
although when they occur, they usually are benign. Early detection of a
malignant MSKT may not only make the difference between life and death,
but also may allow for successful limb salvage surgery rather than
amputation of the limb. The primary bone and soft tissue tumors of
childhood can be classified based on their tissue origin (Table 23-1).
This chapter is not intended as an exhaustive survey of childhood
musculoskeletal neoplasm. It will cover general principles of MSKT
diagnosis and treatment, and some of the more common individual
entities.
physical examination are the basis for determining the correct
diagnosis and therapy. That type of tumors seen in children with an
MSKT are presented in Box 23-1. The age of the
patient is important in establishing a differential diagnosis, because
certain tumors tend to occur in certain age groups (Table 23-2).
|
TABLE 23-1 CLASSIFICATION OF PEDIATRIC MUSCULOSKELETAL TUMORS BASED ON TISSUE OF ORIGIN
|
||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
-
Plain radiographs give the most detailed information about skeletal lesions.
-
□ Orthogonal views showing the entire lesion are necessary.
-
□ 30% to 40% of a bone must be destroyed before lytic changes can be seen in plain radiographs.
-
□ It is often difficult to see soft tissue tumors and soft tissue extension from bony neoplasms with plain radiographs.
-
-
Magnetic resonance imaging (MRI) best demonstrates the soft tissue anatomy and intramedullary extension of the tumor.
-
MRI remains the modality of choice for
staging, for evaluating response to preoperative chemotherapy, and for
long-term follow-up of most bone and soft tissue sarcomas. -
Computed tomography (CT) can demonstrate
bone destruction and mineralization and is particularly helpful for
bone tumors involving axial skeleton. -
A total body radionuclide bone scan will
evaluate the biologic activity of the primary bone lesion and search
for other lesions within the skeletal system.
-
Duration
-
Localization
-
Severity
-
Character
-
Relief and how obtained
-
Duration
-
Size
-
Consistency
-
Mobility
-
Prior symptoms and signs
-
Mechanism of fracture
-
Characteristics of fracture
-
Prior symptoms and signs
-
Why radiograph obtained
|
TABLE 23-2 PEAK AGE OF COMMON PEDIATRIC MUSCULOSKELETAL TUMORSa
|
||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||
-
Location (Box 23-2)
-
□ Epiphyseal, metaphyseal, or diaphyseal
-
□ Central or eccentric
-
-
Destruction of bone (Fig. 23-1)
-
□ Geographic: typical of slow-growing, benign lesions
-
□ Moth-eaten (multiple, small, often
clustered lytic areas) and permeative (ill-defined, very small oval
radiolucencies or lucent streaks) typical of rapidly growing,
infiltrating tumors
-
-
Replacement of bone
-
Response of bone
-
□ “Walled off” by cortex: static lesion
-
□ Destroyed cortex and soft tissue mass: aggressive lesion
-
-
Periosteal reaction (Fig. 23-2)
-
□ Continuous: benign
-
□ Interrupted: malignant
-
□ Sunburst (“hair-on-end”)
-
□ Lamellated (onion-skin).
-
□ Reactive cuff (Codman triangle)
-
outlines staging recommendations. Biopsy should be the last step in the
evaluation of a patient with a bone or soft tissue sarcoma and should
be performed following completion of the radiographic staging and
preoperative consultation with the oncologist, radiologist,
pathologist, and surgeon. After staging studies are completed, a
differential diagnosis can be formulated. Box 23-4 summarizes the basic principles that should be followed in performing an incisional biopsy.
|
Epiphysis |
Multiple |
Anterior elements of spine |
||||
|
Chondroblastoma |
Leukemia (metastasis) |
Langerhans cell histiocytosis |
||||
|
Brodie abscess of the epiphyses |
Multiple hereditary exostoses |
Leukemia |
||||
|
Giant cell tumor |
Langerhans cell histiocytosis |
Metastatic |
||||
|
Fibrous dysplasia |
Polyostotic fibrous dysplasia |
Giant cell tumor |
||||
|
Metaphysis |
Enchondromatosis |
Posterior elements of spine |
||||
|
Any tumor |
Pelvis |
Aneurysmal bone cyst |
||||
|
Diaphysis (FAHEL) |
Ewing sarcoma |
Osteoblastoma |
||||
|
Fibrous dysplasia |
Osteosarcoma |
Osteoid osteoma |
||||
|
Osteofibrous dysplasia/adamantanoma |
Osteochondroma |
Metastasis |
||||
|
Langerhans cell histiocytosis |
Metastasis |
Rib |
||||
|
Ewing sarcoma |
Fibrous dysplasia |
Fibrous dysplasia |
||||
|
Leukemia, lymphoma |
Langerhans cell histiocytosis |
|||||
|
Occasional diaphyseal: |
Ewing sarcoma |
|||||
|
Osteoid osteoma |
Metastasis |
|||||
|
Unicameral bone cyst |
||||||
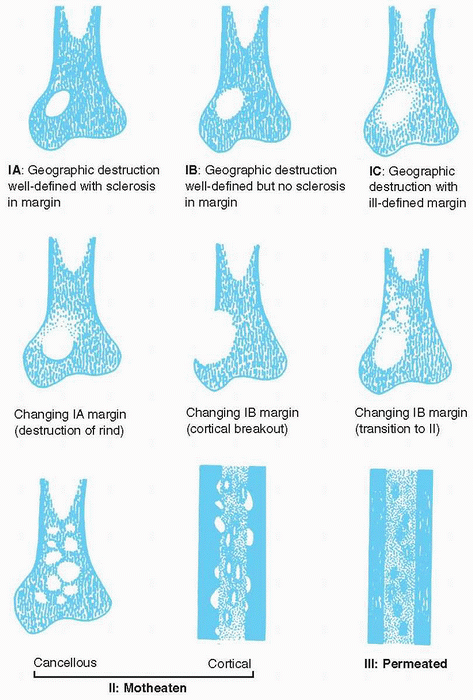 |
|
Figure 23-1
Different patterns of bone destruction. (Adapted from Madewell JE, Ragsdale BD, Sweet DE. Radiologic and pathologic analysis of solitary bone lesions. Part I. internal margins. Radiol Clin North Am 1981;19:715-748.) |
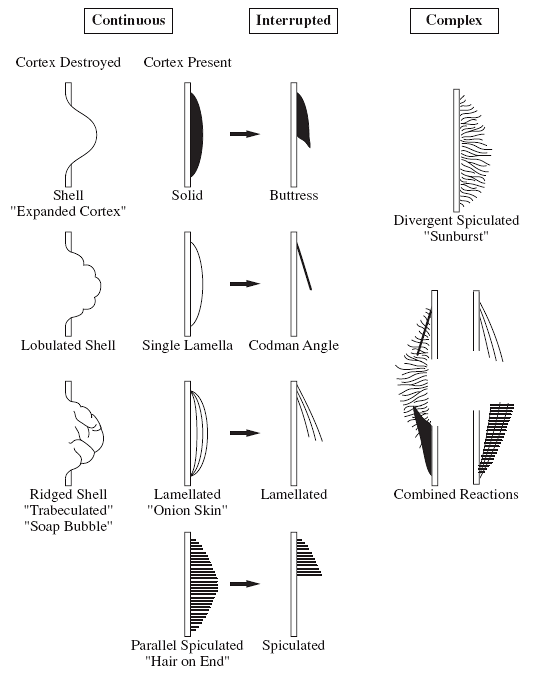 |
|
Figure 23-2
Different patterns of periosteal reaction. (Adapted from Ragsdale BD, Madewell JE, Sweet DE. Radiologic and pathologic analysis of solitary bone lesions. Part II. periosteal reaction. Radiol Clin North Am 1981;19:749-783.) |
-
Magnetic resonance imaging of the primary site including the joint above and below
-
Total body radionuclide bone scan; to search for bone metastases and skip lesions
-
Computed tomography of the chest; to search for lung metastases
-
Intraoperative frozen section
-
Bone marrow aspiration/biopsy for Ewing sarcoma
-
Broviac placement for chemotherapy
-
Radiographic evaluation of the tumor
response to chemotherapy (change in size of tumor, change in amount of
tumor edema, involvement of neurovascular structures) -
Surgical planning
-
Limb-salvage surgery; resection of the
tumor with wide surgical margins and reconstruction; currently possible
in most patients with extremity sarcoma -
Amputation
-
Histologic evaluation of the tumor response to chemotherapy (>90% tumor necrosis demonstrates good response)
-
Verification of wide surgical margins
-
Usually same protocol with neoadjuvant chemotherapy if tumor response to chemotherapy is good
-
Usage limited to lesions that cannot be resected, or that have recurred
-
Keep dose <60 Gy to prevent a secondary sarcoma
-
Every 3 months first year, then 6- to 12-month intervals
-
Longitudinal incisions should be used in the extremities.
-
Smallest incision compatible with obtaining an adequate tumor specimen should be used.
-
The most direct route from the skin to
the tumor should be taken; to prevent tumor spreading, the biopsy
should go through muscle rather than through intermuscular planes and
should avoid exposure of neurovascular bundle or joint. -
Intraoperative frozen section should be done to confirm the adequacy of the specimen.
-
Meticulous hemostasis should be obtained for the prevention of tumor spread by hematoma.
-
Drains should be placed close and in line with the incision.
margins are useful for evaluation, planning, and treatment in the care
of MSKTs. There is usually a capsule (or pseudocapsule) surrounding
soft tissue tumors or a rim of reactive bone around bone tumors. Within
the reactive zone is a variable amount of microscopic tumor extension
and beyond the reactive zone is normal tissue. High-grade tumors may
have neither a capsule nor reactive rim of bone. Four types of surgical
margins of resection have been
defined: intralesional (or intracapsular), marginal, wide, and radical (Fig. 23-3 and Table 23-3).
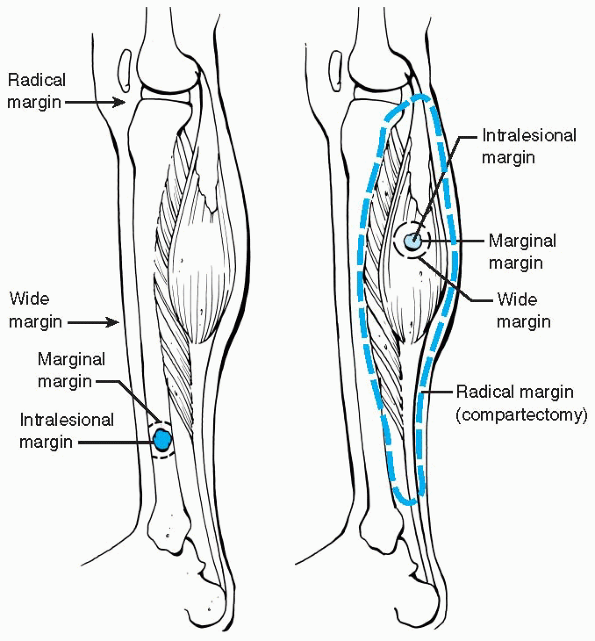 |
|
Figure 23-3 Surgical margins for bone (left) and soft tissue (right)
lesions. Intralesional margin; within the lesion, marginal margin; through the reactive zone of the lesion, wide margin; beyond the reactive zone through normal tissue within compartment, radical margin; normal tissue extracompartmental. (Adapted from Himelstein BP, Dormans JP. Malignant bone tumors of childhood. Pediatr Clin North Am 1996;43:967-984.) |
|
TABLE 23-3 SURGICAL MARGINS
|
||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
-
Nidus of osteoid tissue is surrounded by dense reactive bone (Fig. 23-4).
-
10% of benign bone tumors.
-
The femur is the single most common site of involvement.
-
More than half are found in either the femur or the tibia.
-
Pain is typically worse at night.
-
Relieved by salicylates and nonsteroidal antiinflammatory drugs (NSAIDs).
-
The posterior elements of the spine may also be involved.
-
Burns out with time (years)
-
Standard treatment is complete removal of the nidus by open surgical or CT-guided percutaneous techniques.
-
“Giant osteoid osteoma” (Fig. 23-5)
-
Less than 1% of primary bone tumors
-
Predilection for the posterior elements of the spine
-
Gradually increasing pain
-
Not totally relieved with salicylates or NSAIDs
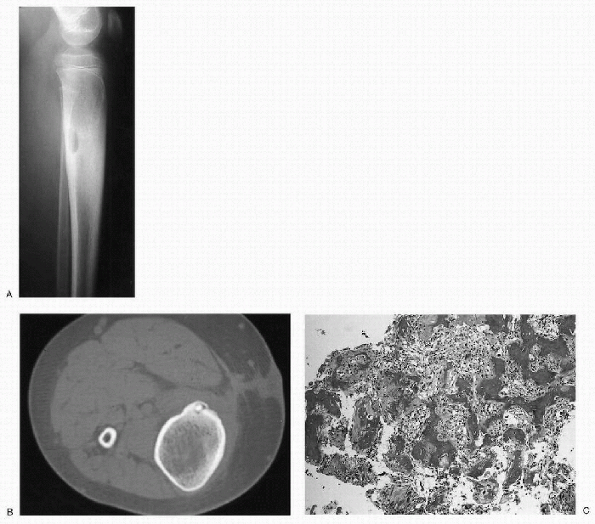 |
|
Figure 23-4 Osteoid osteoma. (A)
Lateral radiograph of the proximal leg shows a well-circumscribed lytic lesion in the posterior cortex of the proximal tibia. There is significant cortical thickening surrounding the lesion. The cortical thickening has caused widening of the tibia. (B) Axial computed tomography image demonstrates the cortical location of the lesion. There is a dense, central nidus surrounded by a lucent rim. Note the extensive cortical thickening. (C) Photomicrograph of the lesion shows a nidus composed of irregular woven bone trabeculae within a background fibroblastic stroma rich in blood vessels. |
-
Excised surgically by extended intralesional curettage or en bloc excision
-
Most common malignant primary bone tumor of childhood
-
Classified by whether it is primary
(occurring with no evidence of a preexisting lesion or prior treatment
of the bone, such as radiation therapy) or secondary, and by the site
of origin, either within the bone (intramedullary) or on the surface
(juxtacortical) -
More than 95% of osteosarcomas involving children and young adults are primary.
-
Osteosarcomas generally have complex
karyotypic abnormalities without chromosomal translocations. Several
nonrandom deletions have been identified, however. The two most obvious
gene deletions in osteosarcomas are located on chromosome 13 and 17,
which are the chromosomes containing tumor suppressor genes—
retinoblastoma (RB1) and p53 genes, respectively.
-
85% of all forms of osteosarcoma
-
Occurs at the metaphyseal ends of the long bones, which have the greatest growth potential
-
50% are located in the distal femur or proximal tibia.
-
The proximal humerus, proximal femur, and pelvis are the next most common sites.
-
Pain and swelling are the most common presenting symptoms.
-
Approximately 15% of patients with
high-grade intermedullary osteosarcoma present with clinically evident
metastases, most commonly in the lungs, but also in other bone
locations and brain. -
Metastases at diagnosis indicate poor prognosis.
|
TABLE 23-4 TYPICAL RADIOGRAPHIC AND HISTOLOGIC FEATURES OF OSTEOID OSTEOMA AND OSTEOBLASTOMA
|
||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
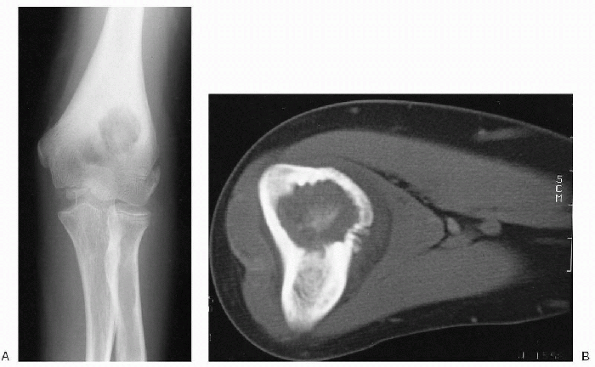 |
|
Figure 23-5 Osteoblastoma. (A)
On anteroposterior radiograph of the elbow, a well-circumscribed lytic lesion with sclerotic borders is seen in the metaphysis of the distal humerus. Note the significant cortical thickening. The location is unusual but the features are classic for osteoblastoma. (B) Axial computed tomography image shows the typical central nidus of bone formation within the lytic lesion. Note the diffuse, reactive cortical thickening. |
-
Plain radiographs: metaphyseal lesion
involving the medullary canal with mixed lytic (radiolucent) and
blastic (radiodense) activity -
Sunburst periosteal reaction with a Codman triangle (Fig. 23-6A)
-
Soft tissue mass which may contain sclerotic foci (tumor bone formation) is also a common finding
-
Bone scan shows increased uptake in the area of the tumor.
-
MRI is the method of choice for evaluating the tumor and its relationship to adjacent structures.
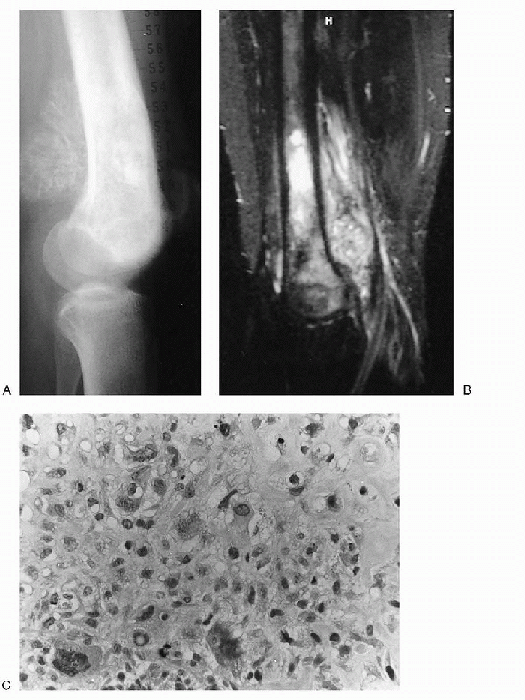 |
|
Figure 23-6 High-grade intramedullary osteosarcoma (conventional osteosarcoma). (A)
Lateral radiograph of the knee shows a lacy, spiculated mass of new bone formation in the metaphysis of the distal femur. Irregular, patchy sclerosis is seen in the metaphysis and epiphysis. (B) On sagittal T2-weighted image a heterogenous mass is seen in the marrow of the metaphysis and epiphysis with extension into the soft tissues posterior to the femur. (C) Photomicrograph (high-power magnification) of the lesion reveals a cellular neoplasm with scattered pleomorphic and bizarre nuclei. Focal osteoid production is evident. |
-
Several histologic subtypes, all with the
same prognosis, and characterized by the formation of osteoid tissue or
new bone by the neoplastic cells -
Osteoblastic subtype is the most common (50%).
-
Highly pleomorphic spindle-shaped and polyhedral tumor cells (see Fig. 23-6C).
-
Nuclear hyperchromasia, abundant mitotic activity, and atypical mitotic figures.
-
Combination of chemotherapy and surgical resection of the tumor.
-
Current standard protocols consist of
preoperative multiagent neoadjuvant chemotherapy, followed by surgical
resection, and subsequent additional chemotherapy (adjuvant
chemotherapy) (Box 23-3). -
Osteosarcoma responds poorly to radiation therapy.
-
Neoadjuvant chemotherapy is to treat
micrometastatic disease, to cause necrosis of the primary tumor and to
decrease the primary tumor size in order to facilitate limb salvage
procedures. -
Following surgical resection, adjuvant chemotherapy is continued to eliminate any micrometastases still present.
-
A good response to chemotherapy, usually
defined as greater than 90% necrosis of the tumor (detected in the
resection specimen), is associated with higher survival rates than a
lesser response. -
Surgery is the mainstay of local control of osteosarcoma.
-
□ Excision of the tumor with wide
surgical margins, which can be achieved through limb salvage or
amputation, is the goal of the surgery. -
□ Currently, limb salvage is possible in most patients with an extremity osteosarcoma.
-
□ Limb salvage reconstruction includes
techniques such as endoprosthetic reconstruction, allograft
reconstruction, or rotationplasty reconstruction. -
□ In some locations, such as the fibula and clavicle, no bony reconstruction is necessary.
-
-
The indications for an amputation are
inability to achieve wide surgical margins, a grossly displaced
pathologic fracture, a tumor that enlarges during preoperative
chemotherapy, and neurovascular bundle involvement that cannot be
appropriately addressed with reconstructive techniques.
general designation for a group of osteosarcomas that arise on the
surface of a bone. Three subtypes are recognized; parosteal,
periosteal, and high-grade. The great majority of juxtacortical
osteosarcomas are low-grade tumors (parosteal osteosarcoma), although
there are moderately (periosteal osteosarcoma) and highly malignant
(high-grade surface osteosarcoma) variants.
-
Low-grade juxtacortical osteosarcomas (parosteal and periosteal) should be treated with wide excision.
-
□ Although the metastatic potential of
these low-grade surface lesions is much lower than conventional
osteosarcoma, these are locally aggressive malignant tumors and
inadequate resection will result in recurrence. -
□ Repeated local recurrence also may result in progression of the tumor to a more aggressive, high-grade lesion.
-
-
High-grade surface osteosarcomas require aggressive treatment similar to that of conventional osteosarcoma.
-
Most common skeletal tumor
-
20% to 50% of benign bone tumors and 10% to 15% of all bone tumors
-
Involves the metaphysis of long bones, particularly around the knee (40% of lesions) and the proximal humerus
-
A firm mass, usually of long duration, adjacent to a joint
-
Pain may result due to irritation of overlying soft tissues by the lesion.
-
Plain radiographs show a bony projection
composed of a cortex continuous with that of the underlying bone and a
spongiosa, similarly continuous. -
The lesions consist of a cartilaginous cap with a broad (sessile osteochondroma) or narrow (pedunculated osteochondroma) base (Figs. 23-7 and 23-8).
-
CT can demonstrate the continuity of
cancellous portions of the lesion and the host bone and the thickness
of the noncalcified cap (it is usually less than 3 mm). -
CT may be useful in differentiating atypical osteochondromas from malignant lesions.
-
Malignant transformation of osteochondroma to chondrosarcoma occurs in less than 1% of solitary lesions.
-
In a skeletally mature patient, a growing
lesion with a thick cartilaginous cap in an axial location (i.e.,
pelvis, scapula) is highly suggestive of this complication. -
Asymptomatic osteochondromas do not require any treatment.
-
For osteochondromas that cause pain or
neurovascular compromise (i.e., lesions in the popliteal fossa) or are
cosmetically unappealing, surgical excision is indicated.-
□ A potential complication of surgery is
injury to an adjacent physis, which may lead to deformity, if the
lesion is in close proximity to the growth plate.
-
-
The incidence of local recurrence after
surgical excision is very low (less than 2%). The entire cartilaginous
cap should be removed to prevent recurrence.
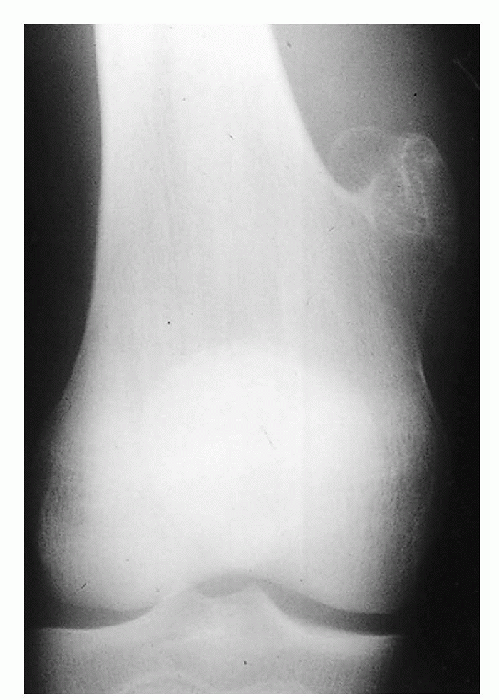 |
|
Figure 23-7 Osteochondroma. A patient with a pedunculated osteochondroma. Note the narrow pedicle.
|
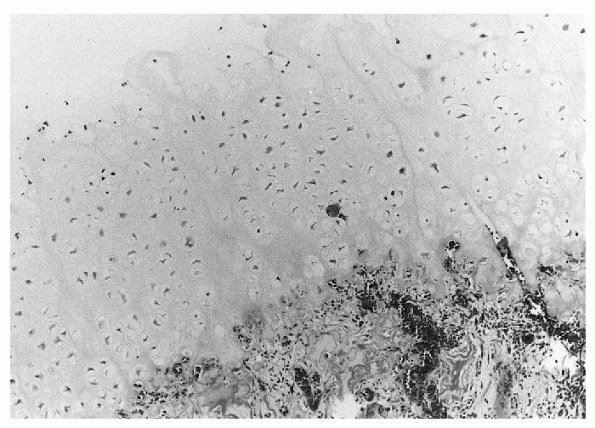 |
|
Figure 23-8 Photomicrograph of osteochondroma, demonstrating a thick cartilaginous cap overlying cancellous bone.
|
-
Autosomal dominant disorder with a variable penetrance
-
Associated with tumor-suppressor genes, termed exostosin (EXT) genes
-
After the age of 30, patients with this disorder have an increased risk of developing a secondary chondrosarcoma.
-
Excision of one or more exostoses often is necessary.
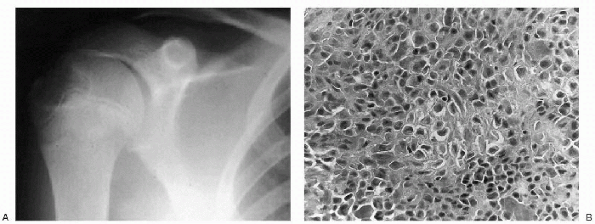 |
|
Figure 23-9 Chondroblastoma. (A)
Anteroposterior radiograph of the shoulder shows a well-circumscribed, lytic lesion in the lateral epiphysis of the humerus. The lesion does not cross the physis. (B) Photomicrograph of the lesion demonstrates tumor cells that are uniform, closely packed and polyhedral, and are focally enveloped by a lace-like, lightly calcified chondroid matrix. |
-
1% of all benign bone tumors
-
Occurs primarily in the epiphysis of the growing skeleton
-
The most common sites are the proximal humerus, proximal and distal femur, and proximal tibia.
-
Pain, swelling, and limited motion are usually localized to the adjacent joint.
-
Plain radiographs show a well-marginated, radiolucent lesion, usually with a sclerotic rim of bone (Fig. 23-9A). The lesion may have small foci or calcifications.
-
Bone scan shows increased uptake.
-
Chest radiography or CT should be performed, because chondroblastoma is one of the benign tumors that can have lung metastasis.
-
Small cuboidal cells (chondroblasts) closely packed together to give the appearance of a cobblestone street (see Fig. 23-9B).
There are areas with varying amounts of amorphous matrix that often
contains streaks of calcification (“chicken-wire” calcification), and
there are numerous multinucleated giant cells.
-
Chondroblastoma is a benign, but locally aggressive lesion (Box 23-5).
-
Most patients are close to skeletal
maturity and damage to the growth plate is not a major concern.
However, the lesions may progress and invade the joint. -
Chondroblastomas should be treated when detected.
-
Intralesional extended curettage is the initial treatment of choice.
-
Recurrence rates of approximately 10% have been reported after intralesional excision.
-
The majority of recurrences are adequately addressed with a second curettage. Rarely, recurrent lesions require en bloc excision.
-
In rare cases of severe bone destruction or recurrence, wide resection and segmental reconstruction sometimes is indicated.
-
Osteoblastoma
-
Chondroblastoma
-
Osteofibrous dysplasia
-
Aneurysmal bone cyst
-
Giant cell tumor
-
Chondromyxoid fibroma
-
Benign tumor of mature hyaline cartilage
-
10% of benign bone tumors
-
Usually present as solitary lesions
-
Short tubular bones of the hand and foot are common sites.
-
Most present with a pathologic fracture
through the lesion, or as an incidental finding on a radiograph taken
for another reason.
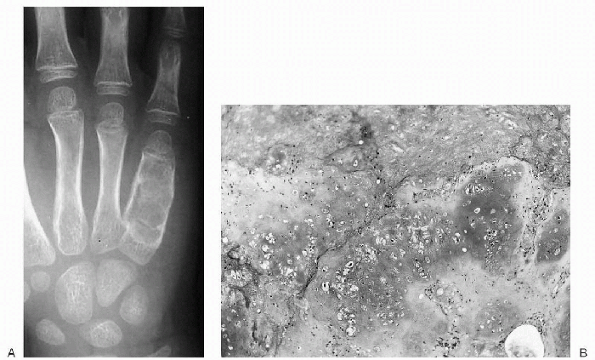 |
|
Figure 23-10 Enchondroma. (A)
Anteroposterior radiograph of the ulnar three digits of the hand shows an expansile, lytic lesion involving the diaphysis of the fifth metacarpal with extension into the distal metaphysis. There is saucerization (scalloping) of the inner cortex. (B) Photomicrograph demonstrates a lobular lesion composed of mature cartilaginous tissue. |
-
Plain radiographs show a sharply circumscribed radiolucent lesion located centrally in the medullary canal (Fig. 23-10A).
-
Grossly, enchondromas are lobular lesions with a bluish color.
-
Histologically, lobules of hyaline cartilage with varying cellularity are seen and are recognized by their blue matrix (see Fig. 23-10B). The chondrocytes are located in rounded spaces called lacunae.
-
Malignant transformation of enchondromas occurs infrequently (less than 1%), and is rare before skeletal maturity.
-
Patients experiencing pain in a
previously asymptomatic lesion without evidence of a pathologic
fracture should be evaluated for this possibility. -
Asymptomatic solitary enchondromas do not require any treatment other than a periodic follow-up evaluation.
-
Symptomatic or large lesions in the short
tubular bones of the hand without a pathologic fracture can be managed
with curettage and bone grafting. -
If a pathologic fracture occurs through
an enchondroma, the fracture should be allowed to heal prior to
curettage and bone grafting. -
Recurrence after curettage and bone grafting is rare.
-
Incisional biopsy usually is contraindicated.
-
□ Pathologists may have difficulty
distinguishing an active enchondroma (most pediatric patients have
active lesions) from a low-grade chondrosarcoma. -
□ An incisional biopsy alters the histologic and radiographic status of the lesion and may make subsequent evaluation difficult.
-
-
Also known as Ollier disease, it is an inherited condition with widespread enchondromas.
-
It is much less common than solitary enchondroma.
-
Patients with Ollier disease have an increased risk of developing secondary chondrosarcoma later in life.
-
Multiple enchondromatosis with vascular anomalies of soft tissues is known as Maffucci syndrome (see “Vascular Tumors”
later in the chapter). Patients with this disorder have an even greater
risk of developing malignant cartilage tumors than patients with Ollier
disease; importantly, they also have a greater risk of developing
carcinoma of an internal organ.
-
Nonossifying fibroma (NOF), fibrous cortical defect, metaphyseal fibrous defect, and fibroma all refer to the same histopathologic process in bone.
-
Fibrous cortical defects are small
asymptomatic lesions that occur in 30% of the population during the
first and second decades of life. -
NOF refers to a lesion that enlarges and encroaches on the medullary bone.
-
Benign proliferation of fibroblast-like mesenchymal tissue that is more likely a hamartomatous process than a true neoplasm
-
Most common in the long bones, especially the distal femur and the tibia
-
Asymptomatic lesion that is often found
only when a radiograph is taken for another reason or when the patient
has a pathologic fracture
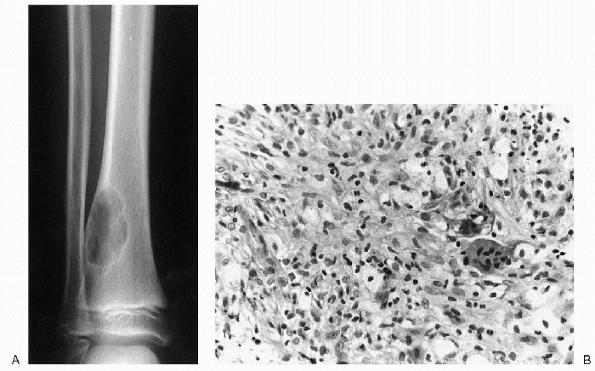 |
|
Figure 23-11 Nonossifying fibroma. (A)
Anteroposterior and lateral radiographs of the distal tibia show a cortically based, lytic lesion with well-defined margins. Note the typical scalloped appearance. Involvement greater than 50% of the diameter of the bone puts this lesion at risk for pathologic fracture. (B) Photomicrograph (high power magnification) of the lesion demonstrates foamy histiocytic cells and giant cells discerned within the fibroinflammatory spindle cell stroma. |
-
Plain radiographs show an eccentric,
metaphyseal lesion, involving the medullary canal, with a loculated
appearance and a radiodense rim (Fig. 23-11A). -
NOFs range in size from 0.5 to 7 cm, with their long axes aligned with the long axis of the affected bone.
-
Histologically, NOF consists of benign, spindle, fibroblastic cells arranged in a storiform pattern (see Fig. 23-11B).
-
Multinucleated giant cells are common, and foam cells containing lipid often can be seen.
-
Hemosiderin within the spindle cells and multinucleated giant cells are usual.
-
Typically, the lesion does not contain bone.
-
Fibrous cortical defects need no treatment, they heal spontaneously.
-
NOFs less than 50% of the diameter of the bone and that are asymptomatic and can be observed.
-
NOFs occupying more than 50% of the
diameter of the bone, have an increased risk of developing pathologic
fractures, and should be considered for curettage and bone grafting. -
Patients who present with pathologic fractures can usually be managed nonoperatively.
-
A nonneoplastic condition of aberrant bone development.
-
Produces a variety of complaints and physical findings.
-
85% of cases are a single skeletal lesion (monostotic fibrous dysplasia).
-
15% have numerous lesions (polyostotic fibrous dysplasia).
-
The most common locations for monostotic fibrous dysplasia are ribs, proximal femur, tibia, and the base of the skull.
-
The patient with monostotic fibrous
dysplasia usually presents without significant symptoms, and the lesion
is usually discovered incidentally on radiographs obtained for other
reasons. -
Occasionally, a child presents with a pathologic fracture or angular deformity (Fig. 23-12A).
-
Fibrous dysplasia is a medullary process usually involving the full width of the bone.
-
Plain radiographs show a radiolucent
lesion with slight expansion and thinning of the cortex, and partial
loss of trabecular pattern in the cancellous bone, which gives the
characteristic ground-glass appearance (see Fig. 23-12A).-
□ Cystic areas can also be seen within the abnormal bone.
-
□ There may be an angular deformity or bowing in the bone, especially when the lesion is large.
-
□ The lesions are usually diaphyseal, and the differential diagnosis may include other common diaphyseal lesions of bone.
-
-
On the bone scan, uptake within the lesion is usually intense.
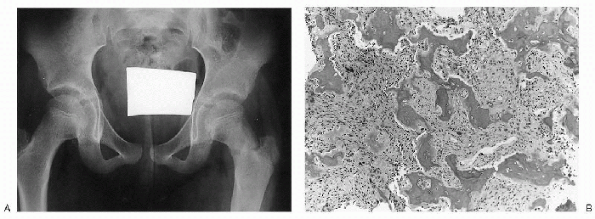 |
|
Figure 23-12 Fibrous dysplasia. (A)
Anteroposterior radiograph of the pelvis shows subtle ground-glass density of the left femoral neck and proximal diaphysis. The cortex blends imperceptibly with the medullary canal. A pathologic fracture is seen at the base of the femoral neck. (B) Photomicrograph of the lesion demonstrates irregular woven bone trabeculae (so-called Chinese letters) in a background of bland fibroblastic stroma. Note absence of osteoblastic rimming. |
-
Histologically, fibrous dysplasia (both
the monostotic and polyostotic variants) is composed of trabeculae of
immature, woven bone within a background stroma of collagen-rich tissue
(see Fig. 23-12B). -
The osteoid and bone appear to arise in a haphazard fashion from the fibrous stroma.
-
The trabeculae often obtain a variety of shapes (C’s and O’s) and are sometimes referred to as alphabet soup or Chinese letters.
-
Monostotic fibrous dysplasia usually does not require any treatment other than observation.
-
Surgical treatment may be required for
lesions that are very large or enlarging, in a high-stress location
(i.e., proximal femur), or have become symptomatic. -
Intralesional extended curettage is
performed for eradication of the lesion; curettage often results in
healing with dysplastic, mechanically deficient bone, similar to the
pattern of fracture healing in fibrous dysplasia. -
Therefore, the goals of surgical
treatment, when indicated, are clearly different from other benign
active or aggressive lesions.P.263-
□ Rather than resect the lesional tissue, the goals are to stabilize the bone, prevent or correct deformity, and relieve pain.
-
□ Prophylactic internal fixation may be required depending on the size and location of the lesion.
-
-
Polyostotic fibrous dysplasia may occur as a part of a condition known as McCune-Albright syndrome which is characterized by a classic triad of polyostotic fibrous dysplasia, café-au-lait skin lesions, and precocious puberty.
-
Activating missense mutations of the guanine nucleotide-binding protein gene (GNAS), encoding the α-sub-unit of the stimulatory G protein, have been identified in patients with this syndrome.
-
Tumor-like cystic lesion of unknown cause, attributed to a local disturbance of bone growth
-
Arises on the metaphyseal side of the growth plate and is displaced from the physis with skeletal growth
-
The most common locations are the proximal humerus and femur, accounting for 90% of lesions, followed by calcaneus
-
Commonly are asymptomatic, and often come to attention only after pathologic fracture has occurred
|
TABLE 23-5 TYPICAL RADIOGRAPHIC AND HISTOLOGIC FEATURES OF ANEURYSMAL AND UNICAMERAL BONE CYSTS
|
|||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
-
Tend to enlarge with skeletal growth
-
Enters a latent phase after skeletal maturity, ceases growing, is resorbed, and is replaced by normal bone
-
Some unicameral bone cysts remain small
and do not present a significant risk of a pathologic fracture.
Observation and activity restriction may be all that is necessary for
these lesions. -
In most cases, a pathologic fracture should be treated nonoperatively until it heals.
-
Healing of the fracture usually does not
result in healing of the bone cyst (less than 10% of cysts heal with
pathologic fracture). -
Surgical treatment options include
curettage and bone grafting, and fluoroscopically guided percutaneous
corticosteroid injection, which are both associated with high
persistence and recurrence rates, and some morbidity. -
Newer techniques include injection with
autologous bone marrow or demineralized bone matrix, and percutaneous
intramedullary decompression, curettage, and grafting using bone graft
substitutes (calcium sulfate pellets) (see Fig. 23-13B and C).-
□ The short-term results of these techniques are very promising, with high healing and low complication and recurrence rates.
-
-
Vascular lesions consisting of widely dilated vascular channels that are not lined by identifiable endothelium.
-
1% to 2% of all benign bone lesions.
-
Etiology is not known.
-
More than 50% of ABCs arise in large tubular bones, and almost 30% occur in the spine.
-
Mild, dull pain; only rarely is there a clinically apparent pathologic fracture.
 |
|
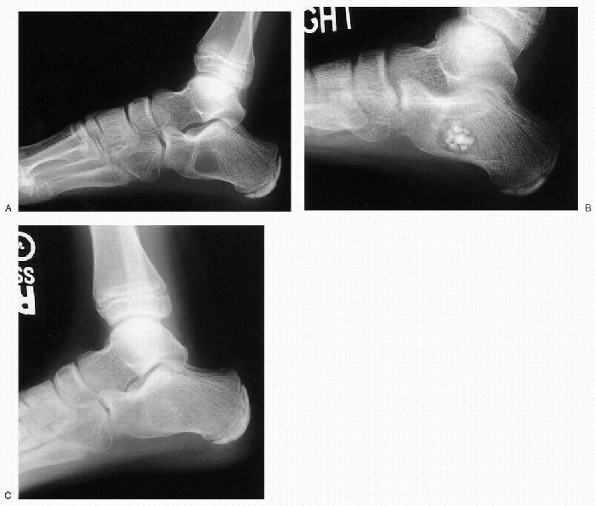
Figure 23-13 Unicameral bone cyst. (A) Lateral radiograph of the foot depicts a lytic lesion in calcaneus with well-defined margins. (B)
This lesion was treated by percutaneous intramedullary decompression, biopsy, curettage, and grafting with calcium sulfate pellets. (C) Postoperative 1-year follow-up radiograph shows complete healing of the lesion. |
-
ABCs are benign by histology, but can be locally aggressive (Box 23-5).
-
They should undergo biopsy to establish the diagnosis, and then be treated surgically.
-
The usual treatment for ABCs is extended curettage and bone grafting.
-
ABCs in expendable bones, such as fibula, ribs, distal ulna, metacarpal, and metatarsal bones, may be treated by en bloc resection.
-
□ Resection is also appropriate for recurrent aggressive lesions.
-
-
An ABC of the spine can present a challenging problem.
-
□ The lesion usually involves the posterior elements, but can also invade into vertebral body.
-
□ Surgery is recommended for most patients as the initial means of treatment.
-
□ Radiotherapy is contraindicated.
-
-
More recently, selective arterial
embolization has been used in conjunction with surgery or as a curative
procedure alone for some pelvic and spinal lesions.
 |
|
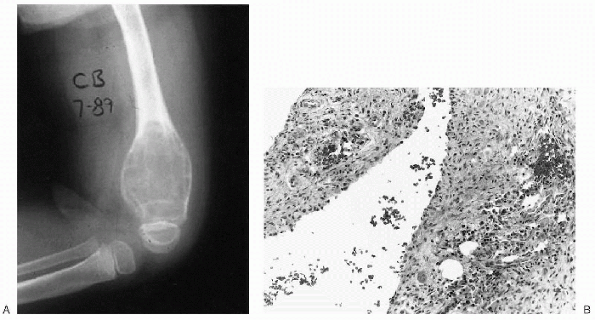
Figure 23-14 Aneurysmal bone cyst. (A)
Lateral radiograph of the knee shows a large lytic lesion expanding the distal femoral metaphysis. Note the trabeculated, nonossified matrix. (B) Photomicrograph of this lesion demonstrates salient features including scattered erythrocytes within the cyst cavity, lack of an endothelial lining, and an occasional giant cell in the underlying stroma. |
-
Second most common primary malignant bone tumor in children
-
Ewing sarcoma (EWS) is closely related to
primitive neuroectodermal tumor (PNET); both these tumors are thought
to arise from the neural crest and at least 90% have a characteristic
chromosomal translocation [t(11:22)(q24:q12)], which leads to a novel
fusion protein called EWS-FLI1 (Table 23-6). -
The femur is the most common site of origin, followed by the pelvis and humerus.
-
Pain and local swelling are the most common presenting symptoms.
-
EWS may present with fever and an elevated sedimentation rate and may mimic bacterial osteomyelitis.
-
Approximately 5% of patients with EWS present with pulmonary metastasis.
|
TABLE 23-6 PEDIATRIC MUSCULOSKELETAL TUMORS WITH NONRANDOM/SPECIFIC TRANSLOCATIONS
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
-
Plain radiographs demonstrate diffuse
destruction of bone, usually occurring in the diaphyseal regions of
long bones or in flat bones of the axial skeleton (Fig. 23-15A).-
□ The lesion usually is associated with a periosteal reaction (Fig. 23-2), which has an “onion-skin” or sunburst appearance, and a large soft tissue mass.
-
-
On bone scan, EWS shows a very increased uptake; it may be multicentric.
-
MRI is essential for definitive
demonstration of extraosseous soft tissue mass, determining
relationship of the tumor to surrounding structures, and determining
medullary canal involvement, both of which are usually more extensive
than what was expected from the plain radiographs (see Fig. 23-15B and C).
-
Histologically, EWS consists of small,
uniform-sized cells characterized by an almost clear cytoplasm and
nuclei that are round and slightly hyperchromatic (see Fig. 23-15D).-
□ Necrotic areas usually are seen.
-
□ There are glycogen granules in the
cytoplasm, and these produce the positive periodic acid-Schiff (PAS)
stain on routine histology.
-
-
Increasingly, genetic analysis is being
done for EWS to identify the 11:22 translocation as a means of
establishing the diagnosis.
 |
|
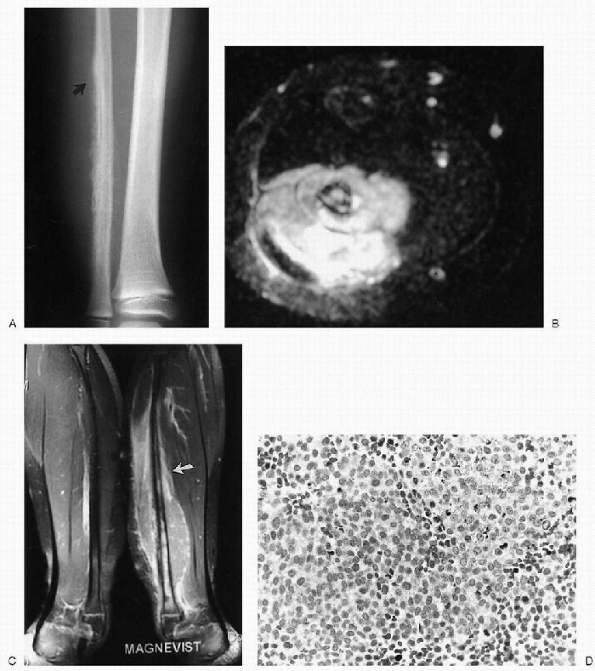
Figure 23-15 Ewing sarcoma. (A)
Anteroposterior radiograph of the distal fibula reveals an irregular, hazy patterned, periosteal new bone formation with fine spiculations and a Codman triangle at the superior edge (arrow). This aggressive pattern of new bone formation is associated with enlargement of the soft tissues and obliteration of the fat planes due to a soft tissue mass. The underlying bone has a permeative pattern of bone destruction. (B) Axial T2-weighted magnetic resonance image shows abnormal, hyperintense signal in the marrow of the fibula. Patches of hypointense signal are scattered throughout the cortex as well. Spicules of new bone formation are well seen. A large, fairly homogeneous, soft tissue mass invades the lateral compartment. (C) Coronal T1-weighted, fat-saturated image with contrast enhancement shows (abnormally) enhancement extending from the distal metaphysis to the mid-fibula (arrow). An enhancing soft tissue mass encompasses the fibula and extends up into the proximal soft tissues above the marrow abnormality. (D) Photomicrograph of the lesion demonstrates primitive, small, and uniform malignant cells with inconspicuous cytoplasm. Darker staining cells are pyknotic. |
-
Multiagent chemotherapy has made a
significant difference in the prognosis for patients with EWS,
improving the 5-year survival rate from 5% to 10% to over 70%. -
Current treatment principles of EWS are summarized in Box 23-3.
-
Treatment of EWS is a combination of chemotherapy and either surgery or irradiation.
neoplastic lesions with endothelial hyperplasia. They are the most
common benign soft tissue tumors of childhood, occurring in 4% to 10%
of all children. They infrequently are present at birth but grow
rapidly during the first 2 to 3 weeks of life. Hemangiomas may be
located in the superficial or deep dermis, subcutaneous tissue,
musculature, bone, or viscera. Head and neck are the most commonly
involved sites.
which are congenital lesions with normal endothelial turnover. They are
subclassified by the predominant vessel found in the lesion (i.e.,
venous, arterial, capillary, lymphatic, mixed) and by the blood flow
within the lesion (high vs. low flow). Vascular malformations are not
seen as commonly as hemangiomas. Many of the common vascular
malformations are present at birth, however some may manifest in
adolescence or adulthood. The majority of these lesions involve the
skin and subcutaneous tissue, but deep or extensive involvement of
structures such as muscle, joint, bone, abdominal viscera, and central
nervous system is not uncommon. The most commonly affected sites are
head and neck.
 |
|
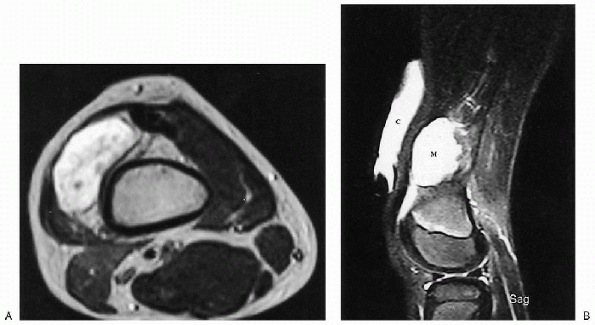
Figure 23-16 Vascular malformation: lymphangioma. (A)
Axial T2-weighted magnetic resonance image of the distal thigh shows a high-signal intensity mass similar to fluid. No vessels or septations are seen within the mass. (B) On sagittal T2-weighted image, a vitamin E capsule (C) is used to mark the site of the palpable mass. Note the high signal of this fluid-filled mass (M). A small suprapatellar effusion, just inferior to the mass, has the same signal intensity. |
nature of vascular anomalies and defining their relationship to
adjacent structures (Fig. 23-16). Ultrasound or color Doppler may also be used.
-
In the absence of intervention, partial
or complete involution of hemangiomas usually occurs, and correction of
cosmetic defects typically follows. -
When intervention for a rapidly expanding
hemangioma is necessary, the first line of treatment is intralesional
or systemic corticosteroids. -
Occasionally, when surgical resection is
indicated for larger, deeper lesions that threaten normal function,
staged excisions may be considered.
-
Compression stockings to prevent
progressive venous dilation, pain, ulceration, and bleeding are the
mainstay of treatment, and low-dose aspirin may also be used to
minimize thrombophlebitis. -
When the patient does not receive
adequate benefit from compression stockings, laser surgery,
sclerotherapy (i.e., 100% ethanol), or surgical resection are all
widely accepted treatment methods. -
Surgical intervention may be required for
deep venous malformations involving muscle or bone, which have led to
pain, functional impairment or pathologic fracture. -
Epiphysiodesis may be performed in patients with extensive venous malformations that have resulted in skeletal overgrowth.
-
AVMs can prove deceptively problematic and even dangerous to treat.
-
Most experts agree that conservative treatment is preferred in the absence of significant symptoms.
-
Deep AVMs may cause significant muscle or bone changes and will necessitate surgical debulking or epiphysiodesis.
-
When surgery is required, angiographic
studies followed by selective embolization is usually done 24 or 72
hours before resection. -
Embolization or ligation of feeding
arteries without surgical resection is contraindicated because
occlusion of the major feeding arteries usually results in rapid
recruitment and dilation of previously microscopic collateral blood
flow.
tumors of peripheral nerves. Both arise from benign proliferation of
periaxonal Schwann cells that embryologically arise from the neural
crest.
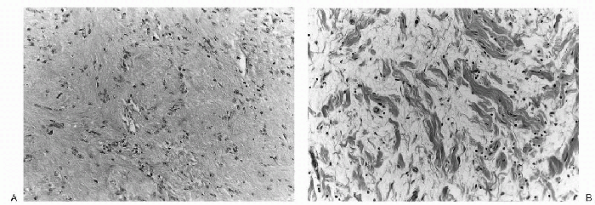 |
|
Figure 23-17 Histopathology of benign peripheral nerve sheath tumors. A: Neurolemmoma. This tumor demonstrates a classic palisading of Schwann cells (Verocay bodies) within a spindlecell background. (B)
Neurofibroma. The lesion is composed of fascicles of Schwann cells and fibroblasts within an edematous and mildly inflamed background stroma, imparting a histologic appearance that has been compared to shredded carrots. |
can be seen in patients of all ages, but they are most often
encountered in early adulthood. They have a predilection for the head,
neck, and flexor surfaces of the extremities (i.e., peroneal, ulnar
nerves). The patient usually presents with a painless, slow-growing,
solitary mass in the subcutaneous tissue. Although they arise from the
nerve sheath, nerve dysfunction is uncommon, and is seen only when the
nerve is compressed between the tumor and an adjacent rigid structure.
A positive Tinel sign with percussion over the mass is not uncommon.
Neurolemmomas rarely are associated with neurofibromatosis.
during young adulthood. They may present as solitary lesions or as
multiple lesions in association with neurofibromatosis, but the
majority (90%) are solitary. Like neurolemmomas, they grow slowly as a
painless mass in the skin, subcutaneous tissue, or the distribution of
a peripheral nerve. Unlike neurolemmomas, they tend to be intimately
associated with the nerve fibers (Fig. 23-17).
solitary neurofibromas is exceedingly rare. In contrast, malignant
transformation of neurofibromas in the face of neurofibromatosis is
well documented; this risk has been reported in about 2% of cases.
Therefore, patients and parents should be aware of the clinical
symptoms leading to suspicion of malignant transformation, such as
enlargement of a neurofibroma and pain. If sarcomatous degeneration
occurs, the prognosis for long-term survival is poor.
Neurofibrosarcomas can also rarely arise de novo.
-
Treatment of neurolemmomas consists of marginal surgical excision.
-
Overlying nerve fibers can usually easily be mobilized and preserved as the lesion is marginally shelled out.
-
Neurolemmoma usually does not recur following marginal excision.
-
Marginal surgical excision is recommended for symptomatic solitary neurofibromas involving a peripheral nerve.
-
Those arising from a major nerve can be resected, but they should be approached cautiously.
-
Most common soft tissue tumor in children
-
5% of all pediatric cancers
-
Arises from the same embryonal mesenchyme that is destined to give rise to striated skeletal muscle.
-
Peak incidence is in the 1- to 5-year age group.
-
There are four histologic patterns: embryonal, botryoidtype, alveolar, and pleomorphic.
-
□ Embryonal and alveolar types are common.
-
□ Embryonal tumors are most often found in the head and neck or the genitourinary tract.
-
□ Alveolar rhabdomyosarcomas are more commonly found in the extremities and trunk.
-
-
Extremity lesions usually present as a painless mass, while paravertebral tumors may cause back pain.
-
Characteristic chromosomal abnormalities have been identified in the alveolar rhabdomyosarcoma (Table 23-6).
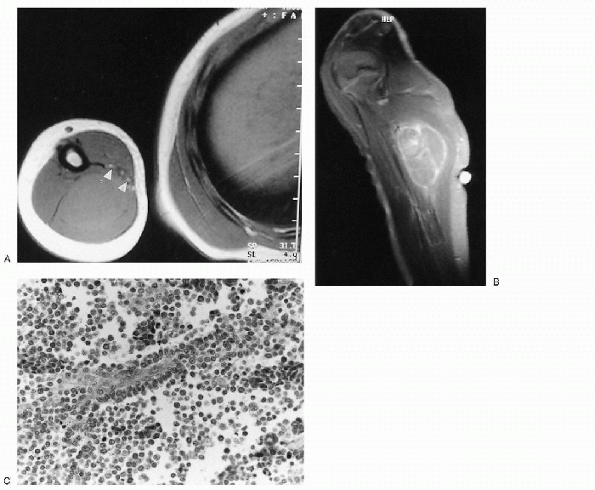 |
|
Figure 23-18 Alveolar rhabdomyosarcoma. (A)
Axial T1-weighted magnetic resonance image through the humerus shows a slightly hyperintense mass replacing the triceps muscles of the arm. The neurovascular bundle (arrowheads) is contiguous with the anterior medial edge of the mass. (B) After contrast administration, there is heterogeneous enhancement of the mass. (C) Photomicrograph of the lesion demonstrates undifferentiated small blue cells lining fibrovascular septae with central discohesion, imparting an alveolar appearance. |
-
Embryonal rhabdomyosarcoma consists of poorly differentiated rhabdomyoblasts with a limited collagen matrix.
-
□ The rhabdomyoblasts are small,
round-to-oval cells with dark-staining nuclei and limited amounts of
eosinophilic cytoplasm (see Fig. 23-18C).
-
-
Alveolar rhabdomyosarcoma is composed of
poorly differentiated small, round-to-oval tumor cells that show
central loss of cellular cohesion and formation of irregular alveolar
spaces.-
□ The individual cellular aggregates are separated and surrounded by irregularly shaped fibrous trabeculae.
-
-
Prognostic variables for
rhabdomyosarcomas include histologic subtype, size of the tumor, site
of the tumor, and age of the patient. -
Alveolar subtype, larger tumors,
extremity location, and patients older than 10 years of age are more
often associated with a poorer prognosis.-
□ Alveolar rhabdomyosarcoma, like the other subtypes, is treated with a combination of chemotherapy and surgery.
-
□ Irradiation can be used if total surgical resection cannot be achieved without excessive morbidity.
-
□ Total resection of the tumor with wide
surgical margins is recommended, and preoperative chemotherapy often
makes total resection of an extremity lesion possible.
-
-
Lymph node biopsy should be considered if the patient has any suggestion of lymph node involvement.
-
Postoperative irradiation may be used
when the surgical margins are positive for tumor or there is poor
necrosis of excised tumor from chemotherapy. -
The overall survival for a patient with extremity rhabdomyosarcoma is approximately 65%.
CM, Dehner LP, O’Shea PA. Pediatric soft tissue tumors: a clinical,
pathological, and therapeutic approach. Baltimore: Williams &
Wilkins, 1997.
JP, Flynn J. Pathologic fractures associated with tumors and unique
conditions of the musculoskeletal system. In: Rockwood and Wilkins
fractures in children, 5th ed., Beatty JH and Kasser JR, eds., 2001,
pp. 139-240.
A, Remagen W. Differential diagnosis of tumors and tumor-like lesions
of bones and joints. Philadelphia: Lippincott-Raven, 1998.
JE, Ragsdale BD, Sweet DE. Radiologic and pathologic analysis of
solitary bone lesions. Part I: internal margins. Radiol Clin North Am
1981;19:715.
BD, Madewell JE, Sweet DE. Radiologic and pathologic analysis of
solitary bone lesions. Part II: periosteal reactions. Radiol Clin North
Am 1981;19:749.