebral palsy patient
with total body involvement. Scoliosis occurs in over half the girls
who are affected with this disorder (283,286,287,288,289).
Orthotic management probably does not alter the progression of the
curve. There is a typical, usually long “c” pattern to the curves.
These can be stabilized surgically when they reach a magnitude that
interferes with sitting or balance. Although case series suggest an
improvement after surgery (285), as is seen in
cerebral palsy, there are no comparative studies showing improved
function after spinal surgery. Spinal instrumentation and fusion should
include the whole curve and any kyphotic segments. Although,
theoretically, walking ability can worsen following extensive fusions,
this has not been reported in the small number of cases in which spinal
surgery was undertaken in ambulatory girls with Rett syndrome (282,285,286).
Coxa valga and lower-extremity contractures can occur, and these should
be managed as in cerebral palsy, with emphasis placed on operative
procedures that will improve function or decrease pain (287,289,290).
are some affected individuals with a normal life span. There are a
variety of nonorthopaedic problems, including cardiac conduction
abnormalities, epilepsy, and vasomotor instability of the lower limbs.
Some of these put the patients at increased risk when undergoing
anesthesia (278). Interestingly, there is a high incidence of left handedness (approximately 40%) in girls with Rett syndrome (291).
and multiple genes are affected. There can be deletions, duplications,
or translocations. Large abnormalities in chromosomes are almost always
associated with some
degree
of mental deficiency. Because there is duplication of multiple genes,
there are multiple abnormalities in multiple organ systems. Since
multiple genes are abnormal in all cells, normal cell functions (such
as the ability to mount an immune response or normal wound healing)
also may be abnormal. Except for rare instances, these disorders are
not inherited, and occur as sporadic events.
Patients have a characteristic facial appearance including
upward-slanting eyes, epicanthal folds, and a flattened profile.
Examination of the hands reveals a single flexion crease, often
referred to as a simian crease. There is also clinodactyly of the small
finger. These hand malformations have no clinical significance (293).
Milestones are delayed, with most children not walking until 2 to 3
years of age. The classic gait pattern is broad based, toed out, and
waddling.
The best-studied changes are in the pelvis, which is characterized by
flat acetabula and flared iliac wings (294).
These pelvic changes are so characteristic that prior to use of
chromosome analysis, pelvic radiographs were used for confirming the
diagnosis. Since the advent of chromosomal diagnosis, routine pelvic
radiographs are not needed. Short stature is a cardinal feature; the
average for men is 155 cm (61 in.), and the average for women is 145 cm
(57 in.) (295). The detection of bone changes
can be useful in prenatal diagnosis. A combination of bone length and
lab tests on the mother (human chorionic gonadotropin and
alpha-fetoprotein levels) may predict the diagnosis, although the
positive and negative predictive values are not as good as had been
initially hoped (296). Cytogenetic study, which identifies complete trisomy 21 in 95% of the cases, remains the best confirmative test.
 |
|
Figure 9.23
Down syndrome. The child has the characteristic face, with upward-slanting eyes, epicanthal folds, open mouth of early childhood, and flattened profile. A: At 1 year of age. B: At 10 years of age. (A, Courtesy of Murray Feingold, MD, Boston, Massachusetts. B, From Goldberg MJ. The dysmorphic child: an orthopedic perspective. New York: Raven Press, 1987, with permission.) |
mosaics and 3% translocations. The overall occurrence is 1 per 660 live
births, and the incidence is closely related to maternal age. If the
mother is younger than 30 years of age, the risk is 1 of 5000 live
births, and if the mother is older than 35 years of age, the incidence
rises to 1 in 250. The critical region for Down syndrome resides in
part of the long arm of chromosome 21. Duplication of a 5-megabase
region of chromosome 21 (located at 21q22.2–22.3) causes the classic
phenotypic features, such as the characteristic facies, hand anomalies,
congenital heart disease, and some aspects of mental retardation (297). This region probably contains a number of genes whose duplication is necessary to produce the syndrome.
There is a characteristic flattened face. Mental retardation is
typical, but performance is far better than expected from standard IQ
testing. Congenital heart disease occurs in about 50% of patients, and
is usually a septal defect (e.g., arteriovenous communis, ventricular
septal defect). Duodenal atresia is found regularly. Leukemia occurs in
about 1% of this population (1,5).
There is a high incidence of endocrinopathies, hypothyroidism in
particular. Infections are common, although the precise molecular
mechanism is not apparent. The appearance of premature aging is
obvious, and there is often an early onset of Alzheimer disease (298).
interval is not associated with symptoms (300,304).
In addition, there is a broad array of other abnormalities in the upper
cervical spine, including instability at occiput and C1 (303,305,306,307), odontoid dysplasia (300,308) (Fig. 9.24C), laminal defects at C1 (309) (Fig. 9.24B), spondylolisthesis (Fig. 9.24D), and precocious arthritis in the midcervical region (310,311) (Fig. 9.24E).
These other abnormalities often complicate decision making about spinal
instability. Although routine screening radiographs often disclose
these cervical spine abnormalities, radiographs are not reliable in
predicting myelopathy (302,304,308,312,313,314,315).
Therefore, their efficacy in the management of the cervical spine in
patients with Down syndrome is uncertain. The management of cervical
instability in Down syndrome is discussed elsewhere in this text.
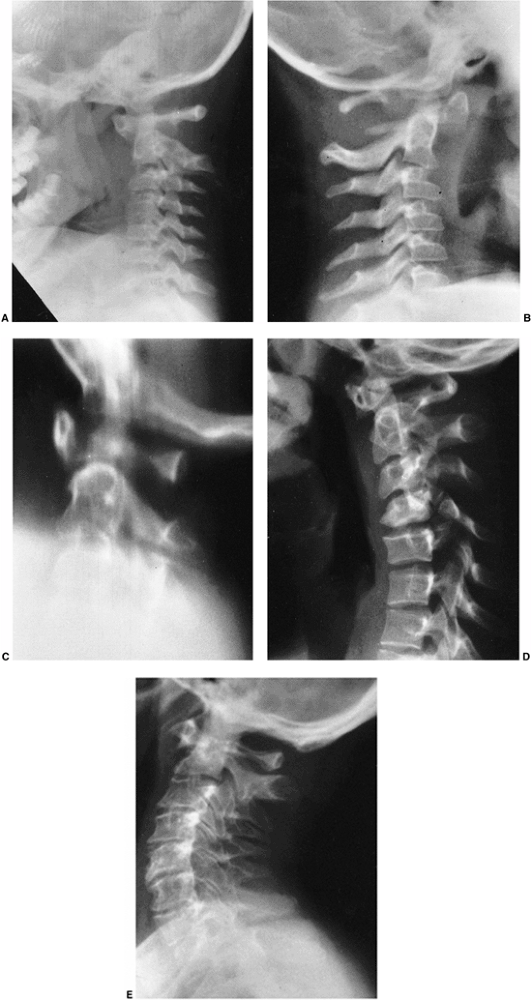 |
|
Figure 9.24 Cervical spine abnormalities in a patient with Down syndrome. A: Atlantodens instability at 8 years of age. B: Hypoplastic posterior elements of C1 at 3 years of age. C: Os odontoideum and increased atlantodens interval at 14 years of age. D: Midcervical spondylolysis at 16 years of age. E: Precocious osteoarthritis of the midcervical spine at 40 years of age. (From Goldberg MJ. The dysmorphic child: an orthopedic perspective. New York: Raven Press, 1987, with permission.)
|
Scoliosis is five times more likely to be detected in a severely
retarded, institutionalized population than in an ambulatory setting.
This finding raises the possibility that factors such as severity of
the phenotype, or other neuromuscular factors contribute to scoliosis.
Management is the same as in idiopathic scoliosis. Similar to the case
of the cervical spine, there is a higher rate of complications from
scoliosis surgery than in the general population (317).
Spondylolisthesis occurs in about 6%, with the lower lumbar spine being
most commonly involved. Spondylolisthesis can also occur in the
cervical spine.
dysplasia may begin during later childhood. This loss of acetabular
containment may lead to an acute or gradual complete dislocation (Figs. 9.25 A,B).
Although the onset of acetabular dysplasia is in late childhood, it can
be progressive even after maturity, leading to dislocations even in
adulthood (318,319,320) (Figs. 9.25 C,D).
Although hip instability and developmental dysplasia are thought to
lead to functional disability (thereby interfering with walking and
reducing independent mobility), there are no studies showing this to be
the case. The etiology of the hip instability is probably
multifactorial, with ligamentous laxity, subtle changes in the shape of
the pelvis and acetabular alignment, and behavior (some children become
habitual dislocators) all contributing. Treatment of the unstable hip
is difficult, and the multiple causative factors also contribute to
higher treatment failure rates. Both operative and nonoperative
treatment have been reported. Prolonged bracing after reduction for the
hip that dislocates acutely has shown success in children younger than
6 years (321). In cases in which there are
repeated dislocations, surgical reconstruction is warranted, especially
in children older than 6 years. Operative treatment is technically
demanding, and requires correction of all the deforming factors.
Reconstruction must take into account the abnormal bone alignment, and
should include femoral and acetabular osteotomies, as well as
imbrication of the redundant capsule. Posterior acetabular deficiency
has been reported, and was not improved following a traditional Salter
style inominate osteotomy (322); therefore,
three-dimensional imaging (such as a CT scan) should be considered
before embarking on surgery. The recurrence rate following hip surgery
is high, suggesting that other factors related to the underlying
disease, but not necessarily related to the hip anatomy itself, are
contributory (323,324,325).
There appears to be a higher-than-expected risk for avascular necrosis.
The reasons are not clear, but factors include more acute slips and
delayed diagnoses. It is tempting to speculate about an association
with the hypothyroid state, which is common in Down syndrome. All
children with Down syndrome should have thyroid function tests.
Symptomatic cases should be initially managed with orthoses and a
physiotherapy program. Individuals who continue to be symptomatic can
be considered for operative treatment. As in hip dysplasia, operative
interventions that correct all of the deformities (bone and soft
tissue) have the best success.
is one of an asymptomatic flexible planovalgus shape, with an increased
space between the great and second toes.
Because
it is important to maintain mobility in adults with Down syndrome,
symptomatic foot problems should be treated. The treatment involves
footwear modification in many cases, but may require surgery in cases
that are symptomatic despite appropriate footwear. Valgus feet with toe
deformities are most likely to become symptomatic. In many, hallux
valgus develops in adolescence, and in adulthood the bunions become
symptomatic. Orthotics will improve foot position, but may actually
slow the walking speed of children with Down syndrome (328).
For that reason, orthodics should be used only in symptomatic cases.
Repair of a hallux valgus and bunion may be needed in late adolescence
or young adulthood. Because of the hindfoot valgus, pronation, and
external tibial torsion, the forces that produce bunions are obvious,
and fusion of the first metatarsophalangeal joint should be considered,
along with osteotomy, to correct hindfoot valgus.
 |
|
Figure 9.25 Down syndrome patient with late-onset developmental dysplasia of the hip and dislocation. A: Pelvicradiograph taken in standing position, at 6.5 years of age. B: At 9.5 years of age, the patient suddenly refused to walk because of hip dislocation. C: Pelvic radiograph of a 31-year-old man with Down syndrome. D: Three years later, dislocation of right hip occurred. (A and B, from Goldberg MJ. The dysmorphic child: an orthopedic perspective. New York: Raven Press, 1987; C and D, from Pueschel SM. Should children with Down syndrome be screened for atlantoaxial instability? Arch Pediatr Adolesc Med 1998;152:123, with permission.)
|
Whether this is true juvenile rheumatoid arthritis or a unique
inflammatory arthritis due to genetic or immune defects is unknown; the
natural history is not documented. Delayed diagnosis is common.
Nonsteroidal antiinflammatory drugs have been the mainstay of
treatment. Foot symptoms are exceptionally frequent with the onset of
polyarthropathy (Fig. 9.28).
able to assume the most intriguing sitting postures. Ligamentous laxity
was traditionally thought to be the cause of joint hypermobility, and
it was assumed that it
predisposes
patients with Down syndrome to orthopaedic pathology. However,
ligamentous laxity correlates poorly with joint hypermobility. This
suggests that other factors, such as subtle malformations in the shapes
of bones and insertion sites of ligaments, play a role in hypermobility
(312,332).
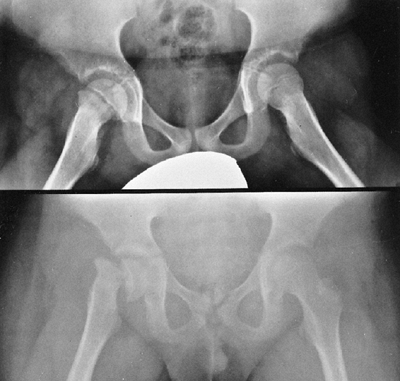 |
|
Figure 9.26
Effects of Down syndrome in a 12-year-old boy with 4 months of knee pain. The grade I slipped capital femoral epiphysis progressed to a total slip while the patient was undergoing preoperative evaluation and bed rest. (From Goldberg MJ. The dysmorphic child: an orthopedic perspective. New York: Raven Press, 1987, with permission.) |
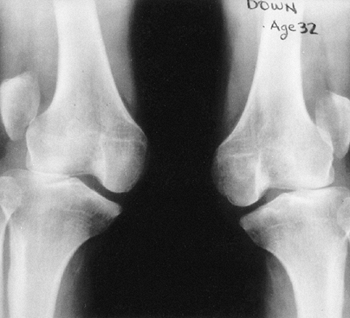 |
|
Figure 9.27
Effects of Down syndrome in a 32-year-old patient. The radiograph shows bilateral dislocated patellae and an oblique orientation of the joint line. The patient is fully ambulatory, but before standing must manually reduce the patellae to the midline. (From Goldberg MJ. The dysmorphic child: an orthopedic perspective. New York: Raven Press, 1987, with permission.) |
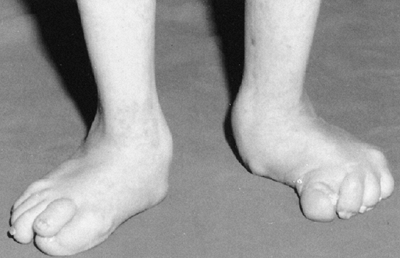 |
|
Figure 9.28 Polyarthritis of Down syndrome and valgus feet led to significant deformity in a 16-year-old patient. (From Goldberg MJ. The dysmorphic child: an orthopedic perspective. New York: Raven Press, 1987, with permission.)
|
changed in the last few decades. Longevity has increased because of the
aggressive surgical approach to congenital heart disease, chemotherapy
for leukemia, and antibiotics for infection. Survival into the sixties
is common. Approximately one of five persons with Down syndrome has
musculoskeletal abnormalities. Many of these, however, are merely
radiographic abnormalities or curious physical findings. These patients
often have excellent functional performance despite the abnormalities.
There is a paucity of well-documented, long-term orthopaedic studies of
patients with Down syndrome. Treatment programs should focus on
functional performance rather than on radiographic findings.
of short stature, sexual infantilism, a webbed neck, and cubitus
valgus. It is a relatively common chromosome disorder affecting 1 in
2500 live births, but the rate of intrauterine lethality is 95%. The
syndrome is caused by a single X chromosome. In two-thirds of cases,
all cells are XO, and parental origin of the single X chromosome is the
mother in 70% of the cases (333). XO mosaicism occurs in about one third of patients, and in 1% there is deletion of only a part of an X chromosome (333,334). Cytogenetic studies will confirm this diagnosis.
depending on whether it is derived from the father or from the mother,
and this is probably the result of imprinting (335).
Recent studies based on individuals with partial loss of the X
chromosome suggest that a critical region at Xp11.2–p22.1 is
responsible for the disease (336).
birth, the child has a webbed neck, widely spaced nipples, and edema of
the hands and feet. The foot edema may persist for several months.
During childhood, the low hairline, webbed neck, cubitus valgus, and
short stature become more apparent. The adolescent has short stature
and sexual infantilism. The most important features that call for
chromosome analysis are edema of the hands and feet at birth, short
stature in childhood, and sexual infantilism as an adolescent.
Bone maturation is normal until 8 to 9 years of age; then, because sex
hormone stimulation is absent, there is neither skeletal maturation nor
pubertal growth spurt. There is no puberty at all, and the girls remain
without secondary sexual characteristics unless exogenous estrogen is
administered.
syndrome, but the cervical spine radiographs are normal. It is a
cutaneous web only, and the cause may be related to an intrauterine
cystic hygroma (338). It is cosmetically unsightly, and plastic surgery is effective (339).
and the curve usually develops in juveniles. The delayed skeletal
maturation allows a long period for curve progression. Growth hormone,
which is almost always administered to girls with this syndrome,
accelerates curve progression. Although the scoliosis can be managed in
the same way as idiopathic scoliosis, patients must be observed more
frequently during growth hormone administration. Kyphosis is present in
a large proportion of individuals with this condition, but its
functional significance is unclear (341).
Genu valgum is also apparent, but the vast majority of cases are
asymptomatic. Osteotomy is performed for the rare symptomatic case.
There is a medial bony protuberance not unlike an osteochondroma,
arising off the proximal tibia in some patients (343).
estrogen and an altered renal vitamin D metabolism, which is
correctable with the administration of growth hormone and sex steroid
supplementation (344,345,346,347). Even in childhood, there may be the sequelae of osteoporosis, with a high incidence of wrist fractures reported (348).
hormone through adolescence, which results in a modest increase in
growth velocity and final height from an average of 140 cm (55 in.) to
just under 149 cm (58.5 in.) (353,354). Limb lengthening is associated with a very high rate of complications, and is therefore not recommended (355).
Cyclic sex hormones are administered during adolescence and throughout
adulthood. Estrogen is necessary for the development of secondary
sexual characteristics, and the estrogens, and possibly the previously
administered growth hormone, help prevent osteoporosis. Many with
Turner syndrome marry, and obstetric techniques of hormone
supplementation and ovum transplantation can result in pregnancy.
abnormality, its phenotype is reminiscent of Turner syndrome, with
short stature, webbed neck, cubitus valgus, and sexual immaturity (356,357),
which is why it is discussed here. Noonan syndrome is an autosomal
dominant disorder, in which approximately half of all cases are caused
by a mutation in the PTPN11 gene, which encodes for a protein—tyrosine phosphatase (358,359,360).
How tyrosine phosphatase causes the observed phenotype has yet to be
elucidated. The incidence is between 1 in 1000 and 1 in 2500 (358).
Many clinical features are shared with the Turner phenotype, but what
distinguishes this syndrome are the normal gonads, a high incidence of
mental retardation, and right-sided congenital heart defects, often
with hypertrophic cardiomyopathy (361,362). Scoliosis is more common (40%) than in patients with Turner syndrome, and more severe (363,364).
Minor to major vertebral abnormalities may be seen on radiographs.
Skeletal maturation is delayed despite normal puberty and menarche.
There is short stature, and the use of growth hormone may be associated
with a modest increase in ultimate height (365);
however, there are no well controlled comparative series on the basis
of which to evaluate the use of growth hormone in these children.
Noonan syndrome is often misdiagnosed, and most frequently confused
with King-Denborough syndrome, a myopathic arthrogryposis syndrome
characterized by short stature, web neck, spinal deformity, and
contractures. Recognizing the difference is important, because a
malignant hyperthermia-like picture is part of the King-Denborough
syndrome. The use of genetic testing for PTPN11 mutations may aid in this differentiation.
confusion, because textbooks describe trichorhinophalangeal syndrome,
trichorhinophalangeal syndrome with exostosis, and Langer-Giedion
syndrome. It is best to think of two relatively distinct
trichorhinophalangeal (TRP) syndromes: types I and II. Despite the
clinical overlaps between the two, there are enough features to
separate them into distinct syndromes.
prominent ears, sparse hair, and cone epiphyses. They have mild growth
retardation. The thumbs are broad, and the fingers are often angled at
the distal interphalangeal and proximal interphalangeal joints. The
hips mimic a Perthes-like disease in radiographs and symptoms (366). There may be lax ligaments.
presence of multiple exostoses, especially involving the lower
extremities. Those with TRP-II have facial features and cone epiphyses
similar to patients with TRP-I. There is a higher chance of mental
retardation in TRP-II. Langer-Giedion syndrome and TRP-II are identical
(367). Patients with TRP-II also have
microcephaly, large and protruding ears, a bulbous nose, and sparse
scalp hair. In infancy, their skin is redundant and loose, and this
condition may be severe enough to mimic EDS. Marked ligamentous laxity
may further support this error in diagnosis. There is a tendency toward
fractures. Similar to TRP-I, the Perthes-like picture, as well as the
hand anomalies, are present in TRP-II (368).
gene is responsible for the facial malformation and cone epiphyses
present in both disorders. Individuals with loss of a large portion of
a chromosome are more likely to have mental retardation. This explains
the mental retardation in some patients with TRP-II, which is
characterized by a larger region of chromosomal deletion. TRP-II is one
of the few disorders actually known to be due to two contiguous genes (5).
TRP-II shows short fourth and fifth metacarpals, cone epiphyses, a
short and broad thumb, and fingers with angled proximal and distal
interphalangeal joints (369) (Fig. 9.29).
The cone epiphyses, so characteristic of this syndrome, are not seen
until after 3 or 4 years of age. The pelvis shows the unilateral or
bilateral changes of Perthes in TRP-I and TRP-II, but rather than
resolution, the Perthes-like picture persists, evolving into a pattern
more like multiple epiphyseal dysplasia with precocious arthritis (Fig. 9.30).
Despite the wealth of radiographic abnormalities, the hands rarely have
functional disturbances. Osteotomy of the thumb is occasionally needed.
If symptomatic, we recommend managing the hips as in symptomatic
Perthes, but there is insufficient information available about
outcomes. Occasionally, an exostosis may be large or symptomatic enough
to require excision.
obesity, hypogonadism, short stature, small hands and feet, and mental
deficiency (370,371,372).
The incidence is 1 in 5000 births, with a prevalence in the population
of 1 in 16,000 to 1 in 25,000. As newborns, those with Prader-Willi
syndrome are floppy babies, having hypotonia, poor feeding, and delayed
milestones (373). The symptoms may mimic those of infants with spinal muscular
atrophy. Approximately 10% of infants have developmental dysplasia of
the hip. The syndrome may be remembered with an “H” mnemonic:
hypotonia, hypogonadism, hyperphagia, hypomentation, and small hands,
all probably based on a hypothalamic disorder.
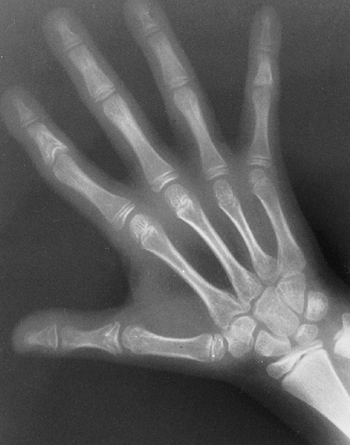 |
|
Figure 9.29
Trichorhinophalangeal syndrome. This 11-year-old patient has cone- or chevron-shaped epiphyses in the hand, and a broad thumb and distal phalanx. |
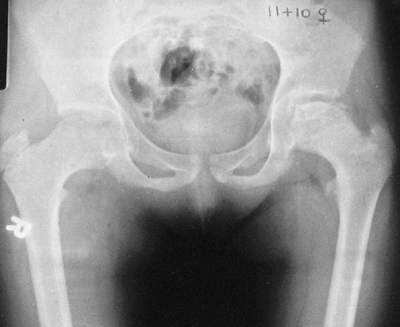 |
|
Figure 9.30
Trichorhinophalangeal syndrome, type I. The changes mimic Legg-Perthes disease, but by 12 years of age they did not resolve. On the right is a small but spherical epiphysis. On the left, the changes are similar to those seen in Perthes disease and in multiple epiphyseal dysplasia. |
Obesity begins, and a Prader-Willi diagnosis is usually suspected
because of the onset of a voracious eating disorder. The patient has a
preoccupation with food and an insatiable appetite (375,376).
Obesity has a central distribution, sparing the distal limbs. Complex
behavioral modification programs are occasionally effective. Although a
trial using fenfluramine reported limited success, it is rarely, if
ever, used today (377). The patient has short
stature, below the tenth percentile, with an ultimate height of 150 cm
(59 in.). There is no adolescent growth spurt. The genitalia are
hypoplastic, and the patient has small hands and feet (378).
Mental retardation is present, but it is extremely variable.
Nevertheless, skills for independent living are almost nonexistent, and
most reside in sheltered homes (375,379).
Genomic imprinting is a process by which genes of maternal origin have
different effects from genes of paternal origin. Angelman syndrome, or
happy puppet syndrome, is phenotypically dissimilar to Prader-Willi
syndrome. Angelman syndrome patients are small and mentally retarded,
and they have athetosis and seizures. However, they have the exact
chromosome deletion that occurs in Prader-Willi syndrome (15q11-13),
except that the deleted DNA is of maternal origin (381).
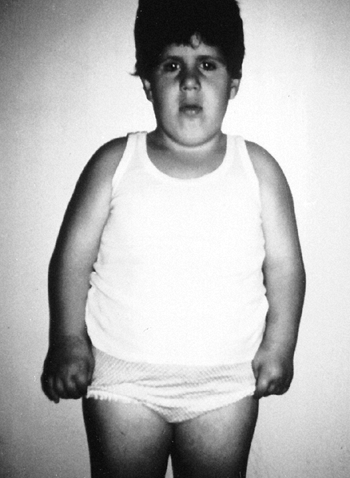 |
|
Figure 9.31
Prader-Willi syndrome in a 7-year-old patient. The features include truncal obesity and a round face with almond-shaped eyes. (From Goldberg MJ. The dysmorphic child: an orthopedic perspective. New York: Raven Press, 1987, with permission.) |
The legs are malaligned, with genu valgum and pes planus, but the
condition has limited or no effect on functional health and physical
performance.
series suggest that growth hormone improves body composition, fat
utilization, and physical strength and agility and as well as growth (388,389).
The lack of controlled trials and ethical issues related to its use in
this patient population makes the use of growth hormone in Prader-Willi
syndrome of uncertain benefit. Such issues have become confounded by
recent reports of deaths of children with Prader-Willi syndrome who
were given growth hormone. However, it is not known whether these
deaths were actually related to growth hormone, or whether the children
succumbed to other manifestations of the syndrome (390,391).
retardation associated with characteristic digital changes, consisting
mainly of broad thumbs and large toes (392). It is relatively common among mentally retarded persons, with an incidence of 1 in 500 (393). Most cases are sporadic, although there is the possibility of autosomal dominant inheritance (394).
Cyrano de Bergerac–like nose with the nasal septum extending below the
nostrils (Fig. 9.33). These facial characteristics may change with time, making this a less reliable finding (395).
Broad terminal phalanges of the thumb are present in 87% of patients,
and the great toe is affected in all patients. One half of the patients
have radially angulated thumbs, and this causes disability. Hallux
varus is common, and the physician should consider Rubinstein-Taybi
syndrome whenever congenital hallux varus is encountered. Patients have
ligamentous laxity and pronated feet, and an increased incidence of
fractures (393).
shows a wide distal phalanx, with soft tissue hypertrophy and a
triangular proximal phalanx (i.e., delta phalanx) that accounts for the
radial deviation (Fig. 9.34). The toe demonstrates duplicated or broad distal phalanx, but true polydactyly is not part of this syndrome (Fig. 9.34B). There is an assortment of other insignificant skeletal anomalies, many in the axial skeleton (396).
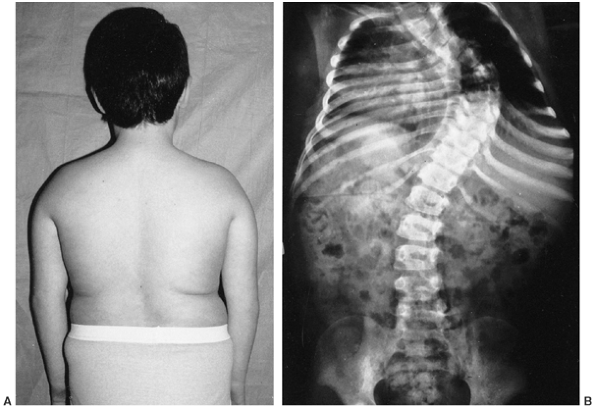 |
|
Figure 9.32 Prader-Willi syndrome in a 6-year-old patient. A: Scoliosis is difficult to detect because of the truncal obesity. B: The roentgenogram of this patient discloses a 50-degree thoracic curve. (B from Goldberg MJ. The dysmorphic child: an orthopedic perspective. New York: Raven Press, 1987, with permission.)
|
to have breakpoints in, and microdeletions of, chromosome 16p13.3. This
region contains the gene for CREB-binding protein, a nuclear protein
participating as a coactivator in cyclic-AMP-regulated gene expression.
This protein plays an important role in the development of the central
nervous system, head, and neck, and this explains the facial
malformation and mental retardation associated with this syndrome. The
propensity to develop tumors in these regions is probably caused by
malregulation of cyclic-AMP-regulated gene expression (397).
 |
|
Figure 9.33
Rubinstein-Taybi syndrome. In a 10-year-girl, the characteristic Cyrano de Bergerac–like nose has a septum that extends below the nostrils. |
is noticed at the end of the first year, and there is no true pubertal
growth spurt (398). The patients are mentally
retarded, many with microcephaly. IQ can range from 35 to 80, with a
delay in acquiring skills. However, these features vary. Associated
medical problems include visual disturbances, congenital heart disease,
and gastrointestinal abnormalities. Later in life, frequent upper
respiratory infections are related to abnormal craniofacial features,
severe dental caries are common, and other infections lead to morbidity
(399,400,401). Individuals with this syndrome are predisposed to certain types of central nervous system and head and neck tumors (402).
with pinch, in which case osteotomy of the proximal phalanx should be
performed. The deformity is progressive, and
recurrence
is common, as with any delta phalanx. The toe rarely requires treatment
unless there is a significant congenital hallux varus. Patellar
dislocation occurs in this syndrome. Although reports suggest that
early surgical intervention might improve function, there are no data
comparing early surgical treatment with other managements to support
this concept. If surgery is performed, the addition of an extensive
quadriceps mobilization seems to decrease the revision rate (403,404).
We reserve surgical intervention for the patella dislocation for
symptomatic cases, or for cases in which the dislocation is clearly
interfering with a patient’s ability to function.
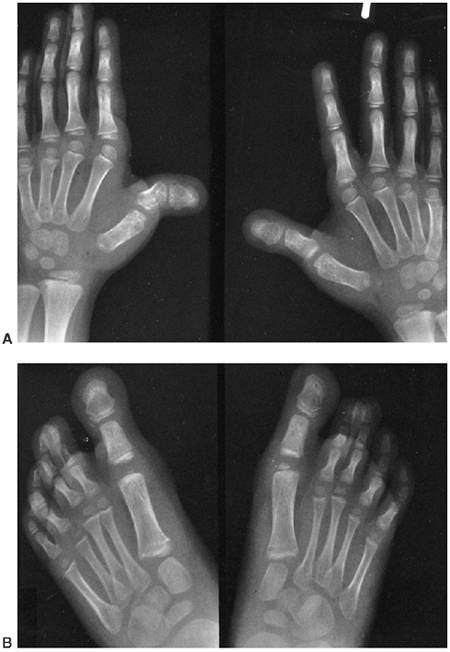 |
|
Figure 9.34 Rubinstein-Taybi syndrome in a 7-year-old patient. A: The thumbs are malformed, with a trapezoid proximal phalanx. The epiphysis extends around the radial side. B: The feet are more symmetric. Notice the broadening of the distal phalanx of the great toe. (From Goldberg MJ. The dysmorphic child: an orthopedic perspective. New York: Raven Press, 1987, with permission.)
|
conductive heart defects. Patients are sensitive to many anesthesia
drugs, including neuromuscular blocking agents, which tend to induce
arrhythmias and prolong awakening from anesthesia (405,406). Keloid formation is common (407).
modify substances for degradation, and cause cell dysfunction when
mutated because of the accumulation of these substances. Mutations in
genes that encode for enzymes can have a wide variety of effects on
cells, resulting in a broad range of abnormalities in cell function and
a wide range of clinical findings. Many of these disorders result in
the excess accumulation of proteins in cells. In these cases, the cells
become larger than normal. This results in increased pressure in bones,
causing avascular necrosis, and in increased extradural material in the
spine, potentially causing paralysis. Multiple systems are almost
always involved in these disorders. Medical treatments to replace the
defective enzyme have been developed for many of these disorders, and
such treatments will often arrest, but
not
reverse, the skeletal manifestations of the disorder. Early diagnosis
and appropriate medical treatment are slowly decreasing the number of
these individuals who present to orthopaedists with musculoskeletal
problems. Most enzyme disorders are inherited in an autosomal recessive
manner.
The mild-to-severe mucopolysaccharidoses (MPS) have similar radiographs
and different clinical features, but each produces a particular sugar
in the urine because of a specific enzyme defect (408,409).
Changes in the naming and numbering of systems over the years have
introduced considerable confusion in understanding the MPS. The
incidence is 1 in 10,000.
especially in the hands. Stiffness is postulated to be the result of
the deposition of mucopolysaccharide in the capsule and periarticular
structures, and is thought to reflect the loss of joint congruity.
Radiographs reveal oval vertebral bodies that are often beaked
anteriorly; a pelvis with wide, flat ilia; capacious acetabuli;
unossified femoral head cartilage; and coxa valga. The radiographic and
clinical features are usually not apparent at birth, but become more
apparent as the child gets older. Thus, it may be difficult to diagnose
a mucopolysaccharidosis during the first year of life.
mucopolysaccharidosis type II (Hunter syndrome), which is X-linked. The
most common MPS are type I (Hurler syndrome) and type IV (Morquio
syndrome).
|
TABLE 9.4 MUCOPOLYSACCHARIDOSES
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
toluidine blue-spot test. If the initial results are positive, specific
blood testing is done for the associated sugar abnormality. Although
spot tests are quick and inexpensive, they have high false-positive and
high false-negative rates. They are the initial tests that should be
ordered.
MPS. Each has a deficiency of a specific lysosomal enzyme that degrades
the sulfated glycosamine glycans: heparan sulfate, dermatan sulfate,
keratan sulfate, and chondroitin sulfate. The incomplete degradation
product accumulates in the lysozymes themselves. The MPS are part of a
larger group of disorders known as the lysosomal storage diseases. The
incomplete product accumulates in the tissues such as the brain, the
viscera, and the joints. This unremitting process leads to the clinical
progression of the disease. The child is normal at birth, but a problem
may be chemically detectable by 6 to 12 months of age, and clinical
progression is apparent by 2 years of age. This accumulation is
responsible for the development of avascular necrosis, presumably
because of too much material in the intramedullary space, and also
contributes to spinal cord compressive symptoms, because of
accumulation of material in the spinal canal.
characterized by a deficiency of L-iduronidase, the enzyme that
degrades dermatan sulfate and heparan sulfate. The Hurler and Scheie
forms represent the severe and mild ends of the clinical spectrum in
MPS I. Children with the Hurler form have progressive mental
retardation, severe, multiple skeletal
deformities,
and considerable organ and soft tissue deformities, and die before the
age of 10 years. The Scheie form is characterized by stiffness of the
joints and corneal clouding, but no mental retardation; the diagnosis
is usually made at approximately 15 years of age, and the patient has a
normal life expectancy. Many patients with MPS I fall in the middle of
this clinical spectrum. The clinical variation is determined by the
location and type of mutation that occurs along the gene for
L-iduronidase (410,411).
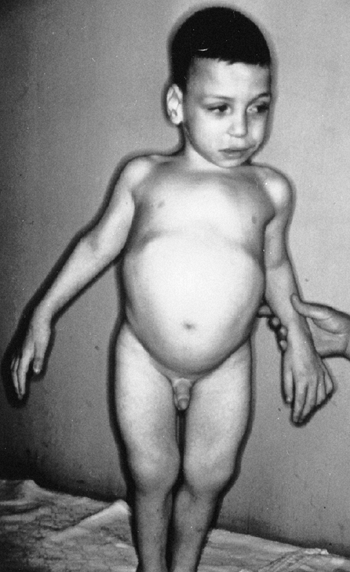 |
|
Figure 9.35
The classic appearance of a mucopolysaccharidosis in a 3-year-old patient includes facial features that are mildly coarsened, an abdominal protuberance from an enlarged spleen and liver, a short trunk, and stiff interphalangeal joints of the fingers. |
more severe forms (Hurler syndrome). However, the results on the bones
are variable (412), with most children still
developing the typical skeletal phenotypic features despite undergoing
successful bone marrow transplant (413,414). This may be due to the poor penetration of the enzyme derived from the transplanted leukocytes to the osseous cells (412).
Initial studies suggested an improvement in most of the nonosseous
manifestations of the disease with marrow transplant. More recent
longer-term studies cast doubt on the long-term effectiveness of marrow
transplantation (415). Despite these
disappointing longer-term results, marrow transplant may provide
short-term improvement, especially in the nonosseous manifestations.
The musculoskeletal deformities that persist after marrow transplant
still require treatment.
Approximately one-fourth of the patients have an abnormality of the
upper cervical spine. Odontoid hypoplasia and a soft tissue mass in the
canal can be managed like those in Morquio syndrome (described in the
following text). The accumulation of degradation products in closed
anatomic spaces, such as the carpal tunnel, causes “triggering” of the
fingers and carpal tunnel syndrome. These can be managed operatively (417,418).
including several types of spondyloepiphyseal dysplasia. Morquio
syndrome is an autosomal recessive disorder with an incidence of 3 per
1,000,000 of the population. Three types of Morquio syndrome are
classified as subtypes of MPS IV. All are caused by enzyme defects
involved in the degradation of keratan sulfate (408,409,410).
dwarfs, although they appear normal at birth. They develop corneal
opacities. The bone dysplasia is radiographically obvious, and the
final height is less than 125 cm (50 in.). Patients have abnormal
dentition. The deficient enzyme is N-acetylgalactosamine-6-sulfate sulfatase, and the chromosomal defect occurs at 16q24.3 (419).
Patients with intermediate MPS IVB have the same but milder phenotypes
as those with type IVA. They are taller, with final heights greater
than 125 cm (50 in.), and they have normal dentition. Here the enzyme
defect is β-D-galactosidase. Patients with mild MPS IVC have very mild
clinical manifestations.
by the severity of symptoms and the patient’s age at detection. All
these patients are normal at birth. For patients with the severe type
IVA, the diagnosis is made between 1 and 3 years of age; those with the
mild type IVC are diagnosed as teens; and those with the intermediate
form (type IVB) are diagnosed somewhere in the middle of this age
range. The three forms may also be separated by the severity of the
radiographic changes.
types, and only rarely are the facial features coarsened. Similarly,
all are short-trunked dwarfs with ligamentous laxity; the laxity is
rather profound in MPS IVA. The degree of genu valgus is significant,
aggravated by the lax ligaments (420,421,422,423).
osseous malalignment and the lax ligaments. Although it is observed
that the fingers and joints are becoming stiff, the medial and lateral
instability of the knee remains. Realignment osteotomies can restore
plumb alignment, but braces may be needed to control the instability
during ambulation. The prophylactic use of braces to prevent initial
valgus or recurrent deformity
after surgery has not been effective (420,421,422,423).
The hips and knees develop early arthritis. The hips show a progressive
acetabular dysplasia. Radiographs may show a small femoral ossific
nucleus, but an MRI or arthrogram will show a much larger cartilagenous
femoral head. The femoral capital epiphyses are initially advanced for
the patient’s age, but between 4 and 9 years of age the femoral heads
grow smaller, then disappear altogether (Fig. 9.36).
The pathophysiology of the progressive hip disease is not completely
understood, and neither medication nor surgery has been shown to
improve the prognosis (420,421,422,423).
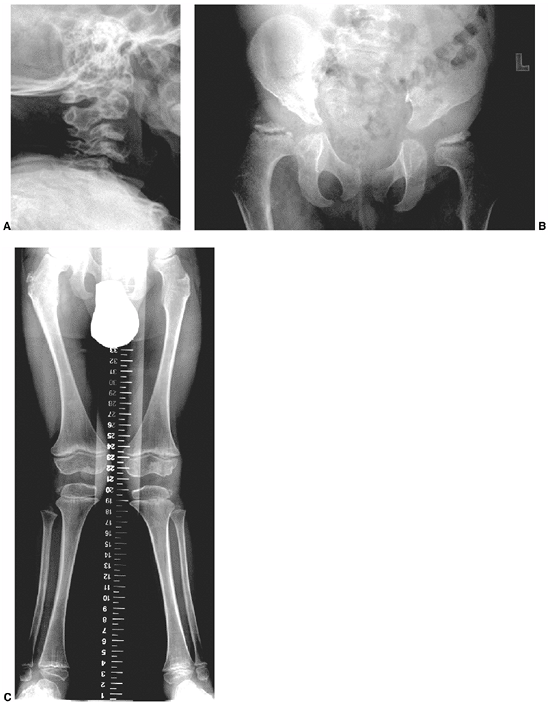 |
|
Figure 9.36 Morquio syndrome. The radiographic features include an absent odontoid (A), a pelvis with capacious acetabuli and coxa valga (B), and marked platyspondyly (C). It is difficult to imagine that these vertebrae were normal at birth. Genu varum is common.
|
cord compression (425,426).
This soft tissue mass can make the space available for the cord smaller
than one would expect on the basis of radiographs alone. Neurologic
function, especially upper-extremity strength and tone, is probably
more important than measuring distances on dynamic cervical spine
films. The upper- and lower-extremity findings are often of flaccidity
rather than spasticity. The onset of the myelopathy can occur as early
as the first decade of life, progressing as the soft tissue
hypertrophies, with the C1–C2 instabilities aggravating the situation.
Sudden deaths of patients with Morquio disease have been reported, and
they are typically attributed to the C1–C2 subluxation. C1–C2 fusion
before the onset of symptoms is controversial, but promoted by some (425,427). Others think the best surgery is occipital cervical fusion because it reduces the anterior soft tissue mass (426,427).
There are no comparative studies evaluating the outcomes of each of the
different management approaches. On the basis of the available
information, it is reasonable to obtain MRI studies on symptomatic
individuals, or on those with radiographic evidence of instability.
C1–C2 fusions are recommended for asymptomatic individuals with MRI
evidence of cord compression. Symptomatic individuals should have
fusions throughout the region of instability and cord compression.
Although decompression is usually performed along with the fusion,
anecdotal evidence suggests that fusion alone may be sufficient,
resulting in the soft tissue mass decreasing in size.
platyspondylia with a thoracic kyphosis. Progressive deformity should
be surgically stabilized. Anterior instrumentation is an effective
surgical technique (428). Despite these
problems, many patients with Morquio disease live for decades.
Cardiorespiratory disease is common, but the problems at the upper
cervical spine account for most disabilities.
arthrodentosteodysplasia, consists of acroosteolysis, with osteoporosis
and hypoplastic changes in the skull and mandible. The osteoporosis
leads to multiple fractures of the skull, spine, and digits. The
cranial sutures persist; wormian bones are seen on the skull
radiographs. Basilar impression is a common finding, often requiring
operative intervention. The terminal digits exhibit gradual loss of
bone mass, sometimes called pseudoosteolysis. Patients tend to have deep voices (429,430,431,432,433).
patellar dislocations, scoliosis, frequent fractures, and basilar
impression (433). The basilar invagination can cause hydrocephalus and an Arnold-Chiari malformation (434). This is usually managed by decompression and an occiput-to-upper-cervical-spine fusion (Fig. 9.37).
Not much data is available on the management of other musculoskeletal
problems. Scoliosis can be managed as in idiopathic scoliosis, although
the underlying osteopenia and associated spinal fractures may make
nonoperative management more difficult. The use of bisphosphonate
therapy to treat the osteopenia has been reported (435), although it is not known whether this therapy will improve the clinical outcome for children with this disorder.
are reported in some Haju-Cheney patients, and thus cardiac and renal
functions should be evaluated before placing the patients under
anesthesia (436,437). The disorder can be inherited in an autosomal dominant manner, but the causative gene is unknown.
of many syndromes characterized by premature aging. It is exceedingly
rare, with fewer than 30 affected children in North America. The cause
is entirely unknown. Autosomal dominant (438) and autosomal recessive (439) inheritance patterns have been proposed, but a sporadic mutation is more likely (440).
and hormone supplementation will increase growth velocity, but not
result in improved survival (441). The cause of
the condition is not known, but a number of reports refer to the use of
tissues from these patients in studying the aging process. Fibroblasts
from tissue cultures derived from these individuals show a variety of
abnormalities, including a decreased ability to clear free radicals (442).
years of age by their clinical features alone. There is severe growth
retardation and an inability to gain weight. If there is survival to
adolescence, there is no pubertal growth spurt. Alopecia and a loss of
subcutaneous fat are dramatic, and account for the distinctive
appearance of a skinny old man or woman (443,444).
These patients have stiffness of the joints that is not arthritis, but
a periarticular fibrosis. Osteolysis occurs in the fingertips,
clavicle, and proximal humerus (438,445,446) (Fig. 9.38). The vertebrae may become osteopenic, creating fish-mouth vertebral bodies on radiographs (438,444,447).
Fractures are common, often with delayed union. There is late
developmental dysplasia of the hip, and the onset of a rather
significant coxa valga (447,448) (Fig. 9.39).
The children do not live long enough to develop arthritis secondary to
the acetabular dysplasia. Not all systems age. There are no cataracts;
there is no senility. Rather than actually aging, the normal tissues
undergo an atrophic or degenerative change that mimics normal aging.
The principal histopathologic atrophic changes occur in the skin,
subcutaneous tissue, bone, and cardiovascular system. Atherosclerosis
with myocardial
infarction by 10 years of age is the rule, and life expectancy rarely exceeds 20 years.
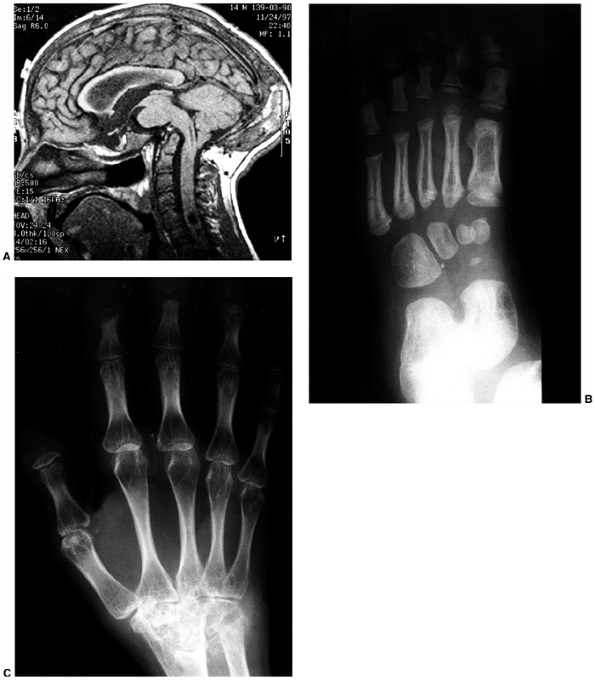 |
|
Figure 9.37 Hadju-Cheney syndrome. A: Magnetic resonance image of the head shows marked basilar invagination with an associated syrinx in the cervical cord. B: Radiographs show osteoporosis with pathologic fractures. C: Loss of bone mass in the terminal digits, termed pseudoosteolysis.
|
myocardial infarction. Despite a short life, it is imperative not to
permit any suffering. Hip surgery is indicated only if there is a
documented functional impairment. Surgery is not indicated to prevent
future arthritis. There is no medical treatment for the basic disease
process.
of orthopaedic conditions ranging from neuromuscular disorders to the
sequelae of injury, there are several disorders in which contractures
are the most prominent phenotypic feature. These syndromes are caused
by a wide variety of etiologies, including mutations causing
developmental
problems,
mutations dysregulating muscle function, and fetal environmental
causes. Many of these are associated with problems in muscle function,
as in the case of distal arthrogryposis, which is caused by mutations
disrupting fast-twitch muscle fiber activity; there is some overlap in
phenotype between these conditions and some of the myopathies. There
are many such disorders of different etiologies but, because the
management for many of these disorders follows similar guidelines, they
are considered together in this chapter.
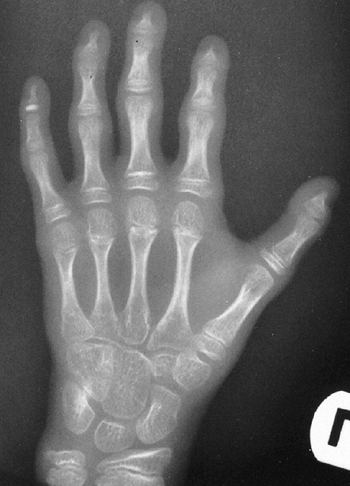 |
|
Figure 9.38 Progeria. The radiograph shows distal acrolysis, with resorption of the distal phalanges. (From Goldberg MJ. The dysmorphic child: an orthopedic perspective. New York: Raven Press, 1987, with permission.)
|
diagnosis, and represents a large group of disorders, all of which
include contractures of joints present at birth. This term is used as a
noun to describe specific diseases, and as an adjective,
“arthrogrypotic,” to refer to rigid joint contractures. There are at
least 65 distinct syndromes coded under the term arthrogryposis in the OMIM (5),
illustrating the large variety of etiologies associated with this term.
Most of the syndromes have different clinical courses, prognoses,
genetics, causes, and pathologic processes, often making it difficult
for the orthopaedist to determine the management of an individual
patient (449,450). A
simple way to think about these disorders is to consider them as
contracture syndromes, which can be grouped into a few general
categories, each of which can be represented by a prototypic disease.
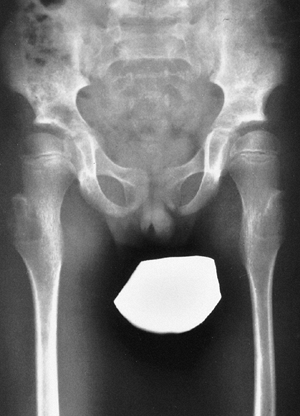 |
|
Figure 9.39 Progeria in an 11-year-old patient. The radiograph shows a marked degree of coxa valgus and some femoral head uncovering.
|
-
Involving all four extremities. This
includes arthrogryposis multiplex congenita and Larsen syndrome, with
more or less total body involvement. -
Predominantly or exclusively involving
the hands and feet. These are the distal arthrogryposes. Facial
involvement can occur with some of these syndromes, and Freeman-Sheldon
whistling face is included. -
Pterygia syndromes in which identifiable
skin webs cross the flexion aspects of the knees, elbows, and other
joints. Multiple pterygias and popliteal pterygia fit into this group.
unknown. It was initially described in 1841 by Adolf Wilhelm Otto, who
referred to his patient as a “human wonder with curved limbs” (453).
The disorder is sporadic, with affected individuals having reproduced
only normal children. Classic arthrogryposis can affect only one of
identical twins (454,455).
The development of arthrogryposis may be influenced by an adverse
intrauterine factor or the twinning process itself. Teratogens have
been suggested, but none are proven, despite the multiple animal models
that lend support to that theory (452,456,457,458,459).
Some mothers of children with arthrogryposis have serum antibodies that
inhibit fetal acetylcholine receptor function. One possibility is that
maternal antibodies to these fetal antigens cause the disorder (460).
fibrosis and fat between the muscle fibers. Myopathic and neuropathic
features are often found in the same muscle biopsy specimen. The
periarticular soft tissue structures are fibrotic and, in essence,
there is a fibrous ankylosis. The number of anterior horn cells in the
spinal cord is decreased, without an increase in the number of
microglial cells (461,462,463).
The pattern of motor neuron loss in specific spinal cord segments
correlates with the peripheral deformities and the affected muscles,
suggesting that a primary central nervous system disorder plays an
important role in causing this condition (464).
They are featureless and tubular. Normal skin creases are lacking, but
there may be deep dimples over the joints. Muscle mass is reduced,
although in infancy there is often abundant subcutaneous tissue.
Typically, the shoulders are adducted and internally rotated, the elbow
more often extended than flexed, and the wrist flexed severely, with
ulnar deviation. The fingers are flexed, clutching the thumb. In the
lower extremities, the hips are flexed, abducted, and externally
rotated; the knees are typically in extension, although flexion is
possible; clubfeet are the rule. Motion of the joints is restricted.
The condition is pain-free, with a firm, inelastic block to movement
beyond a very limited range. In two-thirds of the patients, all four
limbs are affected equally, but in one-third, lower-limb deformities
predominate. Only on rare occasions do the upper extremities
predominate. Deformities tend to be more severe and more rigid
distally. The hips may be dislocated unilaterally or bilaterally.
although gastroschisis has been reported. As a consequence of the
general muscle weakness, there is a 15% incidence of inguinal hernia.
Major feeding difficulties, caused by a stiff jaw and an immobile
tongue, are frequently encountered in infancy, and lead to respiratory
infections and failure to thrive (465). The
face is not particularly dysmorphic. A few subtle features, such as a
small jaw, narrowing of the face and, occasionally, limited upward gaze
(secondary to ocular muscle involvement). A frontal midline hemangioma
may help with the diagnosis (Fig. 9.40).
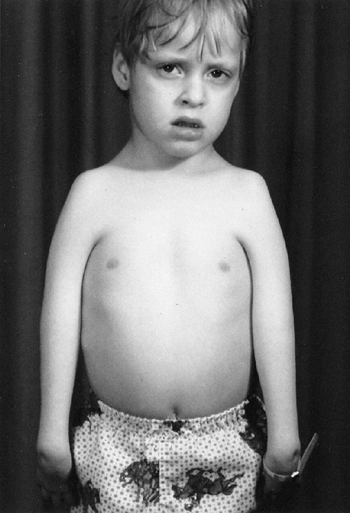 |
|
Figure 9.40 Arthrogryposis multiplex congenita. The picture shows the classic limb position and fusiform limbs lacking flexion creases.
|
changes are adaptive and acquired over time as a consequence of their
fixed position (Fig. 9.41). There is evidence
of a loss of subcutaneous fat and tissue. Electromyograms and muscle
biopsies are of questionable diagnostic value. They have been used to
separate patients with primarily neuropathic changes from those with
myopathic changes, but the clinical implications of such distinctions
are not clear. A diagnosis of arthrogryposis can be suspected when
prenatal ultrasound detects an absence of fetal movement, especially if
seen in combination with polyhydramnios (456).
patients with arthrogryposis, the natural history and long-term
outcomes are not well known (466,467).
Some contractures seem to worsen with age, and the joints become
stiffer. No new joints become involved. At least 25% of affected
patients are nonambulatory, and many others are limited household
walkers (468). As a rule, those with
arthrogryposis who are very weak as infants stay weak, and those who
appear stronger as infants stay strong. The dependency of adults seems
to be related to education and coping skills more than to the magnitude
of contractures of the joints.
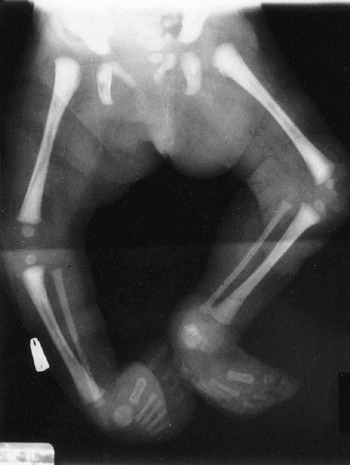 |
|
Figure 9.41
Arthrogryposis multiplex congenita at birth. Features include club feet, knee-flexion deformity, and dislocated right hip. The articular surfaces are normal. Adaptive changes occur as a consequence of the fixed position. (From Goldberg MJ. The dysmorphic child: an orthopedic perspective. New York: Raven Press, 1987, with permission.) |
unique opportunities for orthopaedic intervention, but an overview of
the total patient must be borne in mind. The overall goals are
lower-limb alignment and stability for ambulation, and upper-extremity
motion for self care (449,469,470,471).
Outcomes seem better if surgery on joints is done when children are
younger, usually before adaptive intraarticular changes occur at ages
four to six. Realignment osteotomies, however, are usually performed
closer to the completion of growth. Early motion, and avoidance of
prolonged casting, may increase joint mobility, thereby improving
function. Many children require long-term bracing or other assistive
devices (452).
Mobilization of joints may be accomplished by early and frequent range
of motion exercises and splinting of the joint in a position of
function with a removable orthotic (452,473).
There are no studies clearly demonstrating that early mobilization
improves outcomes in these patients, but such a program may improve the
passive range of motion, although the active range of motion does not
improve very much (452). In our experience,
early mobilization seems to be useful primarily for the upper
extremities. Fractures may accompany an overly vigorous range of motion
program.
At birth, the hips are flexed and abducted. There is considerable
controversy about the management of the hips in these children. Closed
reduction is rarely, if ever, successful. Operative reduction of a
dislocated hip should be performed if it will improve function or
decrease pain. Studies to date have not found pain to be a problem with
these hips; however, only relatively short-term follow-up is reported.
There is significant variability in functioning ability in these
individuals because of the underlying severity of the disease, and this
variability makes it difficult to determine any change in function from
treating the hips. The range of motion of the hips may be important for
functioning, because hip contractures, especially those that cause
flexion deformity, adversely affect the gait pattern. Operative
procedures to locate dislocated hips, therefore, have the potential to
worsen function if they produce significant contractures (475,477).
concluded that those with bilateral dislocations frequently had
satisfactory range of motion; their hips did not prevent them from
walking, although rarely around the community, and pain was uncommon (474,476,477).
Those with unilateral dislocations fared less well. More of them were
limited to the household with walkers, and, although scoliosis was
present in most patients, it was worse and more frequent in those with
unilateral dislocations (452). In both groups,
limitation of ambulation resulted more from the severe involvement of
all four extremities than from the dislocated hips (452).
These data, and case series suggesting little functional improvement
with surgery for bilateral hip dislocations, supports the concept of
leaving bilaterally dislocated hips alone (452,474,475,477).
However, in these studies, hip surgery was delayed until the knees were
mobilized, and reductions did not occur until at least 1 year of age.
This later age at reduction may be associated with higher rates of
contractures, and worse function. Reports of early open reduction of
unilateral and bilateral dislocated hips, with a reduced period of
immobilization, show improved postoperative range of motion (476,478,479).
Hip reduction may not benefit the child who is not an ambulator;
however, there is no way to comfortably predict which children will
become ambulators at the age when early surgical treatment is
contemplated. Longer-term studies show similar results with both
operative and nonoperative approaches, and a reduced range of motion in
hips that have been surgically relocated. However, the numbers of
patients reported in these studies were small (480).
It therefore seems reasonable to perform early open reduction in most
children. The exception may be a child with hips that are quite stiff.
Both medial and
anterior approaches are advocated for early hip reduction (476,478,479).
More than the specific operative approach, the key factor may be to
perform the hip reduction early in life, with minimal immobilization.
This may be more easily accomplished using a medial approach.
The precise plane of motion is often difficult to determine, and
although physical therapy is recommended, medial lateral instability
may result. Hyperextension deformity responds better to physical
therapy and splinting than do flexion deformities. If the flexion
deformity remains more than 30 degrees, ambulation is difficult because
of the associated overall muscle weakness, especially of the quadriceps
and gluteus maximus. Sometime before 2 years of age, soft tissue
surgery, including posterior capsulotomy, should be performed. The
actual procedure needs to be individualized, because each knee has a
different degree of deformity. Posterior soft tissue procedures often
need to be repeated later in life, but before nearing skeletal
maturity. Supracondylar osteotomies of the femur are recommended toward
the end of growth to correct residual deformity (452,482,483,484).
Femoral shortening may need to be added to the osteotomies, especially
in cases where the neurovascular structures will be stretched by
correcting the deformity.
treated without surgery, but quadricepsplasty may be needed in cases
with residual lack of motion. Traditional teaching advocates correction
of the knee deformity before treating a dislocated hip, in order to
allow stretching out of the muscles that cross both joints. However,
with early operative intervention, using a short period of
immobilization, the hip may be operated upon at the same time as a
surgical procedure to correct a hyperextended knee deformity. In this
case, the hamstring muscles are relaxed by both procedures, and the
knee can be immobilized in a flexed position in the hip spica cast. A
flexion deformity of the knee cannot be easily managed at the same time
as hip surgery, because it is impossible to appropriately immobilize
the hip with the knee held extended. Despite good initial nonoperative
results in the hyperextended knee, there may be recurrence of the
contracture over time, with surgery often needed later in life. An
alternative technique of correction of the knee deformity is by using
an external fixator, with gradual correction (485,486);
however, in most cases, an open procedure to release the contracted
structures will be adequate, and the deformity may recur after
treatment with gradual distraction. Late osteoarthritis seems more
common in those with persistent hyperextension contracture.
It is rare for the arthrogrypotic clubfoot to respond to physical
therapy and casts, and surgical intervention is usually necessary.
However, as in the knee deformity, there is a wide range of severity.
Some feet, therefore, respond better to surgery than others. Surgery
for clubfoot is sometimes delayed until 1 year of age or later, as
other joints, especially the knees, are attended to first. However, as
in management of the hip, performing combined procedures, with minimal
immobilization earlier in life, is gaining in popularity. Primary
talectomy has been recommended because of the high incidence of failed
soft tissue surgery (489,490); however, recent reports show good outcomes with circumferential release alone if performed before 1 year of age (491,492).
This is probably a better initial approach, because salvage talectomy
can always be performed later if necessary. The positioning of the
calcaneus is the key to achieving a good result after talectomy (493).
Residual deformity in the teen years can be treated using a triple
arthrodesis, or with multiple osteotomies, to maintain motion of the
subtalar joints, while producing a plantigrade foot. Gradual correction
using an external fixator is also possible (494),
but recurrence after gradual distraction is not unusual. A vertical
talus is an unusual foot deformity in arthrogryposis multiplex
congenita and, if it is encountered, the physician must think of the
distal arthrogryposes or pterygia syndromes.
procedures. The physician should never think of an individual joint in
the upper extremity, but only of the whole arm (495,496).
Analysis needs to include each hand separately, and also how the two
hands work together as an effective functional unit; that is, a
functional assessment should be made before deciding on an operation.
Because of this, surgical procedures on the upper extremity are usually
delayed until the children are old enough for the surgeon to make such
an assessment. There are two key goals in treatment of the upper
extremities: self-help skills, such as feeding and toileting, and
mobility skills, such as pushing out of a chair and using crutches.
For the elbow, it is ideal to achieve flexion to 90 degrees from the
fixed extended position. However, when both elbows are involved,
surgery to increase flexion should be done only on one side. The
fibrotic joint capsule and the weak muscles make the prospect of
achieving active elbow flexion unlikely. Passive elbow flexion to a
right angle is a prerequisite for considering a tendon transfer for
active elbow flexion. Restoration of elbow motion by capsulotomy and
triceps lengthening has had only fair success, diminishing the
likelihood for success even more when an arthrogrypotic muscle is used
to motor the joint (497). The triceps brachii
and pectoralis have been the most frequently tried muscles. Success is
best in children older than 4 years, and who have at least grade four
strength of the muscle to be transferred (496,498,499,500).
Distal humeral osteotomy, designed to place the elbow into flexion and
correct some of the shoulder internal rotation deformity, may be
performed toward the end of
the first decade (477,496).
It is designed to improve hand-to-mouth function. Care must be taken
not to externally rotate the distal humerus excessively. The hand and
wrist are usually flexed and the ulna deviated, but variations within
this pattern exist (501,502).
In general, the ulna-side digits are more involved. Proximal
interphalangeal flexion deformities rarely respond to physical therapy
or surgery. The thumb is flexed and adducted into the palm, and
responds better to surgery than do the other digits.
There are no comprehensive natural history studies of scoliosis in
arthrogryposis, but the curves usually have a C-shaped, neuromuscular
pattern. The use of braces has been reported (504),
although in our experience these children respond poorly to orthoses.
Surgery is indicated for progressive curves interfering with balance or
function. There are reports of patients regaining their ability to
ambulate after surgical correction of large, rigid curves (504); surgery should be considered in patients who lose their ability to ambulate as they develop such curves.
natural ability to learn substitution techniques. There is, however, a
strong association between initial feeding difficulties and subsequent
language development, which should not be mistaken for retardation (465).
congenital dislocations of large joints, a characteristic flat face,
and ligamentous laxity (505) (Fig. 9.42).
The cause of the facial flattening is unclear, but it is especially
noticeable when observed in profile, and is associated with some
hypertelorism and a broad forehead. Dislocation of multiple joints
appears in a characteristic pattern that includes bilateral dislocated
knees, with the tibia anterior on the femur, bilateral dislocated hips,
bilateral dislocated elbows, and bilateral clubfeet (506,507,508,509,510).
The physician should think of this syndrome whenever dislocated knees
are detected. The ligaments are lax or entirely absent. The ligamentous
laxity is often so substantial that Larsen syndrome may be confused
with EDS.
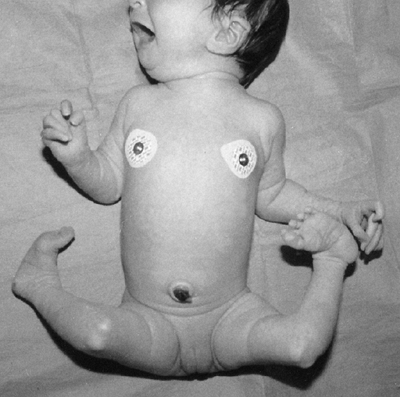 |
|
Figure 9.42 Larsen syndrome in a 1-week-old patient who has bilateral dislocated knees and clubfeet. (From Goldberg MJ. The dysmorphic child: an orthopedic perspective. New York: Raven Press, 1987, with permission.)
|
The elbows have complex radial–humeral, ulnar–humeral, and radial–ulnar
dislocations. Radial–ulnar synostosis is common and usually associated
with ulnar–humeral dislocation (Fig. 9.44B). A
spheroid ossicle frequently occurs anterior to the elbow joint; its
origin is unknown. There are more carpal centers than are normal (Fig. 9.44A), and extra ossification centers in the foot, with a curious double ossification pattern of the calcaneus (Fig. 9.44C).
This double ossification pattern can help confirm the diagnosis in
cases in which the diagnosis is not clear. Abnormal cervical spine
segmentation, with instability, is typical, as is kyphosis, a
complication often associated with myelopathy. Some cases are inherited
in an autosomal dominant manner, and this aids in the diagnosis.
This is an intracellular protein that serves as scaffolding on which
signaling and protein trafficking pathways are organized. It is
expressed in the growth-plate and vertebrae, and
as
such, the mutation likely acts to disrupt normal patterning and
development of the joints and vertebral bodies, resulting in the
typical phenotypic features. There are cases in which only one side of
the body has a Larsen syndrome phenotype, suggesting that some cases
are due to somatic, or mosaic, mutations (515).
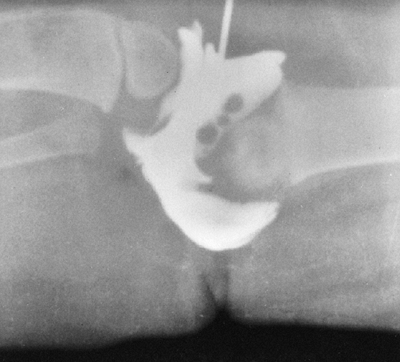 |
|
Figure 9.43
Larsen syndrome in a 5-month-old patient. The arthrogram of a knee shows anterior dislocation of the tibia on the femur and no suprapatellar pouch. |
 |
|
Figure 9.44 Characteristic roentgenograms of a 4-year-old patient with Larsen syndrome. A: The hands show more carpal centers and interphalangeal joint subluxations than is normal. B: The elbow demonstrates total dislocation but full functional ability. C: The foot has an abnormal os calcis containing two ossification centers. (A and B from Goldberg MJ. The dysmorphic child: an orthopedic perspective. New York: Raven Press, 1987, with permission.)
|
extremities require treatment in order to achieve stable, located
joints. Knee stability is necessary to achieve ambulation. Reduction
almost always requires surgery. The knee may remain unstable after
reduction because of the lack of major stabilizing ligaments, such as
the anterior cruciate ligament. Long-term orthoses are often needed.
Successful anterior cruciate ligament reconstruction has not been
reported. The knee is usually reduced before the hips, although
simultaneous procedures are possible (508,516).
Although most knees do not respond to attempts at manipulation and cast
correction, traditionally an initial trial of cast treatment is
attempted. Too-vigorous manipulations result in distal femoral
metaphyseal–physeal fractures. Because manipulation has not been found
to be helpful for true dislocations, we believe that it can be
abandoned once a dislocation is confirmed. Surgery may be undertaken as
early as 3 to 4 months of age. Restoration of the range of motion must
be cautious (gaining full extension is often a problem), and a flexion
splint or brace may be required after operative reduction to guard
against redislocation.
normal-appearing acetabulum. There is a sense of a good range of
motion, although the hip may prove to be irreducible (510).
The evolution of hip management in Larsen syndrome mirrors that in
arthrogryposis multiplex congenita, and there is a trend toward earlier
treatment. The relative rarity of this syndrome, however, accounts for
the lack of good comparative data on how best to manage the hip
dislocations. Reduction of the hip is associated with a high
redislocation rate and revision surgery (508,509,510).
For this reason, some specialists advocate either leaving bilateral
dislocated hips alone, or waiting until after 1 year of age and
performing femoral and pelvic osteotomies, along with the open
reduction. However, we prefer an approach similar to that in
arthrogryposis, with early surgical relocation (usually through a
medial approach). Because the knees are hyperextended when dislocated
and cast in a flexed position after surgical relocation, both knees and
hips can be operated upon at the same time. Secondary osteotomy of the
pelvis and femur can be performed later, if necessary.
serial casting, but long-term results suggest less residual deformity when the foot is treated surgically (508).
The foot may need to be braced to control ankle instability. Despite
the dislocations of the elbow or shoulder, the arms remain functional
and rarely require treatment. Crutches or walkers can be used despite
the dislocations.
This manifestation may occur more frequently than previously
recognized, and children should have cervical spine films taken in the
first year of life to identify this deformity. Kyphosis is often due to
hypoplasia of the vertebral bodies. A combination of cervical kyphosis
and forward subluxation may result in quadriplegia and death. Posterior
stabilization early (within the first 18 months of life) may prevent
the significant problems associated with treatment after myelopathy has
occurred and allow for correction of a kyphotic deformity with growth (519).
infolding arytenoid cartilage creates airway difficulties. The
associated tracheomalacia can be especially problematic in the newborn,
and may delay surgery for the hips and knees (520).
The anesthesiologist should be aware of possible cervical spine
instability, and a preoperative lateral radiograph is recommended.
generally good with aggressive orthopaedic treatment if the child
survives the first year of life. The mortality figures for the first
year may be as high as 40%. During the neonatal period, the
cartilage-supporting structure of the larynx and trachea is soft, and
there may be alarming elasticity of the thoracic cage at the
costochondral junction, leading to respiratory failure and death.
Cervical spine problems may also contribute to early mortality.
Congenital cardiac septal defects, elongation of the aorta, and
acquired lesions of the mitral valve and aorta, similar to those found
in Marfan syndrome, further complicate medical and anesthesia
management (521,522).
fixed hand contractures and foot deformities, but the major large
joints of the arms and legs are spared (523,524,525).
Because different craniofacial abnormalities are often associated with
distal arthrogryposis, the condition has been categorized as several
eponymic syndromes (e.g., Gordon syndrome), a situation that leads to
confusion (526). The cardinal features of
distal arthrogryposis are the hand deformity with ulnar deviation of
the fingers at the metacarpophalangeal (MCP) joint, flexion deformities
at the proximal interphalangeal and MCP joints, and a cup-like palm
with a single palmar crease (Fig. 9.45). The thumb is flexed and adducted, with a web at its base (527).
Distal arthrogryposis is common, and is sometimes incorrectly called
multiple camptodactyly. The inheritance pattern of distal
arthrogryposis is autosomal dominant, but there may be considerable
variation in families, and this can lead to missing the diagnosis (526,527,528,529).
Distal arthrogryposis is divided into type I and type II on the basis
of the absence or presence of facial findings, respectively.
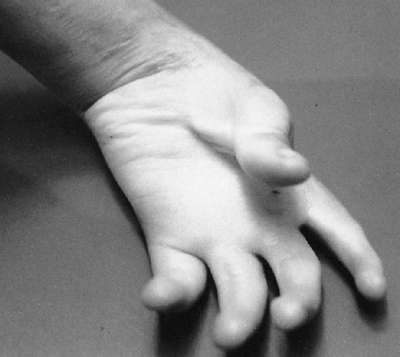 |
|
Figure 9.45
Distal arthrogryposis. Characteristic hand is the result of ulnar deviation at the metacarpophalangeal joints. Notice the deeply cupped palm and webbing of the MCP joint of the thumb. |
Type II distal arthrogryposis (Freeman-Sheldon syndrome) is caused by
mutations in an isoform of troponin I that is specific to the
troponin-tropomyosin complex of fast-twitch myofibers (530).
Both these mutations result in abnormal activity of fast-twitch muscle
fibers, suggesting that dysregulation of these muscle fibers is the
common pathophysiologic cause of distal arthrogryposis. Because there
are a variety of subtypes of distal arthrogryposis, it is likely that a
number of causative genes will be identified, and perhaps all of these
will play a role in the dysregulation of fast-twitch muscle fibers.
constant, the feet may be clubbed, have stiff metatarsus adductus, and
have a vertical talus. The major joints in the upper and lower
extremities are otherwise normal, although a minor knee-flexion
deformity may be found. Intelligence is normal. The associated
craniofacial anomalies are cleft lip or cleft palate and, in such
patients, the syndrome of distal arthrogryposis may have an eponymous
name, such as Gordon syndrome (531,532).
Radiographs show normal bony architecture, and only with persistence of
deformities in the hands and feet are articular changes detected.
This
syndrome can be diagnosed prenatally in the fetus by detecting an
unchanged position and lack of motion of the hands in contrast to the
normal activity of the large uninvolved joints (533).
function. The hands function well because the shoulders, elbows, and
wrists are normal. Thumb surgery to lengthen the flexor pollicis longus
and rebalance the extensor is the most common surgery (534).
The feet more frequently require surgery. Some clubfeet can be
corrected with manipulation and serial casts. Most are treated with
circumferential releases. The outcome of treatment of clubfoot is
better in this syndrome than in other arthrogrypotic clubfeet.
because the hand and foot deformities are similar to those of distal
arthrogryposis. It is recognized by its most characteristic feature, a
“whistling face” (Fig. 9.46). The original name, craniocarpotarsal dystrophy, is misleading because it does not involve the cranium (535,536). This syndrome is usually sporadic, although there is evidence of autosomal dominant and autosomal recessive inheritance (537,538,539).
The eyes are deeply set. The cheeks are fleshy, and pursed lips
simulate whistling. There is a small mouth and a curious H-shaped
dimple in the chin.
feature, but it affects more than one-half of the patients. The onset
is in the first decade. It is often severe, with a left thoracic
pattern reported regularly. The vertebrae are normally shaped. Although
the scoliosis can be managed as in idiopathic scoliosis, the curves are
more rigid and may not respond well to brace treatment (538,540).
 |
|
Figure 9.46
Freeman-Sheldon syndrome in a 3-year-old patient. Notice the small chin and mouth, long philtrum, puffy cheeks, deeply set eyes, and small chin cleft. (From Goldberg MJ. The dysmorphic child: an orthopedic perspective. New York: Raven Press, 1987, with permission.) |
There are other contractures, including flexion deformities of the
elbow and knee, decreased range of motion of the shoulder, decreased
range of motion of the neck, and dislocated hips (542).
Operative management principles for the upper extremity are similar to
those in distal arthrogryposis. The hands are treated with physical and
occupational therapy, but there is less improvement than is seen in the
other distal arthrogryposis syndromes (543). Most of the other associated contractures can be treated like those in the other arthrogrypotic syndromes.
to thrive, and even to death. Surgery to permit adequate mouth opening
for feeding may be necessary (544). Children
who survive the neonatal period do well and have normal intelligence.
Anesthesia complications are common; some are the result of
abnormalities related to the laryngeal cartilages (544,545,546,547).
The cause is unknown, but the buccinator muscle is hypoplastic, and
electromyograms and muscle biopsies are identical to the peripheral
muscle studies in classic arthrogryposis multiplex congenita (548), suggesting some similarity in pathophysiology.
word meaning “little wing.” A pterygium is a web. It can be seen as an
isolated malformation in some syndromes, such as the pterygium colli in
the neck of patients with Klippel-Feil syndrome.
The web syndromes are separated genetically as autosomal recessive
(i.e., lethal pterygium syndrome and multiple pterygium) and autosomal
dominant (i.e., popliteal pterygium) (552).
However, they often overlap. Lethal pterygium syndrome may be diagnosed
prenatally by detecting hydrops and cystic hygroma colli (553).
syndrome are caused by mutations in the gene encoding interferon
regulatory factor-6 (554). Van der Woude syndrome is a dominantly inherited developmental disorder
characterized by pits or sinuses of the lower lip, and cleft lip or
cleft palate. It is unclear how a mutation in this interferon
regulatory factor causes these seemingly dissimilar syndromes.
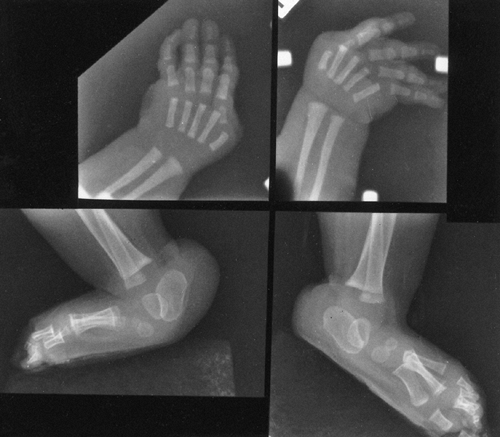 |
|
Figure 9.47
Freeman-Sheldon syndrome in a 5-year-old patient. Radiographs of the hands demonstrate ulnar deviation at the metacarpophalangeal joint, typical of a distal arthrogryposis syndrome. The feet show bilateral congenital vertical tali. All other joints in this patient were normal. (From Goldberg MJ. The dysmorphic child: an orthopedic perspective. New York: Raven Press, 1987, with permission.) |
characterized by a web across every flexion crease in the extremities,
most prominently across the popliteal space, the elbow, and in the
axilla (555,556) (Fig. 9.48).
There also are webs across the neck laterally and anteriorly from
sternum to chin, drawing the facial features down. The fingers are
webbed. The webs can be obvious, but if they are not, the affected
children can look very much like those with arthrogryposis multiplex
congenita. The two features that differentiate this syndrome from
classic arthrogryposis are vertical talus and congenital spine
deformity. The vertical talus is fairly constant in multiple pterygium
syndrome and can be managed only by surgery. Circumferential release
and prolonged protection, as in managing any arthrogrypotic foot
deformity, are necessary. The spine deformity is significant, with
multiple segmentation abnormalities and lordoscoliosis (557) (Figs. 9.49 and 9.50).
The lordoscoliosis may be substantial enough to interfere with trunk
and chest growth, leading to respiratory death during the first or
second year of life (Fig. 9.50). Mobility
depends much on the magnitude of the lower-extremity webs and the
residual motion of the joints, with many patients limited to
wheelchairs for locomotion. The children have normal intelligence, and
efforts should be maximized to enable them to function independently.
Surgery is rarely needed for the upper extremities.
fascial-genital-popliteal syndrome) has recognizable characteristics in
the face, the genitals, and the knee (558,559,560,561,562). The features include a cleft lip and palate, lip pits, and intraoral adhesions (563,564). A fibrous band crosses the perineum and distorts the genitalia (565). A popliteal web is usually present bilaterally (562).
It runs from ischium to calcaneus, resulting in a severe knee-flexion
deformity. Tibia hypoplasia may be associated. Within the popliteal web
is a superficial fibrous band, over which lies a tent of muscle running
from the os calcis to the ischium, and is known in the older literature
as a “calcaneoischiadicus muscle.” The popliteal artery and vein are
usually deep, but the sciatic nerve is superficial in the web, just
underneath the fibrous band (Fig. 9.51). There is a distinctive foot abnormality in this syndrome: a bifid great toenail and syndactyly of the lesser toes.
pterygium syndromes were clearly defined, there is more phenotypic
variation in both than was originally thought. For example, mild webs
in joints of the upper extremity may be found in patients with
popliteal pterygium syndrome. Adaptive changes in the joints occur over
time. On
radiographic examination, the patella look elongated, and the femoral condyles flattened, because of knee-flexion deformity.
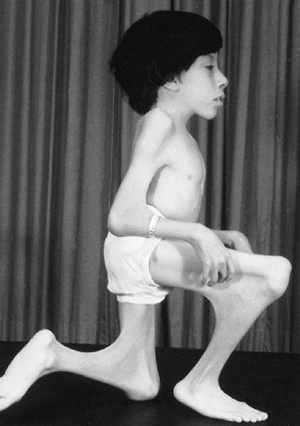 |
|
Figure 9.48
Multiple pterygium syndrome in a 12-year-old patient. Antecubital webs fix the elbows, and popliteal webs prevent ambulation. The patient had normal intelligence and became a college graduate. (From Goldberg MJ. The dysmorphic child: an orthopedic perspective. New York: Raven Press, 1987, with permission.) |
are the magnitude of scoliosis and the size of the web crossing the
knee. The thoracic vertebral dysplasia, thoracic lordosis, and the
small chest impair lung development, resulting in death in the first
years of life in those with multiple pterygium syndrome. For the
longer-term survivors, management of the spine deformity is identical
to those with nonsyndromic congenital scoliosis. Preoperative MRI
evaluation of intraspinal contents and ultrasound of the kidney are
indicated.
Traditionally, treatment of the knee begins with physical therapy, but
the effectiveness of this therapy is doubtful. Early popliteal web
surgery is recommended before the onset of adaptive changes in the
articular surfaces, and before further vascular shortening. The nerve
is usually located just under the skin and web, and care must be taken
to avoid nerve damage. The web is resected, and Z-plasty of the skin is
performed. There is a high recurrence rate despite use of braces.
Femoral shortening with an extension osteotomy is often required. If
almost-full knee extension cannot be achieved at surgery, femoral
shortening should be considered, even if during infancy or childhood (569). Gradual distraction techniques can be used, but an advantage over traditional techniques has not been demonstrated (570).
Posterior soft tissue procedures can be combined with distraction
techniques to gradually extend the knee. Femoral shortening techniques
are associated with low recurrence rates of the deformity, and have the
advantage of reducing tension on the neurovascular structures. These
techniques are therefore our treatment of choice.
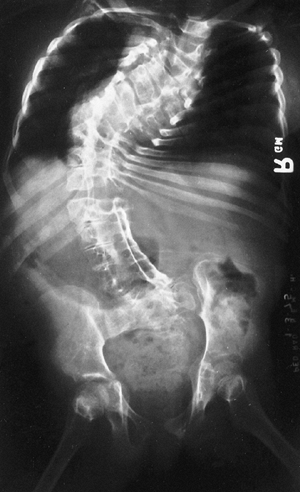 |
|
Figure 9.49
Multiple pterygium syndrome in a 13-year-old patient. Radiograph shows severe scoliosis, vertebral abnormalities, and an unsegmented bar from T9 to T12 and from L1 to S1, with an apparent gap between the bars. (From Goldberg MJ. The dysmorphic child: an orthopedic perspective. New York: Raven Press, 1987, with permission.) |
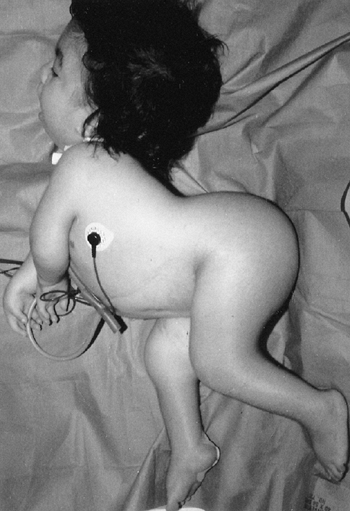 |
|
Figure 9.50
Multiple pterygium syndrome. Severe limitation of trunk growth was caused by vertebral fusions and lordoscoliosis. Death occurred at 24 months of age because of respiratory failure. |
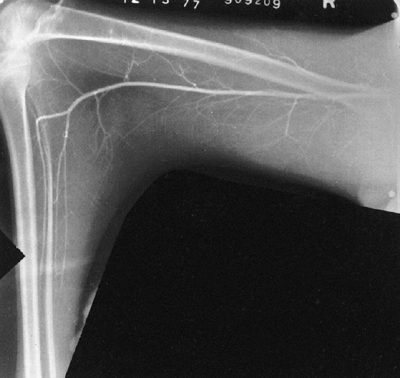 |
|
Figure 9.51
Popliteal pterygium in a 13-year-old patient. Arteriogram shows that the popliteal artery has been drawn up from its normal position. At the margin of the web is the sciatic nerve. (From Goldberg MJ. The dysmorphic child: an orthopedic perspective. New York: Raven Press, 1987, with permission.) |
Mendelian Inheritance in Man, OMIM. Center for Medical Genetics, Johns
Hopkins University and National Center for Biotechnology Information,
National Library of Medicine (Bethesda, MD), 1999. Available at: http://www.ncbi.nlm.nih.gov/omim. Accessed February 24, 2000.
Regemorter N, Dodion J, Druart C, et al. Congenital malformations in
10,000 consecutive births in a university hospital: need for genetic
counseling and prenatal diagnosis. J Pediatr 1984;104:386.
PS, Levy HP, Ahn NU, et al. A comparison of the Berlin and Ghent
nosologies and the influence of dural ectasia in the diagnosis of
Marfan syndrome. Genet Med 2000;2:278–282.
ZA, Grifo JA, Dietz HC. Marfan syndrome as a paradigm for
transcript-targeted preimplantation diagnosis of heterozygous
mutations. Nature Med 1995;1:798–803.
L, Andrikopoulos K, Tian J, et al. Targetting of the gene encoding
fibrillin-1 recapitulates the vascular aspect of Marfan syndrome. Nature Genet 1997;17:218–222.
ER, Frischmeyer PA, Arking DE, et al. Dysregulation of TGF-beta
activation contributes to pathogenesis in Marfan syndrome. Nat Genet 2003;33(3):407–411.
AY, Kim YH, Kim BK, et al. Unexpected tracheomalacia in Marfan syndrome
during general anesthesia for correction of scoliosis. Anesth Analg 2002;95(2):331–332.
T, Giampietro PF, Burke SW, et al. The incidence of protrusio acetabuli
in Marfan’s syndrome and its relationship to bone mineral density. J Pediatr Orthop 2000;20(6):718–721.
G, Sturman JA, Schaffner F. Homocystinuria due to cystathionine
synthase deficiency: enzymatic and ultrastructural studies. J Pediatr 1974;84:381.
GHJ, Smals AGH, Trijbels FJM, et al. Heterozygosity for homocystinuria
in premature peripheral and cerebral occlusive arterial disease. N Engl J Med 1985;313:709.
NP, Nicholls AC, Yates JRW, et al. The gene encoding collagen
alpha-1(V) (COL5A1) is linked to mixed Ehlers-Danlos syndrome type
I/II. J Invest Derm 1996;106:1273–1276.
RJ, Langland GT, Willing MC, et al. A splice-junction mutation in the
region of COL5A1 that codes for the carboxyl propeptide of
pro-alpha-1(V) chains results in the gravis form of the Ehlers-Danlos
syndrome (type I). Hum Molec Genet 1996; 5:1733–1736.
SR, Krane SM, Kenzora JE, et al. Heritable disorder with
hydroxylysine-deficient collagen: hydroxylysine-deficient collagen
disease. New Eng J Med 1972;286:1013–1020.
M, Schwarze U, Superti-Furga A, et al. Clinical and genetic features of
Ehlers-Danlos syndrome type IV, the vascular type. New Eng J Med 2000;342:673–680.
AS, Hill GA, Abela M, et al. Recurrent instability of the elbow in the
Ehlers-Danlos syndrome. A report of three cases and a new technique of
surgical stabilisation. J Bone Joint Surg Br 2000;82(5):702–704.
SM, Compston DAS, Harper PS. A genetic study of von Recklinghausen
neurofibromatosis in southeast Wales. II. Guidelines for genetic
counseling. J Med Genet 1989;26:712.
DH, Collins FS. Recent progress toward understanding the molecular
biology of von Recklinghausen neurofibromatosis. Brief review. Ann Neurol 1992;31:555.
RH, Shiu MH, Senie RT, et al. Malignant peripheral nerve sheath tumors
of the buttock and lower extremity. A study of 43 cases. Cancer 1990;66:1253.
H, Winter RB, Lonstein JB, et al. Pathophysiology of spinal deformities
in neurofibromatosis. An analysis of 71 patients who had curves
associated with dystrophic changes. J Bone Joint Surg Am 1994;76:692.
BG, Pill SG, Drummond DS. Irreducible thoracic spondyloptosis in a
child with neurofibromatosis: a rationale for treatment. Spine 2002;27(14):E342–E347.
KI, Ellis RD. Paraparesis after posterior spinal fusion in
neurofibromatosis secondary to rib displacement: case report and
literature review. J Pediatr Orthop 2000;20(6):799–801.
AH, Dietemann JL, Kastler B, et al. Neurofibromatosis with dural
ectasia and bilateral symmetrical pedicular clefts: report of two
cases. Neuroradiology 1992;34:107.
CP. Lateral thoracic meningocele associated with neurofibromatosis:
total excision by posterolateral extradural approach. A case report. Spine 1989;14:129.
D, Bollini G, Dungl P, et al. The fibula in congenital pseudarthrosis
of the tibia: the EPOS multicenter study. European Paediatric
Orthopaedic Society (EPOS). J Pediatr Orthop B 2000; 9(2):69–74.
FA, Gillespie R. Pseudarthrosis of the radius after fracture through
normal bone in a child who had neurofibromatosis. A case report. J Bone Joint Surg Am 1989;71:1419.
O, Ogawa R. Pseudarthrosis of the radius associated with
neurofibromatosis: report of a case and review of the literature. J Pediatr Orthop 1990;10:128.
DM, Eilert RE, Waldsten G. Congenital pseudarthrosis of the ulna: a
report of two cases and a review of the literature. J Pediatr Orthop 1985;5:463.
GP, Slongo T, Tschappeler H, et al. Congenital pseudarthrosis of the
ulna and radius in two cases of neurofibromatosis type 1. Pediatr Surg Int 2001;17(2-3):239–241.
SA, Mulvihill JJ, Nielsen A. Long-term follow-up of von Recklinghausen
neurofibromatosis. Survival and malignant neoplasms. N Engl J Med 1986;314:1010.
M, Rembeck B, Akesson HO. Life expectancy, mortality and prognostic
factors in neurofibromatosis type 1. A twelve-year follow-up of an
epidemiological study in Goteborg, Sweden. Acta Derm Venereol 1995;75:140.
R, Charrow J, Greenwald M. Emergence of optic pathway gliomas in
children with neurofibromatosis type 1 after normal neuroimaging
results. J Pediatr 1992;121:584.
y Martinez R, Martinez-Carboney R, Ocampo-Campos R, et al.
Wiedemann-Beckwith syndrome: clinical, cytogenetical and radiological
observations in 39 new cases. Genet Couns 1992;3:67.
KW, Gardner A, Williams JC, et al. Paternal origin of 11p15
duplications in the Beckwith-Wiedemann syndrome. A new case and review
of the literature. Cancer Genet Cytogen 1992;58:66.
JG, Byers JW III, Chuipek S, et al. Prenatal diagnosis and perinatal
management of the Beckwith-Wiedemann syndrome: a case and review. Am J Perinatol 1989;6:446.
PL, Siegel MJ, Craft AW. Screening for Wilm tumor in children with
Beckwith-Wiedemann syndrome or idiopathic hemihypertrophy. Med Pediatr Oncol 1999;32:200.
JM, Lejarraga H, Cameron N. The natural history of the Silver-Russell
syndrome: a longitudinal study of thirty-nine cases. Pediatr Res 1975;9:611.
SM, Stanhope R, Garrett C, et al. The spectrum of Silver-Russell
syndrome: a clinical and molecular genetic study and new diagnostic
criteria. J Med Genet 1999;36:837–842.
SH, Switzer HE. Congenital hypoplastic thumb in the Silver syndrome: a
case report and review of upper extremity anomalies in the world
literature. J Hand Surg 1983;8:480.
R, Ackland F, Hamill G, et al. Physiological growth hormone secretion
and response to growth hormone treatment in children with short stature
and intrauterine growth retardation. Acta Paediatr Scand 1989;349:47–52; discussion 53–54.
LG, Happle R, Milliken JB, et al. Proteus syndrome: diagnostic
criteria, differential diagnosis, and patient evaluation. Am J Med Gene 1999;84:389.
AS, Friedmann PA, Powers SK, et al. New observations with genetic
implications in two syndromes: (1) father to son transmission of the
Nager acrofacial dysostosis syndrome and (2) parental consanguinity in
the Proteus syndrome. Am J Hum Genet 1987;41:A43.
HR, Burgio GR, Alenjoff P, et al. The Proteus syndrome: partial
gigantism of the hands and/or feet, nevi, hemihypertrophy, subcutaneous
tumors, macrocephaly or other skull anomalies and possible accelerated
growth and visceral affections. Eur J Pediatr 1983;140:5.
MD, Hager J, Nikolaides N, et al. Proteus syndrome: musculoskeletal
manifestations and management: a report of two cases. J Pediatr Orthop 1992;12:106.
D, Hawkins CF. Nail-patella syndrome with iliac horns and hereditary
nephropathy. Necropsy report and anatomical dissection. J Bone Joint Surg Br 1967;49-B:164.
SD, Zhou G, Baldini A, et al. Mutations in LMX1B cause abnormal
skeletal patterning and renal dysplasia in nail-patella syndrome. Nat Genet 1998;19:47.
J, Macnical MF. Foot deformities associated with onycho-osteodysplasia.
A familial study and a review of associated features. Int Orthop 1985;9:135.
K, Fujii K, Tanaka T, et al. Surgical management of congenital
permanent dislocation of the patella in nail patella syndrome by
Stanisavljevic procedure. J Orthop Sci 1999;4(6): 446–449.
EJ, Hamel BC, te Slaa RL. Nephropathy in hereditary osteo-onycho
dysplasia (HOOD): variable expression or genetic heterogenity. Prog Clin Biol Res 1989;305:157.
PJ, Mulholland HC, Craig BG, et al. Cardiovascular abnormalities in the
oculo-auriculo-vertebral spectrum (Goldenhar syndrome). Am J Med Genet 1992;44:425.
Bever Y, van den Ende JJ, Richieri-Costa A. Oculo-auriculo-vertebral
complex and uncommon associated anomalies: report on 9 unrelated
Brazilian patients. Am J Med Genet 1992;44:683.
R, Trikha A, Venkataraman RK, et al. Goldenhar’s syndrome: an analysis
of anaesthetic management. A retrospective study of seventeen cases. Anaesthesia 1990;45:49.
ID, McCallum J, DeScipio C, et al. Cornelia de Lange syndrome is caused
by mutations in NIPBL, the human homolog of Drosophila melanogaster
Nipped-B. Nat Genet 2004;36:631–635.
ET, Wang T-J, Lisgo S, et al. NIPBL, encoding a homolog of fungal
Scc2-type sister chromatid cohesion proteins and fly Nipped-B, is
mutated in Cornelia de Lange syndrome. Nat Genet 2004;36:636–641.
JA, O’Hara AE, Carpenter GG. Spurs of the mandible and supracondylar
process of the humerus in Cornelia de Lange syndrome. AJR Am J Roentgenol 1977;129:156.
WI, Campbell BM. A study of six cases of de Lange Amsterdam dwarf
syndrome, with special attention to voice, speech and language
characteristics. Dev Med Child Neurol 1978;20:189.
TD, Alman BA. Developmental pathways in musculoskeletal neoplasia:
involvement of the Indian Hedgehog-parathyroid hormone-related protein
pathway. Pediatr Res 2003;53(4):539–543.
JP, Gaffield W, Kapur RP, et al. The teratogenic Veratrum alkaloid
cyclopamine inhibits sonic hedgehog signal transduction. Development 1998;125(18):3553–3562.
DF, Sandor GG, MacLeod PM, et al. Intrinsic defects in the fetal
alcohol syndrome: studies on 76 cases from British Columbia and the
Yukon Territory. Neurobehav Toxicol Teratol 1981;3:145.
L, Smith DW. The VATER association: vertebral defects, anal atresia,
T-E fistula with esophageal atresia, radial and renal dysplasia: a
spectrum of associated defects. J Pediatr 1973;82:104.
S A.; Blumenfeld, A.; Gill, S. P, et al:Tissue-specific expression of a
splicing mutation in the IKBKAP gene causes familial dysautonomia. Am. J. Hum. Genet. 68: 598–605,2001.
J, Pytel BA, Grover-Johnson N, et al. Quantitative studies of dorsal
root ganglia and neuropathologic observations on spinal cords in
familial dysautonomia. J Neurol Sci 1978;35:77.
RN, MacEwen GD. Spinal deformity associated with heritable neurological
conditions: spinal muscular atrophy, Friedreich’s ataxia, familial
dysautonomia, and Charcot-Marie- Tooth disease. J Bone Joint Surg Am 1976;58:13.
E, Floman Y, Sagiv S, et al. Orthopaedic manifestations of familial
dysautonomia. A review of one hundred and thirty-six patients. J Bone Joint Surg Am 2000;82-A(11):1563–1570.
B, Aicardi J, Dias K. A progressive syndrome of autism, dementia,
ataxia, and loss of purposeful hand use in girls: Rett’s syndrome:
report of 35 cases. Brain Dev 1985;14:479.
H, Thomson M, Glasson E. Metacarpophalangeal pattern profile and bone
age in Rett syndrome: further radiological clues to the diagnosis. Am J Med Genet 1999;83:95.
RE, Van den Veyver IB, Wan M, et al. Rett syndrome is caused by
mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 1999;23:185.
NJ, Watt HC, Hackshaw AK. Integrated screening for Down’s syndrome on
the basis of tests performed during the first and second trimesters. N Engl J Med 1999;341:461.
KA, Newton RW, Gupta S, et al. Clinical predictors and radiological
reliability in atlantoaxial subluxation in Down’s syndrome. Arch Dis Child 1991;66:876.
KS, Ball WS, Prenger EC, et al. Evaluation of the craniocervical
junction in Down syndrome: correlation of measurements obtained with
radiography and MR imaging. Radiology 1993;186:377.
T, Izawa T, Kuroki Y, et al. Follow-up study of atlanto-axial
instability in Down’s syndrome without separate odontoid process. Spine 1989;14:1149.
V, Ben-Ami T, Yousefzadeh DK, et al. Hypoplastic posterior arch of C-1
in children with Down syndrome: a double jeopardy. Radiology 1992;183:125.
SM, Scola FH, Pezzullo JC. A longitudinal study of atlanto-dens
relationships in asymptomatic individuals with Down syndrome. Pediatrics 1992;89:1194.
L, Hillstrom HJ, Palisano RJ. The effect of foot orthoses on standing
foot posture and gait of young children with Down syndrome. Neurorehabilitation 2001;16(3):183–193.
HH, Pasquariello PS, Watters WC. Multiple dislocations of the cervical
spine in a patient with juvenile rheumatoid arthritis and Down’s
syndrome. Clin Orthop 1982;162:37.
PA, Betts PR, Cockwell AE, et al. A cytogenetic and molecular
reappraisal of a series of patients with Turner’s syndrome. Ann Hum Genet 1990;54:209.
S, Weber G, Guarneri MP, et al. Effect of estrogen replacement therapy
on bone mineral content in girls with Turner syndrome. Obstet Gynecol 1992;79:747.
G, Federico G, Bertelloni S, et al. Mineral metabolism in Turner’s
syndrome: evidence for impaired renal vitamin D metabolism and normal
osteoblast function. J Clin Endocrinol Metab 1992;75:998.
JL, Long LM, Feuillan P, et al. Normal bone density of the wrist and
spine and increased wrist fractures in girls with Turner’s syndrome. J Clin Endocrinol Metab 1991;73:355.
A, Fryns JP, Kleczkowska A, et al. Intelligence, behaviour and
psychosocial development in Turner syndrome. A cross-sectional study of
50 pre-adolescent and adolescent girls (4–20 years). Genet Couns 1993;4:7.
L, Hagenfeldt K, Brondum-Neilsen K, et al. Middle-aged women with
Turner’s syndrome. Medical status, hormonal treatment and social life. Acta Endocrinol 1991;125:359.
RG, Frane J, Attie KM, et al. Six year results of a randomized,
prospective trial of human growth hormone and oxandrolone in Turner
syndrome. J Pediatr 1992;121:49.
M, Kalidas K, Shaw A, et al. PTPN11 mutations in Noonan syndrome:
molecular spectrum, genotype-phenotype correlation, and phenotypic
heterogeneity. Am J Hum Genet 2002;70:1555–1563.
HJ, Johnson C, Wagner MJ, et al. Molecular definition of the shortest
region of deletion overlap in the Langer-Giedion syndrome. Am J Hum Genet 1991;49:1197.
M, Fryns JP, Van den Berghe H. Psychological profile and behavioral
characteristics in 12 patients with Prader-Willi syndrome. Genet Couns 1990;1:141.
AL, Myers SE, Whitman BY. Growth hormone improves body composition, fat
utilization, physical strength and agility, and growth in Prader-Willi
syndrome: A controlled study. J Pediatr 1999;134:221.
CT, Curfs LM, Sastrowijoto P, et al. Prader-Willi syndrome: causes of
death in an international series of 27 cases. Am J Med Genet 2004;124A(4):333–338.
S, Tomatsu S, Masue M et al., Mucopolysaccharidosis type IVA. N-acetyl
galactosamine-6-sulfate sulfatase exonic point mutations in classical
Morquio and mild cases. J Clin Invest 1992;90:1049.
WD, Jackson CE, Desnick RJ. Mucopolysaccharidosis type VI:
identification of three mutations in the arylsulfatatase B gene of
patients with severe and mild phenotypes provides molecular evidence
for genetic heterogeneity. Am J Hum Genet 1992;50:795.
G, Guffon N, Maire I, et al. Outcome of 27 patients with Hurler’s
syndrome transplanted from either related or unrelated haematopoietic
stem cell sources. Bone Marrow Transpl 2003;31(12):1105–1117.
E, Peters C, Krivit W, et al. Genu valgum deformity in Hurler syndrome
after hematopoietic stem cell transplantation: correction by surgical
intervention. J Pediatr Orthop 1999;19:270.
J, Thomas PS. Clinical findings in 12 patients with MPS IVA (Morquio’s
disease): further evidence for heterogeneity. Part III: odontoid
dysplasia. Clin Genet 1988;33:126.
JM, Kendall BE, Crockard HA, et al. The odontoid process in
Morquio-Brailsford’s disease. The effects of occipitocervical fusion. J Bone Joint Surg Br 1991;73:851.
JM, Kendall BE, Crockard HA. The odontoid process in Morquio-Brailsford
disease. The effects of occipitocervical fusion. J Bone Joint Surg Br 1991;73:851.
F, McLaren AT, Slowie DF. Report on a case of Hutchinson-Gilford
progeria, with special reference to orthopedic problems. Eur J Pediatr Surg 1992;2:378.
JG, Reed SD, McGillvray BC, et al. Part II. Amyoplasia: twinning in
amyoplasia—a specific type of arthrogryposis with an apparent excess of
discordantly affected identical twins. Am J Med Genet 1983;15:591.
WL, Glinski LP, Kirkpatrick SJ, et al. Further evidence that
arthrogryposis multiplex congenita in the human sometimes is caused by
an intrauterine vascular accident. Teratology 1992;45:345.
L, Polizzi A, Morriss-Kay G. Plasma from human mothers of fetuses with
severe arthrogryposis multiplex congenita causes deformities in mice. J Clin Invest 1999;103:1038.
IH, Yang MS, Chung CY, et al. The treatment of recurrent arthrogrypotic
club foot in children by the Ilizarov method. A preliminary report. J Bone Joint Surg Br 2001;83(5):731–737. PMID: 11476315 [PubMed – indexed for MEDLINE].
T, Terashima Y, Murachi S, et al. Clinical features and treatment of
joint dislocations in Larsen’s syndrome. Report of three cases in one
family. Clin Orthop 1976;119:206.
D, Robertson SP, King LM, et al. Mutations in the gene encoding filamin
B disrupt vertebral segmentation, joint formation and skeletogenesis. Nat Genet 2004;36(4):405–410 [Epub ahead of print].
P, De Borre L, De Smet L, et al. Asymmetrical Larsen syndrome in a
young girl: a second example of somatic mosaicism in this syndrome. Genet Couns 2003;14(1):95–100.
SS, Brassington A-ME, Grannatt K, et al. Mutations in genes encoding
fast-twitch contractile proteins cause distal arthrogryposis syndromes.
Am J Hum Genet 2003;72:681–690.
MM, Hertz M, Goodman RM. A new syndrome with camptodactyly, joint
contractures, facial anomalies, and skeletal defects: a case report and
review of syndromes with camptodactyly. Clin Genet 1984;26:342.
H, Tarawneh M. The whistling face syndrome, or craniocarpaltarsal
dysplasia. Report of two cases in a father and son and review of the
literature. J Pediatr Orthop 1983;3:364.
JJ, Delaney JR, Reaume C, et al. Electromyography of oralfacial
musculature in craniocarpotarsal dysplasia (Freeman-Sheldon syndrome). Clin Genet 1974;6:132.
Die-Smulders CE, Vonsee MJ, Zandvoort JA, et al. The lethal multiple
pterygium syndrome: prenatal ultrasonographic and postmortem findings. Eur J Obstet Gynecol Reprod Biol 1990;35:283.
NG, Vermeij-Keers C, Bruijn JA, et al. Case of lethal multiple
pterygium syndrome with special references to the origin of pterygia. Am J Med Genet 1989;33:537.