
Editors: Tornetta, Paul; Einhorn, Thomas A.; Damron, Timothy A.
Title: Oncology and Basic Science, 7th Edition
Copyright ©2008 Lippincott Williams & Wilkins
> Table of Contents > Section III – Specific Soft Tissue Neoplasms and Simulators > 11 – Benign Soft Tissue Tumors
11
Benign Soft Tissue Tumors
Timothy A. Damron
Gustavo De La Roza
Benign soft tissue tumors outnumber soft tissue sarcomas
by approximately 100:1. In adults, lipomas are the most common soft
tissue tumor; in children, hemangiomas are the most common. Recognition
of these relatively more common benign tumors is important not only to
avoid overtreatment but also to allow distinction from their malignant
counterparts. Soft tissue tumors, both benign and malignant, are
classified according to the purported cell of origin or resemblance,
and the organization of this chapter will follow the latest World
Health Organization (WHO) scheme in that regard.
by approximately 100:1. In adults, lipomas are the most common soft
tissue tumor; in children, hemangiomas are the most common. Recognition
of these relatively more common benign tumors is important not only to
avoid overtreatment but also to allow distinction from their malignant
counterparts. Soft tissue tumors, both benign and malignant, are
classified according to the purported cell of origin or resemblance,
and the organization of this chapter will follow the latest World
Health Organization (WHO) scheme in that regard.
Fatty Benign Soft Tissue Tumors
Lipoma
Lipoma is the most common soft tissue tumor in adults.
However, because it is, soft tissue sarcomas are sometimes assumed to
be lipomas, unnecessarily delaying the diagnosis. Hence, recognition of
the distinction between benign lipomas and soft tissue sarcomas is very
important.
However, because it is, soft tissue sarcomas are sometimes assumed to
be lipomas, unnecessarily delaying the diagnosis. Hence, recognition of
the distinction between benign lipomas and soft tissue sarcomas is very
important.
Pathogenesis and Pathophysiology
-
Etiology is unknown.
-
More common in obese patients
-
Peak age: 40 to 60 (rare in children)
-
Multiple lipomas in 5%
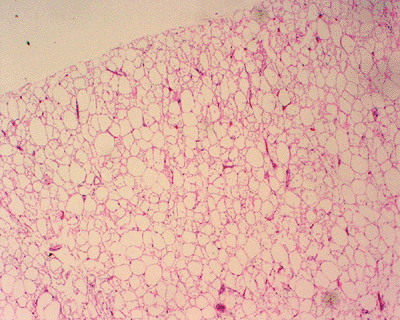
Histopathology
-
Lobules of mature adipocytes
-
Occasionally other areas of tissue formation
-
Bone = osteolipoma
-
Cartilage = chondrolipoma
-
Myxoid change = myxolipoma
-
Genetics
-
Chromosomal aberrations in up to 75%
-
Three patterns
-
12q13-15 aberrations
-
6p21-23 aberrations
-
Loss of material from 13q
-
Classification
-
Histologic variant subtypes have no prognostic significance.
-
Infiltrative
P.264
Intermuscular Versus Intramuscular
-
Intermuscular: easily shelled out between muscles
-
Intramuscular: two types within muscle
-
Well demarcated
-
Infiltrative: infiltrates and encases atrophic muscle fibers
-
Lipoma Arborescens
-
Subsynovial located lipoma
-
One type of intra-articular lipoma
Diagnosis
Clinical Features
-
Most common: painless mass with characteristic doughy feel
-
Most superficial lipomas are small (<5 cm).
-
Most deep lipomas are large (>5 cm).
-
Lipoma arborescens presents as articular swelling.
Radiologic Features
-
Superficial lipomas: difficult to see on radiographs or magnetic resonance imaging (MRI)
-
Deep lipomas (Fig. 11-1)
-
Fatty intramuscular shadow
-
Homogeneous fatty signal on all MRI sequences
-
No gadolinium enhancement
-
Occasional entrapped muscle fibers or fibrous strands
-
-
Lipoma arborescens
-
Fatty infiltration throughout affected synovium with fat-distended villi
-
 |
|
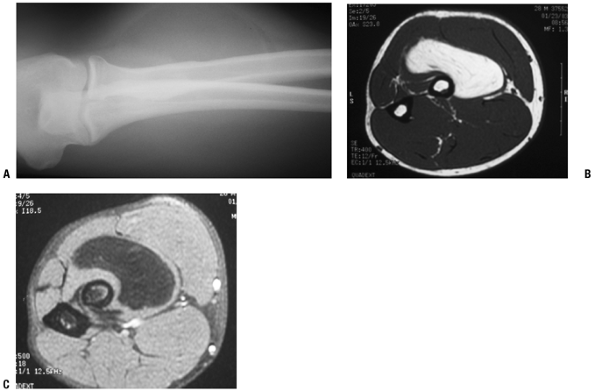
Figure 11-1 Inramuscular lipoma of the proximal forearm. (A) The fatty soft tissue shadow is shown within the muscle of the proximal forearm by plain radiograph. Axial T1-weighted (B) and T2-weighted (C) MR imaging studies show homogenous fatty signal characteristics identical to that of the subcutaneous fat.
|
Diagnostic Work-up
-
Most superficial lipomas are
distinguished by their doughy characteristics on physical examination
and do not warrant radiographic evaluation. -
Larger or deep masses warrant plain radiographic and MRI evaluation, which is usually diagnostic.
-
Biopsy is rarely indicated.
-
Also see Chapter 2, Evaluation of Soft Tissue Tumors.
Treatment
Surgical and Nonoperative Options
-
Superficial lipomas
-
Observation favored
-
Excision only if symptomatic
-
-
Deep lipomas
-
Observation if radiology identifies clearly as benign lesion (not atypical lipoma or liposarcoma)
-
Excision for most
-
Surgical Goals and Approaches
-
Marginal excision (complete excision through pseudocapsule)
-
Avoid transaction of nerves running within deep lipomas (piecemeal excision preferred).
Results and Outcome
-
Rarely recur following marginal excision
Lipomatosis
Lipomatosis is a diffuse systemic or regional overgrowth of mature adipose tissue that differs from normal fat only in
P.265
its distribution. Lipomatosis should be distinguished from lipomas, as
the former conditions may be correctable by addressing the underlying
condition and tend to recur after attempted excision.
Pathogenesis and Pathophysiology
-
Etiology is poorly understood.
-
Possibly due to point mutations in mitochondrial genes
-
See Table 11-1 for epidemiology.
-
Histopathology: mature fat in poorly circumscribed lobules or sheets infiltrating surrounding tissues
Classification
-
Classified by distribution for idiopathic types (diffuse, pelvic, symmetric) or by etiology (steroid, HIV lipodystrophy)
Diagnosis
-
Notable accumulations of fat in affected areas may resemble neoplasms (see Table 11-1).
-
Radiologic studies document extent of fatty deposition.
-
Neither radiological evaluation nor biopsy is usually necessary to diagnosis the lipomatoses.
-
See Chapter 2.
Treatment and Outcome
-
Palliative surgery is rarely indicated
unless life-threatening fat accumulation (such as that causing
laryngeal compression in Madelung’s disease). -
Correction of steroid lipomatosis follows lowering of steroid levels.
|
Table 11-1 Epidemiology and Clinical Features of Lipomatosis Subtypes
|
||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Lipomatosis of Nerve (Lipofibromatous Hamartoma)
Lipofibromatosis, also referred to variously as
lipofibromatous hamartoma, fibrolipomatosis, and intraneural lipoma,
among other names, is a fatty and fibrous infiltrative process
affecting the epineurium and leading to enlargement of the affected
nerve. The median and ulnar nerves are most commonly affected.
lipofibromatous hamartoma, fibrolipomatosis, and intraneural lipoma,
among other names, is a fatty and fibrous infiltrative process
affecting the epineurium and leading to enlargement of the affected
nerve. The median and ulnar nerves are most commonly affected.
Pathogenesis and Pathophysiology
-
Etiology is unknown.
-
Peak age: 10 to 40
-
Frequently evident at birth or early childhood
-
Female > male if macrodactyly present; male > female if none
-
Gross histopathology: yellow fibrofatty infiltration of nerve (Fig. 11-2)
-
Microscopic histopathology: epineurial and perineural fibrofatty infiltration isolating individual nerve bundles
Diagnosis
Clinical Features
-
Gradually enlarging mass with variable neurologic deficits
-
Median nerve or branches > ulnar nerve
-
Foot, brachial plexus less common sites
-
Macrodactyly in one third
P.266
 |
|
Figure 11-2 A
Fibrous hamartoma of infancy. Organoid pattern composed of intersecting fascicles of spindle cells separated by collagen, mature fat, and islands of immature mesenchymal cells with myxoid matrix. B Close-up view of characteristic island of immature mesenchyma in fibrous hamartoma of infancy. |
Radiologic Features
-
MRI findings pathognomonic
-
Fusiform neural enlargement following branching pattern of nerve
-
Hypointense serpentine nerve bundles on both T1- and T2-weighted images
-
Variable intramuscular fatty deposition
-
Diagnostic Work-up
-
Because MRI is diagnostic, biopsy is usually not needed.
Treatment
-
Goal of surgical intervention is to decompress nerve if symptomatic.
-
Avoid attempted excision, which may damage nerve.
Lipoblastoma
Lipoblastoma is a tumor resembling fetal adipose tissue
that occurs predominately in infants and may be localized
(lipoblastoma) or diffuse (lipoblastomatosis). Since lipomas generally
do not occur in this age group, lipoblastoma should be considered
highly in the differential diagnosis of any tumor with fatty imaging
characteristics in a child.
that occurs predominately in infants and may be localized
(lipoblastoma) or diffuse (lipoblastomatosis). Since lipomas generally
do not occur in this age group, lipoblastoma should be considered
highly in the differential diagnosis of any tumor with fatty imaging
characteristics in a child.
Pathogenesis and Pathophysiology
-
Etiology is unknown.
-
Peak age: birth to age 3; less common in older children
-
Males > females
Histopathology
-
Variable mixture of mature adipocytes and immature fat cells (lipoblasts)
-
Variable myxoid background with plexiform vascular pattern suggestive of myxoid liposarcoma
-
Pronounced lobular pattern and lack of atypia distinguishes lipoblastoma from liposarcoma
Genetics
-
8q11-13 rearrangement in most cases
-
Fusion gene products: HAS2/PLAG1, COL1A2/PLAG1
Classification
-
Solitary (lipoblastoma) versus diffuse (lipoblastomatosis)
Diagnosis
-
See Chapter 2 for diagnostic work-up.
Clinical Features
-
Sites: extremities predominate
-
Typically small (2 to 5 cm), superficial mass
-
Lipoblastomatosis often involves muscle as well.
Radiologic Features
-
Fatty signal characteristics
-
Bright on T1-weighted MRI, dark on fat-suppressed sequences, identical to surrounding fat
-
-
Indistinguishable from lipoma, well-differentiated liposarcoma (atypical lipoma) by radiology
Treatment and Outcome
-
Lipoblastoma: marginal en bloc excision
-
Lipoblastomatosis: wide surgical excision
-
Recurrence rare in lipoblastoma; up to 22% in lipoblastomatosis
P.267
|
Table 11-2 Histological Lipoma Variants
|
||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Histologic Lipoma Variants
The histologic variants of lipoma are summarized in Table 11-2, and examples are shown in Figures 11-3 and 11-4.
Fibroblastic Benign Soft Tissue Tumors
Nodular Fasciitis
Nodular fasciitis is a tumor predominately of the upper
extremity and neck region that is distinguished by its rapid growth and
its tissue culture–like histology pattern.
extremity and neck region that is distinguished by its rapid growth and
its tissue culture–like histology pattern.
Pathogenesis and Pathophysiology
-
Etiology: unknown, but history of trauma common
Epidemiology
-
Peak age: young adults, but may involve any age
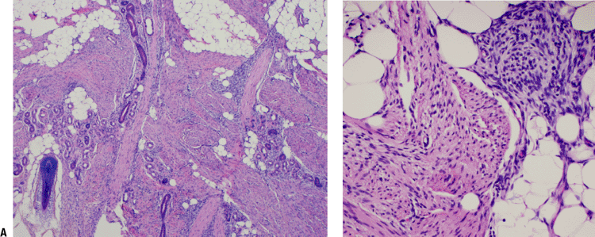
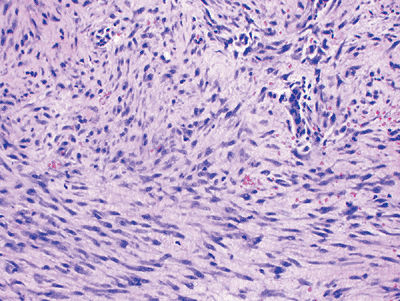
Histopathology (Fig. 11-5)
-
Plump, regular myofibroblasts
-
Frequent mitoses but not atypical mitoses
-
Tissue culture–like, “torn” or “feathery” appearance at low power
-
Prominent small vessels may resemble granulation tissue.
-
Nodular infiltrative pattern of organization
-
Extravasated red blood cells, chronic inflammatory cells, and even giant cells may be seen.
-
SMA and MSA positive
-
Desmin, cytokeratin, and S100 negative
 |
|
Figure 11-3
Angiolipoma composed of mature adipose tissue intermixed with numerous vascular channels. The vascularity is often more prominent in subcapsular areas. |
P.268
 |
|
Figure 11-4
Hibernoma: rare fatty tumor composed of mature adipose tissue and a large number of multivacuolated brown fat cells with abundant granular cytoplasm and a centrally located nucleus. |
Genetics
-
Some clonality suggesting neoplastic nature demonstrated, but may represent artifact of culture conditions
Diagnosis
-
See Chapter 2 for diagnostic work-up.
Clinical Features
-
Subcutaneous >> intramuscular
-
Upper extremity, trunk, head and neck most common
-
Rapid growth is characteristic.
-
Clinical history usually <1 to 2 months
-
Pain and local tenderness common
-
Small size, usually <2 cm
 |
|
Figure 11-5
Nodular fasciitis: cellular proliferation of stellate cells with vague storiform pattern and extravasated red blood cells. Inflammatory cells and mitoses are also commonly seen, but there is no cytologic atypia. |
Radiologic Features
-
Nonspecific soft tissue mass (Fig. 11-6)
Treatment and Outcome
-
Marginal excision
-
Rare recurrence should warrant reconsideration of diagnosis.
Proliferative Fasciitis and Myositis
These two processes are similar to nodular fasciitis in
their rapid growth, predilection for the upper extremity, and plump
myofibroblastic cells, but they are distinguished by the large
ganglion-like cells that are also present. Proliferative fasciitis (PF)
involves the fascia, while proliferative myositis (PM) involves the
muscle.
their rapid growth, predilection for the upper extremity, and plump
myofibroblastic cells, but they are distinguished by the large
ganglion-like cells that are also present. Proliferative fasciitis (PF)
involves the fascia, while proliferative myositis (PM) involves the
muscle.
Pathogenesis and Pathophysiology
-
Etiology is unknown, although a history of trauma may be elicited.
-
Peak age: middle-aged or older adults, older than nodular fasciitis
-
Much less common than nodular fasciitis
Histopathology
-
Two cellular types: plump myofibroblasts and ganglion-like cells
-
Plump myofibroblasts resemble those of nodular fasciitis.
-
Ganglion-like cells: large with one to three rounded nuclei, prominent nucleoli, and abundant cytoplasm
-
Numerous mitoses but not atypical mitoses
-
Infiltrative growth pattern (along fascial planes for PF and between muscle groups in PM)
-
Checkerboard pattern of infiltration between muscle fibers for PM
-
SMA and MSA positive
-
Desmin, cytokeratin, and S100 negative
Genetics
-
Similar to nodular fasciitis
Diagnosis
-
See Chapter 2 for diagnostic work-up.
Clinical Features
-
PF: upper extremity (especially forearm) > lower extremity > trunk
-
PM: trunk > shoulder girdle > upper arm > thigh
-
Rapid growth is characteristic.P.269
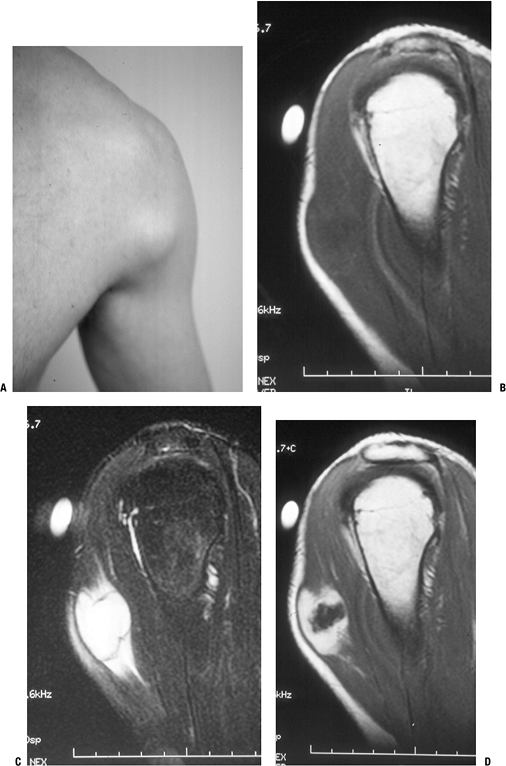
![]() Figure 11-6 Nodular fasciitis of the shoulder. (A)
Figure 11-6 Nodular fasciitis of the shoulder. (A)
The patient presented with a 3-week history of a rapidly enlarging,
painful soft tissue mass over the anterior deltoid. Sagittal MR studies
show nonspecific findings of a heterogeneous intramuscular mass
hypointense to muscle on T1-weighted sequences (B), hyperintense on T2-weighted images (C), and enhancing on post-gadolinium sequences (D). -
Clinical history usually <1 or 2 months
-
Pain and local tenderness more common with PF
-
Small size, usually <5 cm
P.270
Radiologic Features
-
Nonspecific soft tissue mass
Treatment and Outcome
-
Marginal excision
-
Recurrence is rare.
Myositis Ossificans
Myositis ossificans is a benign reparative process
characterized by bone formation within the soft tissues in response to
trauma. On the one hand, it may simulate malignancy, but conversely,
sarcomas that give a similar appearance may be mistaken for this
benign, self-limited condition.
characterized by bone formation within the soft tissues in response to
trauma. On the one hand, it may simulate malignancy, but conversely,
sarcomas that give a similar appearance may be mistaken for this
benign, self-limited condition.
Pathogenesis and Pathophysiology
-
Trauma history elicited in up to 75% of patients; repetitive trauma in others
-
Peak age: young, physically active adults (rare in infants or elderly)
-
Males > females
Histopathology
-
Zonal proliferation with central fibroblasts and peripheral osteoblast-rimmed bone trabeculae
-
Progression from initially fibrous tissue to peripheral woven bone and then eventually lamellar bone
-
Peripheral ossification usually evident by 3 weeks
-
Mitoses frequent but no atypical mitoses
Diagnosis
-
See Chapter 2 for diagnostic work-up.
Clinical Features
-
Initial 1 to 2 weeks after injury: swollen, tender
-
From 2 to 6 weeks after injury: tenderness and pain resolve, and painless, very firm mass forms
-
Chronically, the mass becomes less prominent over time.
-
Any location in body susceptible to trauma
-
Most common locations: thigh, shoulder, buttock, elbow
Radiologic Features
-
Initial 1 to 2 weeks after injury: x-ray,
computed tomography (CT), MRI shows soft tissue shadow, heterogeneous
MRI signal with edema -
From 2 to 6 weeks after injury: x-ray, CT best show peripheral rim of calcifications (Fig. 11-7)
-
Chronically, MRI eventually shows low-signal rim with fatty marrow signal centrally.
Treatment and Outcome
-
Observation is best, but marginal excision is appropriate if sufficiently symptomatic.
-
May recur if excised incompletely early in course
Elastofibroma
Elastofibroma (elastofibroma dorsi) is a unique process
that occurs almost exclusively in a subscapular location in adults and
is characterized by histologic evidence of elastic fibers.
that occurs almost exclusively in a subscapular location in adults and
is characterized by histologic evidence of elastic fibers.
Pathogenesis and Pathophysiology
-
Etiology is unknown, although repetitive trauma has been implicated.
-
Peak age in seventh and eighth decades (nearly always after age 50)
-
Females > males
Histopathology
-
Low-cellularity collagenous tissue with intermixed elastic fibers (Fig. 11-8)
-
Immunohistochemistry positive for elastin
Genetics
-
Familial occurrence reported in Okinawa
Diagnosis
Clinical Features
-
Slowly growing mass, typically painless and nontender
-
May cause popping scapula or local discomfort
-
Classic location
-
Nearly exclusively subscapular, applied to chest wall at the lower portion of the scapula
-
Deep to latissimus dorsi and rhomboid major
-
Often attached to periosteum of ribs
-
Rare musculoskeletal sites: upper arm, hip
-
May be bilateral (especially subclinical) in up to one third
-
Radiologic Features
-
Fibrous and fatty elements create layered picture.
-
MRI may be highly suggestive (Fig. 11-9).
-
Fatty areas: bright areas on T1-weighted images, intermediate on T2-weighted images
-
Fibrous areas: similar to muscle
-
Diagnostic Work-up
-
Biopsy is generally recommended to
confirm diagnosis, although the combination of classic location and
radiologic appearance is highly suggestive. -
Also see Chapter 2.
P.271
 |
|
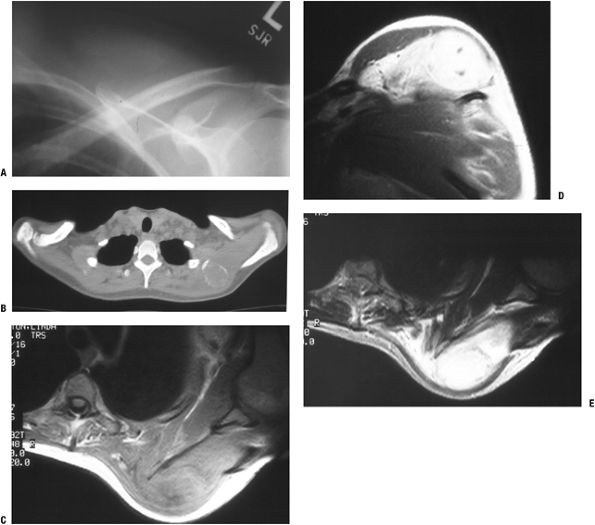
Figure 11-7 (A)
Initial plain radiographs within the first 2 to 3 weeks may not show the characteristic peripheral rim of calcification associated with myositis ossificans. (B) Subsequently, the peripheral rim is best demonstrated on computed tomography. (C) MR studies show a heterogeneous mass that is sometimes confused with a soft tissue sarcoma. |
Treatment and Outcome
-
Marginal excision indicated for symptomatic patients but may be observed if asymptomatic and histologically proven
-
Rarely recurs after complete excision
Superficial Fibromatoses
The superficial fibromatoses include palmar fibromatosis
(Dupuytren’s disease or contracture) and plantar fibromatosis
(Ledderhose disease), both fibroblastic proliferations with
infiltrative growth that are usually easily recognizable clinically and
have a high rate of local recurrence if excised.
(Dupuytren’s disease or contracture) and plantar fibromatosis
(Ledderhose disease), both fibroblastic proliferations with
infiltrative growth that are usually easily recognizable clinically and
have a high rate of local recurrence if excised.
Pathogenesis and Pathophysiology
-
Etiology is multifactorial.
-
Familial component
-
Trauma
-
Associated diseases: epilepsy, diabetes, alcohol- induced liver disease
-
-
Peak age: adults with increasing frequency in advanced ages (rare before age 30)
-
Males >> females (3 to 4:1)
-
Pathophysiology is shown in Figure 11-10.
 |
|
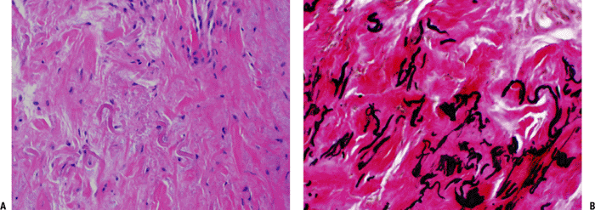
Figure 11-8 (A) Elastofibroma composed of intertwining, swollen collagen and elastic fibers with fibroblasts. (B) Elastic fibers highlighted by EVG stain in elastofibroma.
|
Diagnosis
Clinical Features
-
Palmar fibromatosis
-
Volar surface, 50% bilateral
-
Typical progression
-
Begins with firm painless nodule
-
Progresses to multiple nodules connected by tight cords (distinguished from pre-existing normal fascial bands)
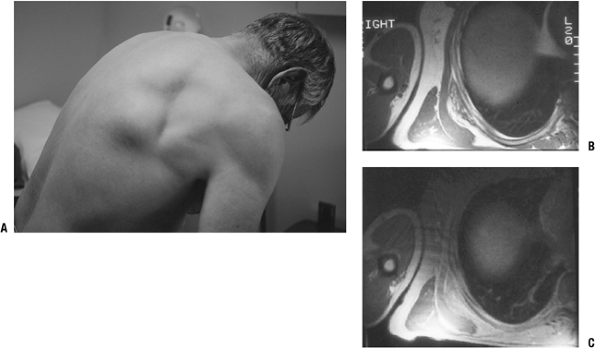 Figure 11-9 (A)
Figure 11-9 (A)
The characteristic location for an elastofibroma is at the inferior
medial border of the scapula in a subscapular position. MR images show
a heterogeneous subscapular soft tissue mass on T1-weighted (B) and proton density (C) axial images. -
Puckering of overlying skin
-
End-stage flexion contractures favoring ring and little fingers
-
-
-
Plantar fibromatosis
-
Plantar aponeurosis
-
Typical course
-
Begins with firm nodule adherent to skin
-
Often painful with shoe wear
-
Progressive enlargement may result.
-
Rarely leads to contracture
-
-
P.273
 |
|
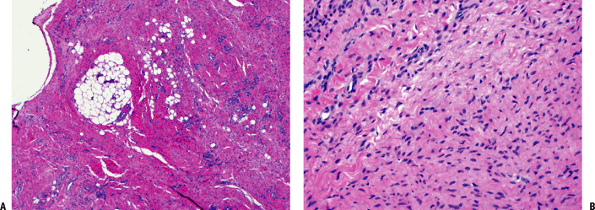
Figure 11-10 (A) Plantar fibromatosis: dense fibrous proliferation infiltrating adipose tissue. (B)
Plantar fibromatosis: early lesions consist of cellular proliferations of bland-looking spindle cells and collagen deposition similar to desmoid tumors. Older lesions tend to be less cellular and contain more collagen. |
Radiologic Features
-
Palmar fibromatosis
-
Cords and hypocellular nodules dark on T1- and T2-weighted images
-
Hypercellular nodules intermediate on T1- and T2-weighted images
-
-
Plantar fibromatosis
-
Isointense to slightly hyperintense always on T1-weighted images and nearly always on T2-weighted images
-
Usually hyperintense on short tau inversion recovery (STIR) sequence
-
Enhancement common
-
Diagnostic Work-up
-
Dupuytren’s contracture and many cases of
plantar fibromatosis are easily recognized clinically and do not
require either further imaging or biopsy. -
If plantar masses are not characteristic, MRI imaging may establish the diagnosis, but biopsy is sometimes needed.
Treatment and Outcome
-
Palmar fibromatosis
-
Flexion contracture is most common indication for operative treatment.
-
Details of operative treatment are beyond the scope of this text.
-
-
Plantar fibromatosis
-
Usually managed nonoperatively unless recalcitrant to adaptive shoe wear and inserts
-
If operative treatment is elected, complete fasciectomy achieves lowest recurrence rates.
-
High recurrence rate for plantar fibromatosis ranging from 50% with complete fasciectomy to 100% with local excision
-
Desmoid-Type Fibromatoses
Extra-abdominal desmoid tumors are the subset of
interest in orthopaedics. They consist of a neoplastic fibroblastic
proliferation that may result in pain, infiltrative growth resulting in
recurrence, and absence of metastatic potential.
interest in orthopaedics. They consist of a neoplastic fibroblastic
proliferation that may result in pain, infiltrative growth resulting in
recurrence, and absence of metastatic potential.
Pathogenesis and Pathophysiology
Etiology
-
Familial component
-
Component of Gardner syndrome (autosomal dominant transmission, variant of familial adenomatous polyposis; Box 11-1)
-
-
Endocrine factors
-
Frequent exacerbation/appearance postpartum
-
Estrogen receptors in desmoid tumors
-
Response to endocrine agents
-
-
Trauma may be contributory factor.
Epidemiology
-
Pediatric patients: extra-abdominal > abdominal
-
Puberty to 40: abdominal > extra-abdominal, female > male
-
Older than 40: abdominal = extra-abdominal
Box 11-1 Components of Gardner Syndrome
-
Gastrointestinal polyps with 100% risk of malignancy
-
Multiple osteomas
-
Epidermoid cysts
-
Desmoid tumors
-
Other benign skin and soft tissue tumors
P.274
 |
|
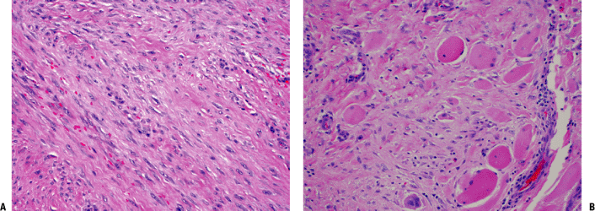
Figure 11-11 (A) Extra-abdominal fibromatosis showing a proliferation of bland-looking spindle cells separated by collagen. (B) Infiltration of surrounding soft tissues in fibromatosis. Note the presence of entrapped skeletal muscle cells.
|
Pathophysiology
Histopathology (Fig. 11-11)
-
Low-cellularity spindle cells in collagenous stroma
-
Extremely infiltrative process arranged in sweeping bundles
Genetics
-
Two mutations with common endpoint of increased beta-catenin expression
-
Inactivation of APC tumor suppressor gene on 5q may be initiating event, especially those with familial occurrence.
-
Activating beta-catenin mutations render beta- catenin resistant to inhibitory effect of APC.
-
-
Trisomies for chromosomes 8 or 20 in 30% of cells
Diagnosis
-
See Chapter 2 for diagnostic work-up.
Clinical Features
-
Deep (as distinguished from superficial fibromatoses)
-
Painful or painless, poorly circumscribed, very hard mass
-
Occasionally multifocal
-
Occasional joint stiffness or neurological symptoms
Radiologic Features
-
MRI nonspecific (Fig. 11-12)
-
Isointense to muscle on T1-weighted images
-
Intense contrast enhancement
-
Hyperintense on STIR and T2-weighted images
-
Treatment
-
Surgical indication
-
Wide resection preferred if adequate margin attainable with acceptable function
-
-
Surgical contraindications
-
Vital structures involved or unresectable disease
-
Consider alternative treatments in this situation:
-
Radiotherapy
-
Pharmacologic treatment
-
Hormonal agents
-
Nonsteroidal anti-inflammatory agents
-
Interferons
-
Cytotoxic chemotherapy
-
-
-
-
Radiotherapy consultation is recommended for positive surgical margins, although efficacy not well established.
Results and Outcome
-
Surgical resection
-
Wide margins: 85% to 90% local control
-
Marginal margins: 50%
-
Intralesional margin: <50%
-
-
Radiotherapy
-
Typically recommended for positive margins, but proof of efficacy not clearly established
-
-
Pharmacologic treatment
-
40% to 50% response rate
-
Solitary Fibrous Tumor and Hemangiopericytoma
According to the latest WHO classification, extrapleural
solitary fibrous tumors (SFT) and hemangiopericytoma (HP) share enough
clinical, histological, and immunohistochemical features to be
classified together and may actually represent a spectrum of the same
entity. They typically behave in a benign fashion, but up to 30% are
malignant.
solitary fibrous tumors (SFT) and hemangiopericytoma (HP) share enough
clinical, histological, and immunohistochemical features to be
classified together and may actually represent a spectrum of the same
entity. They typically behave in a benign fashion, but up to 30% are
malignant.
Pathogenesis and Pathophysiology
-
Etiology is unknown.
-
Peak age 20 to 70 years (median 50)
P.275
 |
|
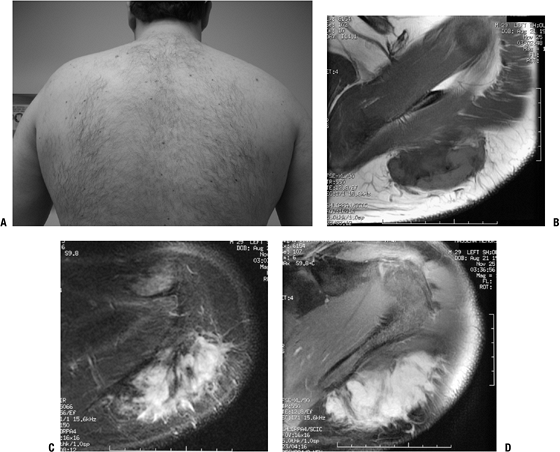
Figure 11-12 (A)
Desmoid tumor typically presents as a slow-growing, sometimes painful soft tissue mass. MR imaging shows nonspecific hypointense signal on T1-weighted sequences (B) and hyperintense signal on T2-weighted sequences (C). (D) Gadolinium enhancement is intense. |
Histopathology
-
SFT
-
Intermixed hypercellular and hypocellular areas separated by bands of hyalinized collagen
-
Round to spindle cells with indistinct cytoplasm
-
Immunohistochemistry
-
Vimentin, CD34, and CD99 positive
-
Variable focal positivity for others
-
-
-
HP
-
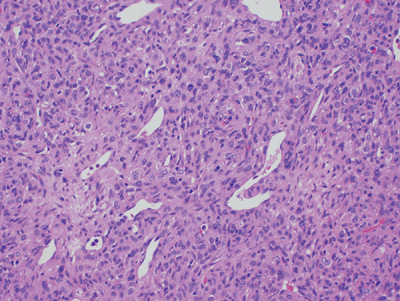
Similar to SFT with addition of prominent variably branching thin-walled vessels with staghorn configuration (Fig. 11-13)
-
Immunohistochemistry
-
Vimentin, CD34, and CD99 positive
-
Cytokeratin, actin, and desmin negative
-
-
 |
|
Figure 11-13
Hemangiopericytoma: cellular neoplasm composed of spindle and round cells with large vascular spaces with a “staghorn” configuration. |
Genetics
-
Inconsistent chromosomal aberrations
Diagnosis
-
See Chapter 2 for diagnostic work-up.
P.276
Clinical Features
-
Superficial or deep, slowly growing, small or large, painless mass in any location
-
Rare paraneoplastic syndrome due to IGF-1
production results in hypoglycemia or acromegaly; oncogenic
osteomalacia is very rare with these tumors.
Radiologic Features
-
Nonspecific radiologic features
-
MRI nonspecific
-
Isointense to slightly hyperintense on T1-weighted images
-
Hyperintense on T2-weighted images
-
Heterogeneous, enhancing
-
Treatment and Outcome
-
Wide surgical resection
-
Postoperative management with adjuvant radiotherapy and/or chemotherapy may play a role in some malignant cases.
-
85% 5-year survival rate including malignant forms
Fibrohistiocytic Benign Soft Tissue Tumors
Giant Cell Tumor of Tendon Sheath
Also referred to as localized tenosynovial giant cell
tumor or nodular tenosynovitis, these common tumors represent the most
common neoplasms of the hand. In some classifications, such as that of
the current WHO, this terminology also includes what in the past has
been referred to as the localized form of pigmented villonodular
synovitis (PVNS).
tumor or nodular tenosynovitis, these common tumors represent the most
common neoplasms of the hand. In some classifications, such as that of
the current WHO, this terminology also includes what in the past has
been referred to as the localized form of pigmented villonodular
synovitis (PVNS).
 |
|
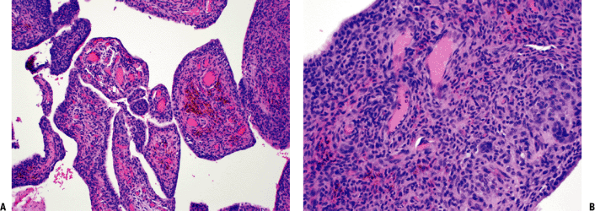
Figure 11-14 (A)
Pigmented villonodular synovitis showing characteristic intra-articular villous pattern. Abundant hemosiderin pigment seen within macrophages in fibrovascular core. (B) Pigmented villonodular synovitis: solid areas composed of oval to spindle-shaped, histiocytoid, mononuclear cells and multinucleated giant cells. |
Pathogenesis and Pathophysiology
-
Current evidence favors a neoplastic etiology over the traditional belief that this was a reactive process.
-
Peak age: 30 to 50 years (but any age may be involved)
-
Female > male
Histopathology (Fig. 11-14B)
-
Grossly well circumscribed with yellow and brown areas
-
Composed of mixture of background
mononuclear cells, variable numbers of giant cells, lipid-laden
xanthoma cells, and hemosiderin-laden macrophages -
Immunohistochemistry
-
Mononuclear cells: CD68 positive
-
Giant cells: positive for CD68, CD45, and tartrate resistant acid phosphatase (TRAP)
-
Genetics
-
Cytogenetic aberrations of chromosome 1p in particular, frequently with translocation, are common.
-
No trisomies of chromosomes 5 or 7, as seen with PVNS
Diagnosis
Clinical Features
-
Predominately in the hand (85%) near tendon sheath or joint > wrist, foot/ankle, knee
-
Small painless nodules
Radiologic Features
-
Approximately 20% erode adjacent bone.
-
Nonspecific MRI features (Fig. 11-15)P.277
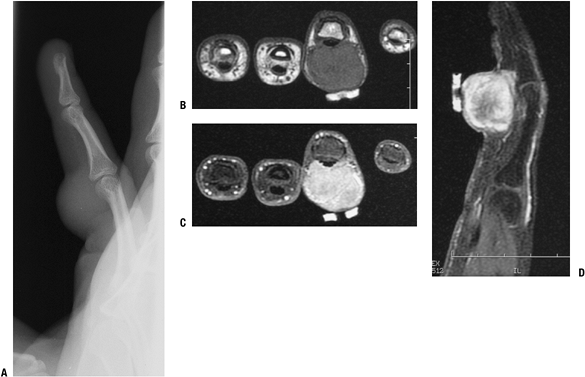 Figure 11-15 Giant cell tumor of tendon sheath shows a soft tissue shadow on plain lateral radiograph (A), hypointense signal on axial T1-weighted MRI (B), hyperintense signal on axial T2-weighted MRI (C), and gadolinium enhancement on post-contrast fat-suppressed T1-weighed sagittal MRI (D).
Figure 11-15 Giant cell tumor of tendon sheath shows a soft tissue shadow on plain lateral radiograph (A), hypointense signal on axial T1-weighted MRI (B), hyperintense signal on axial T2-weighted MRI (C), and gadolinium enhancement on post-contrast fat-suppressed T1-weighed sagittal MRI (D).-
T1-weighted images: hypointense to slightly hyperintense to muscle
-
T2-weighted images: hypointense to hyperintense to muscle
-
Heterogeneous due to hypointense areas
-
Diffuse contrast enhancement
-
Diagnostic Work-up
-
Usually plain radiographs and MRI are sufficient for these small masses.
-
Also see Chapter 2.
Treatment and Outcome
-
Marginal excision
-
Up to 30% recurrence rate, easily controlled by re-excision
Diffuse-Type Giant Cell Tumor (Pigmented Villonodular Synovitis)
Pigmented villonodular synovitis (PVNS) is a destructive
proliferative synovial process predominately seen affecting the knee.
It was formerly subdivided into localized and diffuse forms; the former
is now classified as localized giant cell tumor.
proliferative synovial process predominately seen affecting the knee.
It was formerly subdivided into localized and diffuse forms; the former
is now classified as localized giant cell tumor.
Pathogenesis and Pathophysiology
-
Current evidence favors a neoplastic etiology over the traditional belief that this was a reactive process.
-
Peak age: <40 years (but any age may be involved)
-
Female > male
Histopathology
-
Intra-articular forms: villous pattern with yellow and brown areas (Fig. 11-14A)
-
Extra-articular forms: grossly well circumscribed with yellow and brown areas (Fig. 11-14B)
-
Expansile sheets of infiltrative cells
with intermixed cellular regions and discohesive areas creating
blood-filled pseudoalveolar spaces -
Composed of mixture of background
mononuclear cells, variable numbers of giant cells, lipid-laden
xanthoma cells, and hemosiderin-laden macrophages-
Giant cells less common than in giant cell tumor of tendon sheath, sparse or absent in up to 20%
-
Bimodal mononuclear cells: small histiocyte-like cells and larger dendritic cells
-
-
Immunohistochemistry
-
Mononuclear cells: CD68 positive
-
Larger dendritic cells: desmin positive
-
Giant cells: CD68, CD45, and TRAP positive
-
P.278
Genetics
-
Chromosome 1p translocations common and similar to giant cell tumor of tendon sheath
-
Trisomies of chromosomes 5 and 7 seen only in PVNS, not in giant cell tumor of tendon sheath
Classification
-
Intra-articular and extra-articular forms
Diagnosis
Clinical Features
-
Usually large and associated with pain, local tenderness, and decreased joint motion
-
Recurrent hemarthroses common
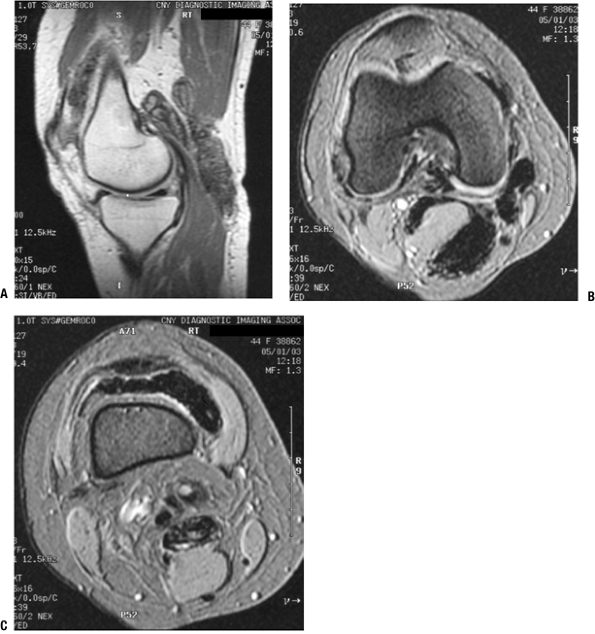
Radiologic Features (Fig. 11-16)
-
Juxta-articular erosions evident in intra-articular form in advanced cases
-
MRI
-
Characteristic hemosiderin attenuation of signal resulting in at least foci of hypointense signal within all sequences
-
Fatty areas due to lipid accumulation
-
Joint effusion
-
 |
|
Figure 11-16
Pigmented villonodular synovitis shows characteristic signal attenuation from the hemosiderin deposition within the synovium on all MRI sequences. In this case, low-signal areas can be seen within the suprapatellar pouch and popliteal region on these sagittal T1-weighted (A), axial proton density (B), and axial T2-weighted (C) MR images. |
Diagnostic Work-up
-
Usually plain radiographs and MRI are sufficient for these processes.
-
Also see Chapter 2.
Neural Benign Tumors
Neurofibroma and Neurofibromatosis
Neurofibromas are benign peripheral nerve sheath tumors
that may occur as solitary lesions or as a component of
neurofibromatosis. Neurofibromas are more characteristically
intertwined with the peripheral nerve and difficult to separate.
that may occur as solitary lesions or as a component of
neurofibromatosis. Neurofibromas are more characteristically
intertwined with the peripheral nerve and difficult to separate.
Pathogenesis and Pathophysiology
-
Etiology is unknown.
-
Peak age: young adults 20 to 30 years
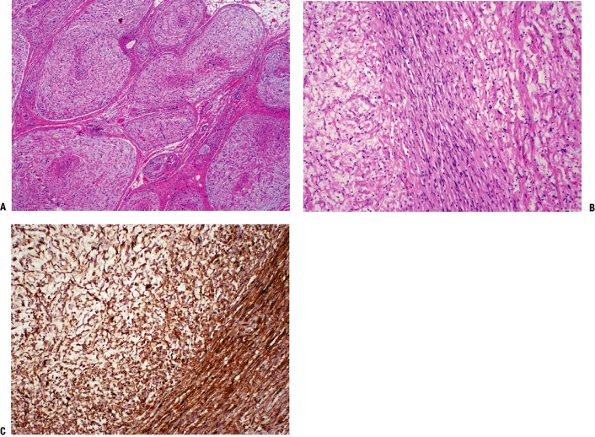 |
|
Figure 11-17 (A) Cross-section of a plexiform neurofibroma showing its distinctive arrangement in large fascicles. (B)
Characteristic neurofibroma with wavy spindle cells in a loose stroma. Toward the center, cells are more tightly apposed, resembling peripheral nerve fibers. (C) Diffuse and strong immunostaining of neurofibroma with S-100 protein stain. |
Histopathology (Fig. 11-17)
-
Mixture of Schwann cells, perineurial-like cells, and fibroblasts
-
Hypocellular, widely spaced cells with thin, elongated nuclei
-
S100 positive
Genetics
-
NF-1 gene mutation in neurofibromatosis (NF)
-
Solitary neurofibromas not as clearly established
Classification (Table 11-3)
-
Localized cutaneous neurofibroma (Fig. 11-18)
-
Most common type
-
10% occur in NF-1
-
-
Diffuse cutaneous neurofibroma
-
Plaque-like thickenings of dermis and subcutaneous tissue
-
-
Localized intraneural neurofibroma (Fig. 11-19)
-
Second most common type
-
Fusiform segmental enlargement of nerve (Fig. 11-20)
P.280Table 11-3 Subtypes of NeurofibromaSubtype Depth Site Size Pain? % in NF-1 Malignant Transformation? Diffuse cutaneous neurofibroma Skin and subcutaneous Any 1 to 2 cm No 10% No Localized cutaneous neurofibroma Skin and subcutaneous Head and neck Several centimeters No 10% Rare Localized intraneural neurofibroma Superficial or deep In NF-1, predilection for cervical spine 1 cm to many centimeters Neural pain if deep Minority Infrequent Plexiform neurofibroma Usually deep Predilection for larger nerves Large Neural pain Almost exclusively Highest Massive soft tissue neurofibroma Skin and deeper tissues Shoulder, pelvis, lower extremity Large Variable 100% Rare -
-
Plexiform neurofibroma (Fig. 11-21)
-
Uncommon tumor usually affecting large nerve
-
“Bag of worms” appearance in plexus regions (Fig. 11-22)
-
Firm, ropy cylinder in nonbranching nerves
-
Highly suggestive of diagnosis of NF-1
-
-
Massive soft tissue neurofibroma (Fig. 11-24)
-
Least common form always seen in NF-1
-
Formerly called “elephantiasis neuromatosa”
-
Localized gigantism or massive regional tissue enlargement
-
Diagnosis
-
See Chapter 2 for diagnostic work-up.
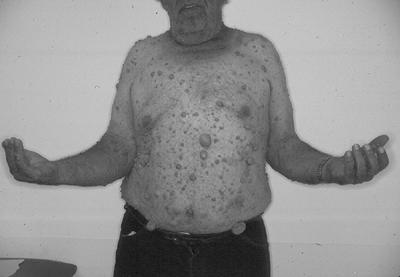 |
|
Figure 11-18
Localized cutaneous neurofibromas, as shown in this patient with type I neurofibromatosis, are the most common type of neurofibroma. |
Clinical Features
-
Solitary painless or painful masses or in the setting of NF-1 (von Recklinghausen’s disease)
-
Intraneural neurofibromas may cause pain or paresthesias along distribution of affected nerve.
-
Tinel’s sign may be positive.
-
More medial–lateral movement than proximal– distal on palpation
-
Radiologic Features
-
MRI features may be highly suggestive of benign peripheral nerve sheath tumors.
-
Target sign more frequent with neurofibromas than schwannomas (Fig. 11-25)
-
Dark on T1-weighted images, bright on T2-weighted images
-
-
Dumbbell tumors (partially intraspinal and partially extraspinal) may cause foraminal enlargement.
Treatment
-
Localized cutaneous neurofibroma and diffuse cutaneous neurofibroma
-
Excision usually curative
-
No neurological deficit expected
-
-
Localized intraneural neurofibroma and plexiform neurofibroma
-
Avoid resection due to need to resect functional nerve fibers.
-
Indications for excision: suspicion for malignant degeneration
-
Rapid increase in size
-
Pain
-
-
-
Massive soft tissue neurofibroma
-
May be amenable only to debulking
-
P.281
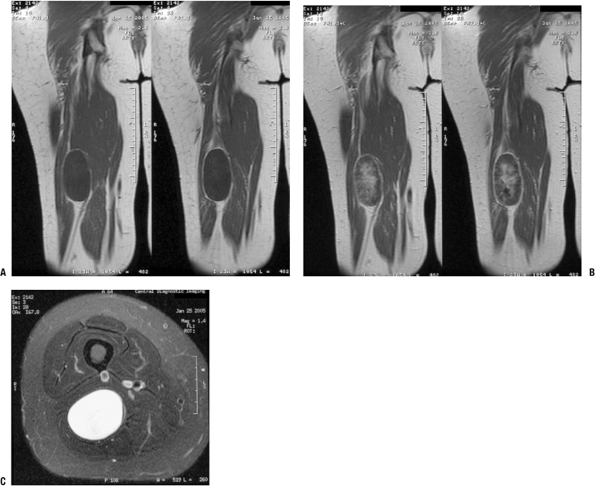 |
|
Figure 11-19
Localized intraneural neurofibroma is usually recognizable as a benign peripheral nerve sheath tumor, but it may be difficult to distinguish from a schwannoma. This case shows a discrete enhancing mass in continuity with deep nerve fibers on coronal T1-weighted (A), post-gadolinium T1-weighted (B), and axial T2-weighted (C) images. Although these features could be seen with either neurofibroma or schwannoma, this tumor was found to be a neurofibroma histologically. |
 |
|
Figure 11-20
Schematic shows appearance of an intraneural neurofibroma, where normal nerve fibers entering and exiting the tumor are intimately intertwined with those of the neurofibroma. |
P.282
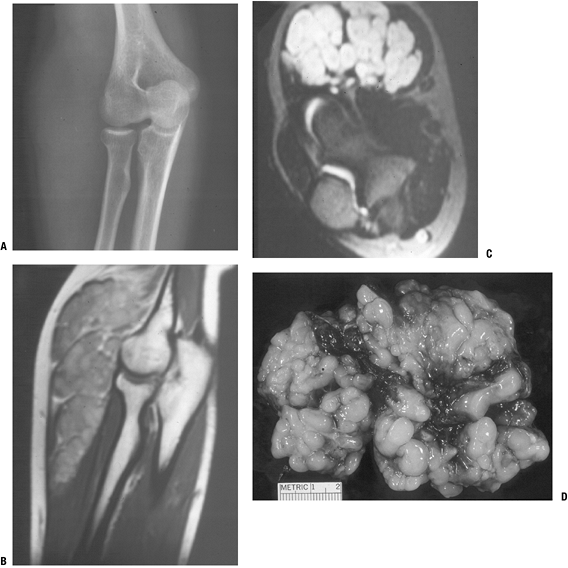 |
|
Figure 11-21
Plexiform neurofibroma, which is highly suggestive of the diagnosis of type I neurofibromatosis, is shown here as the classic “bag of worms” appearance on plain radiograph (A), MR images (B,C), and gross resected specimen (D). |
 |
|
Figure 11-22 Schematic shows typical “bag of worms” appearance of a plexiform neurofibroma.
|
P.283
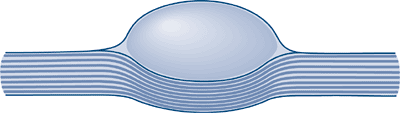 |
|
Figure 11-23 Schematic shows typical eccentric position of a neurilemoma, where normal nerve fibers are displaced peripherally by the mass.
|
 |
|
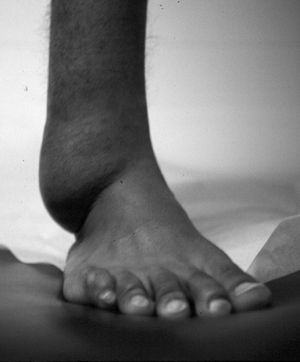
Figure 11-24 Massive soft tissue neurofibroma is manifest in this leg as a bag-like collection.
|
 |
|
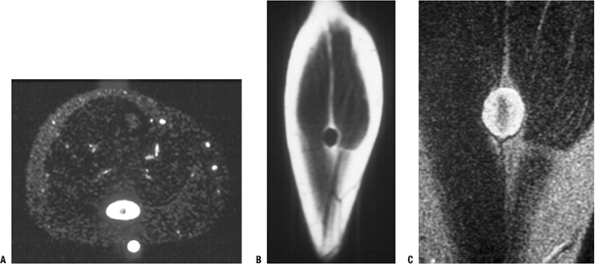
Figure 11-25 (A)
The target sign of peripheral nerve sheath tumors on an axial MR image for a neurofibroma, which more commonly shows this sign than does schwannoma. Coronal T1-weighted (B) and T2-weighted (C) images. |
P.284
Schwannoma (Neurilemoma)
Schwannoma is, along with neurofibroma, one of the two
most common benign peripheral nerve sheath tumors. While there are four
histological variants, this section will concentrate on the
conventional schwannoma.
most common benign peripheral nerve sheath tumors. While there are four
histological variants, this section will concentrate on the
conventional schwannoma.
Pathogenesis and Pathophysiology
-
Etiology is unknown.
-
Peak ages: third to sixth decades
-
A schwannoma is a neoplasm composed of Schwann cells, and its immunohistochemical and ultrastructural profile reflects that.
Histopathology (Fig. 11-26)
-
Biphasic Antoni A and B areas
-
Antoni A pattern
-
Collagenous background
-
Compact, elongated spindle cells aligned in bundles
-
Verocay bodies: palisaded arrangement of peripheral aligned nuclei surround central eosinophilic cell processes
-
-
Antoni B pattern
-
Mucopolysaccharide myxoid background
-
Less organized cells with plump nuclei and delicate cobweb-like interconnecting processes
-
-
-
Cystic degeneration and hemorrhage frequent (formerly termed “ancient schwannoma”)
-
Immunohistochemistry: S100 positive
Genetics
-
Not associated with NF-1 but may be associated with NF-2
 |
|
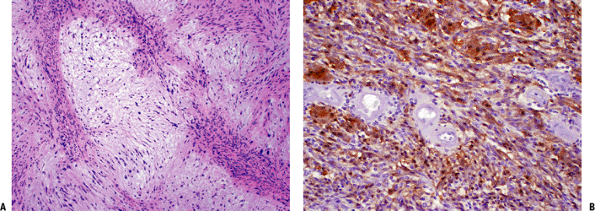
Figure 11-26 (A)
Schwannoma with characteristic areas of high cellularity (Antoni A) and low cellularity (Antoni B). Note the palisading of spindle cells known as Verocay bodies. (B) Strong immunoreactivity of schwannoma cells with S-100 protein stain. |
Classification
-
Conventional: most common form; characterized by Antoni A and Antoni B areas
-
Cellular: higher cellularity, mostly Antoni A areas, absent Verocay bodies
-
Plexiform: rare variant with intraneural pattern but not usually associated with NF-1
-
Melanotic: unusual variant with melanin-producing cells
Diagnosis
-
See Chapter 2 for diagnostic work-up.
Clinical Features
-
Head and neck, flexor surfaces of extremities (Fig. 11-27)
-
Slowly growing masses <10 cm
-
May elicit neural symptoms of pain or paresthesias
-
Physical examination may show positive Tinel sign, limited proximal–distal mobility.
-
Spinal schwannomas usually affect sensory nerves.
Radiologic Features (Fig. 11-28)
-
Well-demarcated soft tissue masses on plain x-ray
-
MRI heterogeneity related to cystic degeneration
-
Hypointense on T1-weighted images, hyperintense on T2-weighted images
-
Enhancing
-
-
Target sign on MRI less common than neurofibroma
-
Foraminal enlargement with dumbbell schwannomas and giant sacral schwannomas
Treatment and Outcome
-
Marginal complete excision usually
possible without resecting nerve due to eccentric growth (unlike
intraneural and plexiform neurofibromas; Fig. 11-23) -
Recurrence unusual except in sacral schwannomas
P.285
 |
|
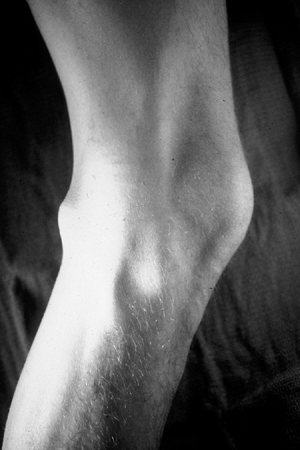
Figure 11-27 Schwannomas of the extremity typically occur on the flexor surface near a joint, such as the knee in this patient.
|
 |
|
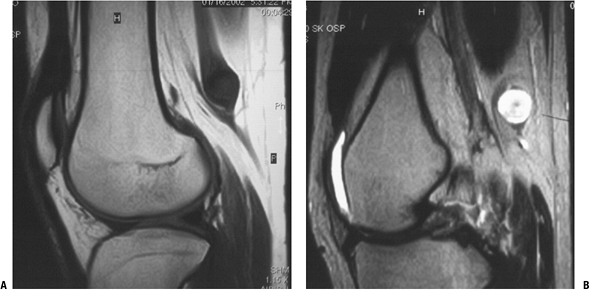
Figure 11-28 Schwannomas are hypointense to muscle on T1-weighted MR image (A) and hyperintense on T2-weighted image (B). Note how the nerve fibers splay around the round to oval mass.
|
 |
|
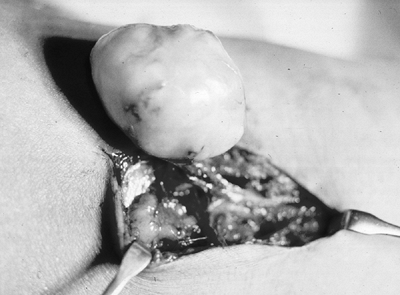
Figure 11-29 The schwannoma is usually easily separable from the associated nerve, unlike the intraneural neurofibroma.
|
P.286
Smooth Muscle Benign Soft Tissue Tumors
Leiomyoma of Deep Soft Tissue
Leiomyoma of the soft tissue is a tumor comprising
normal-appearing smooth muscle cells, but it is extremely rare in the
extra-abdominal soft tissues.
normal-appearing smooth muscle cells, but it is extremely rare in the
extra-abdominal soft tissues.
Pathogenesis and Pathophysiology
-
Etiology is unknown.
-
Extremely rare
-
Cells resemble smooth muscle cells with
eosinophilic cytoplasm and blunt-ended, cigar-shaped nuclei arranged in
intersecting bundles (Fig. 11-30). -
Immunohistochemistry
-
Positive for actin, desmin, and h-caldesmon
-
S100 negative
-
Diagnosis
-
Clinical features: superficial or deep soft tissue masses
-
Radiologic features: frequently calcified, but otherwise non specific
-
See Chapter 2 for diagnostic work-up.
Treatment and Outcome
-
Marginal excision
-
Local recurrence rate ~3%
 |
|
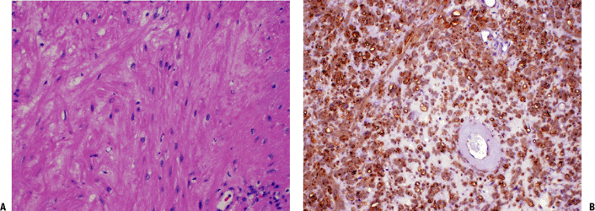
Figure 11-30 (A)
Leiomyoma. Fusiform cells with eosinophilic cytoplasm arranged showing a vague fascicular pattern closely resembling normal smooth muscle. (B) Strong and diffuse reactivity for smooth muscle actin stain in leiomyoma. |
Skeletal Muscle Benign Tumors
Rhabdomyoma
Analogous to leiomyomas, rhabdomyomas are extremely rare tumors of the soft tissue.
Pathogenesis and Pathophysiology
-
Etiology is unknown.
-
Very rare
-
Peak ages:
-
Adult form: median 60 years (33 to 80 years)
-
Fetal form: median 4 years
-
Histopathology (Fig. 11-31)
-
Lobules of closely packed cells
-
Spider cells (large, polygonal cells with abundant eosinophilic cytoplasm)
-
Cytoplasmic inclusion bodies, cross-striations
-
Immunohistochemistry: MSA, desmin, and myoglobulin positive
Classification
-
This section refers only to extracardiac rhabdomyomas at the exclusion of cardiac rhabdomyomas.
-
The extracardiac rhabdomyomas are subdivided into adult and fetal types.
Diagnosis
-
Clinical features: head and neck (90%), soft tissue mass
-
Radiologic features are nonspecific.
-
See Chapter 2 for diagnostic work-up.
P.287
 |
|
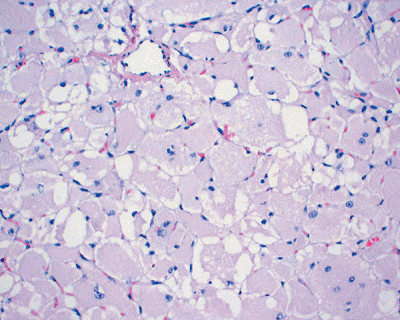
Figure 11-31
Rhabdomyoma. Solid growth of closely packed, large polygonal cells with abundant eosinophilic or granular cytoplasm and eccentrically located nuclei. |
Treatment and Outcome
-
Marginal excision
-
Up to 42% local recurrence
Vascular Benign Soft Tissue Tumors
Hemangiomas
Hemangiomas, which are most commonly seen in the skin,
are seen in the orthopaedic context predominately as intramuscular
angiomas or less commonly as synovial hemangiomas.
are seen in the orthopaedic context predominately as intramuscular
angiomas or less commonly as synovial hemangiomas.
 |
|
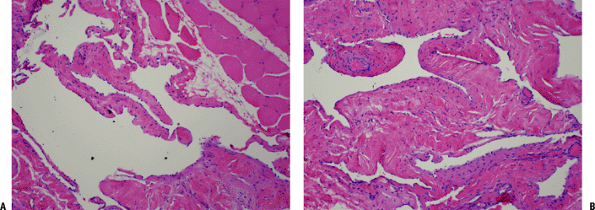
Figure 11-32 (A) Intramuscular hemangioma. Large vascular channels lined by flat endothelial cells surrounded by skeletal muscle. (B) Hemangioma. Cavernous vascular spaces lined by flat endothelial cells and focally containing red blood cells.
|
Pathogenesis and Pathophysiology
-
Although theories of neoplasm, reactive
processes, and malformations have been put forth, these are currently
thought to most likely be simply malformations.
Epidemiology
-
Intramuscular angiomas are among the most
common of soft tissue tumors, particularly in adolescents and young
adults, while the other variants are uncommon to rare. -
Intramuscular angiomas have a female predominance.
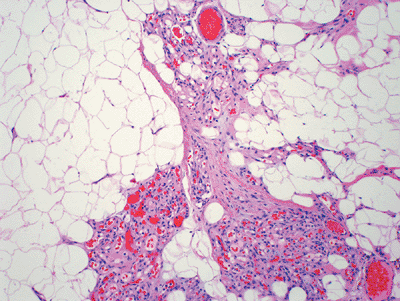
Histopathology (Fig. 11-32)
-
Various combinations of variably thick-walled vascular channels with hemosiderin deposition
-
Variable amounts of fatty tissue within intramuscular angiomas
Classification (Table 11-4)
-
Synovial hemangioma
-
Intramuscular angioma
-
Venous hemangioma
-
Arteriovenous hemangiomas
Diagnosis
Clinical Features
-
Intramuscular angioma causes classic activity-related fluctuation in size and pain even over the course of a single day.
-
Spongiform, compressible mass
-
Slowly enlarging
-
-
Arteriovenous malformations result in shunting.P.288Table 11-4 Features of Hemangioma Subtypes
Hemangioma Subtype Peak Age Occurrence Most Common Location Clinical Symptoms Synovial hemangioma Children and adolescents Rare Knee Joint swelling and pain, mechanical symptoms Intramuscular angioma Adolescents and young adults Common Deep, thigh Activity-related pain and swelling Venous hemangioma Adults Uncommon Superficial or deep, extremities Soft tissue mass Arteriovenous hemangioma Children and young adults Uncommon Superficial or deep, head and neck > extremities Shunting manifestations -
Physical examination reveals audible bruit, palpable thrill.
-
Clinical manifestations of shunting: limb hypertrophy, heart failure, consumption coagulopathy (Kasabach-Merritt syndrome)
-
Radiologic Features (Fig. 11-33)
-
Plain radiographs may reveal phleboliths.
-
MRI findings typically diagnostic
-
Characteristic serpentine pattern of blood vessels
-
Intermediate on T1-weighted images, bright on T2-weighted images; heterogeneous
-
Intermixed fatty signal on established lesions
-
Diagnostic Work-up
-
Clinical examination combined with plain
radiographs and MRI is usually diagnostic and obviates the need for
biopsy except in unusual circumstances. -
Also see Chapter 2.
Treatment and Outcome
-
Only sufficiently symptomatic lesions
warrant treatment consideration; the remainder should be observed or
treated with nonsteroidal anti-inflammatory medications and compressive
devices. -
Surgical excision
-
Small synovial and intramuscular hemangiomas amenable to marginal en bloc excision
-
Especially if single expendable muscle involved
-
-
Embolization and sclerotherapy
-
Larger lesions for which surgical excision is not feasible but sufficiently symptomatic to warrant treatment
-
Contraindicated if neurologic symptoms or compression
-
-
Recurrence rate 30% to 50% following excision unless entire involved muscle excised
Angiomatosis
Angiomatosis represents involvement of a large segment
of the body by hemangioma(s). This may manifest as extensive muscle
involvement or involvement of multiple tissue levels, including skin,
subcutaneous tissue, muscle, and bone.
of the body by hemangioma(s). This may manifest as extensive muscle
involvement or involvement of multiple tissue levels, including skin,
subcutaneous tissue, muscle, and bone.
Pathogenesis and Pathophysiology
-
Etiology is unknown.
-
Peak age: adolescents and young adults
-
Histopathology is same as for hemangioma.
Diagnosis
-
See Chapter 2 for diagnostic work-up.
Clinical Features
-
Common sites: lower extremity > chest wall > upper extremity
-
Skin involvement may be evident by varicosities.
-
Same symptoms as described for hemangioma
Radiologic Features
-
Same as for hemangiomas but more extensive (Fig. 11-34)
Treatment and Outcome
-
Often too extensive for adequate excision
-
High rate of local recurrence following attempted excision
Lymphangioma
Lymphangioma, also referred to as cystic hygroma, is similar to hemangioma except in its absence of blood components.
Pathogenesis and Pathophysiology
-
Etiology is unknown.
-
Peak age: birth to childhood
-
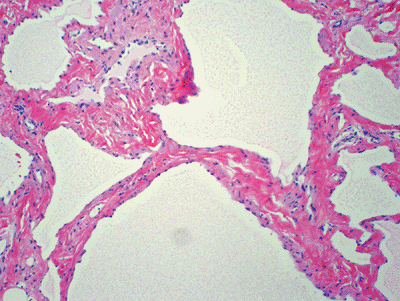
Histopathology: variably sized, thin-walled lymphatic vessels (Fig. 11-35)
-
Genetics: associated with Turner syndrome
P.289
 |
|
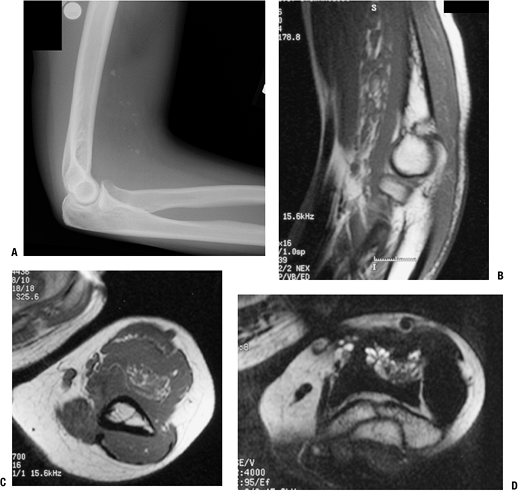
Figure 11-33
This intramuscular hemangioma of the proximal forearm shows classic radiographic features. Phleboliths can be seen on the plain radiograph (A), and the sagittal (B) and axial (C) T1-weighted MR images and axial T2-weighted (D) MR image show serpiginous vascular structures intermixed with fat signal. |
Classification
-
Cystic versus cavernous subtypes
Diagnosis
-
See Chapter 2 for diagnostic work-up.
Clinical Features
-
Cystic: neck, axilla, groin
-
Cavernous: upper trunk, extremities
Radiologic Features
-
Cystic signal characteristics with nonenhancing, fluid-filled, well-defined structures on MRI
-
Cystic structures on ultrasound
Treatment and Outcome
-
Marginal excision
-
High rate of local recurrence
Perivascular Benign Soft Tissue Tumors
Glomus Tumors
Glomus tumors are tumors of the smooth muscle cells that
make up the normal glomus body and are characterized best by their
clinical presentation as small painful nodules predominately found in a
subungual location.
make up the normal glomus body and are characterized best by their
clinical presentation as small painful nodules predominately found in a
subungual location.
Pathogenesis and Pathophysiology
-
Etiology unknown
-
Peak age: young adults predominate
-
Subungual lesions more common in women
P.290
 |
|
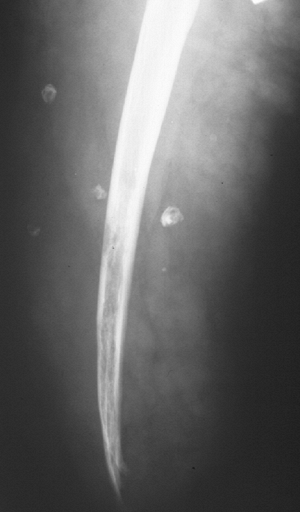
Figure 11-34
Angiomatosis involves multiple tissue levels within a large segment of or an entire limb. This patient had undergone amputation for complications of angiomatosis of the lower extremity. Note the phleboliths within the soft tissues and involvement of the bone as well. |
 |
|
Figure 11-35 Lymphangioma. Thin-walled vascular channels lined by flat endothelial cells and filled by proteinaceous material.
|
 |
|
Figure 11-36 (A)
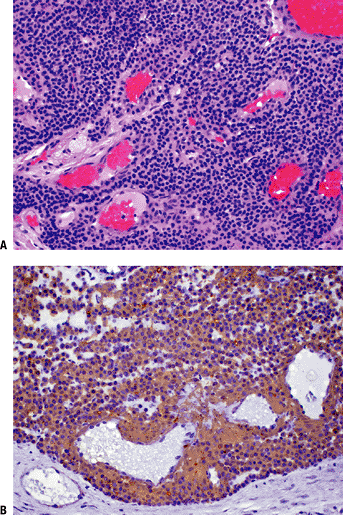
Glomus tumor. Nests of small, uniform, round cells with centrally located nucleus and amphophilic or slightly eosinophilic cytoplasm surrounded by capillary blood vessels. (B) Glomus tumor showing strong immunoreactivity with smooth muscle actin stain. |
Histopathology (Fig. 11-36)
-
Three components in varying amounts
-
Glomus cells
-
Small, uniformly round cells
-
Centrally placed round nuclei
-
Prominent cell membranes define cytoplasm.
-
-
Vascular structures
-
Smooth muscle cells
-
Genetics
-
Familial cases: autosomal dominant pattern of inheritance
-
Subungual glomus tumors associated with NF-I
Classification
-
Typical glomus tumors have three histologic variants:
-
Glomangiomatosis: extremely rare clinical variant similar to angiomatosis but characterized by glomus histology
-
Symplastic glomus tumors: rare histologic variant with atypia but benign behavior
-
Malignant glomus tumors: 1% of glomus tumors
Diagnosis
-
See Chapter 2 for diagnostic work-up.
Clinical Features
-
Subungual region (hand, wrist, foot) predominates.
-
Superficial in vast majority
-
Small reddish-blue nodules
-
Associated with chronic pain in severe paroxysms
-
Exposure to cold or minor pressure often exacerbates pain.
Radiologic Features
-
Ultrasound may detect tumors as small as 3 mm.
-
MRI: bright on T2-weighted images, dark on T1-weighted images; enhancing
Treatment and Outcome
-
Complete excision is the only treatment.
-
Local recurrence in 5% to 50%
Chondro-Osseous Benign Soft Tissue Tumors
Soft Tissue Chondroma
Soft tissue chondroma, otherwise known as chondroma of
the soft parts, is an extraosseous, extrasynovial mature hyaline
cartilage tumor.
the soft parts, is an extraosseous, extrasynovial mature hyaline
cartilage tumor.
Pathogenesis and Pathophysiology
-
Etiology is unknown.
-
Peak age: middle-aged (may involve any age)
-
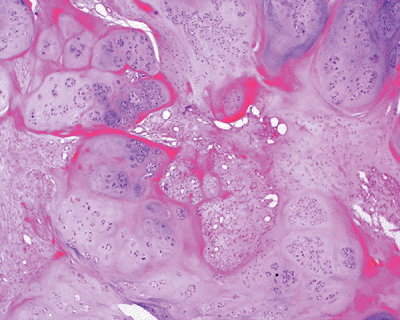
Histopathology: mature hyaline cartilage lobules, S100 positive (Fig. 11-37)
-
Genetics: 12q13-15 structural rearrangements involving HMGA2 gene locus
Classification
-
Chondroblastic chondroma: chondrocytic cells in lacunae predominate
-
Fibrochondroma: prominent fibrosis
-
Osteochondroma of soft tissue: prominent ossification centrally
-
Myxochondroma: prominent myxoid change
 |
|
Figure 11-37 Soft tissue chondroma composed of well-defined lobules of mature cartilage embedded in a fibrous stroma.
|
Diagnosis
-
See Chapter 2 for diagnostic work-up.
Clinical Features
-
Fingers (two thirds), hands, toes, feet predominate.
-
Painless small lumps rarely >3 cm
Radiologic Features
-
Plain films show variable mineralization.
-
MRI shows hyaline cartilage signal (lobulated, dark on T1-weighted images, bright on T2-weighted images).
Treatment and Outcome
-
Marginal excision
-
Recurrence unusual
Tumors of Uncertain Differentiation
Intramuscular Myxoma
Intramuscular myxoma is a benign, low-cellularity myxoid tumor characterized clinically by its somewhat distinctive
P.292
radiographic appearance in showing very hypointense signal compared to
muscle on T1-weighted MRI. It may occur as a solitary soft tissue mass
or may be accompanied by fibrous dysplasia lesions, as in Mazabraud
syndrome.
Pathogenesis and Pathophysiology
-
Etiology is unknown.
-
Peak age: 40 to 70 years
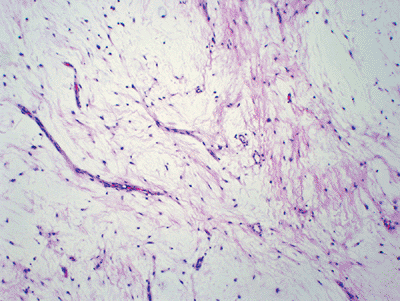
Histopathology (Fig. 11-38)
-
Uniformly bland cellularity with small bland spindle cells
-
Bland extracellular myxoid background
-
Infiltrating borders
Genetics
-
Point mutations in GNAS1 gene common both in Mazabraud syndrome and isolated myxomas
Classification
-
Cellular myxoma: histologic variant with increased cellularity and vascularity
Diagnosis
Clinical Features
-
Deep, painless soft tissue mass
-
Thigh, shoulder, buttocks, upper arm
-
Usually 5 to 10 cm
-
Mazabraud syndrome: myxomas and fibrous dysplasia (monostotic, polyostotic, or Albright syndrome)
 |
|
Figure 11-38 Intramuscular myxoma. Hypocellular neoplasm composed of very bland-looking spindle cells in an abundant myxoid background.
|
Radiologic Features (Fig. 11-39)
-
Homogenous, hypointense to muscle on T1-weighted images, hyperintense on T2-weighted images
-
Variable patterns of enhancement (peripheral and/or central) dependent on cellularity
-
Peritumoral edema and fat cap
Diagnostic Work-up
-
This process may be highly suspected based on MRI, so biopsy may be unnecessary.
-
Also see Chapter 2.
Treatment and Outcome
-
Marginal excision
-
Recurrence unlikely
Juxta-Articular Myxoma
The juxta-articular myxoma arises close to large joints and frequently has associated cystic changes.
Pathogenesis and Pathophysiology
-
Etiology is unknown.
-
Peak age: range 9 to 83 years
Histopathology
-
Similar to cellular myxoma variant of intramuscular myxoma
-
Bland spindle cells
-
Myxoid background
-
-
Cystic-like ganglion spaces
Genetics
-
Lacks the GNAS1 mutations seen in intramuscular myxomas
Diagnosis
-
See Chapter 2 for diagnostic work-up.
Clinical Features
-
Frequently painful or tender soft tissue mass
-
Present for weeks to years
-
May be associated with meniscal tear
-
May be incidental finding during knee or hip arthroplasty
Radiologic Features
-
Similar to intramuscular myxoma
-
Distinguished by juxta-articular location
Treatment and Outcome
-
Marginal excision
-
Local recurrence in up to one third
P.293
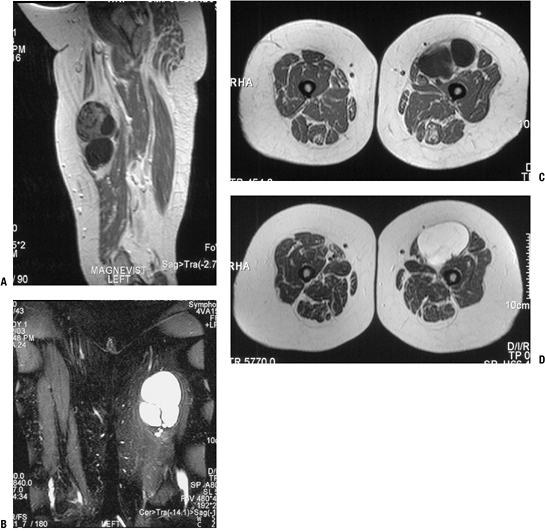 |
|
Figure 11-39 This intramuscular myxoma within the quadriceps shows characteristically hypointense signal on T1-weighted sagittal (A) and axial (C) MRI sequences and hyperintense signal on T2-weighted coronal (B) and axial (D) sequences.
|
Suggested Reading
Aflatoon K, Aboulafia AJ, McCarthy EF Jr, et al. Pediatric soft tissue tumors. J Am Acad Orthop Surg 2003;11(5):332–343.
Ballo
MT, Zagars GK, Pollack A, et al. Desmoid tumor: prognostic factors and
outcome after surgery, radiation therapy, or combined surgery and
radiation therapy. J Clin Oncol 1999;17(1):158–167.
MT, Zagars GK, Pollack A, et al. Desmoid tumor: prognostic factors and
outcome after surgery, radiation therapy, or combined surgery and
radiation therapy. J Clin Oncol 1999;17(1):158–167.
Bos GD, Esther RJ, Woll TS. Foot tumors: diagnosis and treatment. J Am Acad Orthop Surg 2002;10(4):259–270.
Butler MG, Fuchigami KD, Chako A. MRI of posterior knee masses. Skeletal Radiol 1996;25(4):309–317.
Carr MM, Mahoney JL, Bowen CV. Extremity arteriovenous malformations: review of a series. Can J Surg 1994;37(4):293–299.
Damron TA, Beauchamp CP, Rougraff BT, et al. Soft tissue lumps and bumps. AAOS Instr Course Lect 2004;53:625–637.
Forthman CL, Blazar PE. Nerve tumors of the hand and upper extremity. Hand Clin 2004;20(3):233–242.
Goy BW, Lee SP, Eilber F, et al. The role of adjuvant radiotherapy in the treatment of resectable desmoid tumors. Int J Radiat Oncol Biol Phys 1997;39(3):659–665.
Kim
DH, Murovic JA, Tiel RL, et al. A series of 397 peripheral neural
sheath tumors: 30-year experience at Louisiana State University Health
Sciences Center. J Neurosurg 2005;102(2):246–255.
DH, Murovic JA, Tiel RL, et al. A series of 397 peripheral neural
sheath tumors: 30-year experience at Louisiana State University Health
Sciences Center. J Neurosurg 2005;102(2):246–255.
Kransdorf MJ, Murphey MD, Smith SE. Imaging of soft tissue neoplasms in the adult: benign tumors. Semin Musculoskelet Radiol 1999;3(1):21–38.
Laor T. MR imaging of soft tissue tumors and tumor-like lesions. Pediatr Radiol 2004;34(1):24–37.
Merchant NB, Lewis JJ, Woodruff JM, et al. Extremity and trunk desmoid tumors: a multifactorial analysis of outcome. Cancer 1999;86(10):2045–2052.
Murphey MD, Carroll JF, Flemming DJ, et al. From the archives of the AFIP: benign musculoskeletal lipomatous lesions. Radiographics 2004;24(5):1433–1466.
Pakos EE, Tsekeris PG, Goussia AC. Desmoid tumours of the extremities and trunk: a review of the literature. Int Orthop 2005;29(4):210–213.
Rogalski R, Hensinger R, Loder R. Vascular abnormalities of the extremities: clinical findings and management. J Pediatr Orthop 1993;13(1):9–14.
Tang P, Hornicek FJ, Gebhardt MC, et al. Surgical treatment of hemangiomas of soft tissue. Clin Orthop Relat Res 2002;(399):205–210.