
IX – PEDIATRIC DISORDERS > CHAPTER 180 – BONE DYSPLASIAS, METABOLIC
BONE DISEASES, AND GENERALIZED SYNDROMES
in which the structure of the bone is inherently abnormal, thus
altering the growth of affected individuals. The trunk and extremities
are abnormally sized, leading to disproportionate short stature,
defined as a height less than the third percentile for the individual’s
chronologic age. Some of the dysplasias are genetically transmitted,
whereas others occur sporadically.
made clinically. Short stature should be noted when present, and body
proportion and trunk and limb shortening may be helpful. The various
definitions of skeletal dysplasias are shown in Table 180.1. If limb shortening is most notable in the proximal segments (i.e., humerus or femur), the term rhizomelic can be applied. Shortening of the midportion of the limb is termed mesomelic, and acromelic
describes distal shortening. The area of the bone most disturbed by the
dysplasia can help establish the diagnosis. For example, multiple
epiphyseal dysplasia (MED) affects the epiphyses, whereas the
metaphysis is most involved in the various forms of metaphyseal
chondrodysplasia. The presence or absence of spinal involvement also is
helpful in reaching a diagnosis.
 |
|
Table 180.1. Definitions
|
in fibrous dysplasia, may aid in the diagnosis. In addition,
identifying a specific dysplasia may lead to the identification and,
therefore, treatment of associated medical conditions. For example,
patients with nail-patella syndrome are at increased risk for renal
failure, the onset
of which is insidious and would go unnoticed if not for the proper surveillance due to the known association with the syndrome (94).
the diagnosis in difficult cases. Genetic counseling may be of interest
to the patient and family. Some dysplasias are unclassifiable and
should be treated on an individual basis.
understanding of the mechanisms of bony abnormalities in these
conditions. Although gene replacement treatment is not yet possible,
research is progressing rapidly in this direction. See Dietz and
Mathews (65) for an excellent review of the genetic basis of the inherited skeletal dysplasias.
It is inherited in an autosomal dominant pattern, although most cases
are the result of spontaneous mutations. Molecular genetic research has
found that there is a point mutation in the gene that encodes
fibroblast growth factor receptor 3 (19,32,229), located on the short arm of chromosome 4 (183).
Achondroplasia is characterized by a rhizomelic pattern of involvement,
with the humerus and femur affected more than the distal extremities.
Prenatal diagnosis is possible by monitoring the growth of the femur
during the second trimester (165).
Intramembranous bone formation is not affected. Pathologic study of the
growth plate shows marked abnormalities in the zone of hypertrophy,
with loss of normal columnation of chondrocytes and accumulation of
excess matrix (171). Periosteal bone formation
is histologically normal. Additionally, the epiphysis itself is not
affected by the dysplasia; therefore, there is no predisposition toward
early degenerative arthritis.
recognizable at birth. The baby is short limbed and has a
disproportionately large head. Trunk length is normal. Facial features
include a flattened nasal bridge, prominent mandible, and enlarged
forehead. The hands characteristically have a space between the long
and ring fingers, referred to as the “trident hand.” Elbow flexion
contractures and radial head dislocation may be present. The lower
limbs may be bowed, and the musculature appears enlarged. Thoracolumbar
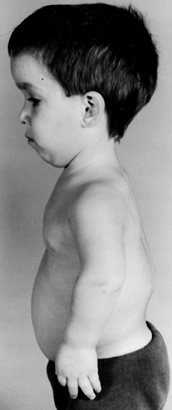
kyphosis may be present in infants (Fig. 180.1).
 |
|
Figure 180.1.
Four-year-old boy with achondroplasia. Note the prominent forehead, flexion contractures of the elbow, trident hand, and thoracolumbar kyphosis. |
The metaphysis is widened, and the epiphysis is relatively normal. The
long bones of the leg may be bowed. The pelvis is wide but short, with
small sciatic notches. The shape of the inner pelvis has been described
as a “champagne glass” appearance. The radiographic hallmark of the
achondroplastic spine is progressive narrowing of the transverse
interpedicular distance as one measures from cephalad to caudad in the
lumbar spine (Fig. 180.3). The pedicles are thickened, and there may be posterior scalloping of the vertebral bodies.
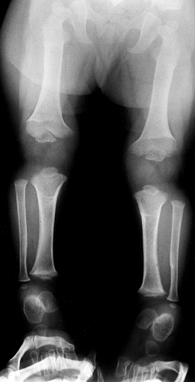 |
|
Figure 180.2. One-year-old girl with achondroplasia. The physis of the distal femur appears as an inverted V.
|
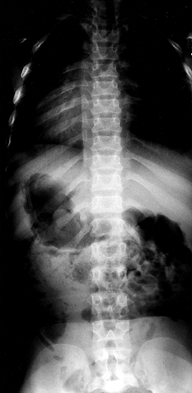 |
|
Figure 180.3. Progressive interpedicular narrowing in the lumbar spine of a 5-year-old girl with achondroplasia.
|
magnum has been described (127,167,265).
Symptoms include sleep apnea (more specifically, central hypoapnea),
and neurologic examination may reveal hypotonia (which is common in all
infants with achondroplasia) or clonus. When compression is noted,
posterior surgical decompression of the foramen magnum is recommended
to prevent sudden death (265,269).
Other neurosurgical concerns include Chiari malformations at the
craniocervical junction and hydrocephalus, which occasionally requires
shunting (76). Sleep apnea can also occur
because of upper airway obstruction, and sleep studies may be able to
identify those children whose respiratory compromise is due to
craniocervical abnormalities and those due to obstructive airway
problems (254,255,272).
Because the orthopaedist often is involved in the care of the infant
with achondroplasia, awareness of these conditions is imperative if
appropriate neurosurgical referral is to be made.
Kyphosis occurs nearly universally in the young baby. Possible causes
include ligamentous laxity, hypotonia, enlarged head size, and hip
flexion contractures. In most cases, the kyphosis resolves as the child
begins to walk. A recent theory, popularized by Hall (100) and Pauli et al. (168),
proposes that unsupported sitting by the hypotonic infant leads to the
development of thoracolumbar kyphosis. The authors advocate prohibiting
unsupported sitting to prevent the kyphosis from occurring, and early
brace treatment with a thoracolumbosacral orthosis (TLSO) for those
babies who do develop kyphosis. Other authors have recommended using a
brace if kyphosis persists beyond 2 years of age. For refractory cases,
surgical treatment consisting of anterior and posterior fusion without
instrumentation may be necessary. Progressive kyphosis and kyphosis
measuring greater than 40° at 5 years of age are indications for
surgery (237). Fusion is generally obtained
without instrumentation, because the narrowed canal and kyphotic
deformity predispose the patient to neurologic injury and paraplegia if
hardware is introduced (see Chapter 158 and Chapter 161).
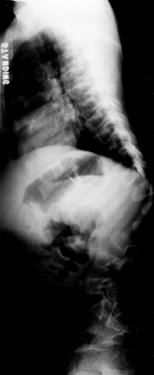 |
|
Figure 180.4. Persistent thoracolumbar kyphosis in a 4-year-old boy with achondroplasia.
|
Symptoms include leg and back pain (neurogenic claudication), lower
extremity weakness, and loss of endurance during ambulation. The
patient attempts to relieve pain during walking by hunching over to
reduce the lumbar lordosis, which
produces
more space within the spinal canal. The decreased space within the
spinal canal is due to narrowing and thickening of the pedicles,
hypertrophy of the facets, and enlargement of the laminae (132).
Magnetic resonance imaging (MRI) is useful in visualizing the extent of
the stenosis. The condition is treated by posterior decompression, with
wide laminectomy. The surgeon should attempt to preserve the facets,
but this may not be possible if the stenosis is severe. Primary fusion
usually is not necessary.
Tibial osteotomy remains the treatment of choice for patients with
symptomatic or cosmetically objectionable varus. Epiphysiodesis of the
fibula has had mixed results (see Chapter 169).
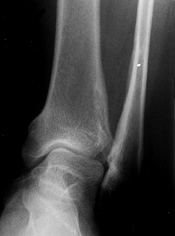 |
|
Figure 180.5. Relative fibular overgrowth in a 13-year-old girl with achondroplasia.
|
controversial. There has been enthusiasm, particularly in Europe, for
the application of the Ilizarov technique for lengthening the limbs in
children with achondroplasia (88,267) (see Chapter 171).
Treatment with recombinant growth hormone has been investigated and
found to increase growth in some individuals with achondroplasia (111,156,257,266).
dwarfism, which is inherited in an autosomal recessive pattern. Its
gene, located on chromosome 5, is responsible for the sulfation of
proteoglycans, a crucial component of cartilage (230). Prenatal DNA testing can establish the diagnosis. Histologic study of the growth plate shows abnormalities in collagen (221) and decreased numbers of chondrocytes.
very short limbs. There is flattening of the nasal bridge and a
puffy-cheeked appearance. Cleft palate is frequently present. Severe
clubfoot deformities are always present, and joint contractures are
common. The thumb, which is radially deviated and short, has been
described as a “hitchhiker thumb.” Within the first months of the
child’s life, the pinnae of the ears become swollen and ossify. The
crumpled and enlarged pinnae have been termed “cauliflower ear” (249).
In some affected children, tracheomalacia may be life-threatening
during the neonatal period. The child’s intelligence is normal.
the epiphyses. When the epiphyses do ossify, they are irregular and may
be flattened (especially at the proximal femur) (244).
Coxa vara is common and may result in hip dislocation. The long bones
are short but appear thickened. Scoliosis is seen in older children (Fig. 180.6).
The first metacarpal is short in relation to the rest of the hand and
is triangular in shape, leading to the development of the hitchhiker
deformity.
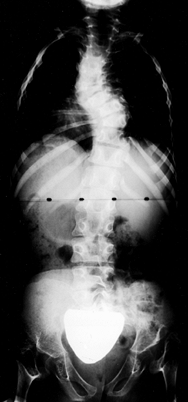 |
|
Figure 180.6. Scoliosis in an 8-year-old girl with diastrophic dysplasia. Note the bilateral hip dislocations.
|
are resistant to casting and are prone to early recurrence after
surgical intervention. Postoperative bracing is
used in these children. Repeated surgical releases of the feet are difficult but should be aggressive (see Chapter 167). Other foot deformities are also seen in patients with diastrophic dysplasia (190).
Joint contractures of the hip and knee may not be treatable with
soft-tissue release. Osteotomy is required for fixed deformity. Hip
dislocation is common and is difficult to treat. Because of the
epiphyseal abnormalities, premature degenerative arthritis does occur.
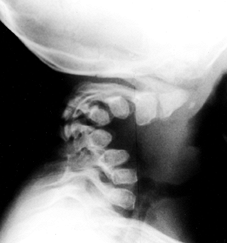 |
|
Figure 180.7. Cervical kyphosis in a 2-year-old girl with diastrophic dysplasia.
|
autosomal dominant and recessive inheritances. The two most common
forms are the Schmid and McKusick types. The McKusick form is also
known as cartilage hair hypoplasia. The primary defect is an
abnormality in the physis of the zone of primary calcification, with
clusters of chondrocytes protruding into the metaphysis. The gene
coding for type X collagen, which is present in hypertrophic
chondrocytes located at the growth plate, is abnormal in Schmid
metaphyseal chondrodysplasia (250,253).
Coxa vara can be seen in the Schmid form, and cervical instability may
be seen in the McKusick form. The long bones may be bowed, leading to
angular deformities. The epiphyses are normal.
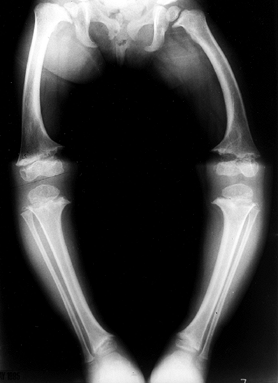 |
|
Figure 180.8.
Radiograph of the lower extremities of a 4-year-old girl with metaphyseal chondrodysplasia. Genu varum and widened physes are seen. |
symptomatic or progressive angular deformities. Patients with the
McKusick form, which has been linked with immunodeficiency, have
recently been treated by bone marrow transplantation (23,245).
common of the bone dysplasias. It is inherited by autosomal dominant
transmission and has been found to be genetically variable among
families (60).
of the epiphyses with a delay in their ossification. Radiographically,
they appear mottled and irregular. The
hips, knees, and ankles are most frequently involved; in the upper extremity, the shoulder is most commonly affected (113). The spine is essentially normal. In the differential diagnosis of bilateral Legg-Calvé-Perthes disease, always include MED (2)
and perform radiographs of the knees and ankles. An abnormal epiphyseal
height-to- metaphyseal width ratio for the distal femur has been found
in most children with MED and has been proposed as helpful in early
diagnosis (246). Another characteristic finding is the double-layered appearance of the patella on lateral radiographs (205). Avascular necrosis of the femoral head frequently occurs in patients with MED (133).
Joint pain may appear in the first decade or remain quiescent until
early adult life. Treatment is the same as that for osteoarthritis,
consisting of joint replacement for advanced cases (238).
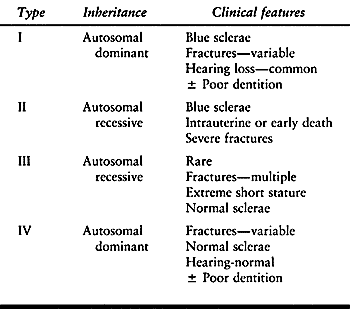
transmitted dysplasias in which the bones are extremely fragile,
predisposing the patient to multiple fractures. Several classifications
of the disease exist (124). In 1906, Looser
divided OI into congenita and tarda forms, with the specific type
determined by whether the patient developed fractures at birth or
later. The most commonly applied classification is that by Sillence (208),
who categorized patients based on how the condition was inherited
(i.e., autosomal dominant or recessive) and on the presence of specific
clinical features (Table 180.2).
 |
|
Table 180.2. Sillence Classification of Osteogenesis Imperfecta
|
Mutations are almost always present, involving either the COL1A1 or
COL1A2 genes, which encode the procollagen. In affected individuals,
the organization of type I collagen is in disarray. In type I OI, there
is a quantitative abnormality in the amount of type I collagen; in the
other forms of OI, there are both quantitative and qualitative
anomalies in type I collagen (44).
Abnormalities can be seen in dermal fibroblasts obtained from skin
biopsies, in which the ratio of type I to type III collagen (which is
uninvolved in patients with OI) is compared with age-matched normal
specimens. Routine use of skin biopsy for diagnosis is discouraged,
however, because the
history and physical examination usually are sufficient to establish the diagnosis (224).
Histologically, there is a relative increase in woven bone that does
not mature to lamellar bone. The osteocyte number is increased.
Trabeculae are thin and poorly arranged, and the haversian canal system
does not develop. Bone mineral density is decreased on dual energy
x-ray absorptiometry (DEXA) scans, even in milder forms of the disease (271).
from that of stillborn babies with numerous fractures and intracranial
bleeds to active adolescents with a history of several fractures
occurring at various times during their childhood. Musculoskeletal
findings include short stature, bowing of the extremities, and
scoliosis. Blue sclerae are seen in Sillence type I and II patients,
and in some type III patients, with the color most intense and
long-lasting in type I patients (209). Dentition may be poor due to defective dentin, a condition known as dentinogenesis imperfecta.
Hearing loss may occur secondary to middle ear involvement. The face is
trefoil shaped. Clinical findings in patients with type IV OI may be
quite subtle, and the differential diagnosis between OI and child abuse
can be vexing.
well. Fractures may be seen throughout the skeletal system. Coxa vara
is a common sequela of femoral neck fracture. Protrusio acetabuli can
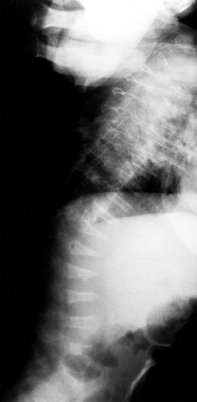
develop. Wormian bones are seen on skull radiographs. The vertebrae may be biconcave, and compression fractures may be present (Fig. 180.9).
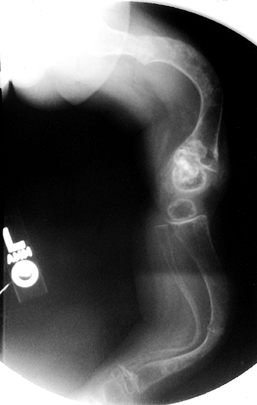
The long bones are thin and osteopenic. The femur may have a
“concertina” appearance, that is, a crumpled shape due to multiple
fractures. There is widening of the metaphyses, and growth arrests may
occur. Irregular calcifications may extend into the metaphysis, a
condition referred to as “popcorn epiphysis” (Fig. 180.10).
 |
|
Figure 180.9. Flattened vertebral bodies throughout the spine of a 1-year-old boy with severe osteogenesis imperfecta.
|
 |
|
Figure 180.10.
Severe bowing, multiple healed fractures, and epiphyseal calcifications in the lower extremity of a child with severe osteogenesis imperfecta. |
delayed. Physical therapy has been useful in helping the patient
develop head and trunk control, and when therapy is used in combination
with surgery and bracing, sitting and ambulation may be improved (27). The ability of the child to sit independently by age 10 months has been correlated
with achieving walking as a primary means of mobility (55).
well as to attempt to prevent fractures. Fractures usually heal within
a normal time interval, but the tendency to refracture persists. Disuse
osteopenia further weakens the bones, so immobilization should be kept
to the minimum time necessary to ensure fracture healing. Orthoses may
be needed on a long-term basis to assist in weight bearing and to
reduce the incidence of fractures (89). Nonunions may occur (867).
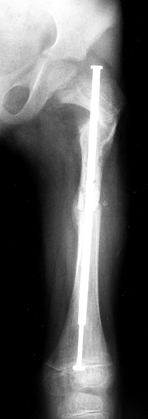
maintain limb alignment. Intramedullary fixation for load sharing, in
combination with multiple osteotomies, was popularized by Sofield and
Miller (218). Telescoping rods are technically
more challenging to insert, but because they grow with the child, the
chances of refracture with growth are lessened (13,14) (Fig. 180.11). Gamble et al. (87)
showed that although the Bailey-Dubow rod was difficult to insert
properly, it was beneficial in providing internal support to growing
bones. Other authors also have published favorable results using the
Bailey-Dubow device but state that the hardware does require periodic
revision during patient growth (154,270).
 |
|
Figure 180.11. Bailey-Dubow elongating rod in the femur of a 7-year-old boy with osteogenesis imperfecta.
|
 |
|
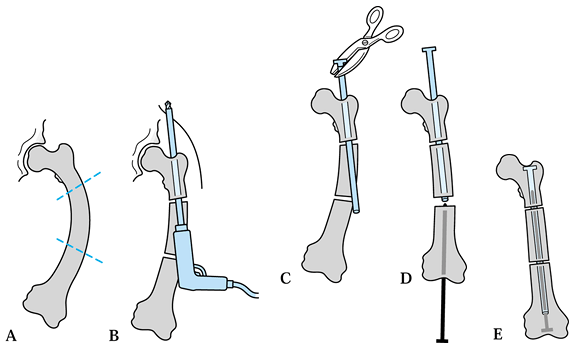
Figure 180.12. Surgical technique for inserting Bailey-Dubow elongation rod. A: Bowed femur and planned osteotomies. B: Hollow rod drilled into proximal fragment and out a small incision in buttock. C: Exchange of drill bit for T-piece; crimp with pliers. D: Solid rod inserted through knee. E: Rods engaged, advanced fully, and buried in femoral epiphysis.
|
-
Preoperatively, check available lengths of rods.
-
Type and crossmatch blood.
-
Position the patient in the lateral decubitus position on a radiolucent table and drape the lower extremity free.
-
Perform a gentle subperiosteal exposure of the femur.
-
Perform multiple osteotomies where needed to pass a straight rod.
-
Ream the medullary canal to allow rod passage.
-
Drill the female end of the rod into
proximal fragment in a retrograde direction through the fracture or
osteotomy, exiting just medial to the greater trochanter. -
Exchange the drill bit for a T-piece through the proximal skin incision.
-
Crimp the T-piece onto the female end of the rod using pliers.
-
Perform a perapatellar exposure of the femoral intercondylar notch at the knee.
-
Insert the male solid rod up through the femoral notch.
-
Engage the male rod into the female rod.
-
Tap each rod in fully.
-
Twist the male rod 90° within the femoral epiphysis to deter distal migration into the knee.
-
Close and dress the incisions and apply a spica cast.
the severely affected groups of patients. Biconcave deformity of the
vertebral bodies has been linked with the early onset of scoliosis (114). Chest cage deformity may result from bracing because of rib fragility (21), and curve progression is not halted by brace wear (102,268).
Treatment is surgical but is fraught with technical problems. Fixation
of the spine with conventional spinal hardware is difficult to achieve
because of osteopenia. Methylmethacrylate augmentation and the use of
mersilene tapes have been proposed as means to assist in fixation (21). Fusion in situ is the norm, because correction is very difficult to achieve.
found in as many as 25% of patients (210)
and is often associated with macrocephaly and hydrocephalus. Clinical
signs of basilar invagination include nystagmus, cranial nerve palsies,
papilloedema, and facial spasm. Surgical treatment is difficult (104). In severely involved patients, basilar invagination may lead to early death due to respiratory arrest (143).
complicated. There is a predisposition to hyperthermia that is
biochemically different from the classic malignant hyperthermia (172). Blood loss during surgery may also be massive (219).
vitamin therapy, and calcitonin have been tried in the past. Current
research is aimed at treating OI at the molecular level (90,139).
characterized by the lack of N-Ac-Gal-6 sulfate sulfatase, a lysosomal
enzyme. Deficiency of this enzyme causes an increase in urinary keratan
sulfate (112). The syndrome is inherited in an autosomal recessive pattern.
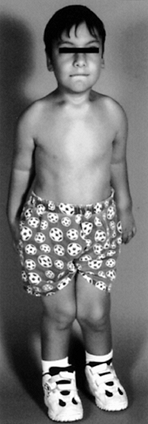
are babies. The clinical features appear later, as glycosaminoglycans
accumulate (Fig. 180.13). Signs of Morquio’s
syndrome include short-trunk dwarfism, ligamentous laxity, and genu
valgum. Facial features are not striking. Kyphosis of the thoracic
spine and pectus carinatum may be present. The patient’s intelligence
is normal. Hepatosplenomegaly is not associated with Morquio’s syndrome.
 |
|
Figure 180.13. Clinical appearance of a 6-year-old boy with Morquio’s syndrome.
|
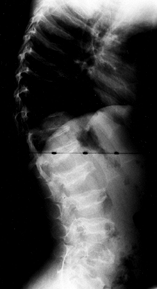
Platyspondyly is present. Odontoid hypoplasia, resulting in
atlantoaxial instability, is common and the surgeon should look for it (152).
The epiphyses are irregular and flattened, and may fragment in the
weight-bearing joints. The bases of the metacarpals are pointed, and
the bones of the hand are short and thick (196). The radiographic appearance is very similar to that seen in spondyloepiphyseal dysplasia.
 |
|
Figure 180.14. Platyspondyly and vertebral irregularities in a 6-year-old boy with Morquio’s syndrome.
|
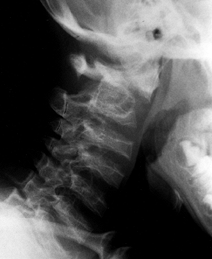
but the surgeon must be cautious and confirm that the cervical spine is
not the cause of the changes in ambulation. Anesthesiology concerns are
related to possible cervical instability. Life expectancy usually is
normal.
 |
|
Figure 180.15. Odontoid hypoplasia and atlantoaxial instability in a 6-year-old boy with Morquio’s syndrome.
|
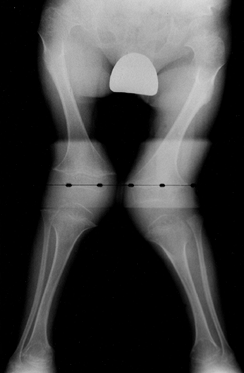 |
|
Figure 180.16. Genu valgum in a child with Morquio’s syndrome.
|
more common forms of dysplasia, occurring in 9 per million people. It
is genetically transmitted as an autosomal
dominant trait. Severity of involvement varies among family members, but penetrance is 100% (260).
Masses develop in those bones that are formed by endochondral
ossification, with a predisposition for the long bones, scapula, ribs,
and pelvis.
continuity with the metaphysis of the parent bone. The exostosis is
covered by a cap of hyaline cartilage, usually less than 3 mm in
thickness. The deep surface of the cartilaginous cap is involved in
endochondral ossification, leading to growth of the lesion.
first decade of life. Multiple bumps, which are usually nontender, may
be palpable throughout the skeleton. The patient tends to be short in
stature. The spine is rarely affected (1,9,157).
Limb-length discrepancy and angular deformity are common because of
disturbance of normal longitudinal growth. Scapular lesions can lead to
“winging” (56).
metaphysis of the long bones, along the borders of the scapula, on the
ribs, or originating from the iliac apophysis. The exostoses may be
sessile, with a broad base or origin, or they may be pedunculated,
arising from the bone on a bony stalk. The medullary canal of the mass
is in continuity with the canal of the long bone. There is a
predilection for the masses to appear at the end of the bone that grows
fastest (e.g., the distal rather than the proximal radius) and for
greater involvement of the smaller bone of two-bone limbs (e.g., the
ulna more so than the radius) (Fig. 180.17). Pedunculated lesions typically point away from the epiphysis.
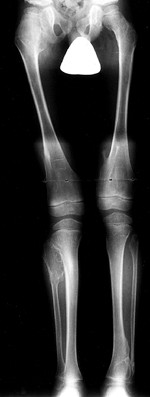 |
|
Figure 180.17. Multiple osteochondromatosis of the femur, tibiae, and fibulae in a 6-year-old boy.
|
Excision may be postponed until adolescence, when multiple symptomatic
lesions can be excised under one anesthetic. Angular deformities may
require correction, especially at the knee, where valgus is common (168). Genu valgum may lead to patellofemoral instability (151).
Compression of surrounding structures may occur owing to the size of
the osteochondromas. The peroneal nerve is particularly prone to
compression from masses and is susceptible to injury during surgical
excision of proximal fibular lesions (38,264). Proximal tibial lesions may irritate the pes anserinus (81). Large exostoses of the distal tibia can compress the adjacent fibula, leading to erosion and deformity (57,214).
quite common, consisting of distal ulnar and radial osteochondromas,
shortening of the ulna, tilt of the distal radial epiphysis, and ulnar
translation of the carpus (Fig. 180.18). Dislocation of the radial head may occur because of the ulnar shortening (117).
At present, treatment of osteochondromatosis of the forearm is
controversial. Some authors advocate early excision of osteochondromas
around the wrist, believing that such treatment prevents subsequent
deformity and radial head dislocation (169,170).
However, a recent study comprising 78 patients with forearm
osteochondromas found that function was not compromised and that
cosmetic concerns were rare in those patients who did not have early,
aggressive treatment of their exostoses (10).
Outcome studies agree that deformities of the upper extremity in
patients who have hereditary multiple exostoses are well tolerated and
cause little loss of function as measured both objectively and
subjectively (222). When osteochondromas result
in progressive discrepancy in length between the radius and the ulna,
and forearm rotation becomes painful, osteotomy of the bowed radius can
be performed with satisfactory but not perfect results. Acute
lengthening and gradual lengthening of the ulna (via the Ilizarov
technique) have both been described (54,256). Radial head excision is reserved for pain relief in skeletally mature patients.
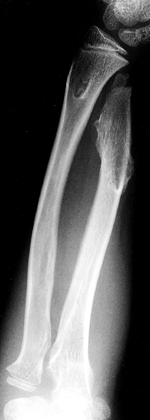 |
|
Figure 180.18. Shortening of the ulna and translation of the carpus due to multiple osteochondromatosis.
|
chondrosarcoma occurs in less than 1% of affected individuals. The
physician should become clinically suspicious when a lesion increases
in size or becomes painful
after
skeletal maturity. The cartilage cap of a benign osteochondroma is
quite thin; a cap greater than 1 cm in thickness suggests malignant
transformation. Accurate measurements of cartilage cap thickness can be
made using MRI or ultrasound scans (135).
Even if a chondrosarcoma occurs, such malignancies are late to
metastasize; usually respond well to wide resection; and have a good
prognosis, particularly in long bone lesions (see Chapter 127 and Chapter 128).
which masses of cartilage form within the metaphyses of tubular bones
and the pelvis. Lesions may be single or multiple and involve only a
single extremity, or they may affect multiple extremities. Asymmetry is
a hallmark of the disease, with one side of the body markedly more
involved than the other side. Multiple enchondromatosis is known as
Ollier’s disease; enchondromatosis with multiple hemangiomas is known
as Maffucci’s syndrome.
lesions. Mitotic figures may be present but should be few in number.
Grossly, the lesions appear as gray-white masses amid the bone and may
feel gritty due to calcification.
Calcification can be seen within the lesion. The cortex of the bone may
expand with the lesion, especially in the phalanges of the hand. The
metaphysis becomes widened and trumpet shaped. Angular deformity and
shortening are seen (179) (Fig. 180.19).
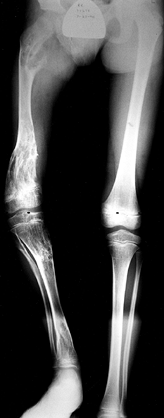 |
|
Figure 180.19. Unilateral enchondromatosis in an 8-year-old boy.
|
deformity and equalizing limb length. Osteotomy addresses angulation,
and lengthening procedures are useful for shortened extremities.
Regenerate formation is adequate during lengthening despite the
presence of enchondromas.
bone is replaced by abnormal fibrous tissue. One bone (monostotic
fibrous dysplasia) or multiple bones (polyostotic fibrous dysplasia)
may be affected. In the polyostotic form, usually one side of the
patient’s body is involved to a greater degree than the other side. The
disease can be associated with endocrine abnormalities and
hyperpigmented skin lesions, known as McCune-Albright syndrome (41,126,129). Precocious puberty is the hallmark of the McCune-Albright variant.
Multiple immature fragments of woven bone are seen within the stroma,
an appearance similar to that of alphabet soup. The bone has widened
osteoid seams and does not mature into lamellar bone.
the patient’s first two decades of life. Presenting signs include pain,
limp, deformity, or pathologic fractures. Involvement of the skull and
facial bones can occur in varying degrees. Leg-length discrepancy is
common.
a ground-glass appearance because of the calcification of immature
bone. The cortex is thinned, and the shaft of the involved bone may
expand. Lesions are metaphyseal or diaphyseal in location. Coxa vara
with “shepherd’s crook” deformity of the proximal femur is a classic
radiographic finding (Fig. 180.20). Isolated
lesions may be mistaken for simple bone cysts, particularly in the
humerus. Spinal involvement, usually associated with neurologic
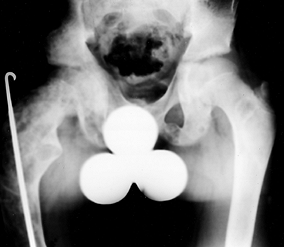
compromise, has been described (73,96,150,174). Because the diagnosis can usually be made from radiographs, biopsy is rarely necessary.
 |
|
Figure 180.20.
Extensive fibrous dysplasia of the femur and pelvis in a 13-year-old boy. Note the “shepherd’s crook” deformity of the proximal femur. |
and on the biomechanical implications of the weakened bone. Pain,
progressive deformity, and impending pathologic fracture are
indications for surgery. Closed treatment of fractures due to fibrous
dysplasia usually is sufficient for the upper extremities but is rarely
successful for the lower extremities, for which the demands of weight
bearing require strong bony support (225). Excision and curettage rarely work because bone graft is quickly replaced by the abnormal fibrous tissue (97).
When bone grafting is necessary, cortical strut grafts (e.g., the
fibula) take longer to resorb and thus provide more structural support (35,75).
Osteotomy to correct coxa vara, combined with curettage and grafting of
the femoral neck (usually with cortical bone), is performed when the
calcar becomes progressively thin, leading to coxa vara and a painful
limp. Internal fixation should be used, but extension of the lesion up
into the femoral neck makes standard fixation tenuous (97).
Osteotomies to correct deformity and intramedullary fixation,
particularly using reconstruction nails, which provide fixation into
the femoral head and neck, can help restore alignment and support in
the femur and tibia (82). Because the medullary
canal in fibrous dysplasia is less distinct and very vascular, the
surgeon should anticipate difficulties when using closed techniques as
well as increased blood loss during intramedullary fixation.
Osteotomies tend to heal reasonably well, and nonunions are uncommon.
require surgical equalization. For moderate discrepancies,
contralateral epiphysiodesis is far simpler than lengthening procedures.
Malignant tumors have been described as having originated from both the
monostotic and the polyostotic forms of fibrous dysplasia (105,189,195).
A 3% rate of malignant transformation has recently been reported by the
Mayo Clinic, with the majority of tumors being osteosarcomas (189). Degeneration of fibrous dysplasia into aneurysmal bone cysts has also been described (153) (see Chapter 127).
most prevalent skeletal dysplasia transmitted by a single gene. It is
inherited in an autosomal dominant pattern with variable expression,
although the disease is believed
to occur due to a new mutation in half of the affected individuals (248). The estimated prevalence is 1 per 1000 live births.
(NF-I) is the more common form and is characterized by peripheral
neurofibromas, skeletal involvement, and “café au lait spots.” The
genetic locus for NF-I has been localized to chromosome 17q11.2, an
area that encodes for the protein neurofibromin (98,206). This protein is present in several organ systems and is believed to be a tumor suppressor (249).
Neurofibromatosis type II (NF-II) manifests as central neurofibromas
with bilateral acoustic neuromas and usually presents in the third or
fourth decade of life (142). The gene for NF-II has been localized to chromosome 22 and encodes for the protein schwannomin (131).
palisading arrangement of densely packed fibrous bundles and spindle
cells. Grossly, the masses are firm and pale.
neurofibromatosis. The National Institutes of Health has developed
specific criteria for the diagnosis of neurofibromatosis (Table 180.3).
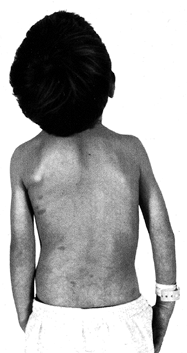
The most common skin lesion is the café au lait spot, a hyperpigmented
irregular area with a smooth border that varies in size and shape (Fig. 180.21).
These spots usually are not present immediately after birth but appear
and increase in number throughout childhood. More subtle skin findings
in patients with neurofibromatosis include axillary or inguinal
freckling. Fibroma molluscum are superficial, raised nodules that
represent dermal neurofibromas. They are not seen in prepubertal
children. Nevi are heavily pigmented lesions that may present
unilaterally. The lesions may be hypersensitive and may overlie
plexiform neurofibromas. Plexiform neurofibromas are large nerve tumors
that are locally invasive, feel like a “bag of worms,” and have the
potential for malignant transformation.
 |
|
Table 180.3. NIH Criteria for Diagnosis of Neurofibromatosis
|
 |
|
Figure 180.21. Multiple café au lait spots in a 3-year-old boy with neurofibromatosis and scoliosis.
|
Three types of scoliosis exist. The first is an idiopathic-appearing
curve without significant bony changes or kyphosis. A second form is
the dysplastic type, a short, sharp curve in which six or fewer
vertebrae are involved. The dysplastic curve occurs in up to 72% of
patients with neurofibromatosis and scoliosis (212).
The vertebrae may assume a wedge shape, rotation usually is severe, and
penciling of the apical ribs is characteristic. The neural foramen may
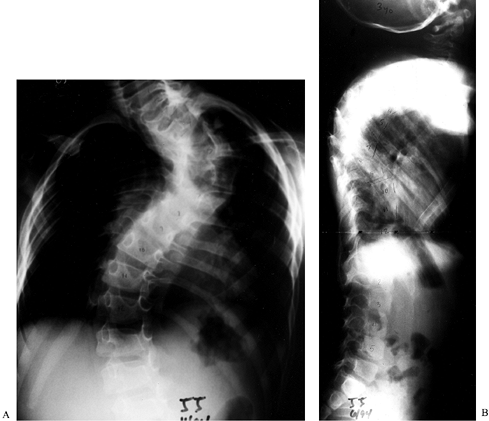
be expanded owing to pressure from dumbbell neurofibromata. A third
form of scoliosis is a dysplastic curve with associated hyperkyphosis (Fig. 180.22A, Fig. 180.22B).
 |
|
Figure 180.22. PA (A) and lateral (B) radiographs of a dysplastic kyphotic curve in a 3-year-old boy with neurofibromatosis.
|
be seen, and the physician should rule out the disease whenever
evaluating a child with hemihypertrophy. Congenital pseudarthrosis of
the tibia and forearm may be present. Protrusio acetabuli is also seen
in patients with neurofibromatosis.
neurofibromatosis most often involves the spine. Treatment of the
idiopathic-type curve is similar to the treatment of adolescent
idiopathic scoliosis (see Chapter 155,
Chapter 156, and Chapter 158).
Bracing is used for progressive curves in growing patients, with spinal
fusion reserved for larger curves. Dysplastic curves have a more
ominous prognosis, with rapid progression commonly seen. Those curves
that are most prone to progression occur in younger patients, are
greater in magnitude at presentation, are associated with kyphosis, and
have vertebral and rib erosive changes (84). Spinal fusion was often complicated by pseudarthrosis (50,212), leading to the current recommendation of combined anterior and posterior spinal fusions to treat these curves (263). Surgery should not be delayed in young children with dystrophic curves (37).
Even with anterior and posterior fusion, some dystrophic curves
continue to progress, requiring difficult subsequent revision
procedures (50,110,118,261).
technically challenging for several reasons. First, the bone itself is
often soft due to erosion from neurofibromas. This compromises the
purchase of hooks or screws used during instrumentation. Second, the
surgical approach can be difficult owing to thoracic neurofibromas
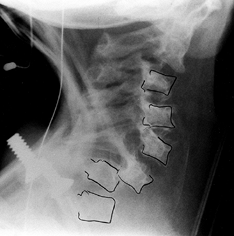
surrounding the spine (198). Last, intradural lesions and outpouchings of dura (known as dural ectasia) are seen in patients with neurofibromatosis, increasing the risk of neurologic complications during surgical correction (72,108). Preoperative MRI studies of the spinal cord are imperative in patients with neurofibromatosis (50).
 |
|
Figure 180.23. Cervical kyphosis in a 3-year-old girl with neurofibromatosis.
|
neurofibromatosis. The treatment of hemihypertrophy is individualized
and ranges from epiphysiodesis for mild cases to ablative amputation
for severely affected persons.
most challenging disorders treated by pediatric orthopaedists. The
condition usually, but not always, is associated with
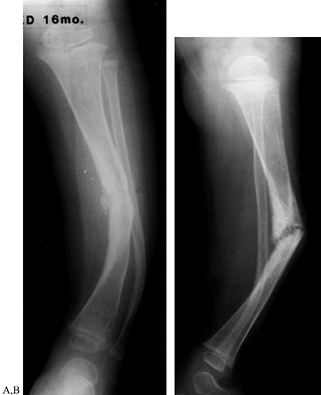
neurofibromatosis. Boyd (33) described four types of congenital pseudarthrosis of the tibia (Fig. 180.24; Table 180.4).
 |
|
Figure 180.24. AP (A) and lateral (B) views of the tibia of a 16-month-old girl with congenital pseudarthrosis of the tibia and neurofibromatosis.
|
 |
|
Table 180.4. Types of Pseudarthrosis of the Tibia
|
bowing of the tibia has been described in which the fibula is stout,
and the bowing remodels without treatment and does not progress to
pseudoarthrosis. This benign form does not occur in patients with
neurofibromatosis (241).
The prepseudarthrosis should be treated with total contact clamshell
orthoses. Once the tibia has fractured, closed treatment will not be
successful (49). The traditional treatment for
congenital pseudarthrosis of the tibia is intramedullary fixation of
the tibia and fibula, with excision of the pseudarthrosis and bone
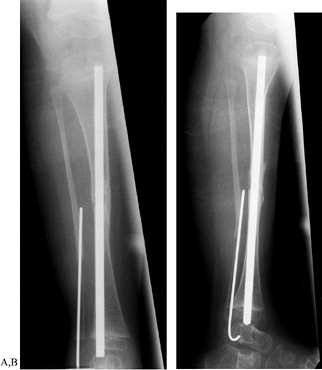
grafting. The Williams rod can be inserted through the calcaneus into
the tibia and usually is left protruding across the ankle joint. With
growth, the rod migrates proximally into the tibia (4) (Fig. 180.25A, Fig. 180.25B).
 |
|
Figure 180.25. A: Intramedullary fixation in congenital pseudarthrosis of the tibia. B: Note the proximal migration of the distal end of the rod.
|
the tibia have been recommended in response to the high rate of
nonunion using intramedullary fixation. Advocates for Ilizarov fixation
and bone transport as well as for vascularized fibula transfer find
that union can be achieved in some, but not all, patients with
congenital pseudarthrosis of the tibia (31,46,62,66,78,91,93,211,242,258) (see Chapter 36, Chapter 169, and Chapter 171).
The long-term ability to maintain union in these patients remains
unknown. Amputation continues to be a feasible option for the patient
who has undergone several operations and still fails to achieve union (115). A long-term study by Crossett et al. (52)
found that approximately half of their patients with congenital
pseudarthrosis of the tibia eventually were treated by amputation.
Regardless of the method used to achieve union, precaution must be
taken to prevent recurrent fractures. Protection of the tibia with
ankle-foot orthoses is usually prescribed. Even when union is achieved,
muscle weakness and gait disturbances are almost always present (122).
congenital pseudarthrosis of the tibia, with the affected leg being
shorter than the normal limb. Equalization surgery is often indicated.
metabolism of calcium and phosphate. Rickets and renal osteodystrophy
are the most common forms of metabolic bone disease in children. A
thorough understanding of calcium metabolism is required to evaluate
affected patients properly (30,138).
particularly the cardiovascular and neurologic systems, in which
irritability, conductivity, and contractility rely on the precise
control of serum calcium levels. Nearly all of the body’s calcium is
stored in bone in the form of hydroxyapatite, with a small proportion
circulating in the bloodstream. Serum calcium levels are regulated by
vitamin D and parathyroid hormone (PTH) through their actions on the
gut, kidneys, and bone. Calcium is absorbed in the distal duodenum and
proximal jejunum by means of a transport mechanism that is activated by
the active form of vitamin D (1,25-dihydroxy vitamin D) and by PTH.
the liver as 7-dehydrocholesterol. When they are exposed to ultraviolet
light, these compounds become calciferol and cholecalciferol. The first
hydroxylation occurs in the liver and the second hydroxylation takes
place in the kidney, with the end result being 1,25-dihydroxy
vitamin D (233). Conversion to this dihydroxy form is stimulated by hypocalcemia and high levels of PTH.
released in response to hypocalcemia. PTH allows for the absorption of
calcium from the gut and renal tubules, frees calcium from
hydroxyapatite in bone, and activates osteoclasts, an event that takes
on importance in renal osteodystrophy.
intake of vitamin D in the growing child. Although the condition is
uncommon in the United States (116), it remains
a problem throughout the rest of the world. Children who are fed a
vegetarian diet or who are breast fed for prolonged periods of time are
at increased risk for nutritional rickets (188).
Malabsorption of vitamin D due to celiac or hepatic disease results in
gastrointestinal rickets, which is more commonly seen in developed
countries. In either form, the lack of vitamin D results in the
inability to absorb calcium or phosphorus. PTH is released in response
to the hypocalcemia, but hypophosphatemia persists. Laboratory studies
reveal normal or mildly decreased serum calcium, decreased serum
phosphate, elevated PTH, and decreased vitamin D.
where loss of columnation occurs in the zone of hypertrophy. Because
there is insufficient calcium to mineralize bone, plump chondrocytes
persist into the zone of provisional calcification and down into the
metaphysis. There is sparse mineralization and elongation of the growth
plates. Within trabecular bone, large amounts of unmineralized osteoid
surround the fragile trabeculae.
The metaphyses are flared or cupped. The long bones may be abnormally
bowed because they are unable to withstand mechanical stress. The
trabecular pattern of the bone is indistinct. Osteopenia is present.
Looser’s lines (radiolucent lines extending transversely across the
axis of the bone) are associated with rickets and are present in
approximately 20% of patients with all forms of rickets (223).
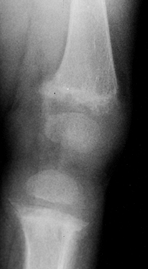 |
|
Figure 180.26. Osteopenia and growth plate widening in the knee of a 1-year-old girl with nutritional rickets.
|
Clinical features include short stature, muscular weakness, and
ligamentous laxity. Genu varum and periarticular enlargement of the
wrists, elbows, and ankles may be noted. The term “rachitic rosary”
refers to the prominence of the costochondral cartilages along the
chest wall. The abdomen may be protuberant. Neurologic involvement,
such as listlessness and irritability, may be present.
supervised by a pediatric endocrinologist, consists of vitamin D
replacement. Osteotomies are rarely required for residual deformity
following medical treatment.
neonates in intensive care units sustain multiple fractures without
antecedent trauma (125). Risk factors include
hepatobiliary disease, total parenteral nutrition, diuretic therapy,
physical therapy with passive motion, and chest percussion therapy. The
fractures require minimal immobilization to heal. The patients are
treated by the neonatal intensivists with supplemental feedings (53).
hypophosphatemic rickets, is a group of diseases in which ordinary
dietary intake of vitamin D is inadequate in maintaining normal mineral
balance (251). The condition usually is inherited in an X-linked dominant pattern, although an autosomal dominant form also exists (69). The genetic locus for the disease has been identified (7,71,184,185). A renal tubular defect leads to the inability to reabsorb phosphate, resulting in phosphate diabetes and hypophosphatemia (39,71,101).
Laboratory studies reveal near-normal levels of calcium, PTH, and
vitamin D but low levels of serum phosphate. Urinary phosphate and
serum alkaline phosphatase are elevated.
The disease becomes clinically apparent after 12 months of age, but it
can be diagnosed earlier by laboratory testing for urine phosphate
levels in suspected infants (146).
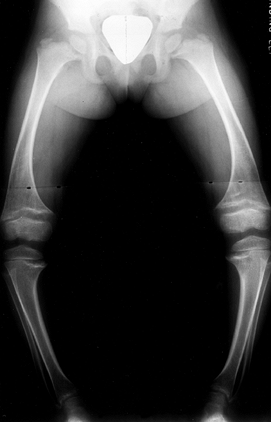 |
|
Figure 180.27. Bowing and physeal widening in the lower extremities of a 4-year-old girl with hypophosphatemic rickets.
|
medical, typically consisting of oral replacement of phosphate and
large amounts of vitamin D (247). Nephrocalcinosis is a known complication of medical treatment (227). Growth hormone therapy has been used to improve phosphate metabolism and increase stature in these patients (164,191,262).
Careful preoperative planning is required because the deformities are
multiplanar and thus may benefit from external fixation that precisely
corrects angulation in each plane (121). Bone
healing is delayed after osteotomy and may take approximately twice as
long as expected in metabolically normal children. Calcification of
ligaments and degeneration of articular cartilage may occur in affected
adults (79).
In addition, certain tumors may produce hypophosphatemic rickets.
Patients with no family history of rickets should be suspect. Tumors
usually are present in bone or skin (103).
Glomerular disease leads to retention of phosphate. The injured kidney
is unable to perform the final hydroxylation of vitamin D. Hypocalcemia
ensues, which stimulates secondary hyperparathyroidism. It is the
secondary hyperparathyroidism that produces most of the skeletal
manifestations of renal osteodystrophy.
osteodystrophy, lysis of bone is extensive, resulting in the
development of osteitis fibrosa (163). A small number of patients may develop osteosclerosis.
nitrogen (BUN), creatinine, alkaline phosphatase, and PTH. Serum
calcium and vitamin D levels are low, whereas the serum phosphate level
is high.
This occurs in children who are younger than those most often seen with
slipped epiphyses, and the condition usually is bilateral. The
previously described features of nutritional rickets may also be
present. Bowing of the long bones and genu valgum are common.
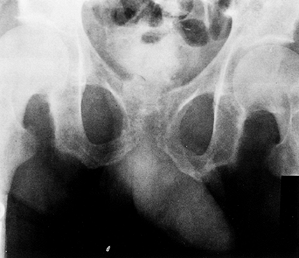
can be seen at the terminal tufts of the distal phalanges, the
symphysis pubis (Fig. 180.28), and the end of
the clavicle. Metaphyseal resorption of bone leads to widened physes
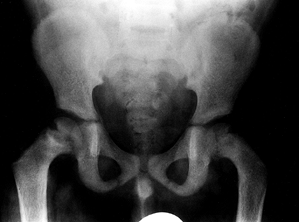
and slipped capital femoral epiphysis (SCFE; see Chapter 172) (Fig. 180.29). Disturbance of the proximal lateral tibial physis with associated genu valgum has been described (161).
Brown tumors appear as large, lucent lesions with indistinct margins
within the pelvis or long bones. These tumors can best be seen on MRI (158).
“Rugger jersey” spine describes the sclerotic appearance of the
vertebral endplates seen on a lateral radiograph. Periarticular
soft-tissue calcification may be seen.
 |
|
Figure 180.28. Resorption of the pubis due to renal osteodystrophy.
|
 |
|
Figure 180.29.
Physeal widening and slipped capital femoral epiphysis in a 7-year-old boy presenting with hip pain. A diagnosis of renal failure was made. |
correcting the metabolic imbalance. Calcitriol, administered in high
doses intravenously, orally, or by dialysis, has been
found to be helpful in the treatment of pediatric renal osteodystrophy by lowering PTH levels (192). Pinning of SCFEs may be necessary (107), but osteopenia and continued metaphyseal resorption may compromise fixation (see Chapter 172).
The patient’s hip pain may resolve simply with medical treatment.
Osteolysis of the distal femoral physis has also been described, with
cast immobilization and medical treatment sufficient to promote healing
and remodeling (231). When an angular deformity
of the lower extremities interferes with gait, osteotomy of the femur
or tibia is indicated. Best results are obtained if the patient’s
metabolic state is optimized before surgery (58,160).
It results from trisomy of chromosome 21 because of maternal
nondisjunction, translocation, or mosaicism. One risk factor for Down
syndrome is advancing maternal age. The most notable feature is mental
retardation, but a variety of other congenital malformations, including
heart defects, commonly occur (120). The IQs of
children with Down syndrome vary considerably. About 30% of patients
develop orthopaedic problems that require hospitalization (63).
upslanting palpebral fissures, prominent epicanthal folds, mild
microcephaly, and a protruding tongue. The child tends to be short.
Ligamentous laxity is universal. Development is delayed, with the onset
of ambulation usually not occurring until 2 to 3 years of age.
appearance of the pelvis. The iliac wings are widened, the ischial rami
are small and tapered, and the acetabulae are horizontal. Coxa valga is
present. Hand radiographs show that all five metacarpals are equal in
length, and there is clinodactyly of the small finger.
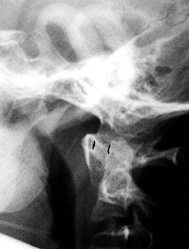
and its effects on the spine, hips, knees, and feet. Atlantoaxial
instability, the most commonly recognized skeletal manifestation of
Down syndrome, is caused by laxity in the transverse ligament. The
radiographic definition of atlantoaxial instability is an increase in
the atlanto-dens interval (ADI), as seen on flexion lateral cervical
radiographs (Fig. 180.30). It has been reported to occur in 9% to 31% of persons with Down syndrome (177).
Patients with atlantoaxial instability have a particularly high rate of
associated congenital cervical spine anomalies (e.g., os odontoideum,
hypoplasia of the posterior arch of C-1, and hypoplasia of the dens)
that may have an impact on surgical treatment (140,176).
 |
|
Figure 180.30. The atlanto-dens interval is increased in flexion in this 12-year-old boy with Down syndrome.
|
cervical instability. The American Academy of Pediatrics Committee on
Sports Medicine and Fitness conducted a review of the literature and
concluded that greater effort should be directed toward “identification
of those patients who already have or who later have complaints or
physical findings consistent with symptomatic spinal cord injury”
rather than obtaining routine radiographs (6). This report pointed out that almost all children who suffered
catastrophic neurologic injury because of atlantoaxial instability had
pre-existing neurologic symptoms, and that children rarely develop
abnormal radiographic findings following normal films (147).
ADI from head trauma by removing them from “at-risk” athletic
activities is the traditional recommendation. A few authors have
proposed that because neurologic abnormalities are so rare even in
affected children, activity restriction may be unnecessary (51).
Others have found that an equal proportion of patients have neurologic
signs with and without radiographic instability of the cervical spine,
and they question the need to screen all patients (186). Once atlantoaxial instability is documented radiographically, rarely does it increase significantly over time (175).
Neurologic injury following nonspinal surgery has been described; thus,
preoperative radiographic evaluation of the cervical spine in patients
with Down syndrome is warranted (59,148,240).
A high complication rate with major complications, including nonunion,
loss of reduction, neurologic deterioration, late subaxial instability,
infection, and wound dehiscence, should be anticipated after posterior
arthrodesis of the upper cervical spine in patients with Down syndrome.
Ligamentous laxity in patients with precarious preoperative neurologic
status increases the neurologic mortality risk during surgery (213).
Nonsurgical management is currently recommended for patients with Down
syndrome who have atlantoaxial instability but no neurologic signs or
symptoms (67,200).
between the occiput and C-1. Posterior occipitoatlantal hypermobility
(POAH), defined as subluxation of the occiput posteriorly during
extension of the neck, has been identified in 8% to 63% of patients
with Down syndrome (85,162).
The discrepancy in prevalence is probably due to the difference in
techniques used to measure motion at this level. Plain radiographs may
suggest instability, but precise measurement of hypermobility is
fraught with error and is best evaluated with MRI (123,259). POAH may be seen in patients with atlantoaxial instability (243). Treatment is based on the child’s neurologic status and on the amount of space available for the spinal cord (239). Posterior occipital-cervical fusion has been recommended (145).
patients with Down syndrome. Spondylolysis of the cervical spine and
early degenerative arthritis have been described (239).
instability of the joints of the lower extremities, resulting in hip
dislocations and patellar subluxation and dislocation. Approximately
7.9% of patients with Down syndrome have some hip abnormality,
including dysplasia, dislocation, avascular necrosis, or SCFE (202).
Dislocation of the hip in patients with Down syndrome is rarely painful
initially but may become so over time. Treatment is difficult, because
redislocation is not uncommon. Nonoperative bracing is rarely
successful, but prolonged spica casting may allow the hip to stabilize
in a reduced position. Open reduction, capsulorrhaphy of the attenuated
capsule, and femoral and pelvic osteotomy may result in a stable hip (20) (see Chapter 166).
Hypothyroidism may be a predisposing factor, so thyroid function tests
should be ordered for children with both Down syndrome and SCFE.
Treatment is in situ fixation, but hardware failure may occur (see Chapter 172).
third of patients with Down syndrome, but it rarely affects their
ability to walk (68). Surgery is advised in
selected cases and consists of aggressive realignment and Galleazzi
transfer of the semitendinosus to the patella (see Chapter 87).
The disorder is nonprogressive, and the cause is unclear. Abnormalities
of muscles, joints, and nervous system; teratogens; infection; and
environmental factors have all been implicated.
Many
genetic syndromes include arthrogrypotic contractures and, thus, should
be considered when evaluating a child with joint rigidity. The disease
can resemble spinal muscular atrophy, and genetic associations between
the two diseases have been proposed (36).
Arthrogryposis usually is not genetically transmitted, but genetic
research is being performed on the less common inherited forms (16,28,36,83,207,217).
Muscle changes include replacement of skeletal muscle by fibrofatty
tissue. Neuropathic findings include a decrease in the number of
anterior horn cells, with sparing of the posterior columns. Sensation
is not affected.
extremities. There is occasional sparing of the upper extremities and
rare sparing of the lower extremities. There is a distal form of the
disease that consists of hand and foot contractures, including clubfeet
(16). Intelligence is usually normal to above normal (217).
creases at the joints. Muscle mass is decreased, but the atrophy may be
difficult to appreciate in young infants. Motion in the joints is
limited but painless, and a solid block is felt at the extremes of
range of motion.
flexed, abducted, and externally rotated. Dislocation is common. The
knees may have flexion or hyperextension contractures. Rigid clubfeet
usually are present, but vertical talus may occur. The upper
extremities, when involved, are adducted at the shoulder and internally
rotated. The elbows may have flexion or extension contractures. The
wrist may be flexed and ulnarly deviated, with flexion of the fingers
and thumb-in-palm deformity. Scoliosis may be seen in young children.
surgery is to improve function and enable ambulation when possible.
With surgery, it has been found that up to 85% of patients with
amyoplasia can walk at least on a limited basis by 5 years of age (201). The prognosis for ventilator-dependent babies is less favorable (26). Recurrence of deformities is common following surgery, and orthoses are usually prescribed.
diagnosis. Include examination by a geneticist and a pediatric
neurologist. Muscle biopsy is occasionally needed. Initiate physical
therapy for range-of-motion and developmental skills. Range of motion
should always be performed with great care, because pathologic
fractures may occur.
in patients with arthrogryposis. Pavlik harness treatment is
unsuccessful, so open reduction is the treatment of choice. The medial
approach has been specifically advocated by Staheli et al. (220)
for use in arthrogryposis. Bilateral dislocations may be addressed
medially during the same surgery, with good results reported in 80% of
a series of 40 dislocated hips in arthrogrypotic children (232) (see Chapter 166).
contractures require surgery more often than hyperextension
contractures (149,215).
Surgery is indicated if physical therapy fails to attain less than 30°
flexion contracture. Hamstring tenotomy and posterior capsulotomy, with
or without femoral shortening osteotomy, are required (234).
Sectioning of the cruciate ligaments and resection of intra-articular
fibrous tissue often is necessary. Femoral extension osteotomy can
increase extension at the knee, but flexion deformity of the distal
femur recurs with growth of the child (61). Chronic contractures lead to bony changes, such as squaring off of the femoral condyles and joint incongruity (94). Ilizarov techniques have been used in difficult cases (34) (see Chapter 171). Many children who have undergone knee flexion contracture surgery stop walking in adolescence (149).
around the age when the child learns to walk, which is nearly always
delayed in babies with arthrogryposis. Posteromedial release, with
resection of tendons and capsules, and lateral column shortening, when
needed, is the initial surgery of choice. Recurrence of deformity has
been seen in 73% of children with arthrogryposis (155).
Talectomy or tarsal medullostomy is usually reserved for recurrent
severe deformity, but the procedure has been performed in primary
releases by some surgeons (64,92,144,216) (see Chapter 167).
complete functional assessment can be performed. Optimally, one elbow
should be extended for toileting and use of walking aids, and the
contralateral elbow should be flexed to enable the child to self-feed (12).
The ability of these intelligent children to adapt to their deformities
can be surprising, so care should be taken not to intervene surgically
when the child is functioning satisfactorily. Surgical procedures that
may be useful for the upper extremities include rotational osteotomy of
the humerus, triceps transfer to gain active elbow flexion,
capsulotomies at the elbow, and wrist stabilization.
disease and “marble bone disease” is apparently a hereditary disease of
bone characterized by abnormal function of osteoclasts. This results in
varying degrees of bone remodeling deficiency, the skeleton being
predominately composed of primary unremodeled trabecular bone and
calcified cartilage. On electron microscopy, there are increased
numbers of osteoclasts but their ruffled border is absent or diminished.
surgeons will not encounter a single case in their lifetime. Pediatric
referral centers may see only a few cases at any given time. A recent
review of the management of osteopetrosis by Armstrong, Newfield, and
Gillespie based upon a review of the literature and a survey of the
members of the Pediatric Orthopaedic Society of North America (10a),
is highly recommended for readers interested in more details on this
rare disorder. They provide an excellent summary of the characteristics
of osteopetrosis, list 63 references, and provide clinical details on
79 patients found in their survey.
malignant type. It is inherited by autosomal recessive transmission and
a more benign adult form, which is autosomal dominant. The latter can
be divided into Type I. This is characterized by marked thickening of
the cranial vault, and a substantially lesser risk of fractures. Type
II is characterized by vertebral end-plate thickening resulting in the
classic appearance of “rugger jersey” spine and “endobones” in the
pelvis. Type II has a higher incidence of fracture and delayed union as
well. There is an extremely rare third form that is autosomal
recessive. It is regarded as an “intermediate” form.
dysfunction of the osteoclast, has not yet been established in humans.
However, mutations have been found in the mouse and rat that produce an
autosomal recessive type of osteopetrosis. In the op mouse and the tl rat, the production of colony-stimulating factor – 1 (CSF-1) appears to be deficient or the factor is inactive.
usually diagnosed during their first year of life and only 30% of
patients survive past the age of 6 years, dying of complications
resulting from myelophthisic anemia which is due to obliteration of
their marrow spaces by unremodeled bone. Clinical characteristics
include blindness, failure to thrive, seizures, repeated infections,
pathologic fractures, hepatosplenomegaly, hypersplenism, hydrocephalus,
mental retardation, cranial nerve palsies, and hemorrhagic diathesis.
macrocephaly, frontal bossing, hypertelorism, exophthalmos, and
flattening of the nasal bridge with chronic rhinitis.
entire skeleton and the complete absence of the medullary canal. There
is metaphyseal widening producing club-shaped long bones, and the “bone
within a bone” phenomenon is seen in the pelvis, vertebra, hands and
feet. Transverse radiolucent bands can be seen in the metaphyseal
regions of the long bones. The skull may progressively thicken and show
a “hair-on-end” appearance. Transverse or short oblique pathologic-type
diaphyseal and metaphyseal fractures are common.
although patients are prone to long-bone fractures. The age of
presentation of these patients can be quite variable depending upon the
severity of the disease. Up to 40% of patients may remain asymptomatic.
There may be a family history of the disease or of multiple fractures.
Laboratory studies show mild or moderate anemia and increased serum
acid phosphatase. Bone involvement is less severe than in the infantile
form but osteomyelitis of the jaw, progressive coxa vara, lateral
bowing of the long-bones, spondylolysis and spondylolisthesis of L4 and
L5 or both, and osteoarthritis of the hips and the knees may develop in
adults. Dental malocclusion and caries can be problems. The diagnosis
is usually obvious from the clinical presentation and characteristic
radiographs. It can be confirmed by bone biopsy.
marrow transplantation (in selected patients) has been successful in
reversing hematologic and skeletal defects of the disease. Variable
results have been obtained with restriction of dietary calcium,
splenectomy, high dose prednisone to increase bone marrow
hematopoiesis, parathormone, calcitriol, and gamma-interferon.
of their fractures (which usually respond to nonoperative closed
treatment), and treatment for coxa vara. In the 79 patients reviewed by
Armstrong et al. (10a) 20 were treated
surgically for coxa vara, all of whom had complications. Valgus
osteotomy provided the most consistent results. Fourteen
subtrochanteric and 31 other fractures of the femur were treated with
traction or casting, with good results in the majority. Twenty-nine
tibial fractures, and upper extremity fractures healed well with closed
reduction and cast immobilization. For the most part, spine problems
were treated non-operatively.
chalky, and brittle. It is difficult to drill. It is easily over heated
causing necrosis, possibly leading to implant loosening or infection.
When placing screws for pins in osteopetrotic bone, it is extremely
important to use sharp, fresh drill points and pins. Clean these often
and exchange them if necessary to maintain their sharpness. The drills
must be cooled constantly by bathing them with sterile saline. The lack
of a medullary canal makes drilling even more difficult. Intramedullary
nailing is nearly always impossible and internal fixation with plates,
because of the multiple screw holes required, is usually best avoided.
The presence of a plate on the diaphysis often results in a stress
riser, which leads to subsequent fractures at the ends of the plate. If
rigid fixation is required for treatment than external fixation is
probably the best choice. These patients seem to heal their fractures
well in most cases of the adult form. The bone they heal with, however,
is defective.
scheme: *, classic article; #, review article; !, basic research
article; and +, clinical results/outcome study.
PE Jr, Schantz K, Bollerslev J, Justesen P. Bilateral Femoral Head
Dysplasia and Osteochondritis. Multiple Epiphyseal Dysplasia Tarda,
Spondylo- epiphyseal Tarda, and Bilateral Legg-Perthes Disease. Acta Radiol 1988;29:705.
DJ, Schoenecker PL, Sheridan JJ, Rich MM. Use of an Intramedullary Rod
for the Treatment of Congenital Pseudarthrosis of the Tibia. J Bone Joint Surg 1992;74A:161.
Atlantoaxial Instability in Down Syndrome: Subject Review. American
Academy of Pediatrics Committee on Sports Medicine and Fitness. Pediatrics 1995;96:151.
A Gene (PEX) with Homologies to Endopeptidases is Mutated in Patients
with X-linked Hypophosphatemic Rickets. The HYP Consortium. Nat Genet 1995;11:130.
DG, Newfield JT, Gillespie R. Orthopaedic Management of Osteopetrosis:
Results of a Survey and Review of the Literature. J Pediatr Orthop 1999;122.
MW, Niethard FU, Doderlein L, Weber M. Principles of Treatment of the
Upper Extremity in Arthrogryposis Multiplex Congenita Type I. J Pediatr Orthop B 1997;6:179.
RW, Dubow HI. Evolution of the Concept of an Extensible Nail
Accommodating to Normal Longitudinal Bone Growth: Clinical
Considerations and Implications. Clin Orthop 1981;159:157.
F, Siegrist CA, Ozsahin H, et al. Bone Marrow Transplantation in
Cartilage-hair Hypoplasia: Correction of the Immunodeficiency but not
of the Chondrodysplasia. Eur J Pediatr 1996;155:286.
PM, Shen N, Rennert H, et al. Arthrogryposis due to Infantile Neuronal
Degeneration Associated with Deletion of the SMNT Gene. Neurology 1997;49:848.
S, Catagni M, Donzelli O, et al. Congenital Pseudarthrosis of the Tibia
Associated with Neurofibromatosis I: Treatment with Ilizarov’s Device. J Pediatr Orthop 1997;17:675.
J, Rousseau F, Legeai-Mallet L, et al. Common Mutations in the
Fibroblast Growth Factor Receptor 3 (FGFR 3) Gene Account for
Achondroplasia, Hypochondroplasia, and Thanatophoric Dwarfism. Am J Med Genet 1996;63:148.
L, Amiel J, Viollet L, et al. Survival Motor Neuron Gene Deletion in
the Arthrogryposis Multiplex Congenita—Spinal Muscular Atrophy
Association. J Clin Invest 1996;98:1130.
JM, Dormans JP, Drummond DS, et al. Proximal Fibular Osteochondroma
with Associated Peroneal Nerve Palsy: A Review of Six Cases. J Pediatr Orthop 1995;15:574.
LR, Marini JC. Communicating Hydrocephalus, Basilar Invagination, and
Other Neurologic Features in Osteogenesis Imperfecta. Neurology 1993;43:2603.
SS, Coleman DA. Congenital Pseudarthrosis of the Tibia: Treatment by
Transfer of the Ipsilateral Fibula with Vascular Pedicle. J Pediatr Orthop 1994;14:156.
MJ, Bol E, de Roos F, van Gijn J. Risk of Sports Activities in Children
with Down’s Syndrome and Atlantoaxial Instability. Lancet 1993;342:511.
LS, Stern LS. Talectomy in the Treatment of Resistant Talipes
Equinovarus Deformity in Myelomeningocele and Arthrogryposis. J Pediatr Orthop 1987;7:39.
JP, Krajbich JI, Zuker R, Demuynk M. Congenital Pseudarthrosis of the
Tibia: Treatment with Free Vascularized Fibular Grafts. J Pediatr Orthop 1990;10:623.
JS, Lauerman WC, Wood KB, Krause DR. Complications and Long-term
Outcome of Upper Cervical Spine Arthrodesis in Patients with Down
Syndrome. Spine 1996;21:1223.
MJ, Francis F. Positional Cloning of the PEX Gene: New Insights into
the Pathophysiology of X-linked Hypophosphatemic Rickets. Am J Physiol 1997;273:F489.
RH, Helders PJ, Keessen W, et al. Intramedullary Rodding in Type III
Osteogenesis Imperfecta: Effects on Neuromotor Development in 10
Children. Acta Orthop Scand 1995;66:361.
LL, Berdon WE, Harcke HT, et al. Severe Mid-cervical Kyphosis with Cord
Compression in Larsen’s Syndrome and Diastrophic Dysplasia: Unrelated
Syndromes with Similar Radiologic Findings and Neurosurgical
Implications. Pediatr Radiol 1995;25:136.
BH, d Bray EW, Meyer LC. Multiple Osteotomies with Zickel Nail Fixation
for Polyostotic Fibrous Dysplasia Involving the Proximal Part of the
Femur. J Bone Joint Surg 1987;69A:691.
H, Winter RB, Lonstein JB, Denis F. Pathophysiology of Spinal
Deformities in Neurofibromatosis: An Analysis of Seventy-one Patients
Who Had Curve Associated with Dystrophic Changes. J Bone Joint Surg 1994;76:692.
JG, Strudwick WJ, Rinsky LA, Bleck EE. Complications of Intramedullary
Rods in Osteogenous Imperfecta: Bailey-Dubow Rods Versus Nonelongating
Rods. J Pediatr Orthop 1988;8:645.
LH, Binder H, Weintrob J, et al. Rehabilitation of Children and Infants
with Osteogenesis Imperfecta: A Program for Ambulation. Clin Orthop 1990;251:254.
A, Brockman R. Congenital Pseudarthrosis of the Tibia: Long-term
Followup of 29 Cases Treated by Microvascular Bone Transfer. Clin Orthop 1995;314:37.
DA, Winter RB, Lutter L, et al. Osteogenesis Imperfecta: Radiographic
Classification, Natural History, and Treatment of Spinal Deformities. J Bone Joint Surg 1992;74:598.
NL, Eilert RE, Davino N, et al. Osteogenic Sarcoma Arising from Bony
Regenerate Following Ilizarov Femoral Lengthening through Fibrous
Dysplasia. J Pediatr Orthop 1994;14:123.
RC, Marchal C, Fall M. Diagnosis of Morquio’s Disease: A Simple
Chromatographic Method for the Identification of Keratosulfate in
Urine. J Pediatr 1972;81:107.
S, Kumar SJ, Takahashi HE, Homma M. Vertebral Body Shape as a Predictor
of Spinal Deformity in Osteogenesis Imperfecta. J Bone Joint Surg 1996;78A:212.
LA, Haideri NF, Halliday SE, et al. Gait Analysis and Muscle Strength
in Children with Congenital Pseudarthrosis of the Tibia: The Effect of
Treatment. J Pediatr Orthop 1998;18:381.
LA, Sheffield EG, Crawford K, et al. Reproducibility in the Measurement
of Atlanto-occipital Instability in Children with Down Syndrome. Spine 1996;21:2463.
WW, Sherman R, Succop P, et al. Fractures and Rickets in Very Low Birth
Weight Infants: Conservative Management and Outcome. J Pediatr Orthop 1993;13:577.
L, Jaffe HL. Fibrous Dysplasia of Bone: A Condition Affecting One,
Several, or Many Bones, the Graver Cases of which May Present Abnormal
Pigmentation of Skin, Premature Sexual Development, or Still Other
Extraskeletal Abnormalities. Arch Pathol 1942;33:777.
J, Vande Berg B, Noel H, Maldague B. Benign Osteochondromas and
Exostotic Chondrosarcomas: Evaluation of Cartilage Cap Thickness by
Ultrasound. Skeletal Radiol 1992;21:33.
V, Ben-Ami T, Yousefzadeh DK, Roizen NJ. Hypoplastic Posterior Arch of
C-1 in Children with Down Syndrome: A Double Jeopardy. Radiology 1992;183:125.
C, Gilbert A, Azze RG. Congenital Pseudarthrosis of the Forearm:
Treatment of Six Cases with Vascularized Fibular Graft and a Review of
the Literature. Microsurgery 1993;14:252.
K, Minagawa M, Yasuda T, Niimi H. Early Detection of Infants with
Hypophosphatemic Vitamin D Resistant Rickets (HDRR). Endocr J 1996;43:339.
ME, Reese ME, Di Gaudio K, et al. Symptomatic Atlantoaxial Instability
Associated with Medical and Rehabilitative Procedures in Children with
Down Syndrome. Pediatrics 1990;85:447.
K, Teshima R, Minamizaki T, Yamamoto K. Knee Deformities in Multiple
Hereditary Exostoses: A Longitudinal Radiographic Study. Clin Orthop 1995;313:194.
J, Thomas PS. Clinical Findings in 12 Patients with MPS IV A (Morquio’s
Disease): Further Evidence for Heterogeneity. Part III: Odontoid
Dysplasia. Clin Genet 1988;33:126.
MF, Bridwell KH, Lenke LG, Schoenecker PL. Intracanalicular
Osteochondroma Producing Spinal Cord Compression in Hereditary Multiple
Exostoses. J Spinal Disord 1994;7:236.
CL, Cross PK, Gensburg LJ, Hughes JP. The Effects of Prenatal
Diagnosis, Population Ageing, and Changing Fertility Rates on the Live
Birth Prevalence of Down Syndrome in New York State, 1983–1992. Prenat Diagn 1996;16:991.
WL, Fischer SR, Salusky IB. Surgical Correction of Angular Deformity of
the Knee in Children with Renal Osteodystrophy. J Pediatr Orthop 1997;17:41.
TA, Bertrand SL, Powers MJ, et al. Posterior Occipitoatlantal
Hypermobility in Down Syndrome: An Analysis of 199 Patients. J Pediatr Orthop 1994;14:304.
L, Clayton PE, Brain C, et al. Acute Biochemical Effects of Growth
Hormone Treatment Compared with Conventional Treatment in Familial
Hypophosphataemic Rickets. Clin Endocrinol 1996;44:687.
MD, Filly RA. Homozygous Achondroplasia: US Distinction Between
Homozygous, Heterozygous, and Unaffected Fetuses in the Second
Trimester. Radiology 1995;196:541.
RM, Horton VK, Glinski LP, Reiser CA. Prospective Assessment of Risks
for Cervicomedullary-junction Compression in Infants with
Achondroplasia. Am J Hum Genet 1995;56:732.
SM, Scola FH, Pezzullo JC. A Longitudinal Study of Atlanto-dens
Relationships in Asymptomatic Individuals with Down Syndrome. Pediatrics 1992;89:1194.
F, Bonaventure J, Legeai-Mallet L, et al. Mutations in the Gene
Encoding Fibroblast Growth Factor Receptor-3 in Achondroplasia. Nature 1994;371:252.
PS, Goulding J, Francis F, et al. The Gene for X-linked
Hypophosphataemic Rickets Maps to a 200-300 kb Region in Xp22.1 and Is
Located on a Single YAC Containing a Putative Vitamin D Response
Element (VDRE). Hum Genet 1996;97:345.
PS, Goulding J, Read A, et al. Refining the Genetic Map for the Region
Flanking the X-linked Hypophosphataemic Rickets Locus (Xp22.1-22.2). Hum Genet 1994;93:291.
M, Said SE, Glorieux FH, et al. Principles and Results of Corrective
Lower Limb Osteotomies for Patients with Vitamin D–Resistant
Hypophosphatemic Rickets. Clin Orthop 1988;237:264.
G, Baroncelli GI, Bertelloni S, Perri G. Long-term Growth Hormone
Treatment in Children with Renal Hypophosphatemic Rickets: Effects on
Growth, Mineral Metabolism, and Bone Density. J Pediatr 1995;127:395.
E, Langman CB, Parfitt AM, et al. Constitutively Activated Receptors
for Parathyroid Hormone and Parathyroid hormone–related Peptide in
Jansen’s Metaphyseal Chondrodysplasia. N Engl J Med 1996;335:708.
LS, Drummond DS, Zanotti RM, et al. Complications of Posterior
Arthrodesis of the Cervical Spine in Patients Who Have Down Syndrome. J Bone Joint Surg 1991;73A:1547.
P, Parham D, Kovnar E, et al. Neurofibromatosis Type I and Malignancy:
Review of 32 Pediatric Cases Treated at a Single Institution. Med Pediatr Oncol 1994;22:78.
M, Lotan R, Magal N, et al. A Gene for Arthrogryposis Multiplex
Congenita Neuropathic Type is Linked to D5S394 on Chromosome 5qter. Am J Hum Genet 1997;61:1139.
MD, Phillips WA, Hensinger RN. Fusion of the Upper Cervical Spine in
Children and Adolescents: An Analysis of 17 Patients. Spine 1991;16:695.
J, Hakamies-Blomqvist L, Sainio K, et al. Arthrogryposis Multiplex
Congenita: Perinatal and Electromyographic Findings, Disability, and
Psychosocial Outcome. J Pediatr Orthop B 1997;6:167.
HA, Miller EA. Fragmentation, Realignment and Intramedullary Rod
Fixation of Deformities of the Long Bones in Children: A Ten-year
Appraisal. J Bone Joint Surg 1959;41A:1371.
RD, Pepin M, Byers PH. Studies of Collagen Synthesis and Structure in
the Differentiation of Child Abuse from Osteogenesis Imperfecta. J Pediatr 1996;128:542.
JM, Kendall BE, Crockard HA, Ransford A. The Odontoid Process in
Morquio-Brailsford’s Disease: The Effects of Occipitocervical Fusion. J Bone Joint Surg 1991;73B:851.
A, Rossi A, Steinmann B, Gitzelmann R. A Chondrodysplasia Family
Produced by Mutations in the Diastrophic Dysplasia Sulfate Transporter
Gene: Genotype/phenotype Correlations. Am J Med Genet 1996;63:144.
BA, Diepstraten AF, vd Heyden BJ. Distal Femoral Physiolysis in Renal
Osteodystrophy: Successful Nonoperative Treatment of 3 Cases Followed
for 5 years. Acta Orthop Scand 1993;64:382.
RW, Cook SD, Thomas KA, et al. Effects of Anticonvulsant Drug Therapy
on Bone Mineral Density in a Pediatric Population. J Pediatr Orthop 1988;8:467.
NJ, Jensen FO, Bankier A, et al. Development of the Hip in Multiple
Epiphyseal Dysplasia: Natural History and Susceptibility to Premature
Osteoarthritis. J Bone Joint Surg 1990;72B:1061.
ER. Myseros JS, Smoker WR, et al. Atlantooccipital Subluxation in a
Neonate with Down’s Syndrome: Case Report and Review of the Literature.
Pediatr Neurosurg 1994;21:55.
der Burgt I, Haraldsson A, Oosterwijk JC, et al. Cartilage Hair
Hypoplasia, Metaphyseal Chondrodysplasia Type McKusick: Description of
Seven Patients and Review of the Literature. Am J Med Genet 1991;41:371.
GA, Rash B, Sykes B, et al. Mutations within the Gene Encoding the
Alpha 1 (X) Chain of Type X Collagen (COL10A1) Cause Metaphyseal
Chondrodysplasia Type Schmid but Not Several Other Forms of Metaphyseal
Chondrodysplasia. J Med Genet 1996;33:450.
BA, Harkey HL, Parent AD, et al. Severe Cervical Kyphotic Deformities
in Patients with Plexiform Neurofibromas: Case Report. Neurosurgery 1994;35:960.
KA, Everett F, Sillence DO, et al. Treatment of Obstructive Sleep Apnea
in Achondroplasia: Evaluation of Sleep, Breathing, and
Somatosensory-evoked Potentials. Am J Med Genet 1995;59:460.
AJ, Weiss AP, Moore JR, Tolo VT. Vascularized Fibular Grafts in the
Treatment of Congenital Pseudarthrosis of the Tibia. J Bone Joint Surg 1990;72A:654.
KS, Ball WS, Prenger EC, et al. Evaluation of the Craniocervical
Junction in Down Syndrome: Correlation of Measurements Obtained with
Radiography and MR Imaging. Radiology 1993;186:377.
KD, Park TS, Tantuwaya VS, Kaufman BA. Posterior Fossa Decompression
without Duraplasty in Infants and Young Children for Treatment of
Chiari Malformation and Achondroplasia. Pediatr Neurosurg 1996;25:221.