
Editors: Tornetta, Paul; Einhorn, Thomas A.; Damron, Timothy A.
Title: Oncology and Basic Science, 7th Edition
Copyright ©2008 Lippincott Williams & Wilkins
> Table of Contents > Section
II – Specific Bone Neoplasms and Simulators > 5 – Benign Bone Tumors
> 5.5 – Fibrous Lesions of Bone
II – Specific Bone Neoplasms and Simulators > 5 – Benign Bone Tumors
> 5.5 – Fibrous Lesions of Bone
5.5
Fibrous Lesions of Bone
Sung Wook Seo
Young Lae Moon
Francis Young-In Lee
Considerable controversy exists with respect to the
terminology for fibrous lesions of bone. Although nonossifying fibromas
(NOFs) and metaphyseal fibrous cortical defects (FCDs) are the most
common lesions of bone, the current World Health Organization does not
even include them in their classification, as they are not neoplastic (Box 5.5-1).
Avulsive cortical irregularity has identical histology but is
distinguished by a very specific location, the posteromedial distal
femoral metaphysis. Benign fibrous histiocytoma, for which there are
several synonyms, also has the same histology as NOF/FCD but is
distinguished by its occurrence in older ages, in an atypical non–long
bone location, with atypical radiographic appearance, or with unusual
clinical presentation. Fibrous dysplasia and osteofibrous dysplasia
have typical clinical presentations and radiographic appearances.
Desmoplastic fibroma is a rare tumor that occurs most commonly in the
mandible.
terminology for fibrous lesions of bone. Although nonossifying fibromas
(NOFs) and metaphyseal fibrous cortical defects (FCDs) are the most
common lesions of bone, the current World Health Organization does not
even include them in their classification, as they are not neoplastic (Box 5.5-1).
Avulsive cortical irregularity has identical histology but is
distinguished by a very specific location, the posteromedial distal
femoral metaphysis. Benign fibrous histiocytoma, for which there are
several synonyms, also has the same histology as NOF/FCD but is
distinguished by its occurrence in older ages, in an atypical non–long
bone location, with atypical radiographic appearance, or with unusual
clinical presentation. Fibrous dysplasia and osteofibrous dysplasia
have typical clinical presentations and radiographic appearances.
Desmoplastic fibroma is a rare tumor that occurs most commonly in the
mandible.
Box 5.5-1 Current Terminology for Fibrous Lesions of Bone
Nonossifying Fibroma, Metaphyseal Fibrous Cortical Defect, and Avulsive Cortical Irregularity
NOFs and their smaller counterparts, metaphyseal FCDs,
are among the most common lesions of bones, particularly in children.
Avulsive cortical irregularity (ACI) is a clinically distinct entity
that shares the same histology but occurs only at the distal femoral
posteromedial metaphysis. Each of these is most often discovered
incidentally on radiographs obtained for other purposes, such as the
evaluation of injuries about the knee. Their clinical presentations as
incidental findings and their radiographic appearance of geographic,
eccentric, radiolucent, partially intracortical lesions with a
soap-bubble appearance are so characteristic that biopsy is usually
unnecessary to establish the diagnosis. Their natural history of
resolution over time strongly favors nonoperative treatment in most
cases.
are among the most common lesions of bones, particularly in children.
Avulsive cortical irregularity (ACI) is a clinically distinct entity
that shares the same histology but occurs only at the distal femoral
posteromedial metaphysis. Each of these is most often discovered
incidentally on radiographs obtained for other purposes, such as the
evaluation of injuries about the knee. Their clinical presentations as
incidental findings and their radiographic appearance of geographic,
eccentric, radiolucent, partially intracortical lesions with a
soap-bubble appearance are so characteristic that biopsy is usually
unnecessary to establish the diagnosis. Their natural history of
resolution over time strongly favors nonoperative treatment in most
cases.
Pathogenesis
Etiology
-
NOF/FCD: focally defective periosteal cortical bone development leads to failure of ossification (developmental defect)
-
ACI: traumatic stress of avulsive
microfracture, healing, and repeat avulsive fracture related to origin
of medial head of gastrocnemius muscle and/or distal adductor magnus
insertion on posteromedial cortical surface of distal femur only
P.150
Epidemiology
-
Incidence: true incidence unknown
-
NOF/FCD
-
NOF/FCD most common bone lesion(s) overall
-
Up to 40% of children with knee radiographs have one or more.
-
-
ACI
-
On x-ray, 10% to 40%, but on MRI up to 60%
-
-
-
Age: 3 to 20 for all three lesions (peak 10 to 15), with some NOFs and ACIs persisting into adulthood
-
Male:female 2 to 3:1 (both NOF/FCD and ACI)
-
Location
-
NOF/FCD
-
Long bone metaphyses predominate.
-
55% around knee
-
Distal femur > proximal tibia > distal tibia
-
Diaphyseal lesions and flat bones less common
-
-
ACI: By definition, these occur only in the posteromedial distal femoral metaphysis and are frequently bilateral.
-
Pathophysiology
Pathology (Fig. 5.5-1)
-
Fibrous tissues arranged in storiform or
cartwheel pattern with foamy histiocytes (30% to 50%, more frequent in
adults), hemosiderin (variable), occasional multinucleated giant cells
Natural History
-
All of these lesions usually regress after skeletal maturity at the latest, sometimes sooner.
-
Most FCDs resolve without residual radiographic evidence, but some progress to NOF size.
-
Some lesions persisting into adulthood become latent or eventually ossify.
-
Assessing fracture risk
-
>50% of diameter of bone on both anteroposterior and lateral radiographs
-
>33 mm vertical dimension in nonfibular lesions
-
Distal tibial lesions most common site with associated fracture
-
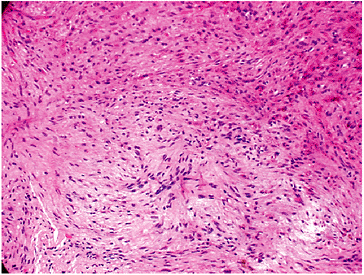 |
|
Figure 5.5-1 Histopathology of NOFs.
|
Classification
-
NOF vs. FCD
-
FCD: typically small (<0.5 cm) radiolucency completely within the cortical bone
-
NOF: involves cortex but also extends into medullary bone
-
Some authors use a 3-cm size delineation.
-
-
ACI: posteromedial distal femoral metaphyseal lesion
-
Jaffe-Campanacci syndrome
-
Forme fruste of type I peripheral neurofibromatosis
-
Key components: multiple NOFs and café-au-lait spots
-
Variably associated: mental retardation, hypogonadism or cryptorchidism, ocular abnormality, cardiovascular malformations
-
-
NOF in neurofibromatosis type I (NF-1)
-
Most common bone lesion of NF-1
-
Diagnosis
Physical Examination and History
-
Most common clinical presentation: incidental asymptomatic finding
-
Other scenarios
-
Pathologic fracture with larger NOF
-
Usually not preceded by pain
-
-
Pain associated with lesions
-
Uncommon in FCD/NOF
-
Occurs more frequently with ACI but dissipates over time
-
Search for another source of pain.
-
Consider chondromyxoid fibroma, periosteal chondroma, and osteomyelitis in differential diagnosis for symptomatic lesions.
-
-
Radiographic Features
Plain Radiographs (Fig. 5.5-2)
-
Usually diagnostic in vast majority of cases
-
Well-marginated (geographic), eccentric,
partially intracortical, multiloculated, soap-bubble radiolucent lesion
with a sclerotic rim -
May be central within smaller bones such as fibula but still partially intracortical
-
Occasional cortical thinning and expansion
-
ACI best visualized on oblique radiographs; often bilateral
-
Magnetic Resonance Imaging
-
Indicated for atypical plain film features, symptoms, or older patients
-
FCD: fibrous signal void within cortex, dark on T1-weighted but brighter on T2-weighted
-
NOF
-
Low-signal (dark) central region on both T1- and T2-weighted sequences due to hemosiderin and/or dense collagen
-
Typically no surrounding edema in absence of pathologic fracture
-
-
ACI: variable manifestations, including discrete lesions and simple irregularity of the posteromedial cortex
P.151 -
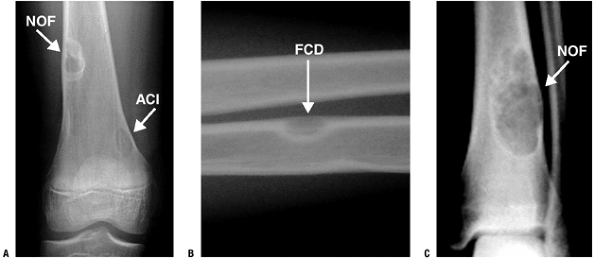 |
|
Figure 5.5-2
Various types of cortically based fibrous lesions: nonossifying fibroma (NOF), avulsive cortical irregularity (ACI), and fibrous cortical defect (FCD). |
Bone Scan
-
Minimal to no uptake seen; bone scan usually not needed
Treatment
-
Observation: treatment of choice for essentially all FCDs and ACIs and the vast majority of NOFs
-
Open biopsy: atypical radiographic
appearance, symptomatic lesions, and lesions in adults for which the
diagnosis cannot be established by clinical and radiographic
presentation (unusual) -
Curettage and grafting with or without
internal fixation: usually reserved for larger lesions after fracture
(see fracture risk above) or in very active patients
Results and Outcomes
-
Local recurrence is nearly nonexistent for completely curetted NOF.
-
Peripheral radiolucencies may indicate residual NOF tissue.
Benign Fibrous Histiocytoma of Bone
This rare neoplasm is the prototypical fibrohistiocytic
benign tumor within the World Health Organization classification
schema. Its importance lies in its distinction from the more common
NOF, so this section will contrast the features of this lesion with NOF.
benign tumor within the World Health Organization classification
schema. Its importance lies in its distinction from the more common
NOF, so this section will contrast the features of this lesion with NOF.
Pathogenesis
Etiology
-
Unknown
Epidemiology
-
Incidence: rare
-
Age: 6 to 74 years (60% are above typical upper age limit for NOF)
-
Unlike NOF, female > male preponderance (slight)
-
Location
-
Any bone may be affected.
-
Like NOF, femur and tibia are most
common, BUT diaphyseal or epiphyseal regions predominate in long bones
(unlike metaphysis for NOF). -
Higher frequency of pelvic bone involvement (25%) than in NOF (especially ilium)
-
Pathophysiology
-
Histologically similar to that of NOF
-
Storiform, whorled pattern of spindle cells with frequent foam (xanthoma) cells and variable benign multinucleated giant cells (Fig. 5.5-3)
-
Variable hemorrhage, hemosiderin deposition
Diagnosis
Physical Examination and History
-
Clinical presentation varies from that of NOF.
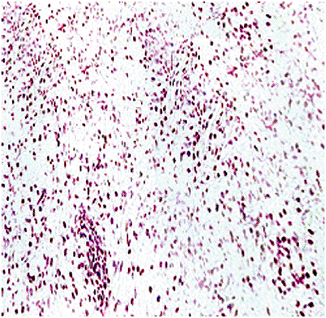 |
|
Figure 5.5-3 Histopathology of benign fibrous histiocytoma.
|
Radiographic Features
Plain Radiographs
-
Lytic medullary lesions with sharply defined margins and a sclerotic rim without matrix mineralization (Fig. 5.5-4)
-
Trabeculation or pseudoseptations
-
May lack well-defined margins
-
Absent soft tissue extension or periosteal reaction
-
Either central or eccentric in epiphyseal location
-
Majority 3 cm or less (up to 7 cm)
|
Table 5.5-1 Distinguishing Features of Benign Fibrous Histiocytoma Compared with Nonossifying Fibroma
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
Differential Diagnosis
-
Metaphyseal or diaphyseal lesion
-
NOF or metaphyseal FCD
-
-
Epiphyseal lesion
-
Giant cell tumor of bone
-
Fibrosarcoma
-
Diagnostic Algorithm
Because benign fibrous histiocytoma of bone cannot be
distinguished histologically from NOF, it must be distinguished based
upon clinical and radiologic features (Table 5.5-1).
distinguished histologically from NOF, it must be distinguished based
upon clinical and radiologic features (Table 5.5-1).
Fibrous Dysplasia
Pathogenesis
Etiology
-
Developmental anomaly of bone formation caused by missense mutation in exon 8 of the GNAS gene (chromosome 20q13.2-13.3)
-
Constitutive activation of the adenyl cyclase signaling pathway and synthesis of cyclic adenosine monophosphate (cAMP)
Epidemiology
-
Incidence
-
Relatively common
-
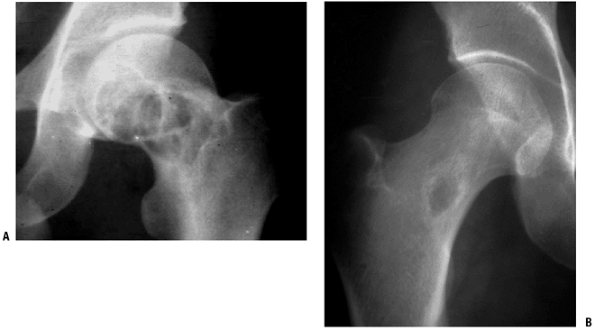
Monostotic six times more common than polyostotic form (Fig. 5.5-5)
-
-
Age
-
Onset as children and adolescents (median onset 8 years)
-
Diagnosis in children and adults
-
-
Male = female
-
Location
-
Most common location in surgical series: jaw
-
Gender differences
-
Women: long bones most common
-
Men: ribs and skull most common
-
-
Differences by type
-
Monostotic: skull > femur and tibia > ribs
-
Polyostotic: femur > pelvis > tibia
-
May be confined to one limb or one side of body
-
-
Pathophysiology
Pathology (Fig. 5.5-6)
-
Bone replaced by firm, whitish fibrous stromal tissue of gritty consistency. It presents immature woven bone
rather than lamellar bone without osteoblastic rimming of trabeculae
(distinguishes from osteofibrous dysplasia, which exhibits osteoblastic
rimming).
 |
|
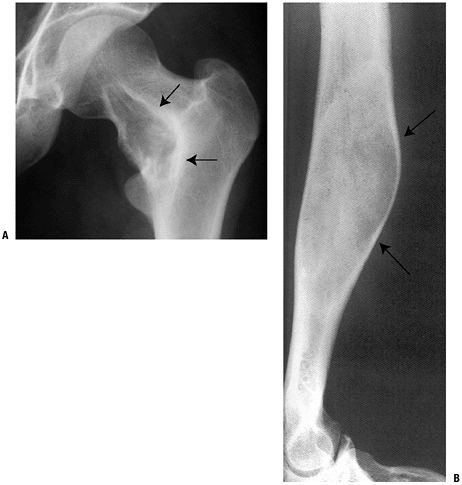
Figure 5.5-5 Radiographs of monostotic fibrous dysplasia showing typical ground glass matrix mineralization.
|
P.154
 |
|
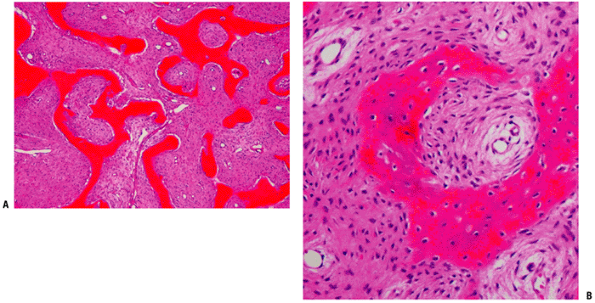
Figure 5.5-6 Histopathology of fibrous dysplasia. Photomicrographs show woven bone trabeculae without osteoblastic rimming.
|
Natural History
-
Normal medullary bone is replaced by variable amounts of fibrous/osseous tissues.
-
More active lesions in children and adolescents; inactive more common in adults
-
Malignant change rarely occurs.
Classification
-
Monostotic: solitary lesion
-
Polyostotic: multiple lesions
-
McCune-Albright syndrome
-
Polyostotic disease, and usually presents earlier
-
Long bones, hands, feet, and pelvis often unilaterally involved (Fig. 5.5-7)
-
Café-au-lait spots with serrated borders (called “coast of Maine”) that tend to stop abruptly at the midline of the body
-
Precocious puberty (endocrinopathy) may be present.
-
Malignant transformation (chondrosarcoma or osteosarcoma) about 4%
-
-
Mazabraud’s syndrome
-
Associated intramuscular myxomas
-
Diagnosis
Physical Examination and History
-
Clinical presentation: often asymptomatic, but may cause pain or fracture
-
McCune-Albright syndrome: polyostotic form associated with endocrine abnormalities and skin lesions (see above)
-
Mazabraud syndrome: associated intramuscular myxomas
Radiographic Features
Plain Radiographs
-
Location: epiphysis, metaphysis, or diaphysis
-
“Long lesion in long bone”
-
Radiolucent lesion with fine, granular, ground glass matrix appearance
-
Variable expansion of cortex
-
No cortical disruption or periosteal reaction
-
Extensive involvement of proximal femur results in a characteristic varus deformity that resembles a shepherd’s crook (see Fig. 5.5-7).
Magnetic Resonance Imaging
-
Dark on T1-weighted images, bright on T2-weighted images, enhancing
-
Cystic degeneration may be seen.
-
Defines extent of disease
Bone Scan
-
Typically shows intense uptake on bone scan
Differential Diagnosis
-
Paget’s disease, fibrous cortical defect, hyperparathyroidism, osteoblastoma, osteosarcoma
P.155
 |
|
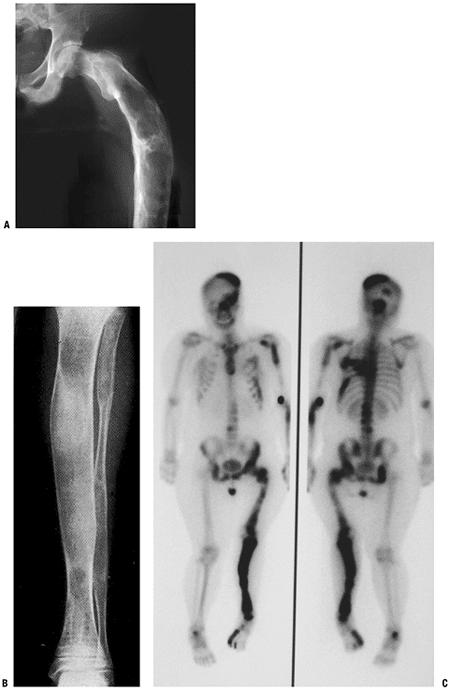
Figure 5.5-7 Radiographs (A,B) and bone scan (C)
of polyostotic fibrous dysplasia. It usually involves unilateral bones. Note the shepherd’s crook deformity of the proximal femur. |
P.156
|
Table 5.5-2 Distinguishing Features of Fibrous and Osteofibrous Dysplasias
|
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Diagnostic Workup Algorithm
-
Radiographic features are often diagnostic; if so, no need for biopsy.
-
If plain films are not diagnostic, additional imaging and biopsy needed
Treatment
-
Observation: diagnostic radiographic features with minimal or no symptoms
-
Curettage and bone grafting
-
Particulate graft material usually resorbed and replaced more rapidly, so not recommended
-
Structural graft material often resorbed more slowly, so recommended
-
-
Prophylactic stabilization: provides structural stability without concern for resorption of bone graft
-
Radiotherapy increases the risk of sarcomatous change and is generally avoided.
-
Bisphosphonate treatment has been used for symptomatic polyostotic fibrous dysplasia.
Results and Outcomes
-
Overall prognosis: good
-
Worst outcomes: higher local recurrence, pain, deformity in younger patients and those with polyostotic dysplasia
-
Malignant transformation: rare
|
Table 5.5-3 Overlapping and Distinguishing Features of Adamantinoma and Osteofibrous Dysplasia
|
|||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Osteofibrous Dysplasia
Despite the similarity in nomenclature with fibrous
dysplasia, osteofibrous dysplasia (formerly referred to as ossifying
fibroma or Campanacci’s lesion) is a distinct clinical, radiographic,
and pathologic entity (Table 5.5-2). It should
be recognized clinically and radiographically so that unnecessary and
potentially damaging operations are avoided prior to skeletal maturity
and so that it can be distinguished from adamantinoma, the low-grade
malignancy with overlapping features (Table 5.5-3).
dysplasia, osteofibrous dysplasia (formerly referred to as ossifying
fibroma or Campanacci’s lesion) is a distinct clinical, radiographic,
and pathologic entity (Table 5.5-2). It should
be recognized clinically and radiographically so that unnecessary and
potentially damaging operations are avoided prior to skeletal maturity
and so that it can be distinguished from adamantinoma, the low-grade
malignancy with overlapping features (Table 5.5-3).
Pathogenesis
Etiology
-
Unknown; NOT related to GNAS gene mutations seen in fibrous dysplasia
-
Occurrence of areas with positive
immunohistochemistry for epithelial elements has been termed “OFD-like
adamantinoma” and suggests a relationship with adamantinoma.
Epidemiology
Pathophysiology
Pathology (Fig. 5.5-8)
-
Osteoblast-lined woven bone fragments separated by bland fibrous stroma
-
Zonal architecture with loose fibrous
tissue in the central zone, woven bone rimmed by osteoblasts in the
fibro-osseous zone, and lamellar bone in loose, fibrous, vascular
tissue in the peripheral zone
Natural History
-
Benign fibrous tumor with potentially locally aggressive behavior
-
Progresses during childhood but not after puberty
-
Frequently recurs after curettage or subperiosteal resection
-
Regression or resolution at skeletal maturity
-
Rare cases may progress to adamantinoma.
Diagnosis
Physical Examination and History
-
Most common: swelling or painless anterior tibial bowing
-
Occasionally: pathologic fractures (usually stress fractures) cause episodic pain
-
Rarely: may exhibit aggressive behavior in terms of causing progressive deformity
 |
|
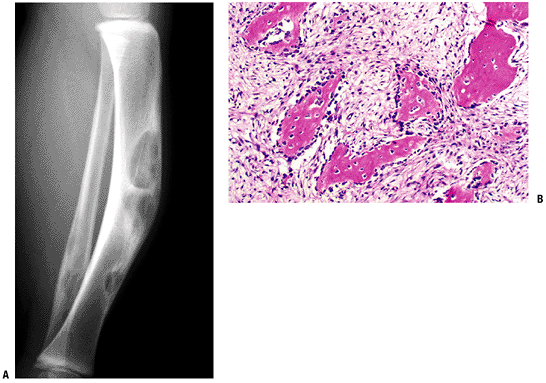
Figure 5.5-8 Radiograph (A) and histopathologic specimen (B)
of osteofibrous dysplasia. Anterior intracortical tibial involvement is characteristic. Osteoblastic rimming is shown here as prominent, in contrast to fibrous dysplasia. |
Radiologic Features
Plain Radiographs (see Fig. 5.5-8)
-
Anterior intracortical involvement of middle third of tibia is classic.
-
Overlying cortex invariably thinned, expanded, or even missing
-
Sclerotic, geographic intramedullary border
-
Saw-toothed or soap-bubble, often multiple lucent areas anteriorly bordered by the deeper sclerotic border
-
No soft tissue extension or periosteal reaction
Magnetic Resonance Imaging
-
Lucent areas intermediate to dark on T1-weighted and bright on T2-weighted images
-
Sclerotic surrounding reactive bone dark on all sequences
Bone Scan
-
Increased uptake
Treatment
-
Up to skeletal maturity
-
After skeletal maturity
-
Curettage and grafting with or without stabilization
-
Results and Outcome
-
High incidence of local recurrence and progression if operated on prior to skeletal maturity
Desmoplastic Fibroma
Among the benign fibrous lesions, desmoblastic fibroma
is notable both for its rarity and its aggressive local behavior. Its
histologic appearance mimics that of desmoid tumor of the soft tissue,
but it may be confused with fibrosarcoma. Its radiographic appearance
may also mimic bone sarcoma, but it does not metastasize.
is notable both for its rarity and its aggressive local behavior. Its
histologic appearance mimics that of desmoid tumor of the soft tissue,
but it may be confused with fibrosarcoma. Its radiographic appearance
may also mimic bone sarcoma, but it does not metastasize.
Pathogenesis
Etiology
-
Unknown, but trisomies of chromosomes 8 and 20 resemble those of soft tissue desmoids.
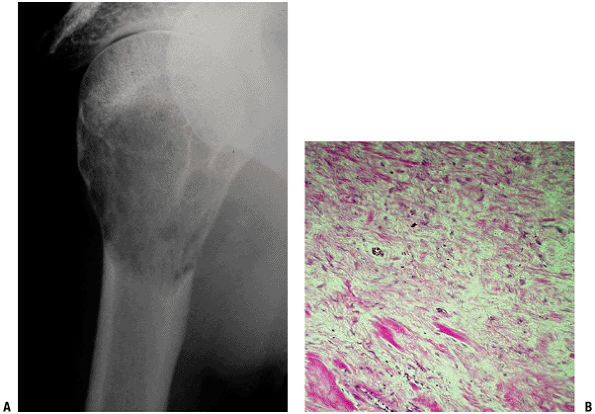 |
|
Figure 5.5-9 Radiograph and histopathology of desmoplastic fibroma.
|
Epidemiology
-
Incidence: rare
-
Age: adolescents and young adults within first three decades of life
-
Male = female
-
Location: long bones (1/2) > mandible (1/4) > pelvis (1/8)
Pathophysiology
Pathology (Fig. 5.5-9)
-
Identical finding to soft tissue fibromatosis
-
Dense and irregularly arranged collagen bundles with infrequent fibroblasts
-
Mitosis, vascularity, and necrosis are unusual.
Natural History
-
Locally aggressive behavior except in rare instances
-
No metastases
Diagnosis
Physical Examination and History
-
Pain, deformity, or loss of function
Radiologic Features (see Fig. 5.5-9)
Plain Radiographs
Magnetic Resonance Imaging
-
Dense fibrous tissue is low signal on T1-weighted and T2-weighted sequences.
Treatment
-
En bloc wide resection: treatment of choice
-
Extended intralesional curettage: recurrence rate unacceptably high
-
Radiotherapy and chemotherapy (tamoxifen, indomethacin): reported anecdotally for inoperable tumors or locations
Results and Outcome
-
Local recurrence rates
-
Extended intralesional curettage: 72%
-
En bloc wide resection: 17%
-
Postoperative Management
-
Local recurrence may result years later.
-
Long-term follow-up recommended
Suggested Reading
Bertoni F, Calderoni P, Bacchini P, et al. Benign fibrous histiocytoma of bone. J Bone Joint Surg [Am] 1986;68(8):1225–1230.
Gebhardt
MC, Campbell CJ, Schiller AL, et al. Desmoplastic fibroma of bone. A
report of eight cases and review of the literature. J Bone Joint Surg [Am] 1985;67(5):732–747.
MC, Campbell CJ, Schiller AL, et al. Desmoplastic fibroma of bone. A
report of eight cases and review of the literature. J Bone Joint Surg [Am] 1985;67(5):732–747.
Kumar S. Bisphosphonate therapy in polyostotic fibrous dysplasia. Indian Pediatr 2004;41(10):1069–1070.
Lane JM, Khan SN, O’Connor WJ, et al. Bisphosphonate therapy in fibrous dysplasia. Clin Orthop Relat Res 2001;(382):6–12.
Marks KE, Bauer TW. Fibrous tumors of bone. Orthop Clin North Am 1989;20(3):377–393.
O’Sullivan
M, Zacharin M. Intramedullary rodding and bisphosphonate treatment of
polyostotic fibrous dysplasia associated with the McCune-Albright
syndrome. J Pediatr Orthop 2002;22(2):255–260.
M, Zacharin M. Intramedullary rodding and bisphosphonate treatment of
polyostotic fibrous dysplasia associated with the McCune-Albright
syndrome. J Pediatr Orthop 2002;22(2):255–260.
Parisi MS, Oliveri MB, Mautalen CA. Bone mineral density response to long-term bisphosphonate therapy in fibrous dysplasia. J Clin Densitom 2001;4(2):167–172.
Pensak ML, Nestok BR, Van Loveren H, et al. Desmoplastic fibroma of the temporal bone. Am J Otol 1997;18(5):627–631.